Back to Journals » Drug Design, Development and Therapy » Volume 14
A First-in-Human Dose Finding Study of Camrelizumab in Patients with Advanced or Metastatic Cancer in Australia
Authors Lickliter JD, Gan HK, Voskoboynik M ![]() , Arulananda S, Gao B, Nagrial A, Grimison P, Harrison M, Zou J, Zhang L, Luo S, Lahn M
, Arulananda S, Gao B, Nagrial A, Grimison P, Harrison M, Zou J, Zhang L, Luo S, Lahn M ![]() , Kallender H
, Kallender H ![]() , Mannucci A, Somma C
, Mannucci A, Somma C ![]() , Woods K
, Woods K ![]() , Behren A
, Behren A ![]() , Fernandez-Penas P
, Fernandez-Penas P ![]() , Millward M
, Millward M ![]() , Meniawy T
, Meniawy T
Received 26 December 2019
Accepted for publication 20 February 2020
Published 18 March 2020 Volume 2020:14 Pages 1177—1189
DOI https://doi.org/10.2147/DDDT.S243787
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Tin Wui Wong
Jason D Lickliter, 1 Hui K Gan, 2–4 Mark Voskoboynik, 1, 5, 6 Surein Arulananda, 2–4 Bo Gao, 7 Adnan Nagrial, 7 Peter Grimison, 8 Michelle Harrison, 8 Jianjun Zou, 9 Lianshan Zhang, 9 Stacey Luo, 9 Michael Lahn, 10 Howard Kallender, 11 Andrea Mannucci, 10 Catello Somma, 10 Katherine Woods, 3 Andreas Behren, 3, 4 Pablo Fernandez-Penas, 12 Michael Millward, 13 Tarek Meniawy 13
1Nucleus Network, Melbourne, Victoria, Australia; 2Department of Medicine, University of Melbourne, Heidelberg, Victoria, Australia; 3Olivia Newton-John Cancer Wellness and Research Centre, Austin Hospital, Melbourne, Victoria, Australia; 4Department of Medical Oncology, La Trobe University School of Cancer Medicine, Bundoora, Victoria, Australia; 5Department of Medical Oncology, Alfred Hospital, Melbourne, Victoria, Australia; 6Department of Medical Oncology, Monash University, Central Clinical School, Alfred Campus, Melbourne, Victoria, Australia; 7Blacktown Cancer and Haematology Centre, Blacktown Hospital, University of Sydney, Sydney, New South Wales, Australia; 8Department of Medical Oncology, Chris O’Brien Life House, Camperdown, New South Wales, Australia; 9Jiangsu Hengrui Medicine Co. Ltd, Shanghai, People’s Republic of China; 10Incyte Biosciences International Sarl, Geneva, Switzerland; 11Incyte Corporation, Wilmington, Delaware, USA; 12Department of Dermatology, The University of Sydney, Westmead Hospital, Westmead, New South Wales, Australia; 13Linear Clinical Research, Nedlands, Western Australia, Australia
Correspondence: Jason D Lickliter
Nucleus Network, Level 5, Burnet Tower, 89 Commercial Road, Melbourne, Victoria, Australia
Tel +61 3 9076 8900
Fax +61 3 9076 8911
Email [email protected]
Purpose: Camrelizumab inhibits PD-1 in non-clinical models and showed typical non-clinical pharmacokinetic (PK) and safety profiles for an IgG4 monoclonal antibody. We report results from the First-in-Human Phase 1 trial of camrelizumab in Australian population.
Methods: Camrelizumab was administered to patients with advanced solid tumors who had failed standard therapies. In the dose-escalation phase (n=23), camrelizumab was administered intravenously at 1 mg/kg, 3 mg/kg, 6 mg/kg, and 10 mg/kg every 2 weeks. In dose expansion (n=26), camrelizumab was given at 200 mg or 600 mg every 4 weeks.
Results: Two dose-limiting toxicities were observed during dose escalation: transaminase elevation and diarrhea (both grade 3). Overall, treatment-related adverse events were consistent with the expected toxicity profile of immune checkpoint inhibition, with the striking exception of the dose-related development of angiomatous skin lesions characterized as reactive cutaneous capillary endothelial proliferation. The PK profile showed a dose-progressive increase in half-life from 3 days at 1 mg/kg to 7 days at 10 mg/kg. Moreover, receptor occupancy assays showed a PD-1 occupancy of > 50% in most patients out to 28 days post-dose. The objective response rate was 15.2% (95% CI 6.3– 28.9).
Conclusion: Camrelizumab has manageable toxicity and encouraging preliminary antitumor activity in advanced solid tumors in Australia.
Clinical Trial Registration: ClinicalTrials.gov Identifier: NCT02492789.
Keywords: PD-1, monoclonal antibody, first-in-human dose study, cancer, reactive cutaneous capillary endothelial proliferation
Background
Programmed cell death protein 1 (PD-1), also known as CD279, is an immune-inhibitory receptor and a member of the immunoglobulin superfamily. It is expressed on a variety of immune cells including CD8+ cytotoxic T cells and is involved in the regulation of peripheral tolerance.1,2 Binding of its ligands PD-L1 and/or PD-L2 can lead to a dampening of T cell activation and immune reactions, and expression of PD-L1 on tumor cells often allows for a successful immune-escape. Inhibition of PD-1 can lead to re-invigoration of T cell activity and can enhance the recruitment of effector T cells in otherwise poorly immunogenic tumors.3–5 These and similar observations led to the development of monoclonal antibodies blocking PD-1 and PD-L1 for the treatment of cancer.6–8 One such monoclonal antibody is camrelizumab, a humanized IgG4 that binds PD-1 at a high affinity of 3 nM as determined by the BiaCore T200 assay. Furthermore, the camrelizumab inhibited the binding interaction of PD-1 and PD-L1 at an IC50 of 0.70 nM. In a T cell proliferation assay using tuberculin treated peripheral blood mononuclear cells, camrelizumab induced a T cell proliferation at an EC50 of 0.11 nM. Finally, in a similar assay measuring IFN-gamma secretion, camrelizumab induced IFN-gamma production at an EC50 of 0.38 nM. Camrelizumab had the appropriate safety and pharmacokinetic properties in non-clinical studies to justify its clinical investigation in cancer patients.
Several studies reported the clinical potential of camrelizumab due to its promising antitumor activity and manageable toxicity profile in multiple cancers, but these clinical evidences were only limited to Chinese population.9–11 Here, we report the results from the First-in-Human (FiH) Phase 1 study of camrelizumab conducted in Australia, which was launched preceding those camrelizumab trials in China, in patients with advanced cancer.
Methods
Experimental Design
The study was conducted at five sites in Australia. Camrelizumab was investigated in a FiH, 3+3 dose-escalation and dose-expansion phase 1 study to evaluate the safety and tolerability of camrelizumab in patients with advanced solid tumors (Supplementary Figure 1). In dose-escalation phase, camrelizumab was given at a dose of 1 mg/kg, 3 mg/kg, 6 mg/kg, and 10 mg/kg every 2 weeks (Q2W) with the intention of determining the maximum tolerated dose (MTD). In dose-expansion phase, cohort was expanded to further explore the clinical safety and preliminary antitumor activity of camrelizumab at the recommended dose identified from dose-escalation phase in four tumor types: endometrial carcinoma, thymic carcinoma, biliary tract carcinoma (BTC), and carcinoma of unknown primary (CUP).
