Back to Journals » International Journal of Nanomedicine » Volume 12
A facile method to prepare superparamagnetic iron oxide and hydrophobic drug-encapsulated biodegradable polyurethane nanoparticles
Received 21 August 2016
Accepted for publication 2 December 2016
Published 2 March 2017 Volume 2017:12 Pages 1775—1789
DOI https://doi.org/10.2147/IJN.S120290
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Kuo-Wei Cheng, Shan-hui Hsu
Institute of Polymer Science and Engineering, College of Engineering, National Taiwan University, Taipei, Taiwan, Republic of China
Abstract: Superparamagnetic iron oxide nanoparticles (SPIO NPs) have a wide range of biomedical applications such as in magnetic resonance imaging, targeting, and hyperthermia therapy. Aggregation of SPIO NPs can occur because of the hydrophobic surface and high surface energy of SPIO NPs. Here, we developed a facile method to encapsulate SPIO NPs in amphiphilic biodegradable polymer. Anionic biodegradable polyurethane nanoparticles (PU NPs) with ~35 nm size and different chemistry were prepared by waterborne processes. SPIO NPs were synthesized by chemical co-precipitation. SPIO NPs were then added to the aqueous dispersion of PU NPs, followed by application of high-frequency (~20 kHz) ultrasonic vibration for 3 min. This method rendered SPIO-PU hybrid NPs (size ~110 nm) suspended in water. SPIO-PU hybrid NPs contained ~50–60 wt% SPIO and retained the superparamagnetic property (evaluated by a magnetometer) as well as high contrast in magnetic resonance imaging. SPIO-PU NPs also showed the ability to provide cell hyperthermic treatment. Using the same ultrasonic method, hydrophobic drug (Vitamin K3 [VK3]) or (9-(methylaminomethyl)anthracene [MAMA]) could also be encapsulated in PU NPs. The VK3-PU or MAMA-PU hybrid NPs had ~35 nm size and different release profiles for PUs with different chemistry. The encapsulation efficiency for VK3 and MAMA was high (~95%) without burst release. The encapsulation mechanism may be attributed to the low glass transition temperature (Tg) and good mechanical compliance of PU NPs. The new encapsulation method involving waterborne biodegradable PU NPs is simple, rapid, and effective to produce multimodular NP carriers.
Keywords: superparamagnetic iron oxide, polyurethane, drug release, hybrid nanoparticles
Introduction
Nanotechnology has evolved rapidly over the past 2 decades. Nanoparticles (NPs) have unique optical, thermal, electric, and magnetic characteristics,1 and are used as platforms carrying various biofunctions.2,3 Iron oxide NPs possess good biocompatibility4,5 and when the size is below ~15 nm, they have strong superparamagnetic property with single magnetic domain.6 These superparamagnetic iron oxide NPs (SPIO NPs) may have the potential to be applied in drug delivery,7–9 hyperthermia therapy,10,11 and magnetic resonance imaging (MRI).12,13 There are many ways to produce SPIO NPs, and the chemical co-precipitation method is relative easy and cost efficient.14 Without surface modification, SPIO NPs tend to aggregate, which leads to an increase in size. Surface modification of SPIO NPs not only decreases toxicity, but also allows the NPs to be dispersed and even localized in specific areas. Encapsulating SPIO NPs in a hydrophilic polymer like polyethylene glycol (PEG) surface helps the SPIO NPs evade the endothelial reticular system and increase the blood circulation time. In addition, PEG shell decreases the adsorption of proteins on the dispersed SPIO NPs.6,15
The multifunctional SPIO NPs can meet the theranostic needs in modern medicine. Zhu et al developed the molecule cystamine tert-acylhydrazine with disulfide and acylhydrazine functional groups to react on the surface of SPIO NPs. The anticancer drug doxorubicin (DOX) and the polymer PEG were bound to SPIO NPs through the acid-responsive acylhydrazone link; therefore, the NPs may respond to the acidic environment by hydrazone bond cleavage, which results in the rapid release of DOX.16 Zhang and Misra developed a drug carrier combined with SPIO NPs and DOX and coated it with the thermoresponsive dextran-g-poly(NIPAAm-co-DMAAm), which had a low critical solution temperature at 37°C. When the temperature was above the low critical solution temperature, the phase transition of the thermoresponsive polymer led to structure collapse and rapid drug release.17
Polyurethane (PU) is synthesized from diisocyanate, oligodiol, and chain extender. PU can have diverse physicochemical properties by changing the composition of hard (diisocyanate, chain extender) and soft (oligodiol) segments. PU is widely used in biomedical applications because of its excellent biocompatibility and mechanical properties.18,19 The environment-friendly waterborne PU is dispersed in water and has advantages of lower toxicity and low viscosity, in general, compared with the traditional organic PU.20,21 As a drug carrier, waterborne PU can encapsulate hydrophilic as well as hydrophobic drugs. Chen and Hsu produced waterborne biodegradable PU NPs to encapsulate SPIO NPs and hydrophobic drugs.22 However, SPIO NPs and drug must be incorporated in the solvent reaction phase. Zhang et al prepared amphiphilic multiblock poly(lactic acid)-PU from hexamethylene diisocyanate, PEG, and poly(lactic acid).23 The PU micelle carrier showed very low drug release rate without burst effect, and the drug release rate could be tuned by changing the environmental pH value. However, the micelle system may not be very stable in the human body.
In this study, we prepared SPIO NPs (~9 nm) by the co-precipitation method and synthesized different types of biodegradable PU NPs (~35 nm) separately by waterborne processes. Using a high-power vibrational sonicator, SPIO NPs and hydrophobic drug may be encapsulated by PU NPs to form hybrid NPs (SPIO-PU NPs, drug-PU NPs, and drug-SPIO-PU NPs). We analyzed the physicochemical properties, superparamagnetic activity, and magnetic heating functions of SPIO-PU NPs, and the drug release behavior of the NPs containing drug. It was expected that the hybrid PU NPs may possess superparamagnetic property and serve as a potential multimodular nanocarrier.
Materials and methods
Synthesis of SPIO NPs
SPIO NPs were synthesized by chemical co-precipitation as previously described.24 Iron(II) chloride tetrahydrate (8.95 g; Alfa Aesar, Lancashire, UK) and iron(III) chloride hexahydrate (18.25 g, Alfa Aesar) were added to 150 mL distilled water and vigorously stirred. NaOH (Showa, Tokyo, Japan) 11.75 g was dissolved in 50 mL distilled water and slowly added dropwise to the iron oxide precursor solution. The solution was allowed to react at room temperature for 30 min. The product was washed with distilled water several times by centrifugation. Finally, the SPIO NPs were washed and redispersed in 95% ethanol several times by centrifugation. SPIO NPs were purged with nitrogen and stored in 95% ethanol at 4°C.
Synthesis of waterborne biodegradable PU
Three types of PU NPs were prepared, abbreviated as PU1, PU2, and PU3. They differed in the soft segment (oligodiol) composition. PU1 used 100 wt% poly(ε-caprolactone) diol (PCL diol, Mn 2,000 g/mol; Sigma-Aldrich, St Louis, MO, USA) as the soft segment. PU2 used 80 wt% PCL diol and 20 wt% polyethylene butylene adipate diol (PEBA diol, Mn 2,000 g/mol; Yong Shun Chemical Corp, Taipei, Taiwan, Republic of China). PU3 used 40 wt% PCL diol and 60 wt% PEBA diol as the soft segment (Figure 1A). PU NPs were synthesized according to the procedures described previously.25 Briefly, prepolymerization was carried out by adding oligodiols, isophorone diisocyanate (IPDI, Evonik Degussa GmbH, Essen, Germany), and the catalyst tin(II)2-ethylhexanoate (T-9, Alfa Aesar) at 75°C for 3 h under nitrogen environment with mechanical stirring at 180 rpm. After this, 2,2-bis(hydroxymethyl)propionic acid (Sigma-Aldrich) and methyl ethyl ketone (MEK) (Avantor Performance Materials, Pennsylvania, PA, USA) were added and made to react for 1 h. The temperature was cooled down to 45°C and triethylamine (Avantor Performance Materials) was added. After 30 min, water was added rapidly with vigorous (1,200 rpm) stirring for 2–3 min. Ethylenediamine (Tedia, Fairfield, OH, USA) diluted in water was added and made to react for chain extension. Residual MEK and triethylamine were removed by vacuum distillation.