Patients
In the dose-escalation phase, patients with histologically or cytologically confirmed advanced or metastatic solid tumors who had relapsed after or were refractory to standard therapies, were intolerant to standard therapies or had refused standard therapy were enrolled. In dose-expansion phase, patients with four tumor types were enrolled. Endometrial carcinoma patients with histologically confirmed advanced or metastatic carcinoma (sarcomas and mesenchymal tumors were excluded) that had relapsed after or were refractory to at least 1 prior standard therapy in the metastatic setting, were intolerant to standard therapies or refused standard therapy were included. In addition, patients with disease recurrence within 12 months of completion of adjuvant therapy were eligible. Thymic carcinoma patients had histologically or cytologically confirmed advanced or metastatic disease12 based on local guidelines. Biliary tract carcinoma patients had histologically or cytologically confirmed advanced or metastatic extrahepatic cholangiocarcinoma or carcinoma of the ampulla of Vater and had relapsed after or were refractory to at least 1 prior standard therapy, were intolerant to standard therapies or refused standard therapy. Patients with cancer of unknown primary per European Society for Medical Oncology (ESMO) guidelines13 were also included.
Eligible patients had an Eastern Cooperative Oncology Group (ECOG) Performance Status of 0 or 1; a life expectancy ≥12 weeks; measurable lesion(s) according to Response Evaluation Criteria in Solid Tumors (RECIST) v1.1; adequate laboratory parameters. Female patients agreed not to become pregnant or to breast-feed from the beginning of the study screening period through at least 3 months after receiving the last dose of study treatment were enrolled. Both men and women of reproductive potential agreed to employ a highly effective method of contraception.
Patients with any active autoimmune disease or history of autoimmune disease or history of a syndrome that required systemic steroids or immunosuppressive medications (except for patients with vitiligo or resolved childhood asthma/atopy) were excluded. Also, patients with a concurrent medical condition requiring the use of immunosuppressive medications, or immunosuppressive doses of systemic or topical corticosteroids were excluded. Patients with active central nervous system (CNS) metastases were excluded, as well as patients who had an uncontrolled clinically significant medical condition, had received prior systemic chemotherapy, radiotherapy, immunotherapy, hormone therapy, surgery or targeted therapy within 4 weeks before the study drug administration or had any unresolved adverse events (AEs) > Common Terminology Criteria for Adverse Events (CTCAE) Grade 1 (with the exception of stable chronic toxicities not expected to resolve).
Treatment: Dose and Dose Levels
Camrelizumab, a new fully humanized IgG4 monoclonal anti-PD-1 antibody, was produced as a lyophilized powder. It was reconstituted in water and diluted in 5% dextrose to a final concentration of between 0.5 mg/mL and 10 mg/mL, before administration as an IV infusion over 30 mins. In the dose-escalation phase, the dose of camrelizumab was determined based on body weight (1 mg/kg, 3 mg/kg, 6 mg/kg, or 10 mg/kg). Camrelizumab was administered on Day 1 of Cycle 1 (each cycle is 4 weeks) and Q2W from Cycle 2. In the dose-expansion phase, camrelizumab was administered at a flat dose of 200 mg every 4 weeks (Q4W), except for the first 3 subjects enrolled, whose initial dose was 600 mg Q4W.
Dose Escalation and Dose-Limiting Toxicities (DLTs)
The safety and tolerability of camrelizumab was assessed by ongoing reviews of clinical laboratory tests, ECOG performance status, physical examination, electrocardiogram (ECG), and adverse events. All immune-related adverse events (irAEs) were classified as AEs of special interest (AESIs) if the investigator deemed that there was a likely causal relationship to camrelizumab. Evaluations of skin toxicities were conducted by skin photography and histopathology as guided by clinical assessment.
In dose-escalation phase, three to six patients were enrolled in each dose cohort and assessed for DLTs from the first infusion of the study drug through the end of Cycle 1 (28 days after the first dose). Any patient withdrawn from the study before the completion of the first cycle for any reason other than a DLT was replaced unless there were already sufficient DLT-evaluable patients in that cohort. If none of the initial three patients at a dose level experienced a DLT, then dose escalation to the next planned level could proceed. If one of three patients in a dose cohort experienced a DLT, three additional patients were enrolled in the cohort and if no DLT was observed in these patients, dose escalation could proceed. If two DLTs were observed at a given dose level, an intermediate dose level could be considered by the Safety Monitoring Committee (SMC) for testing as the MTD.
Severity of AEs was graded according to National Cancer Institute (NCI)-CTCAE version 4.0. In dose-escalation and expansion phase, the occurrence of any of the following toxicities during Cycle 1 (28 days from the first infusion) was considered a DLT, if judged by the investigator to be probably or definitely related to study drug administration: (a) Grade ≥2 uveitis; (b) Grade ≥2 pneumonitis that persisted for >7 days despite treatment with systemic corticosteroids (subjects with Grades 3 and 4 pneumonitis were to be discontinued from study treatment); (c) Grade ≥3 nonhematologic AE with Grade 3 AEs considered dose-limiting when they required medical intervention, resulted in hospitalization, lasted >3 days (Grade 3 fatigue >7 days) despite optimal supportive care or were a clinically significant laboratory abnormality; (d) Grade ≥3 hematologic toxicity.
AEs were not considered DLTs if there was a clear, well-documented, alternative explanation for the AE. The SMC could also consider AEs as DLTs if they occurred after Cycle 1 and appeared to result from delayed or cumulative toxicities.
Safety and Efficacy Assessments
Safety was evaluated in patients who received at least one dose of camrelizumab. All reported adverse events were coded according to MedDRA, version 18.1 and graded as per CTCAE version 4.0. All concomitant medications were documented throughout the patient’s participation in the study. The anti-tumor responses were assessed per RECIST 1.1.
Interim Analyses
While no formal interim analyses were conducted, the SMC reviewed the comprehensive data on an ongoing basis. During this review, safety and emerging preclinical and efficacy data were reviewed to decide the dose and dose regimen during the conduct of the trial. At each dose level, the SMC reviewed all data and decided on commencing the next dose level as well on the recommended Phase 2 dose and dose regimen.
Bioanalytical Methods
Serum samples to determine the levels of camrelizumab and its pharmacokinetic (PK) profile were collected during Cycle 1 at Days 1, 2, 3, 8, 15, and 22 and during the remaining cycles at Days 1 and 15. Camrelizumab was directly detected with a specific immune assay, the results were graphically analyzed and then pooled for final analyses using nonlinear mixed-effect modeling (NONMEM, version 7.3).
PD-1 receptor occupancy (RO) assessment using PD-1–expressing JURKAT cells (ATCC® Number: TIB-152TM): We first assessed the serum of patients for the presence of camrelizumab and its ability to bind to PD-1 receptors on JURKAT cells. Serum samples were collected at the same time points as for the PK samples (see above) and incubated with a fixed amount of PD-1–expressing JURKAT cells for 60 mins, followed by incubation with 3 μg/mL fluorescein-labeled camrelizumab for 30 mins, and RO was determined by flow cytometry.
Whole Blood Processing
The fresh human blood samples (3 mL) were aliquoted into a 50 mL centrifuge tube. A 30 mL 1× BD FACS Lysing (Cat No.: 555899, San Jose, CA) was added and the tube was inverted several times to confirm the effective mixing of the blood and FACS Lysing solution. Samples were incubated at room temperature for 10–12 mins. Samples were centrifuged 400× g, 5 mins at room temperature and the supernatant was discarded. The tube was filled with 30 mL DPBS, and centrifuge for 5 mins at 400× g. The supernatant was discarded, the cells re-suspended in 1.5 mL DPBS, counted and used immediately in the assays below.
Receptor Occupancy Assay
Approximately 4 × 105 cells were plated in a 96-well U-bottom plate, and either human IgG4 protein (Abcam Cat. No.: Ab90286, Cambridge, MA) or camrelizumab-Fluorescein (provided by Incyte, Wilmington, DE) was added at 20 μg/mL final concentration. Samples were incubated at room temperature for 30 mins. The plate was centrifuged at 300× g for 5 mins and the supernatant was discarded. Samples were stained: Stain I at the following dilutions in PBS/0.5% FCS: 1/1000 eFluor 710 live dead (Invitrogen Cat. No.: 65-0865-18, Carlsbad, CA); 1/150 anti-huCD4 (OCT4)-BV421 (Biolegend Cat. No.: 317434, San Diego, CA); 1/150 anti-huCD3-(SK7)-BV510 (Biolegend Cat. No.: 344828); 1/150 anti-huCD8-PeCy7 (Biolegend Cat. No.: 344712); 1/100 anti-IgG4-PE (Southern Biotech Cat. No.: 9200–09, Birmingham, AL). Samples were incubated at 4°C for 15 mins in the dark. Samples were washed with DPBS followed by centrifugation at 300× g for 5 mins and the supernatant was discarded. Samples were re-suspended in 100 µL DPBS and immediately run on the FACs Canto. Analysis was performed using FlowJo software (version 10; Treestar, Ashland, OR). Assays were performed at least in duplicate or in triplicate where sufficient cells were available.
Tumor Tissue
Where available, tumor tissue was assessed for PD-L1 by immunohistochemistry (22C3 clone for IHC; Indivumed, Hamburg, Germany).
Statistical Analysis
In the dose-escalation phase, no sample size calculation for efficacy was used. The enrollment was based on standard safety and tolerability evaluation. In the dose-expansion phase, approximately 52 patients were to be enrolled (approximately 13 patients in each of four tumor-expansion cohorts). This was determined from a Bayesian analysis based on a prior probability of 32% that the response rate is at least 20% (prior response rate as Beta [0.2, 0.8]), and assuming 3 responses are observed in a cohort of 13 patients, then there is a 56% probability that the true response rate is at least 20%.
The objective response rate (ORR) and disease control rate (DCR) per RECIST 1.1 along with the 95% Clopper–Pearson confidence interval (CI) were tabulated by dose levels in dose-escalation phase and expansion phase combined, and by tumor type in expansion phase. Progression-free survival (PFS) was analyzed using the Kaplan–Meier method, along with median and 95% confidence intervals for the median.14 Laboratory parameters were reported as percentages, cells/μL, GI/L, U/L, pg/mL, where appropriate and indicated on tables/figures.
Results
Patient Disposition and Characteristics
Patients were enrolled and treated in this FiH study between October 2015 and February 2017 (with one patient still on treatment in May 2019) at five centers in Australia (three centers participating in dose-escalation phase only). A total of 52 patients were screened, of which 49 were enrolled and treated with camrelizumab. Of the 49 patients, 23 patients were treated in the dose-escalation phase and 26 in dose-expansion phase (Supplementary Figure 1). The dose exposure to camrelizumab in each sub-group was presented in Supplementary Table 1. As of data cutoff, four (8.2%) patients were still receiving treatment. The reasons for treatment discontinuation were radiographic progression (n=26, 53.1%), clinical progression (n=13, 26.5%), intolerable toxicity/AE (n=3, 6.1%), investigator discretion (n=2, 4.1%), and death (n=1, 2.0%).
Baseline characteristics were presented in Table 1. Thirty-two of 49 patients were female (65.3%) and the majority of patients were Caucasian (77.6%). The median age was 60 years (range, 32–80), with 67.3% of patients being younger than 65 years (33/49). An ECOG Performance Status of 1 was reported for 63.3% of all patients (31/49). Metastatic sites included lung, liver, lymph nodes and other sites. The median time since diagnosis across all patients was 1.5 years (range, 0.3–10.4). Patients received a median of three (range, 1–20) prior anti-cancer therapies. In the dose-escalation cohort, all patients were diagnosed with advanced solid tumors, including ovarian, breast, head and neck, colorectal and other carcinomas. In the dose-expansion cohort, there were six endometrial carcinomas, 11 biliary tract carcinomas, six other carcinomas and three CUP.
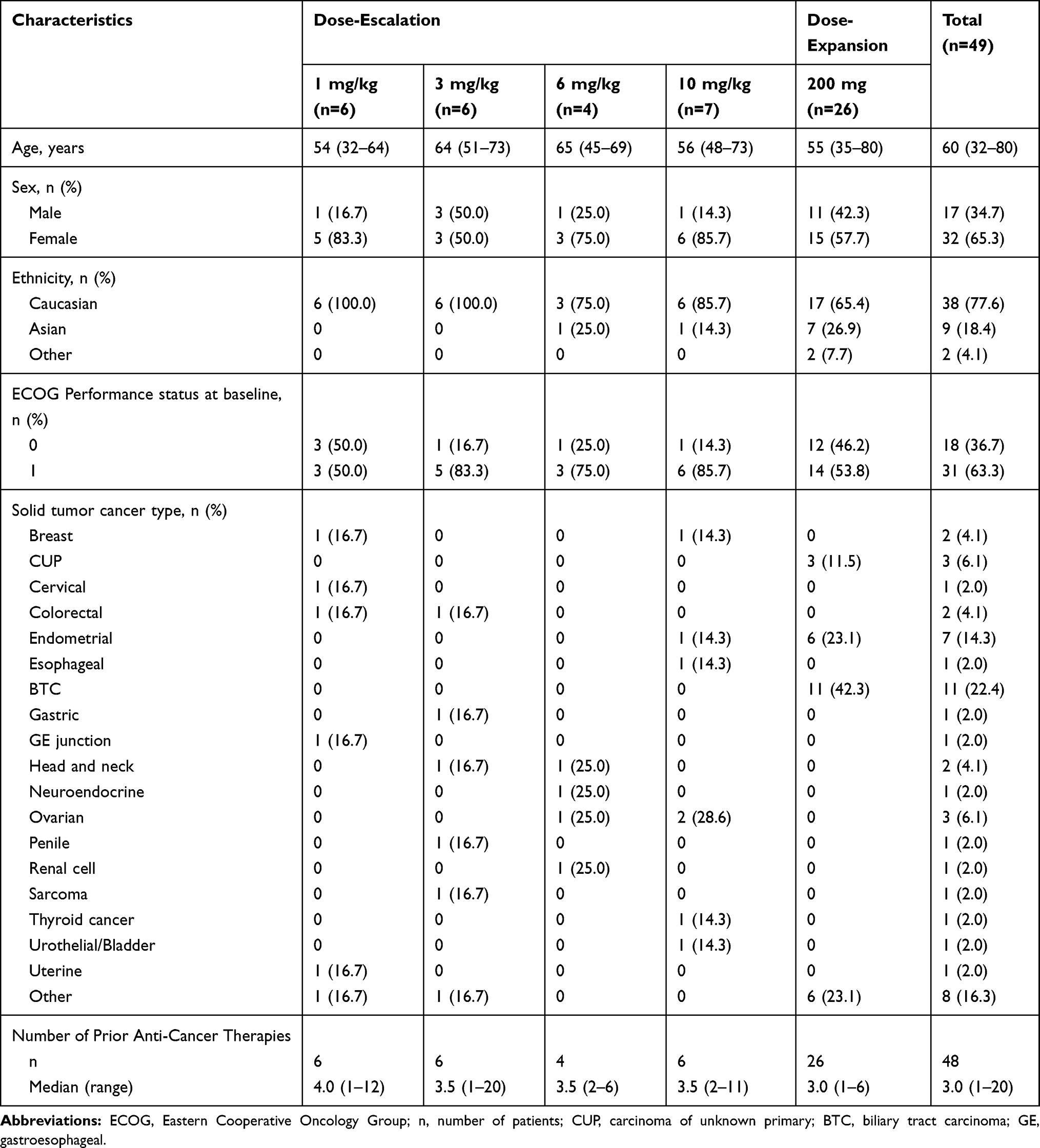 |
Table 1 Demographics and Other Baseline Characteristics |
Safety Outcomes
All 49 treated patients were evaluated for safety and tolerability. During the dose-escalation phase, there were two instances where DLTs were observed requiring the expansion of the cohort: during the first cohort (1 mg/kg) and during the second cohort (3 mg/kg). The DLT at 1 mg/kg occurred after the first dose and was Grade 3 aspartate aminotransferase (AST) increased and alanine aminotransferase (ALT) increased (Supplementary Table 2). Glucocorticoid treatment was commenced and transaminase levels promptly returned to normal. Re-treatment with camrelizumab did not result in additional liver AEs. A DLT of diarrhea was observed in a patient with gastric cancer after the first dose of camrelizumab at 3 mg/kg. This resolved after starting glucocorticoid therapy. The patient was not re-treated with camrelizumab because of tumor progression. Because of these DLTs, the first and second cohorts were expanded to 6 patients and it was decided to add an intermediate dose level at 6 mg/kg before proceeding to the 10 mg/kg dose level.