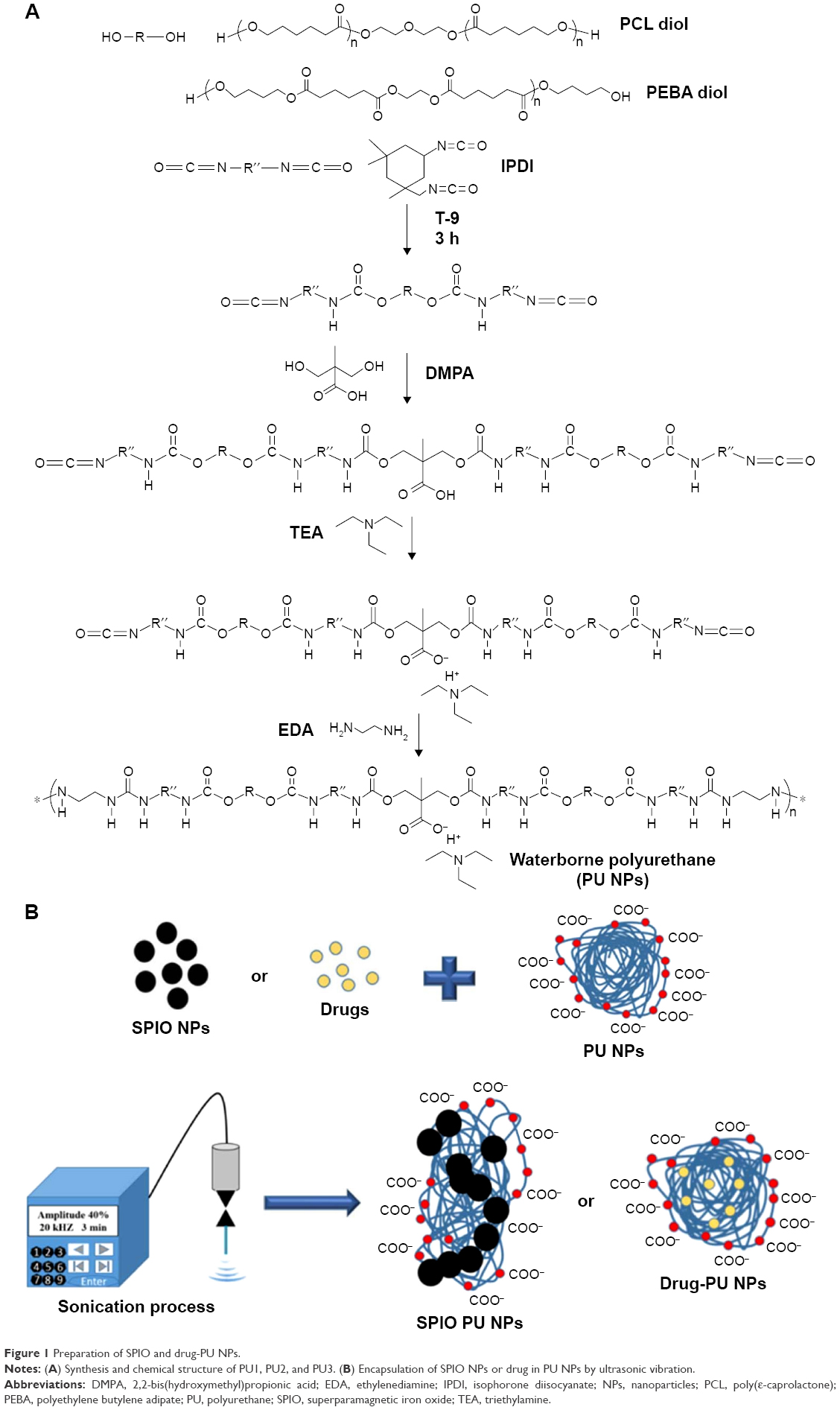 | Figure 1 Preparation of SPIO and drug-PU NPs. |
Preparation of SPIO-PU NPs by ultrasonic vibration
SPIO NPs were washed by centrifugation and redispersed in distilled water. The dispersion of PU NPs was mixed with distilled water and adjusted to have a solid content 15 wt% in a 100 mL beaker. SPIO NPs (~0.08 g) were dropped into PU NPs (~20 g). A vibration sonicator (Vibra cell, VCX750; Sonics & Materials, Newtown, CT, USA) was applied to the mixture for 3 min. The vibration parameters were amplitude 40%, pulse (on 10 sec and off 1 sec), and frequency 20 kHz. After the procedure (Figure 1B), the solution became homogeneously black in color. Free SPIO NPs and aggregate particles were removed by centrifugation (3,000 rpm). The hybrid NPs were purified and redispersed in distilled water by magnetic attraction. For comparison, the vibration time was reduced from 3 min to 20 sec. Subsequent characterization of and experiments were conducted for the SPIO-PU NPs prepared by an ample period of sonication (ie, 3 min).
Preparation of Vitamin K3-PU and 9-(methylaminomethyl)anthracene-PU NPs by ultrasonic vibration
Vitamin K3 (VK3, Menadione; Acros Organics, Pittsburgh, PA, USA) or 9-(methylaminomethyl)anthracene (MAMA, Sigma-Aldrich) was dissolved in MEK (10 mg in 0.5 g) in a sonic bath for 10 min. The drug solution was added to 20 g PU dispersion (solid content 15 wt%). Vibration sonicator was applied to the mixture for 2.5 min (amplitude 30%, pulse [on 10 sec and off 1 sec], and frequency 20 kHz). The hybrid NPs were diluted with distilled water, keeping the drug concentration at 250 ppm.
Characterization of NPs
The hydrodynamic diameter and zeta potential of various NPs were measured by a particle analyzer (Delsa™ Nano, Beckman Coulter) which employed dynamic light scattering (DLS) to determine the size and electrophoretic light scattering to determine the zeta potential. The morphology of SPIO-PU NPs was characterized by transmission electron microscopy (TEM). Samples were dropped (8 μL) onto carbon-coated copper grids and examined by TEM (JEM-1200EX II, 80 keV; JEOL, Tokyo, Japan). The NP size obtained from TEM was based on >50 NPs in multiple images. PU NPs and hybrid NPs were cast and dried in vacuum for analyses by attenuated total reflection-infrared spectroscopy (Spectrum 100 model, Perkin Elmer). Each sample was scanned 16 times at a resolution of 2 cm−1 over the frequency range of 500-4000 cm−1. Thermal analyses were carried out by a thermogravimetric analyzer (TGA, TGA Q50; TA Instruments, New Castle, DE, USA). The thermal condition was isothermal for 10 min at 100°C and then the temperature was increased from 100°C to 800°C at a rate of 10°C/min under a nitrogen atmosphere. TGA data were used to determine the fraction of SPIO in hybrid NPs. The water swelling ratio of different PU films was evaluated by cutting the film into rectangular shape of 1×1 cm and the dry weight W0 of each film was recorded. After immersing in distilled water for 24 h at 37°C, the film was taken out and water wiped from the film surface to record the weight We. The water swelling ratio of different PU films was obtained from the equation (We−W0)/W0.
Magnetic measurement and MRI analysis
SPIO NPs and hybrid NPs were dried in vacuum and the magnetic hysteresis curves were measured at 300 K with a magnetic field between −30 and 30 kOe by a superconducting quantum interference device magnetometer (MPMS-7; Quantum Design, San Diego, CA, USA). The iron concentration of SPIO-PU NPs was quantified using atomic absorption spectroscopy (ICE 3000; Thermo Fisher Scientific, Waltham, MA, USA). Various SPIO-PU NPs were then diluted to contain 0–60 ppm of iron. A high-resolution 7 T MRI equipment (Bruker BioSpec 70/30) was employed to acquire T2-weighted images. The imaging parameters were spin echo sequence, repetitive time/echo time =2,500/33 ms, field of view =8×8 cm, thickness =1 mm, slice sep =1 mm, matrix =256×256, number of excitations =3, and flip angle =180°.
Magnetic heating test
The magnetic heating test was conducted in a high-frequency oscillating magnetic field (LT-15-80, Lantech Industrial Co., Taichung, Taiwan, Republic of China). A volume of 1 mL of the dispersion of SPIO NPs or SPIO-PU3 NPs, each containing 1 wt% SPIO NPs, was heated using high-frequency oscillating magnetic field. PU3 was selected in this experiment because of its excellent biocompatibility25 and higher inorganic iron ratio after sonication with SPIO NPs. The dispersion of SPIO NPs or SPIO-PU3 NPs in a glass tube was placed in the middle of the coil and the five-turn coil generated a magnetic field (~20 kA/m). An alcohol thermometer was used to measure the temperature of the dispersion and the temperature was recorded per minute for a period of 17 min.
Drug release experiment
VK3-PU NPs or MAMA-PU NPs were diluted to 250 ppm drug concentration with distilled water. The drug release behavior in distilled water was observed at 2 different temperatures (37°C and 50°C) for VK3-PU NPs and MAMA-PU NPs. At a specific time point, the medium was collected by centrifugation (6,500 rpm) for 10 min in a centrifugal ultrafilter (50 kDa molecular weight cut-off; EMD Millipore, Billerica, MA, USA). The VK3-PU NPs or MAMA-PU NPs remained in the upper tube, and the released VK3 or MAMA that dissolved in the medium was ultrafiltered to the lower tube. The optical absorption of VK3 or MAMA in the lower tube was read by a microplate reader at 340 and 385 nm (SpectraMax® M5 Multimode Plate Reader, Molecular Devices, Sunnyvale, CA, USA). Using the calibration curve of optical density versus drug concentration, the amount of drug in the medium was obtained. The drug loading content (DLC) and drug entrapment efficiency (DEE %) were defined as the initial amount of encapsulated drug divided by the total carrier or the total drug added (Equations 1 and 2). The amount of encapsulated drug was determined by subtracting the remaining drug in water from the total amount of drug. The percent drug release was calculated based on the amount of drug released over that of initially encapsulated drug.
|
|
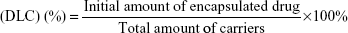
|
|
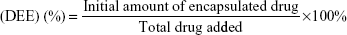
Preparation of MAMA-SPIO-PU NPs by ultrasonic vibration
First, the SPIO-PU NPs prepared from SPIO-PU solution were purified by magnet attraction to remove blank PU NPs (without SPIO NPs) and the SPIO-PU NPs were redispersed in distilled de-ionized water. Here, we chose PU3 as the basic material because of its excellent biodegradability and high inorganic iron ratio after sonication with SPIO NPs. MAMA was dissolved in MEK (1 mg in 0.3 g) in a sonic bath for 10 min. MAMA solution was added to 20 g of purified SPIO-PU3 NPs solution. The vibration sonicator was applied to the mixture for 2.5 min (amplitude 30%, pulse [on 10 sec and off 1 sec], and frequency 20 kHz) to produce MAMA-SPIO-PU3 NPs.