All patients (49/49, 100%) experienced at least one all-causality adverse event during this study (Table 2). The most commonly reported AEs included reactive cutaneous capillary endothelial proliferation (RCCEP; 31/49, 63.2%), fatigue (18/49; 36.7%), diarrhea (11/49; 22.4%), nausea (10/49; 20.4%), and vomiting (9/49; 18.4%). Nineteen (38.8%) patients reported AEs of Grade ≥3. Subjects with any serious AE (SAE) occurred in 21 (42.9%) patients, with the most common being disease progression (3; 6.1%), cellulitis (2; 4.1%), hepatic failure (2; 4.1%), respiratory tract infection (2; 4.1%), and small intestinal obstruction (2; 4.1%). Treatment-related AEs occurred in 39 (79.6%) of 49 patients and 11 (22.4%) patients discontinued treatment due to an AE. Six of the 20 subject deaths were due to a fatal AE, but no deaths were deemed to be related to camrelizumab and considered to be a result of disease progression.
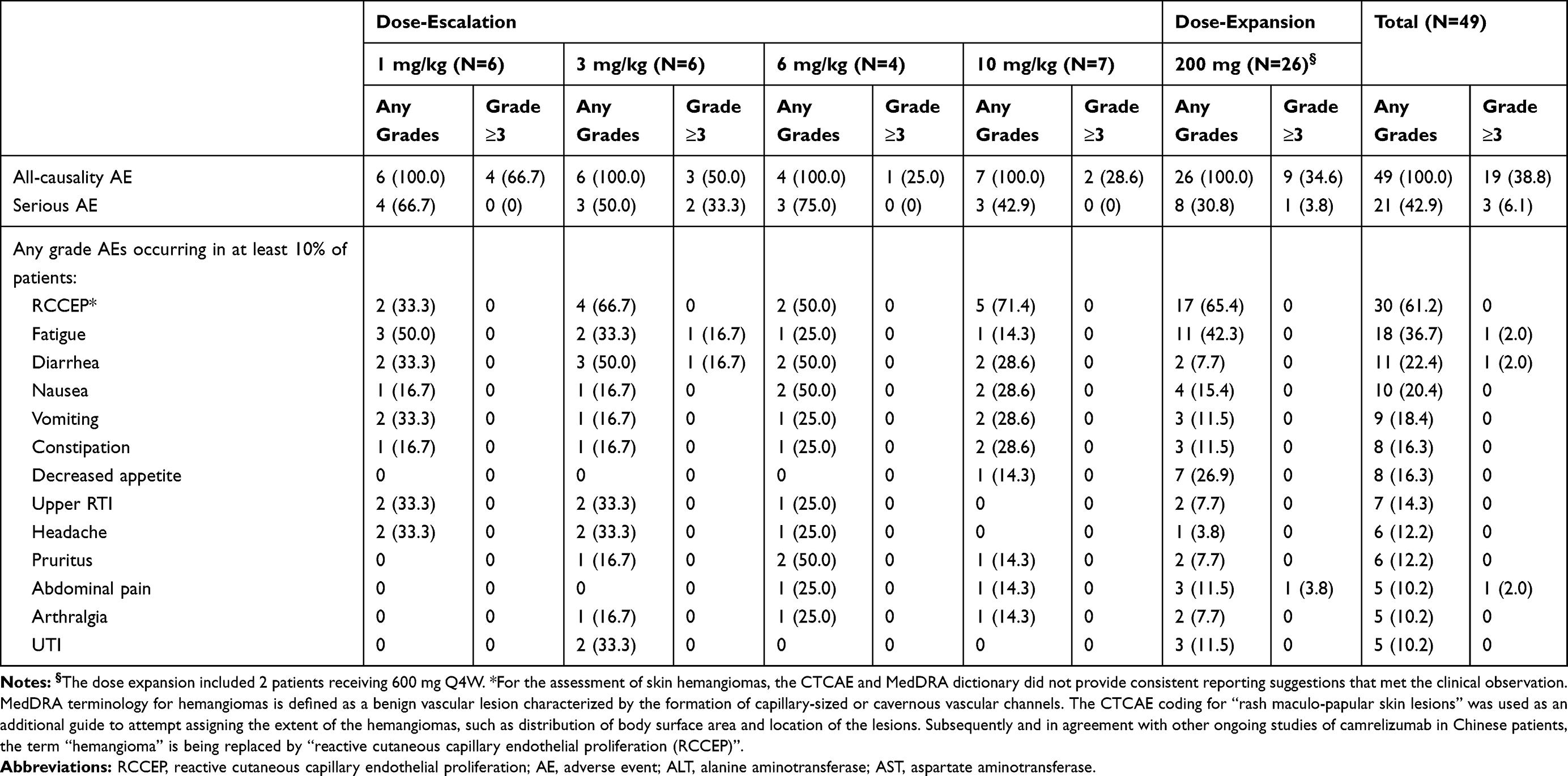 |
Table 2 Adverse Events |
Six (12.2%) patients reported a total of eight immune-related AEs (irAEs), which were collected as AEs of special interest (AESI), after administration of camrelizumab, including arthropathy (Grade 2), diarrhea (Grade 3), autoimmune hepatitis (Grade 3), pneumonitis (Grade 3), hypopituitarism (Grade 3), ALT elevation (Grade 3), AST elevation (Grade 3) and pancreatitis (Grade 2) (Supplementary Table 2). There was no clear association between the incidence and severity of irAEs and camrelizumab dose.
In contrast to the reported effects of other anti-PD-1 antibodies, camrelizumab treatment was associated with the development of focal vascular lesions of the skin characterized as Grade 1 or 2 RCCEP. In a minority of patients, RCCEP also involved the oral mucosa. Although numerically higher dose levels of camrelizumab were not associated with a higher incidence and severity of RCCEP, the overall presentation of RCCEP and extent over several areas of the body surface appeared to be worse as the doses progressed. Patients were particularly concerned at their physical appearance if they received the highest doses of camrelizumab. Patients reported the lesions within the first 10 weeks of starting camrelizumab treatment and lesions persisted for many months in some cases. The lesions mostly occurred on the upper body of study patients (torso, arms, head and neck). In three patients administered camrelizumab at 1 or 3 mg/kg Q2W for more than 6 months, the RCCEP spontaneously regressed despite ongoing study drug treatment. The RCCEP affecting three other patients (treated at 1 mg/kg, 3 mg/kg or 6 mg/kg Q2W) largely resolved within 4 weeks of discontinuing study drug. One of these patients subsequently restarted camrelizumab and RCCEP re-growth was observed at one of the previous sites while the other lesions gradually regressed further (Figure 1). No internal bleeding was reported, and hemoglobin levels remained stable or increased despite the development of RCCEP.