Cell cytotoxicity and cellular uptake of SPIO-PU NPs
Lung cancer cells (A549, human lung adenocarcinoma epithelial cell line) were cultured in Roswell Park Memorial Institute 1640 medium supplemented with 10% fetal bovine serum in an incubator maintained at 37°C in the presence of 5% CO2. Fibroblasts (L929, mouse dermal fibroblast cell line) were cultured in Dulbecco’s Modified Eagle’s Medium-low glucose supplemented with 10% fetal bovine serum. To conduct the cytotoxicity experiment, A549 cells and L929 cells were seeded at a density of 1.5×104 cells per well in 96-well culture plates and incubated for 12 h in the incubator for cell adhesion. MAMA-SPIO-PU3 NPs were diluted with culture medium at different iron concentrations (25–400 ppm) and incubated with A549 or L929 cells for 24 h. Phosphate-buffered saline (PBS) was used to wash the cells 3 times to remove free NPs. The cytotoxicity of MAMA-SPIO-PU3 NPs was determined by 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. MTT was prepared in PBS solution at 0.5 mg/L concentration. A volume of 75 μL MTT solution was added to each well and allowed to react for 4 h in the incubator. The supernatant solution was removed and then, dimethyl sulfoxide (150 μL) was added to dissolve the formazan crystals. A multimode plate reader (SpectraMax M5) was used to read the absorbance of each well at 550 nm for cell viability analysis. For the cellular uptake study, A549 cells and L929 cells were seeded at a density of 5×104 cells per well in 24-well culture plates and cultured for 12 h. The culture medium was replaced with that containing MAMA-SPIO-PU3 NPs with different iron concentrations and incubated for another 24 h. After the treatment, the cells were washed with PBS for 3 times to remove extra NPs. Prussian blue staining was used to identify the presence of SPIO inside the cells. Before staining, the cells were fixed by 2.5% glutaraldehyde solution for about 15 min and washed with PBS for 3 times before the Prussian blue agent was added for 1 h. The stained cells were observed with an inverted microscope (Leica DMIRB; Leica Microsystems, Wetzlar, Germany).
Cell hyperthermia experiment
The in vitro cell hyperthermia study was conducted by following the method reported in literature with some modifications.26 The suspended A549 cells (1×106) were centrifuged at 1,500 rpm for 5 min to obtain the cell pellet. A volume of 1 mL SPIO-PU3 NPs (0.4 wt% SPIO NPs) was used to resuspend the cell pellet. On the other hand, cells of the same number were treated with 1 mL of fresh medium as a control group. The sterile tubes containing SPIO-PU3 NPs were placed in the middle of the coil which generated a magnetic field of 20 kA/m for 40 min. After the magnetic heating, the cells was washed with PBS and centrifuged for 2 times to remove SPIO-PU3 NPs. The groups included cells with SPIO-PU3 NPs and magnetic field, cells with SPIO-PU3 NPs only, and cells with the fresh medium only. To assess the cell viability, 1×104 cells per well were seeded in 96-well culture plates and incubated for 24 h for evaluation by the MTT assay.
Statistical analysis
Experimental results are expressed as average ± standard deviation (n≥3). Statistical differences were analyzed by Student’s t-test. Data were considered statistically significant when P<0.05.
Results
Size and zeta potential of NPs
The hydrodynamic sizes and zeta potentials of various SPIO-PU NPs prepared by the ultrasonic vibration are shown in Table 1. The average sizes after vibration for 3 min were 140.4±8.45, 117.9±0.7, and 111.5±3.61 nm for SPIO-PU1, SPIO-PU2, and SPIO-PU3, respectively. The sizes of SPIO-PU NPs prepared by sonication for only 20 sec were 119.33±17.17, 135.31±10.39, and 66 nm for SPIO-PU1, SPIO-PU2, and SPIO-PU3, respectively. These sizes were not very different from those of SPIO-PU NPs prepared by sonication for 3 min, except for SPIO-PU3. The zeta potentials of all SPIO hybrid NPs were in the range of −30 to −40 mV, which was less negative than the pristine PU NPs (−55 mV).
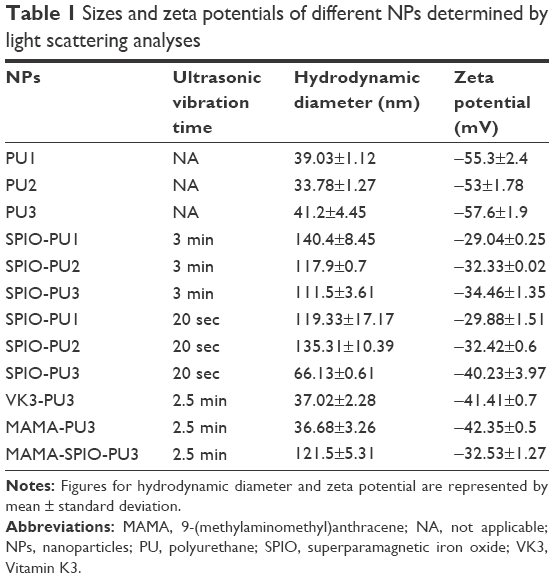 | Table 1 Sizes and zeta potentials of different NPs determined by light scattering analyses |
The TEM images of various SPIO-PU NPs are shown in Figure 2. SPIO-PU NPs demonstrated an irregular shape and appeared to be aggregated from smaller clusters. The black dots in the images are SPIO NPs, while the gray spots are PU. The original PU NPs seemed to be broken into small clusters. SPIO and smaller PU clusters were loosely assembled. The size defined from TEM images was about 100 nm. SPIO-PU NPs produced by sonication for 20 sec looked more homogeneous in appearance, but still irregular in shape.
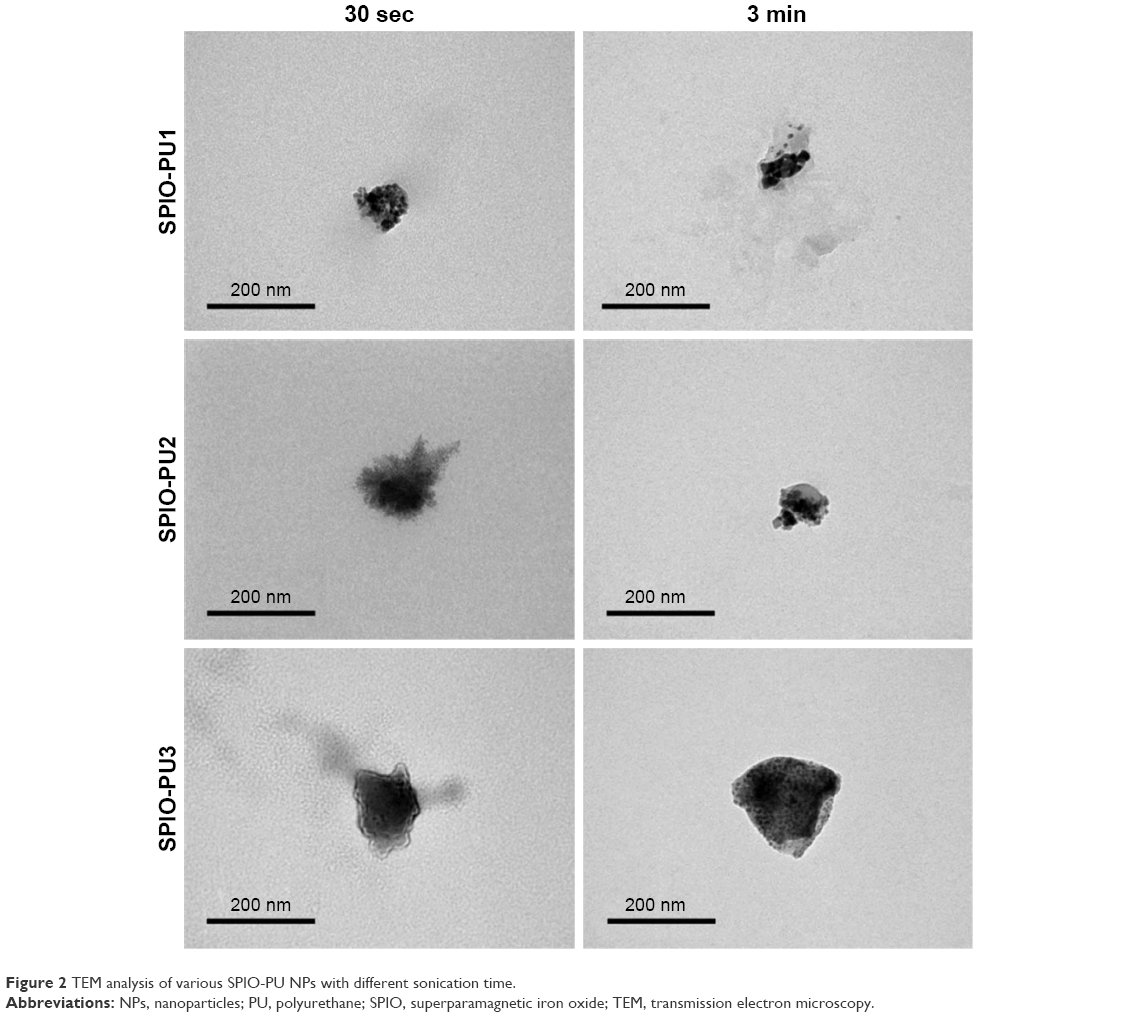 | Figure 2 TEM analysis of various SPIO-PU NPs with different sonication time. |
Physicochemical characterization of SPIO-PU NPs
Attenuated total reflection-infrared spectroscopy spectra for SPIO-PU NPs are shown in Figure 3A. The original PU NPs demonstrated absorption peaks at about 3,373 cm−1 (N–H stretching), 2,947 cm−1 (C–H3), 2,867 cm−1 (C–H2), 1,729 cm−1 (C=O), 1,556 cm−1 (N–H bending), and 1,236 cm−1 (C–O). The SPIO NPs had a peak at 560 cm−1 (Fe–O). Various SPIO-PU NPs showed the peaks of PU as well as the peak of Fe–O at 560 cm−1, indicating that SPIO NPs were encapsulated in PU by the ultrasonic vibration method. The N–H stretching absorption peak shifted to a lower wavenumber in various SPIO-PU NPs, suggesting the increase in bonded N–H as a result of the interaction between N–H and SPIO. This shift also occurred for N–H bending (1,556 cm−1 for PU). The extent of peak shift associated with N–H for various PU NPs is summarized in Table 2.