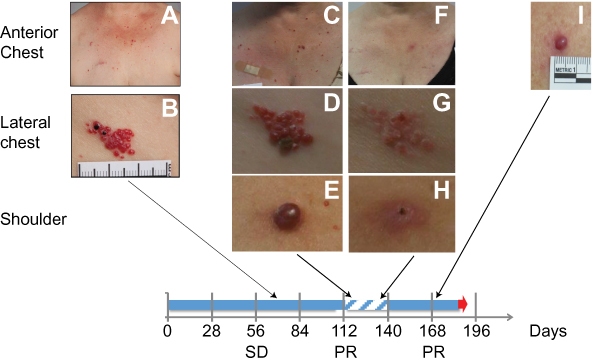 |
Figure 1 Development of RCCEP in a breast cancer patient treated with 1 mg/kg of camrelizumab. (A, B) The initial RCCEP lesion was observed at approximately 2 weeks after the first dose of study drug on the lateral chest followed by lesions on the anterior chest. (C–E) Since this patient showed stable disease (SD) at Day 56 and subsequently a partial response (PR) at Day 112, she continued to receive camrelizumab and additional RCCEP appeared on her upper trunk and neck. (F–H) After interrupting camrelizumab treatment for 28 days in response to cellulitis requiring hospitalization, there was substantial regression of the RCCEP. (I) Upon re-starting camrelizumab, one of the previous RCCEP lesions re-grew without progression of RCCEP lesions elsewhere. Blue solid arrow represents treatment time, while hatched blue color represents the treatment pause. Black arrows connect the timing of RCCEP photographs and the treatment timeline. Abbreviations: SD, stable disease; PR, partial response. |
Pharmacokinetics
The pharmacokinetic profile of camrelizumab is consistent with typical therapeutic antibodies (Figure 2A and B). Non-compartmental analysis indicated a half-life of three to 10 days from 1 mg/kg to 10 mg/kg after a single IV infusion. While Cmax increased proportionally to dose from 1 mg/kg to 10 mg/kg, AUC0-∞ showed supra-linearity over the same dose range. Low inter-subject variability was observed for PK exposures up to 6 mg/kg Q2W, and higher interpatient variability was observed at 10 mg/kg Q2W (Supplementary Table 3). The mean exposure profile for the 200 mg flat dose is essentially superimposable on that of the 3 mg/kg profile.
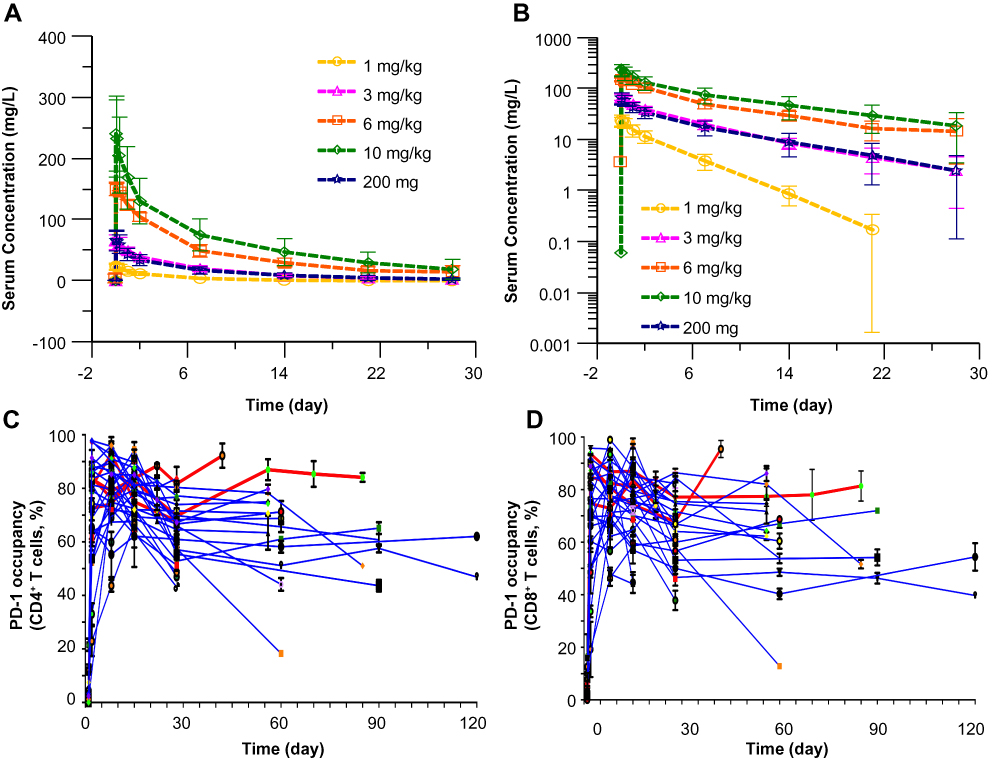 |
Figure 2 Serum camrelizumab concentration–time curve and receptor occupancy (RO). (A, B) Serum camrelizumab concentration–time curve after single IV infusion in linear (A) and semi-logarithmic (B) scale during the first 28 days after the initial infusion. (C, D) Camrelizumab RO on CD4+ (C) and CD8+ (D) T Cells at the 10 mg/kg dose level and 200 mg flat dose level for the entire treatment period. Blood samples were obtained at the time points shown out to 28 days after the first dose and then prior to the second and subsequent dose (every 28 days). Each line represents an individual patient, with red lines indicating 10 mg/kg treated patients and blue lines indicating 200 mg flat dosing. |
Receptor Occupancy (RO) of PD-1
In the first three dose-escalation cohorts, we assessed RO of circulating camrelizumab in JURKAT cells and tracked with plasma half-life in patients. In addition, starting with dose level 10 mg/kg Q2W (dose-escalation phase) and in all subsequently treated patients (dose-expansion phase), we measured PD-1 RO on patient circulating CD8+ and CD4+ T cells. This showed that RO remained greater than 50% during the 28 days after the initial camrelizumab dose in most patients, with higher levels at earlier post-dose timepoints (Figure 2C and D). After completing Cycle 1, the RO evaluation was performed predose every 4 weeks and these levels were generally similar to those at day 28.
Efficacy Outcomes
Among the 46 patients included in the efficacy analysis, one (2.2%) subject achieved a complete response (biliary tract carcinoma). Six (13.0%) additional patients had a partial response (one breast carcinoma, one parotid carcinoma, one renal cell carcinoma, one urothelial carcinoma, one ovarian carcinoma and one endometrial carcinoma), eight (17.4%) patients had stable disease, and 25 (54.3%) patients had disease progression as their best response (Table 3). In additional 6 patients, there was no scan to perform a RECIST assessment. Thus, the ORR and DCR for the entire study population were 15.2% (95% CI 6.3–28.9) and 32.6% (95% CI 19.5–48.0), respectively. A waterfall plot of best reductions from baseline in target-lesion size is shown in Figure 3A. No clear association between camrelizumab dose and tumor response was evident. A spider plot of changes from baseline in the size of target lesions over time is presented in Figure 3B. The estimated median PFS was 1.9 months (95% CI 1.8–3.1) and the occurrence of RCCEP across all doses appeared not to be correlated with tumor responses (Figure 3C).