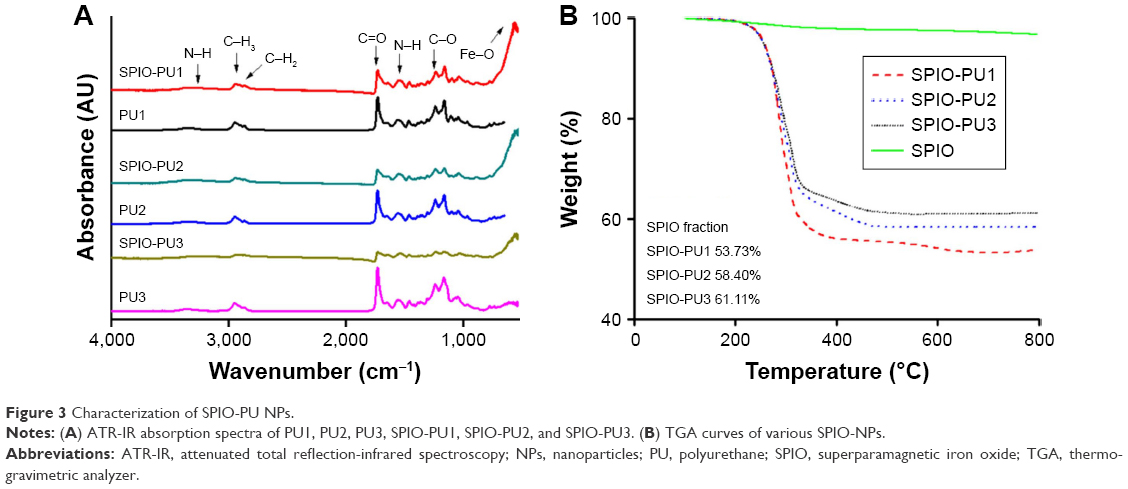 | Figure 3 Characterization of SPIO-PU NPs. |
 | Table 2 The extent of N–H stretching peak shift of SPIO-PU NPs |
TGA curves of SPIO-PU NPs are shown in Figure 3B. Based on the curve, the organic/inorganic fractions of hybrid NPs could be obtained. The weight fraction of SPIO in the SPIO-PU NPs was 53.73%, 58.40%, and 61.11% for SPIO-PU1, SPIO-PU2 and SPIO-PU3, respectively.
The magnetic properties of SPIO NPs, SPIO-PU1 NPs, SPIO-PU2, and SPIO-PU3 NPs measured between −30 and 30 kOe are shown in Figure 4A. The saturation magnetization (Ms) values of SPIO, SPIO-PU1, SPIO-PU2, and SPIO-PU3 were 60.81, 69.96, 63.58, and 65.57 emu/g, respectively. There was no hysteretic effect in the curves. The Ms values of SPIO-PU NPs were very close to those of pure SPIO NPs and even slightly higher. This indicated that SPIO-PU NPs prepared by the new encapsulation method retained the superparamagnetic property of SPIO NPs. The T2-weighted images of SPIO-PU NPs are shown in Figure 4B. The magnetic resonance (MR) image turned darker as the iron concentration increased. SPIO-PU NPs retained the ability to enhance the MR signal and still functioned as imaging NPs.
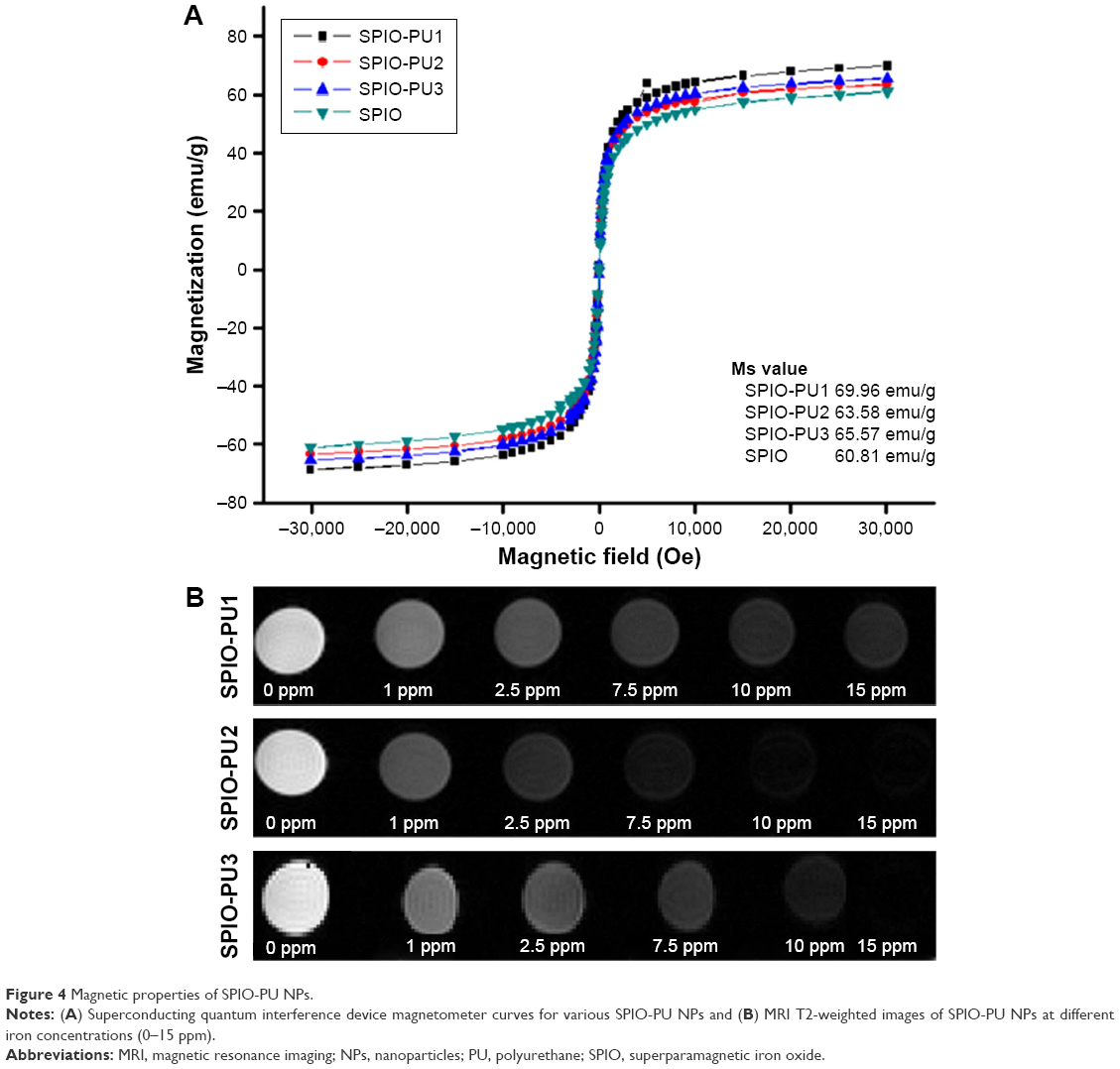 | Figure 4 Magnetic properties of SPIO-PU NPs. |
The magnetic heating curves of SPIO NPs and SPIO-PU3 NPs containing 1 wt% SPIO NPs in the aqueous dispersion are shown in Figure 5. In 3 min, the temperature of SPIO-PU3 NPs dispersion increased from 34°C to 41.83°C, while that of SPIO NPs dispersion increased only to 38.83°C. After 15 min, the temperature of SPIO-PU3 NPs dispersion increased to 47.86°C, while that of SPIO NP dispersion increased to 42.66°C.
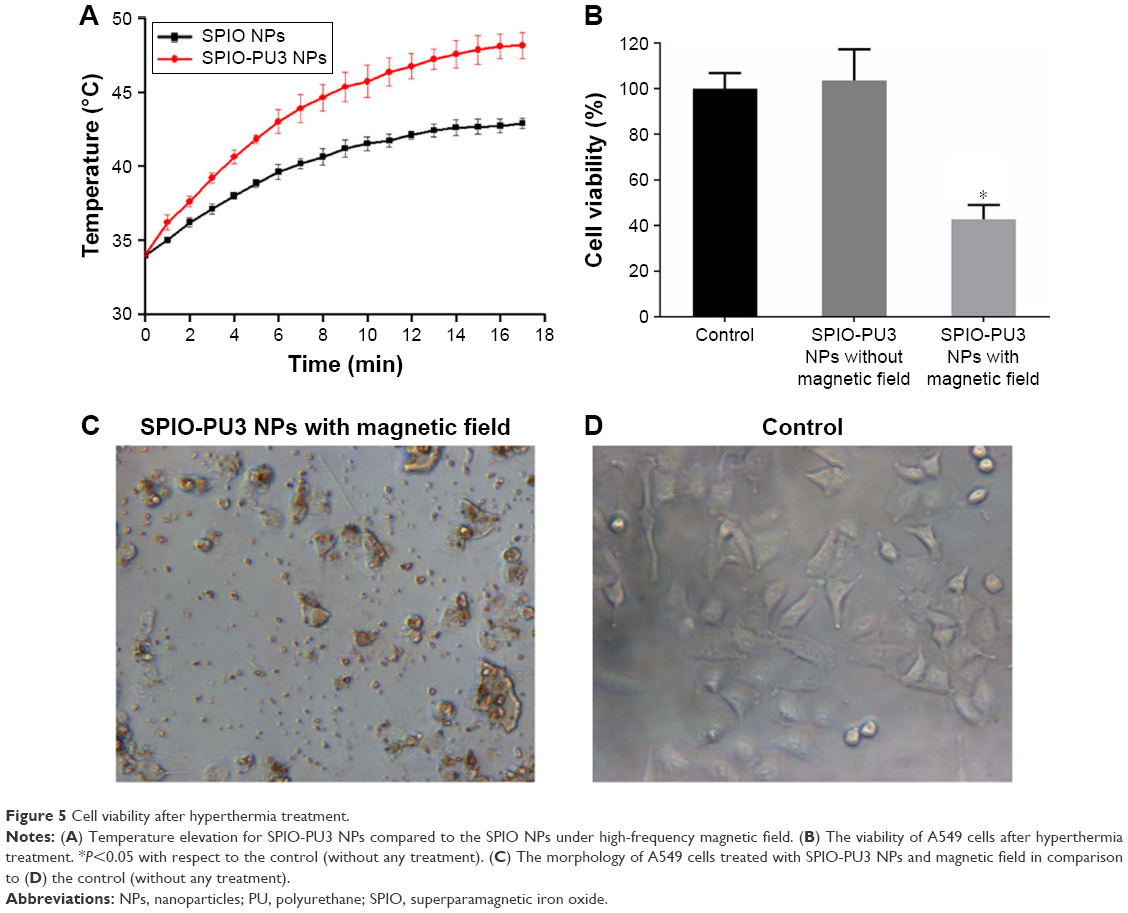 | Figure 5 Cell viability after hyperthermia treatment. |
Drug release
The VK3-PU NPs were diluted by distilled water to contain 250 ppm of VK3. The DLC and DEE of different drug-PU NPs are shown in Table 3. The DLC of VK3-PU1, VK3-PU2, and VK3-PU3 was about 0.32% (0.32 g drug/100 g PU). The measured DEE valuse of VK3-PU1, VK3-PU2, and VK3-PU3 were 96.36%±0.30%, 96.21%±0.19%, and 95.51%±0.25%, respectively. The drug release profiles from VK3-PU NPs are shown in Figure 6. At 37°C, the release ratios of VK3-PU1, VK3-PU2, and VK3-PU3 were still <20% in 5 days and the release from VK3-PU1 was faster than from VK3-PU2 and VK3-PU3. The percent drug release for VK3-PU1, VK3-PU2, and VK3-PU3 at 5 days was 19.11%, 15.02%, and 5.91%, respectively (Figure 6A). The drug release rate at 50°C was higher than that at 37°C. The cumulative release within 2 days at 50°C was beyond that within 5 days at 37°C, for each type of NPs. The percent drug release from VK3-PU1 at 50°C was over 20% after 2 days (Figure 6B). At either 37°C or 50°C, VK3-PU1 had the most rapid drug release.
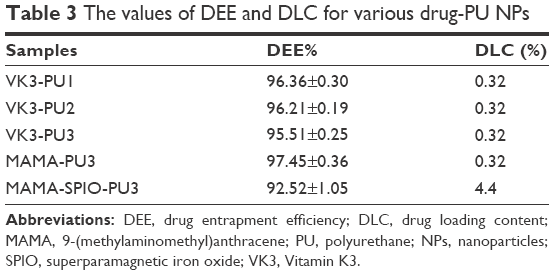 | Table 3 The values of DEE and DLC for various drug-PU NPs |
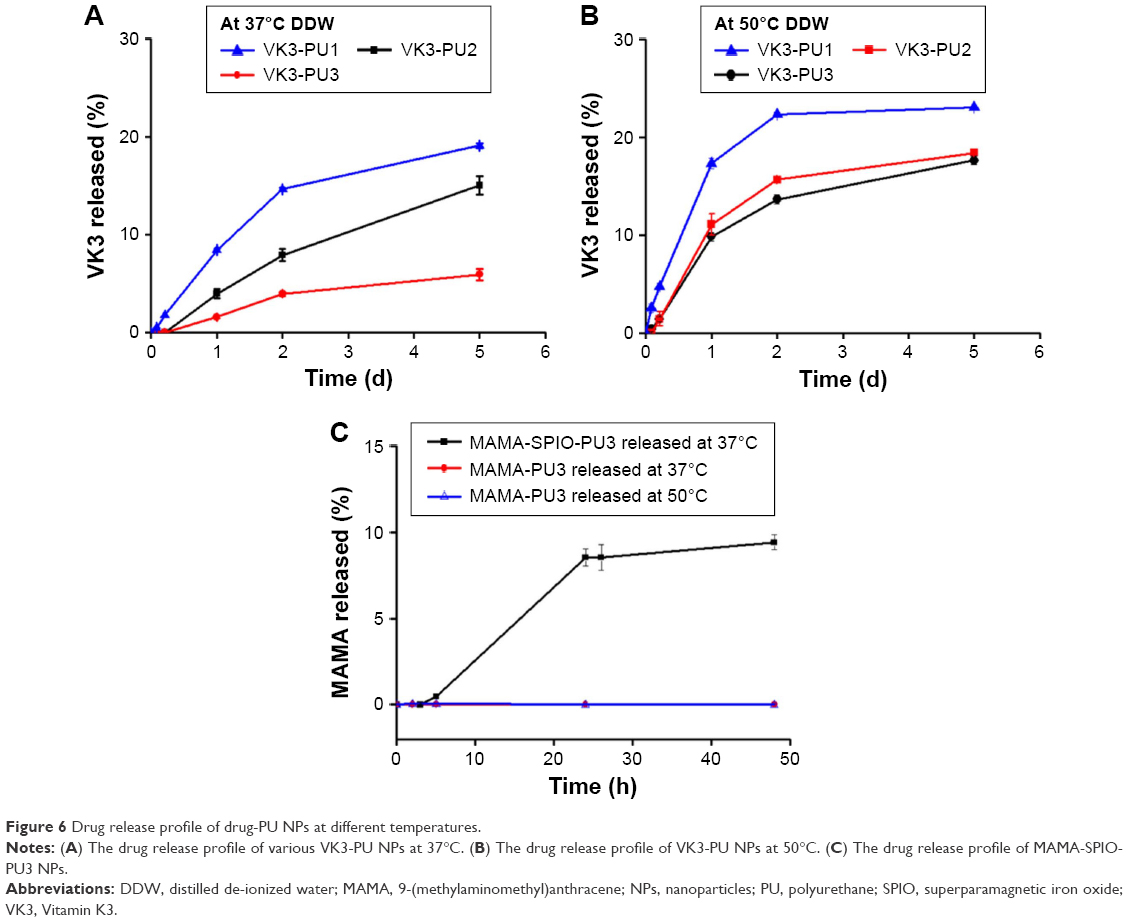 | Figure 6 Drug release profile of drug-PU NPs at different temperatures. |
PU films were obtained by casting PU NPs emulsion at room temperature. The water swelling ratios of PU films are shown in Table 4 and were used to correlate with drug release. The water swelling ratios of PU1, PU2, and PU3 were 9.78%, 12.79%, and 14.65%, respectively.
 | Table 4 The water swelling ratios of different PU films |
MAMA-SPIO-PU3 NPs preparation and drug release
SPIO-PU3 NPs were sonicated to encapsulate MAMA to produce MAMA-SPIO-PU3 NPs successfully. The DEE was ~93% and the DLC was ~4.4% for MAMA-SPIO-PU3 NPs. From Figure 6C, the drug release ratio obtained for MAMA-SPIO-PU3 was ~8.5% after 24 h at 37°C. If the drug MAMA only was encapsulated in PU3 NPs without SPIO, the DEE was ~99% and the DLC was 0.32%. However, the hydrophobic MAMA was so tightly encapsulated and nearly no release from MAMA-PU NPs was observed at 37°C or 50°C.