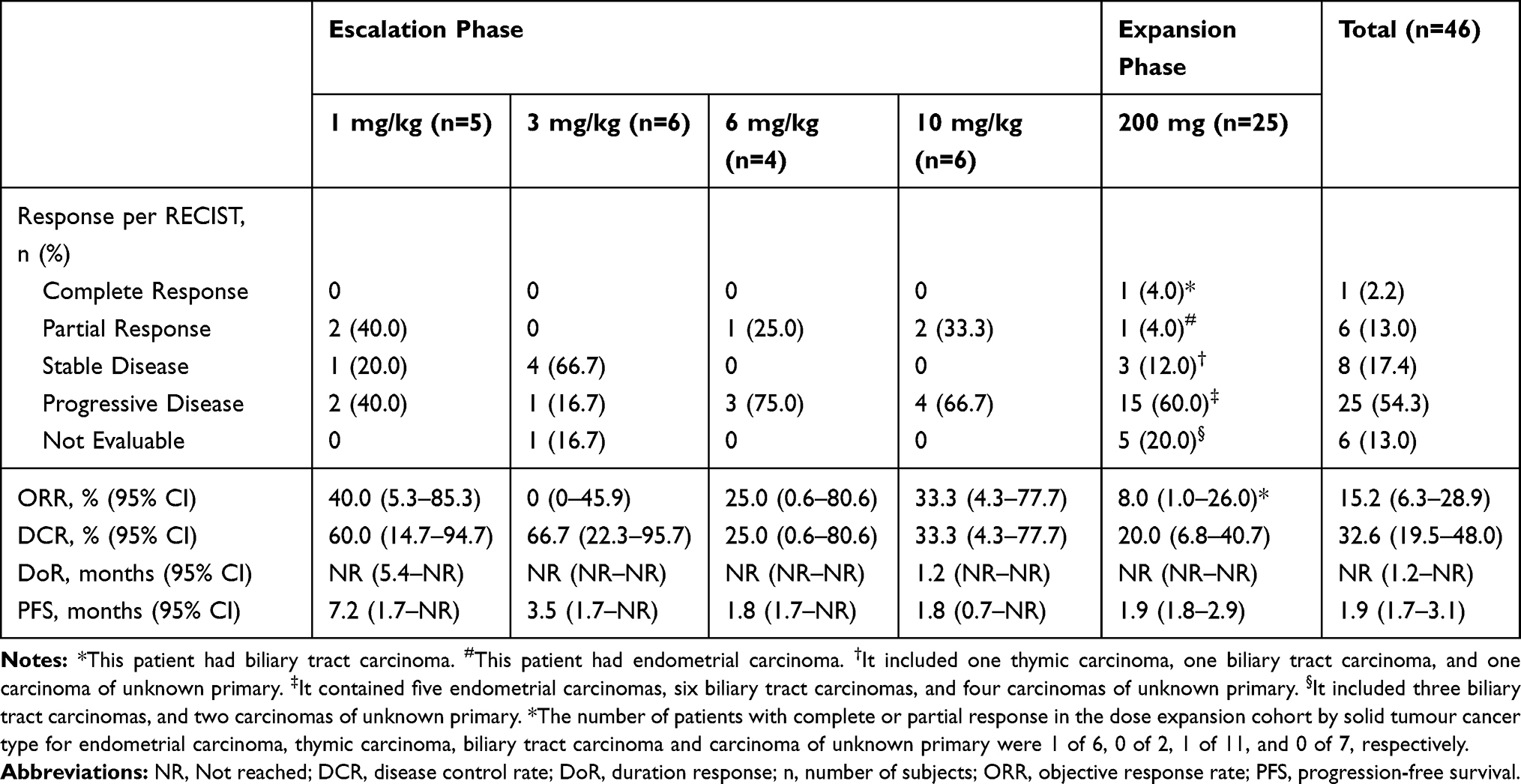 |
Table 3 Response and Survival Data |
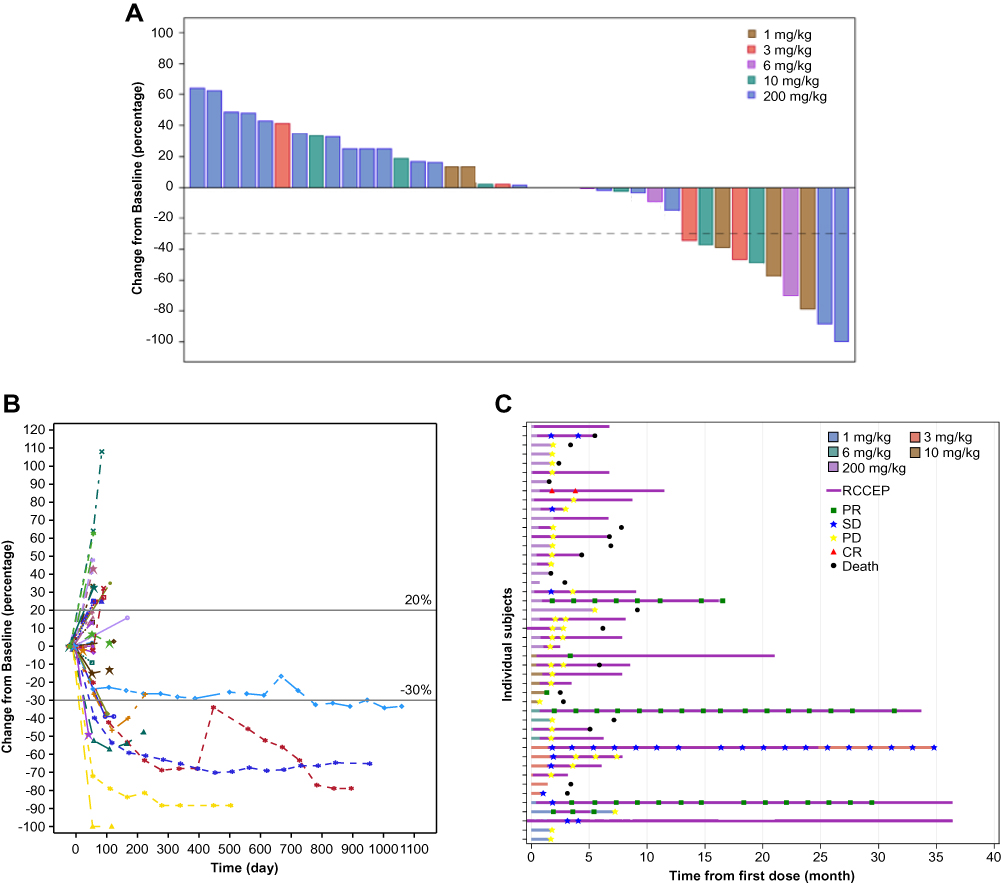 |
Figure 3 Clinical response. (A) Waterfall plot of the maximal percentage reduction from baseline of the sum of longest diameters of target lesions for both dose-escalation and expansion phases. (B) Spider plot of the percentage change from baseline in the sum of longest diameters of target lesions over time. (C) Duration of treatment (n=46). Each individual patient treatment time is plotted from time of first dose (x-axis) based on the dose level and dose regimen. Patients with partial responses (green square), stable disease (blue star), progressive disease (yellow star), complete response (red triangle) and death (black circle) are shown along with the start of RCCEP. Abbreviations: CR, complete response; PD, progressive disease; PR, partial response; SD, stable disease; RCCEP, reactive cutaneous capillary endothelial proliferation. |
Discussion
We reported the first FiH trial conducted outside of China. In this FiH study, we evaluated the clinical safety, PK profile, and initial efficacy of camrelizumab, a novel monoclonal antibody against PD-1 with a binding affinity of 3 nM, which is comparable to that of other monoclonal antibodies directed against PD-1 such as pembrolizumab.15,16 The Australian patients in this study had exhausted all standard therapies and most had received at least two prior lines of therapy. Hence, the ORR of 15% was consistent with response rates reported for other PD-1 inhibitors that were investigated in a wide range of different tumor types and in FiH dose studies.15,16
Notwithstanding these similarities, there were some differences compared to other PD-1 inhibitors. First, responses were observed in patients with no or very low expression of PD-L1 expression in their tumor samples (Supplementary Table 4). Because of the availability of tumor tissue from 12 patients, this observation will need to be further investigated. Second, even as camrelizumab was declining in concentration due to its short half-life, the concomitantly measured RO remained elevated at above 50% in most patients. In one patient who stopped taking camrelizumab, the RO remained unchanged for close to 3 months before RO levels started to decline. Third, we observed RCCEP, a unique feature so far not reported for other PD-1 inhibitors. While camrelizumab had rates of all-cause AEs Grade 1/2 of 75% and Grade 3/4 of 25% and an AESI (or irAEs) rate of 19% (8/42) similar to other PD-1 inhibitors investigated in FiH studies,15,17 we observed an increasing number of patients reporting RCCEP. These RCCEPs were distinct from the types of skin-related toxicity reported for approved PD-1 inhibitors.18–20 While the RCCEPs were not considered DLTs or life-threatening, patients were often concerned about their physical appearance. In search of understanding the mechanism of these RCCEPs, we found that similar RCCEP had been reported for patients treated with the VEGFR2 inhibitors tanibirumab (about 50% of patients) and CDP749 (23% of patients)21,22 and in a single case for ramucirumab.23 Because of this clinical similarity to tanibirumab or CDP749, we investigated the binding affinity of camrelizumab to VEGFR2. In contrast to tanibirumab, camrelizumab bound to VEGFR2 at approximately 2.8 μM in a BiaCore assay, and it showed no significant binding in cellular assays. Alternatively, we considered that camrelizumab may target PD-1 expressing cells in the skin, which in turn may produce VEGF via chemokine release.24 For example, infantile or toxicity-induced RCCEP are thought to be partially induced by such an immune-related activation, but a role of PD-1 has not been reported.24–26 A treatment with topical propranolol as indicated in some patients with infantile RCCEP was not attempted in this trial.27 We also considered the possibility that genetic polymorphisms of VEGFR2 may pre-dispose Caucasian patients to developing RCCEP. However, Chinese patients treated with camrelizumab had similar skin toxicities, including similar times from the start of treatment to onset and the disappearances of RCCEP after longer treatment periods, including patients who were still receiving camrelizumab.11,28 Given these observations, we also cannot exclude that camrelizumab may interact with an unknown ligand. Recent studies have suggested that despite similarities in binding affinity, PD-1 inhibitors are folded differently and this may have subtle implications on their clinical profile.29 Recently, a group investigated the possibility of whether the protein folding of camrelizumab may expose sequences that activate VEGFR2.30 As these RCCEP are histologically similar to pyogenic granulomas,31 then the origin of the RCCEP may be the result of an initial mechanical injury (for example, friction), and camrelizumab might block the subsequent healing process.