Cell cytotoxicity and cellular uptake of SPIO-PU NPs
The cell viability of A549 and L929 cells is shown in Figure 7A. MAMA-SPIO-PU3 NPs at different concentrations of iron (Fe 0–400 ppm) were used to treat cells. There was no cytotoxicity for A549 and L929 cells at low iron concentrations (50–100 ppm). When the iron concentration increased to 100 ppm, the viability decreased to about 85%–90% for L929 and A549 cells. At higher concentrations of 200 and 400 ppm, the cell viability of A549 decreased to ~74% and 50% and that of L929 decreased to ~68% and 45%, respectively. The uptake of MAMA-SPIO-PU3 NPs by A549 cells and L929 cells was confirmed by Prussian blue staining, as shown in Figure 7B, C. It is obvious that MAMA-SPIO-PU3 NPs were internalized by cells. When the iron concentration was elevated, the amount of cellular uptake also increased. At lower iron concentrations (25 ppm), the uptake of MAMA-SPIO-PU3 NPs was still observed. The morphology of A549 cells at higher concentrations (100–400 ppm) revealed some filamentous projection with irregular shape, which may suggest an abnormal or unhealthy state. At lower iron concentrations (<50 ppm), there was no obvious change in cell morphology.
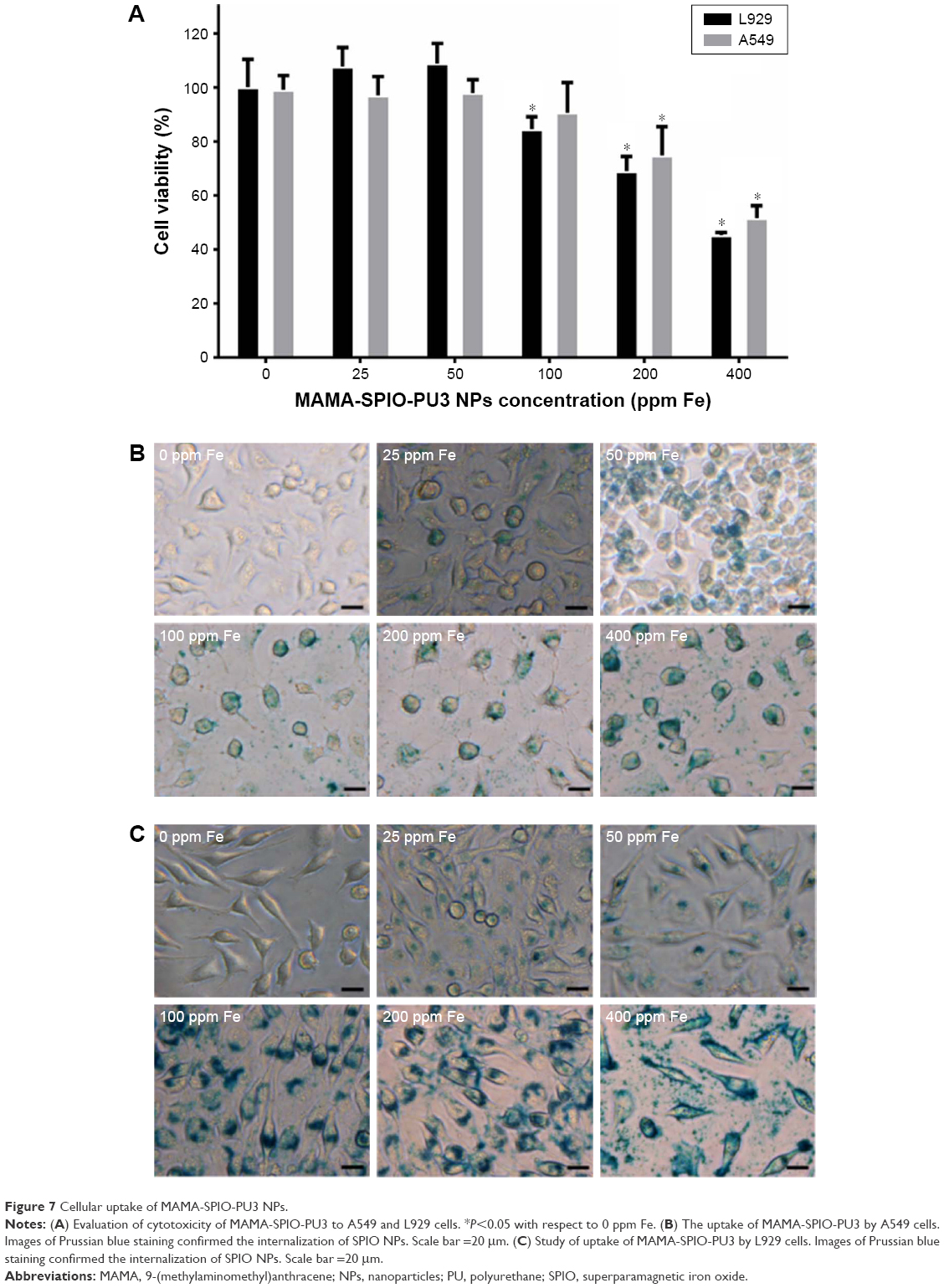 | Figure 7 Cellular uptake of MAMA-SPIO-PU3 NPs. |
Cell hyperthermia treatment
Results from cell hyperthermia studies are shown in Figure 5B. The viability of A549 cancer cells exposed to the magnetic field and SPIO-PU3 NPs was significantly reduced to 42%. The viability of A549 cells exposed to SPIO-PU3 NPs, but without magnetic heating was very close to that of control group. These results indicate that magnetic hyperthermia was effective in killing cancer cells. In addition, after the magnetic hyperthermia treatment, cells showed abnormal morphology and did not attach to the culture plate (Figure 5C).
Discussion
In this study, we used a facile sonication method for PU NPs to encapsulate hydrophobic drugs and SPIO NPs. We developed these techniques based on the characteristics of our polymer NPs (low glass transition temperature Tg). These procedures are reproducible for the current systems. DLS and electrophoretic light scattering analyses showed that the SPIO-PU NPs existed in the form of NPs and had enough capability to disperse in the solution state. Waterborne PU was a dispersion of compact NPs, each consisting of hundreds of polymer chains.27 We speculated that the elasticity25 and low Tg of PU chain segment may allow SPIO NPs to be embedded into PU NPs under ultrasonic high frequency and temperature operation (close to 60°C). Because PU NPs accommodated foreign SPIO NPs (~10 nm), the chain segment of PU NPs stretched and their zeta potential decreased, and the primary SPIO-PUs may further aggregate to reach a stable suspension state. On the basis of our suggestion, SPIO-PU NPs had a larger size and lower zeta potential than the pristine PU NPs. The speculated encapsulation mechanism is shown in Figure 8. On comparison, SPIO-PU NPs prepared using the self-assembly of PU chains during the synthesis had a slightly smaller size than the pristine PU NPs.22 For the more convenient sonication method, we considered that the high density and hard structure of SPIO NPs was a key to penetrate into PU NPs (SPIO NPs: bullets and PU NPs: matrix). On the other hand, the size of drug-PU produced by the sonication method was ~35 nm, similar to that of the original PU NPs. We proposed that the smaller molecular size of hydrophobic drugs did not destroy the structure of PU NPs and secondary aggregation did not occur. During sonication, the hydrophobic drug may be encapsulated inside the PU NPs, while the structure of PU may be slightly disrupted by ultrasound. The hydrophobic interaction may lead to smaller size of NPs.