The limitations of this FiH study include that no pre-treatment PD-L1 status assessment was mandatory. Available tumor tissue was assessed in a retrospective manner and therefore these data are hypothesis-generating. In addition, the underlying mechanism of the pathogenesis of RCCEP needs further investigation.
In summary, camrelizumab appears to be an active PD-1 inhibitor with acceptable skin and other toxicities when dosed at 200 mg Q4W in Australian population. However, the more frequent dosing of 200 mg Q2W appears to be an acceptable alternative as several studies in Chinese patients have shown.10,32-38 This larger experience of camrelizumab in Chinese patients has been the basis of its approval in China for the treatment of patients with Hodgkin’s Disease.10,34,39 Since all studies were conducted in Chinese population so far, our study provided clinical evidence in Australian and expanded the generalizability of camrelizumab in solid tumors in patients outside of China.
Ethics Approval and Consent to Participate
Each institution’s review board approved the study and all patients signed an informed consent document before study participation (Supplementary Table 5). The study was conducted according to the principles of Good Clinical Practice, applicable laws and regulations, and the Declaration of Helsinki.
Data Sharing Statement
The datasets used and/or analyzed during this study are available from the corresponding author on reasonable request.
Acknowledgments
The study team thanks patients and families for their willingness to participate in this study. We thank Atridia for the management of the entire study. Further, we thank all site staff and investigators at the institutions, the trial personnel at Incyte, Tigermed, IQVIA, and Hengrui.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Funding
This study was sponsored by Jiangsu Hengrui Medicine Co. Ltd, Shanghai, China.
Disclosure
Jason Lickliter and Mark Voskoboynik are employees of Nucleus Network (Australia). Jianjun Zou, Lianshan Zhang and Stacey Luo are employees of Jiangsu Hengrui Medicine Co. Ltd (China). Michael Lahn, Andrea Mannucci and Catello Somma were or are employees of Incyte Biosciences International Sarl (Switzerland). Howard Kallender is employee of Incyte Corporation (USA). Michael Millward and Tarek Meniawy are employees of Linear Clinical Research (Australia). Hui K. Gan reports receiving honoraria from speaker bureau of Bristol-Myers Squibb, Ignyta, Eisai and Merck Serono and is a consultant/advisory board member of AbbVie and Bristol-Myers Squibb. Andrea Mannucci has ownership interests (including stock, patents, etc.) at Incyte Biosciences International Sarl (Switzerland). Michael Millward has been or currently is a member of consultant/advisory board of Merck Sharp & Dohme, Novartis, Bristol-Myers Squib, Roche, AstraZeneca, and Pfizer, and has received travel support from Bristol-Myers Squibb and Roche. Adnan Nagrial is a member of consultant/advisory board of Bristol-Myers Squibb, Merck Sharpe Dohme, Astra Zeneca and Roche, and reports receiving speaking fees from Bristol-Myers Squibb, Merck Sharpe Dohme and Roche. Andreas Behren receives research support from Incyte (Switzerland) for the RO assay. Michael Lahn has ownership interests (including stock, patents, etc.) at Incyte Biosciences International Sarl (Switzerland), AstraZeneca and Eli Lilly. Surein Arulananda reports receiving personal fees from Merck Sharpe Dohme, Astra Zeneca, Roche and Boehringer-Ingelheim. Pablo Fernandez Penas is member of advisory committee (AC) or lecture (L, only educational, non-promotional lectures) of Janssen (AC, L), Lilly (AC, L), Abbvie (AC, L), Novartis (AC, L), UCB (L), MSD (AC), La Roche Posay (L), Merck (AC, L), Roche (AC, L), Amgen (L), Sun Pharma (AC, L), Sanofi (AC), Leo (AC, L), Avene (L), Celgene (AC), Galderma (L) and Schering Plough (L), and reports receiving clinical trial supports from Boehringer Ingelheim, Pfizer, CSL, OncoSec, Lilly, CBP, Abbvie, miRagen, Galderma, Regeneron, BMS, Eisai, Jiangsu Hengrui, Sun Pharma, Novartis, UCB, Leo, Janssen, Arena, Akaal Pharma, Roche, Xoma, Kyowa Hakko Kirin, GSK, Amgen and Merck Sharpe Dohme. No potential conflicts of interest were disclosed by other authors.