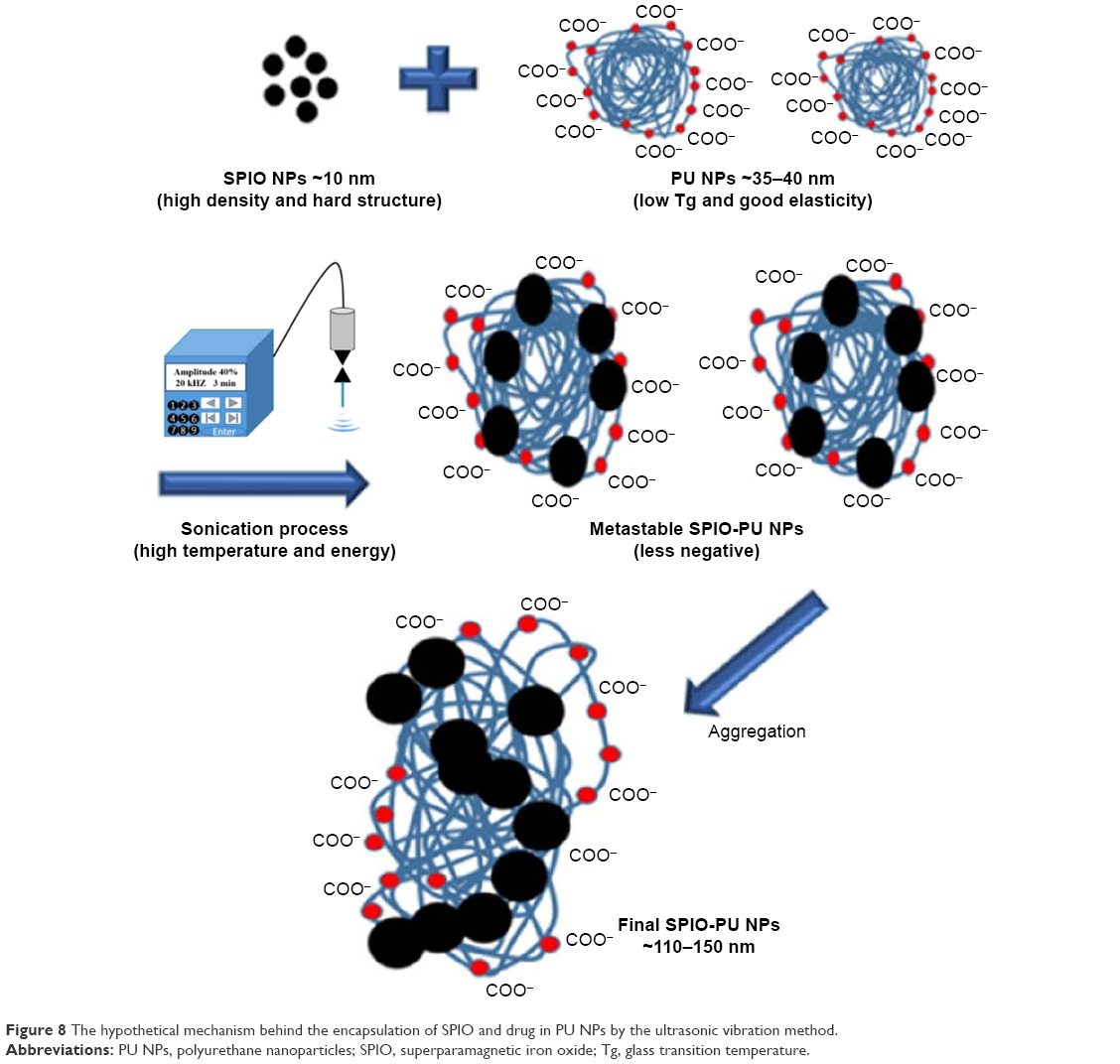 | Figure 8 The hypothetical mechanism behind the encapsulation of SPIO and drug in PU NPs by the ultrasonic vibration method. |
The images of SPIO-PU NPs as found in TEM analysis showed diverse and irregular morphology. Because of the high electron density of SPIO NPs compared to PU NPs, the black part of SPIO NPs could be distinguished from the light gray PU NPs in TEM images. Some SPIO-PU NPs were close to 200 nm by DLS size analysis. This may result from aggregation of SPIO-PU NPs when air-dried during TEM sample preparation. The shape and size polydispersity of SPIO NPs may result from the process of random and high-intensity sonication.
Compared with the original PU NPs, the absorption peak for N–H stretching and N–H bending shifted in SPIO-PU NPs. According to the literature, the N–H stretching of PU can be classified into the N–H stretching absorption of hydrogen bond (3,320 cm−1), the free (3,420 cm−1) as well as trans and cis (3,180 cm−1).28 The absorption peak of 3,370 cm−1 in PU was composed of free N–H and the hydrogen bonded N–H.29 The prepared PU1 NPs had an absorption peak at 3,373 cm−1, consistent with the literature data.29 After SPIO NPs were encapsulated in PU1 NPs by sonication, the N–H stretching absorption peak of SPIO-PU1 NPs shifted from 3,373 to 3,347 cm−1, representing more bonded N–H through the interaction of SPIO with the N–H bond of PU NPs.22 SPIO-PU2 NPs and SPIO-PU3 NPs also showed such a shift in the N–H absorption peak.
The melting point of PEBA diol is ~25°C and that of PCL diol is ~50°C.27 Therefore, the soft segment chain PEBA diol is more mobile than PCL diol and more SPIO NPs can be embedded into PU NPs under sonication operation. This may explain why SPIO-PU3 NPs composed of 60% PEBA diol in the soft segment had the highest inorganic SPIO ratio. The high ratio of SPIO NPs in SPIO-PU NPs could elaborate their special magnetic properties in MRI and magnetic heating functions.
After the SPIO NPs were encapsulated in PU NPs, they still retained nearly all of the high saturation magnetization value of SPIO NPs without hysteresis effect. The encapsulated SPIO-PU NPs showed superparamagnetic properties sufficient for applications in the biomedical field.6 In the MRI study, the prepared SPIO-PU NPs retained the ability to accelerate the decline of magnetic resonance signals to produce darker contrast images. They could serve as MRI NPs.
Various VK3-PU NPs demonstrated high DEE (~95%), that is, they effectively encapsulated hydrophobic drug by the sonication method. The hydrodynamic size of PU NPs increased from ~35 to 42 nm as the temperature increased from 37°C to 50°C.22 This temperature had already reached the softening temperature of PCL and PEBA segments, so the volume of various VK3-PU NPs expanded with faster drug release. The drug release ratios for various VK3-PU NPs at both 37°C and 50°C were somewhat unexpected. The softer and more degradable PU3 revealed a slower drug release rate. We explain this situation from the aspect of water swelling ratio of various PU NPs because degradation had not occurred at this time.25 The high water swelling ratio of PU3 NPs may introduce more water to the polymer chains and interfere with the diffusion of hydrophobic drug. At 50°C, the release rate of VK3-PU3 NPs was still lower but closer to that of VK3-PU2, compared to that at 37°C. We suggest that the molecular chain may become more mobile at the higher temperature to facilitate drug release and reduce the effect from water swelling. Based on the above analysis, the drug-PU NPs had no burst release and were sensitive to temperature.
The prepared MAMA-SPIO-PU3 NPs had good DEE (~93%) and high DLC (~0.04 g/g). MAMA itself had a highly hydrophobic structure and was encapsulated in the tight chain structure of PU NPs. In contrast to VK3, MAMA was difficult to be released from MAMA-PU3 NPs. In addition, MAMA had a secondary amine (−NH2) functional group that may interact with the high-molecular-weight PU chains to form intermolecular hydrogen bonds, making it even more difficult to be released. In case of MAMA-SPIO-PU3 NPs encapsulating MAMA and SPIO NPs simultaneously, MAMA could be more easily released from the NPs, probably because of the more stretched polymer chain and less interaction of MAMA with PU3 in the presence of SPIO. In addition, there was no significant increase in MAMA drug release after magnetic heating. The MAMA-SPIO-PU3 NPs were gathered together from a few NPs, and this slight aggregation state may increase the difficulty of drug release while it is being subjected to oscillating magnetic field.
The results of cell viability revealed that the encapsulated PU NPs had good biocompatibility. At extremely high iron concentrations (200–400 ppm), the cell viability started to decrease to 45%–50%. The biocompatibility was better than SPIO-PU NPs produced by in situ synthesis method.22 NPs of smaller size are considered to have greater toxicity to cells.30 In this study, the particle size was about 120 nm, which was larger than that produced in situ (~32 nm).22 This larger particle size may account for the greater biocompatibility. In the cellular uptake study, MAMA-SPIO-PU3 NPs were effectively taken up by cells. The effective cellular uptake without significant toxicity may make SPIO-PU NPs a suitable drug-carrying platform.
In the magnetic heating study, both SPIO NPs and SPIO-PU NPs showed the ability to elevate the temperature. Compared with SPIO NPs, SPIO-PU NPs had better magnetic heating ability and the temperature could meet the need for cell hyperthermic treatment.11 The better heating ability of SPIO-PU NPs may be attributed to the friction of the Brownian relaxation of SPIO NPs10,31 and the PU coating under the oscillating magnetic field. Moreover, the tendency of bare SPIO NPs to aggregate may restrict their magnetic heating ability.
The cell hyperthermia experiment showed that SPIO-PU NPs have good heating effect and could kill cancer cells. The cancer cells treated with SPIO-PU NPs only showed similar viability and cell morphology to the control group, agreeing with the literature.26 This result supports that cancer cells were killed by magnetic heating but not by the NPs, and that SPIO-PU NPs may serve as a viable magnetic hyperthermia carrier.
PU-based drug delivery systems have been reported recently.32–34 These systems, either waterborne or not, contained PEG as the soft segment, which is soluble but not biodegradable. Some of the PU developed was in the form of micelles,34 which may not have long-term stability. Our system has the advantage of highly efficient loading and the process does not involve toxic solvent. Therefore, we consider that the current SPIO-PU NPs and drug-SPIO-PU NPs may have the potential to be further developed as a multimodular theranostic platform.
Conclusion
In this study, we prepared environment-friendly and biocompatible PU NPs by a waterborne process. Using the high-frequency sonication method, SPIO NPs and drugs were facilely encapsulated in PU NPs to form biomedical nanocarriers. The magnetic properties of SPIO-PU NPs retained well, and they showed the ability to perform magnetic heating and cell hyperthermia treatment effectively. The drug (VK3 or MAMA) encapsulation efficiency in drug-PU NPs was high. Various VK3-PU NPs showed a broad range of drug release rates because of different PU chemistry. Multifunctional MAMA-SPIO-PU NPs had favorable biocompatibility and cellular uptake and exhibited faster drug release rate than MAMA-PU NPs. Therefore, various NPs were successfully produced by a simple sonication method. SPIO-PU NPs may possess sufficient potential to become multimodular theranostic platform and drug-SPIO-PU NPs may be further incorporated to offer drug release function.
Acknowledgments
This research was supported by the Ministry of Science Council and Technology (103-2923-E-002-003-MY3), Taiwan, Republic of China. We are grateful to Professor Ting-Yu Liu for kindly providing the magnetic heating equipment. We are also obliged to the Precious Instrument Center of National Taiwan University and Ms Chia-Ying Chien for providing technical assistance with TEM.
Disclosure
The authors report no conflicts of interest in this work.
References
Sanvicens N, Marco MP. Multifunctional nanoparticles properties and prospects for their use in human medicine. Trends Biotechnol. 2008;26(8):425–433. | ||
Bhattacharyya S, Kudgus RA, Bhattacharya R, Mukherjee P. Inorganic nanoparticles in cancer therapy. Pharm Res. 2011;28(2):237–259. | ||
Huang HC, Barua S, Sharma G, Dey SK, Rege K. Inorganic nanoparticles for cancer imaging and therapy. J Control Release. 2011;155(3):344–357. | ||
Ahmed N, Fessi H, Elaissari A. Theranostic applications of nanoparticles in cancer. Drug Discov Today. 2012;17(17–18):928–934. | ||
Wu W, He Q, Jiang C. Magnetic iron oxide nanoparticles: synthesis and surface functionalization strategies. Nanoscale Res Lett. 2008;3(11):397–415. | ||
Gupta AK, Gupta M. Synthesis and surface engineering of iron oxide nanoparticles for biomedical applications. Biomaterials. 2005;26(18):3995–4201. | ||
Pankhurst QA, Connolly J, Jones SK, Dobson J. Applications of magnetic nanoparticles in biomedicine. J Phys D Appl Phys. 2003;36(13):R167–R181. | ||
Jain TK, Morales MA, Sahoo SK, Leslie-Pelecky DL, Labhasetwar V. Iron oxide nanoparticles for sustained delivery of anticancer agents. Mol Pharm. 2005;2(3):194–205. | ||
Yu MK, Park J, Jeong YY, Moon WK, Jon S. Integrin-targeting thermally cross-linked superparamagnetic iron oxide nanoparticles for combined cancer imaging and drug delivery. Nanotechnology. 2010;21(41):415102. | ||
Fortin JP, Wilhelm C, Servais J, Ménager C, Bacri JC, Gazeau F. Size-sorted anionic iron oxide nanomagnets as colloidal mediators for magnetic hyperthermia. J Am Chem Soc. 2007;129(9):2628–2635. | ||
Laurent S, Dutz S, Häfeli UO, Mahmoudi M. Magnetic fluid hyperthermia: focus on superparamagnetic iron oxide nanoparticles. Adv Colloid Interface Sci. 2011;166(1–2):8–23. | ||
Lam T, Pouliot P, Avti PK, Lesage F, Kakkar AK. Superparamagnetic iron oxide based nanoprobes for imaging and theranostics. Adv Colloid Interface Sci. 2013;199–200:95–113. | ||
Sun C, Sze R, Zhang M. Folic acid-PEG conjugated superparamagnetic nanoparticles for targeted cellular uptake and detection by MRI. J Biomed Mater Res Part. 2006;78(3):550–557. | ||
Laurent S, Forge D, Port M, et al. Magnetic iron oxide nanoparticles: synthesis, stabilization, vectorization, physicochemical characterizations, and biological applications. Chem Rev. 2008;108(6):2064–2110. | ||
Zhang Y, Kohler N, Zhang M. Surface modification of superparamagnetic magnetite nanoparticles and their intracellular uptake. Biomaterials. 2002;23(7):1553–1561. | ||
Zhu L, Wang D, Wei X, et al. Multifunctional pH-sensitive superparamagnetic iron-oxide nanocomposites for targeted drug delivery and MR imaging. J Control Release. 2013;169(3):228–238. | ||
Zhang J, Misra RD. Magnetic drug-targeting carrier encapsulated with thermosensitive smart polymer: core-shell nanoparticle carrier and drug release response. Acta Biomater. 2007;3(6):838–850. | ||
Guelcher SA, Gallagher KM, Didier JE, et al. Synthesis of biocompatible segmented polyurethanes from aliphatic diisocyanates and diurea diol chain extenders. Acta Biomater. 2005;1(4):471–484. | ||
Tong F, Chen X, Chen L, et al. Preparation, blood compatibility and anticoagulant effect of heparin-loaded polyurethane microspheres. J Mater Chem B. 2013;1(4):447–453. | ||
Yang CH, Yang HJ, Wen TC, Wu MS, Chang JS. Mixture design approaches to IPDI–H6XDI–XDI ternary diisocyanate based waterborne polyurethanes. Polymer. 1999;40(4):871–885. | ||
Kuan HC, Ma CCM, Chang WP, Yuen SM, Wu HH, Lee TM. Synthesis, thermal, mechanical and rheological properties of multiwall carbon nanotube/waterborne polyurethane nanocomposite. Compos Sci Technol. 2005;65(11):1703–1710. | ||
Chen YP, Hsu SH. Preparation and characterization of novel water-based biodegradable polyurethane nanoparticles encapsulating superparamagnetic iron oxide and hydrophobic drugs. J Mater Chem B. 2014;2(21):3391–3401. | ||
Zhang J, Wu M, Yang J, Wu Q, Jin Z. Anionic poly (lactic acid)-polyurethane micelles as potential biodegradable drug delivery carriers. Colloids Surf A. 2009;337(1):200–204. | ||
Hsu SH, Ho TT, Tseng TC. Nanoparticle uptake and gene transfer efficiency for MSCs on chitosan and chitosan-hyaluronan substrates. Biomaterials. 2012;33(14):3639–3650. | ||
Hsu SH, Hung KC, Lin YY, et al. Water-based synthesis and processing of novel biodegradable elastomers for medical applications. J Mater Chem B. 2014;2(31):5083–5092. | ||
Chandrasekharan P, Maity D, Yong CX, Chuang KH, Ding J, Feng SS. Vitamin E (D-alpha-tocopheryl-co-poly(ethylene glycol) 1000 succinate) micelles-superparamagnetic iron oxide nanoparticles for enhanced thermotherapy and MRI. Biomaterials. 2011;32(24):5663–5672. | ||
Ou CW, Su CH, Jeng US, Hsu SH. Characterization of biodegradable polyurethane nanoparticles and thermally induced self-assembly in water dispersion. ACS Appl Mater Interfaces. 2014;6(8):5685–5694. | ||
Srichatrapimuk VW, Cooper SL. Infrared thermal analysis of polyurethane block polymers. J Macromol Sci, Part B Phys. 1978;15(2):267–311. | ||
Zhang S, Lv H, Zhang H, Wang B, Xu Y. Waterborne polyurethanes: spectroscopy and stability of emulsions. J Appl Polym Sci. 2006;101(1):597–602. | ||
Mahmoudi M, Simchi A, Milani AS, Stroeve P. Cell toxicity of superparamagnetic iron oxide nanoparticles. J Colloid Interface Sci. 2009;336(2):510–518. | ||
Liu TY, Hu SH, Liu DM, Chen SY, Chen IW. Biomedical nanoparticle carriers with combined thermal and magnetic responses. Nano Today. 2009;4(1):52–65. | ||
Yang J, Chen H, Yuan Y, Sarkar D, Zheng J. Synthesis and characterization of biocompatible polyurethanes for controlled release of hydrophobic and hydrophilic drugs. Front Chem Sci Eng. 2014;8(4):498–510. | ||
Ganivada MN, Rao NV, Dinda H, Kumar P, Sarma JD, Shunmugam R. Biodegradable magnetic nanocarrier for stimuli responsive drug release. Macromolecules. 2014;47(8):2703–2711. | ||
Yao Y, Xu H, Liu C, et al. Biodegradable multi-blocked polyurethane micelles for intracellular drug delivery: the effect of disulfide location on the drug release profile. RSC Adv. 2016;6(11):9082–9089. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.