References
1. Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 1999;11(2):141–151. doi:10.1016/S1074-7613(00)80089-8
2. Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992;11(11):3887–3895. doi:10.1002/embj.1992.11.issue-11
3. Iwai Y, Terawaki S, Honjo T. PD-1 blockade inhibits hematogenous spread of poorly immunogenic tumor cells by enhanced recruitment of effector T cells. Int Immunol. 2005;17(2):133–144. doi:10.1093/intimm/dxh194
4. Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A. 2002;99(19):12293–12297. doi:10.1073/pnas.192461099
5. Yost KE, Satpathy AT, Wells DK, et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat Med. 2019;25(8):1251–1259. doi:10.1038/s41591-019-0522-3
6. Boussiotis VA, Longo DL. Molecular and biochemical aspects of the PD-1 checkpoint pathway. N Engl J Med. 2016;375(18):1767–1778. doi:10.1056/NEJMra1514296
7. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264. doi:10.1038/nrc3239
8. Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–2454. doi:10.1056/NEJMoa1200690
9. Song Y, Wu J, Chen X, et al. A single-arm, multicenter, Phase II study of camrelizumab in relapsed or refractory classical hodgkin lymphoma. Clin Cancer Res. 2019;25(24):7363–7369. doi:10.1158/1078-0432.CCR-19-1680
10. Fang W, Yang Y, Ma Y, et al. Camrelizumab (SHR-1210) alone or in combination with gemcitabine plus cisplatin for nasopharyngeal carcinoma: results from two single-arm, phase 1 trials. Lancet Oncol. 2018;19(10):1338–1350. doi:10.1016/S1470-2045(18)30495-9
11. Mo H, Huang J, Xu J, et al. Safety, anti-tumour activity, and pharmacokinetics of fixed-dose SHR-1210, an anti-PD-1 antibody in advanced solid tumours: a dose-escalation, phase 1 study. Br J Cancer. 2018;119(5):538–545. doi:10.1038/s41416-018-0100-3
12. Girard N, Ruffini E, Marx A, Faivre-Finn C, Peters S, Committee EG. Thymic epithelial tumours: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2015;26(Suppl 5):v40–v55. doi:10.1093/annonc/mdv277
13. Fizazi K, Greco FA, Pavlidis N, et al. Cancers of unknown primary site: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2015;26(Suppl 5):v133–v138. doi:10.1093/annonc/mdv305
14. Crowley J, Breslow N. Statistical analysis of survival data. Annu Rev Public Health. 1984;5:385–411. doi:10.1146/annurev.pu.05.050184.002125
15. Patnaik A, Kang SP, Rasco D, et al. Phase I study of pembrolizumab (MK-3475; Anti-PD-1 monoclonal antibody) in patients with advanced solid tumors. Clin Cancer Res. 2015;21(19):4286–4293. doi:10.1158/1078-0432.CCR-14-2607
16. Brahmer JR, Drake CG, Wollner I, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. 2010;28(19):3167–3175. doi:10.1200/JCO.2009.26.7609
17. Michot JM, Bigenwald C, Champiat S, et al. Immune-related adverse events with immune checkpoint blockade: a comprehensive review. Eur J Cancer. 2016;54:139–148. doi:10.1016/j.ejca.2015.11.016
18. Belum VR, Benhuri B, Postow MA, et al. Characterisation and management of dermatologic adverse events to agents targeting the PD-1 receptor. Eur J Cancer. 2016;60:12–25. doi:10.1016/j.ejca.2016.02.010
19. Hwang SJ, Carlos G, Wakade D, et al. Cutaneous adverse events (AEs) of anti-programmed cell death (PD)-1 therapy in patients with metastatic melanoma: a single-institution cohort. J Am Acad Dermatol. 2016;74(3):455–461 e451. doi:10.1016/j.jaad.2015.10.029
20. Sanlorenzo M, Vujic I, Daud A, et al. Pembrolizumab cutaneous adverse events and their association with disease progression. JAMA Dermatol. 2015;151(11):1206–1212. doi:10.1001/jamadermatol.2015.1916
21. Ton NC, Parker GJ, Jackson A, et al. Phase I evaluation of CDP791, a PEGylated di-Fab’ conjugate that binds vascular endothelial growth factor receptor 2. Clin Cancer Res. 2007;13(23):7113–7118. doi:10.1158/1078-0432.CCR-07-1550
22. Lee SJ, Ham JS, Kim HK, et al. Phase I trial and pharmacokinetic study of Tanibirumab, a fully human monoclonal antibody to the vascular endothelial growth factor receptor 2 in patients with refractory solid tumors. Paper presented at: 2015 ASCO annual meeting. J Clin Oncol. 2015;33:2522. doi:10.1200/jco.2015.33.15_suppl.2522
23. Lara PE, Medina-Puente C, Oliveira AR, Jimenez-reyes J. Eruptive cherry angiomas developing in a patient treated with ramucirumab. Acta Oncol (Madr). 2017;57(5):709–711.
24. Sun ZJ, Zhao YF, Zhang WF. Immune response: a possible role in the pathophysiology of hemangioma. Med Hypotheses. 2007;68(2):353–355. doi:10.1016/j.mehy.2006.07.013
25. D’Arcangelo D, Nicodemi EM, Rossi S, Giampietri C, Facchiano F, Facchiano A. Identification of serum regression signs in infantile hemangioma. PLoS One. 2014;9(3):e88545. doi:10.1371/journal.pone.0088545
26. Askari N, Vaez-Mahdavi MR, Moaiedmohseni S, et al. Association of chemokines and prolactin with cherry angioma in a sulfur mustard exposed population–Sardasht-Iran cohort study. Int Immunopharmacol. 2013;17(3):991–995. doi:10.1016/j.intimp.2012.12.016
27. Price A, Rai S, McLeod RWJ, Birchall JC, Elhassan HA. Topical propranolol for infantile haemangiomas: a systematic review. J Eur Acad Dermatol Venereol. 2018;32(12):2083–2089. doi:10.1111/jdv.2018.32.issue-12
28. Chen X, Ma L, Wang X, et al. Reactive capillary hemangiomas: a novel dermatologic toxicity following anti-PD-1 treatment with SHR-1210. Cancer Biol Med. 2019;16(1):173–181. doi:10.20892/j.issn.2095-3941.2018.0172
29. Tan S, Zhang H, Chai Y, et al. An unexpected N-terminal loop in PD-1 dominates binding by nivolumab. Nat Commun. 2017;8(1):1.
30. Finlay WJJ, Coleman JE, Edwards JS, Johnson KS. Anti-PD1 ‘SHR-1210ʹ aberrantly targets pro-angiogenic receptors and this polyspecificity can be ablated by paratope refinement. MAbs. 2019;11(1):26–44. doi:10.1080/19420862.2018.1550321
31. Patrice SJ, Wiss K, Mulliken JB. Pyogenic granuloma (lobular capillary hemangioma): a clinicopathologic study of 178 cases. Pediatr Dermatol. 1991;8(4):267–276. doi:10.1111/j.1525-1470.1991.tb00931.x
32. Huang J, Mo H, Zhang W, et al. Promising efficacy of SHR-1210, a novel anti-programmed cell death 1 antibody, in patients with advanced gastric and gastroesophageal junction cancer in China. Cancer. 2019;125(5):742–749. doi:10.1002/cncr.31855
33. Huang J, Xu B, Mo H, et al. Safety, activity, and biomarkers of SHR-1210, an Anti-PD-1 antibody, for patients with advanced esophageal carcinoma. Clin Cancer Res. 2018;24(6):1296–1304. doi:10.1158/1078-0432.CCR-17-2439
34. Nie J, Wang C, Liu Y, et al. Addition of low-dose decitabine to Anti–PD-1 antibody camrelizumab in relapsed/refractory classical hodgkin lymphoma. J Clin Oncol. 2019;37(17):1479–1489. doi:10.1200/JCO.18.02151
35. Shen L, Peng Z, Zhang Y-Q, et al. Camrelizumab combined with capecitabine and oxaliplatin followed by camrelizumab and apatinib as first-line therapy for advanced or metastatic gastric or gastroesophageal junction cancer: updated results from a multicenter, open label phase II trial. Abstract 4031.
36. Xie L, Guo W, Xu J, et al. Apatinib plus camrelizumab (SHR-1210) for unresectable high-grade osteosarcoma (APFAO) progressing after chemotherapy: A prospective, open label, phase II trial. J Clin Oncol. 2019;37(15_suppl):11013.
37. Xu J, Zhang Y, Jia R, et al. Anti-PD-1 antibody SHR-1210 combined with apatinib for advanced hepatocellular carcinoma, gastric, or esophagogastric junction cancer: an open-label, dose escalation and expansion study. Clin Cancer Res. 2019;25(2):515–523. doi:10.1158/1078-0432.CCR-18-2484
38. Zhu H, Yang X, Zhao Y, Yi C. Efficacy of anti-PD-1 antibody SHR-1210 as second-line treatment in hepatocellular carcinoma patient with sorafenib resistance: a case report. Medicine (Baltimore). 2019;98(20):e15755. doi:10.1097/MD.0000000000015755
39. Markham A, Keam SJ. Camrelizumab: first global approval. Drugs. 2019;79(12):1355–1361.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.