Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 21
A Cross-Tissue Transcriptome Association Study Revealed Novel Susceptibility Genes for Chronic Obstructive Pulmonary Disease
Authors Luan H ![]() , Wang T, Su X, Zhao J, Yuan Z, Ding S, Yang Y
, Wang T, Su X, Zhao J, Yuan Z, Ding S, Yang Y
Received 19 November 2025
Accepted for publication 13 February 2026
Published 7 March 2026 Volume 2026:21 578900
DOI https://doi.org/10.2147/COPD.S578900
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
Hao Luan,1,* Tianhua Wang,2,* Xinjie Su,3 Junde Zhao,1 Zi Yuan,3 Shengkai Ding,1 Yikun Yang1
1Innovation Institute of Chinese Medicine and Pharmacy, Shandong University of Traditional Chinese Medicine, Jinan, Shandong, People’s Republic of China; 2Faculty of Chinese Medicine, Macau University of Science and Technology, Macau SAR, People’s Republic of China; 3College of Traditional Chinese Medicine, Shandong University of Traditional Chinese Medicine, Jinan, Shandong, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yikun Yang, Innovation Institute of Chinese Medicine and Pharmacy, Shandong University of Traditional Chinese Medicine, Jinan, People’s Republic of China, Email [email protected]
Background: Chronic obstructive pulmonary disease (COPD) is characterized by persistent airflow limitation, with its pathogenesis remaining elusive. Genetic factors are recognized as crucial factors in the pathogenesis of COPD.
Methods: This study employed transcriptome-wide association studies (TWAS) and single cell transcriptome analysis to identify predisposition genes and potential mechanisms for COPD. Cross-tissue TWAS analysis was performed using the FinnGen R10 database and sCCA weights built from the GTEx v8. Single-tissue and single-cell validations were conducted using a FUSION method based on functional summaries. SMR and colocalization analysis were carried out. C57BL/6 mice and Beas-2b cells were exposed to smoke to simulate COPD inflammation and verify the function of the genes. The regulatory effect of genes was verified by overexpression of plasmid and siRNA. Eventually, single-cell transcriptomics was conducted to investigate the expression of susceptibility genes in lung tissue cells, while GeneMANIA analysis enhanced our insights into the functional significance of these genes.
Results: A total of 125 susceptibility genes associated with COPD were identified by cross-tissue TWAS analysis. Single-tissue and single-cell TWAS, along with MAGMA validation, revealed two novel susceptibility genes, DNAJA4 and IREB2. SMR and colocalization analysis further confirmed these findings. Both mouse and cell experiments can prove that the occurrence of COPD is related to two genes. Both genes exhibit specific cell type enrichment in the lung tissue of COPD patients. The GeneMANIA analysis revealed that DNAJA4 and IREB2 potentially influence COPD risk by regulating protein folding and modification and metabolic processes, respectively.
Conclusion: We identified two novel susceptibility genes (DNAJA4 and IREB2) that are causally associated with COPD. These results provide a new perspective on the genetics of COPD.
Keywords: COPD, susceptibility gene, cross-tissue TWAS, single-cell transcriptomics
Introduction
Chronic obstructive pulmonary disease (COPD) is a chronic respiratory condition characterized by persistent respiratory symptoms and limited airflow, leading to irreversible lung tissue damage as the disease progresses.1 Statistically, COPD ranks as one of the four major chronic diseases, places fifth in terms of global disease economic burden, and is the third leading cause of death worldwide.2,3 The pathogenesis of COPD involves oxidative stress, proteinase-antiproteinase imbalance, increased inflammatory cells and mediators, and pulmonary fibrosis.4 This disease results from the combined effects of environmental (particularly cigarette smoke) and genetic factors.5 Nevertheless, the progression of COPD among smokers demonstrates substantial heterogeneity, as merely 25% of long-term smokers advance to COPD. This implies that genetic factors might exert a significant impact on the susceptibility to COPD.6
Previous studies have shown that individuals with a family history of COPD have a 2.7-fold higher risk of developing the disease.7 Based on twin studies, the estimated heritability of COPD is approximately 60%, which reflects the proportion of phenotypic variation attributable to genetic factors.8 Another genetic force estimation algorithm based on population samples with low but non-zero correlation levels demonstrated that the genetic force of forced expiratory volume in the first second (FEV1) was approximately 40%.9 With the advancement of genome-wide association studies (GWAS), research has identified single nucleotide variants (SNPs) significantly associated with COPD susceptibility, pinpointing regions of interest near or within target genes.10 Major candidate genes linked to COPD identified through gene expression analysis include HHIP, FAM13A, DSP, AGER, and TGF-β.11–13 The discovery of these genes can provide new target support for COPD drug development, prediction of acute exacerbation and construction of COPD animal models.
While GWAS plays a pivotal role in broadly identifying genetic risk loci, its focus on SNPs without considering gene function often fails to provide mechanistic insights. Transcriptome-wide association studies (TWAS) integrate expression quantitative trait loci (eQTLs) with aggregated GWAS statistical data to effectively screen candidate genes and investigate gene-trait associations.14 Besides, compared to single-tissue TWAS, the cross-tissue TWAS method enables gene-level association analysis across multiple tissues, facilitating the discovery of shared eQTL effects while retaining strong tissue-specific effects.15 Currently, TWAS has been widely applied in the identification of candidate susceptibility genes for complex multisystem diseases, such as neurodegenerative diseases and rheumatoid arthritis.16,17
In this study, we combined COPD GWAS data from FinnGen R10 with eQTL data from the Genotype-Tissue Expression (GTEx) v8 project to conduct both cross-tissue and single-layer (whole blood, tissue, and cell) TWAS analyses. Functional Summary-based Imputation (FUSION) was employed to evaluate associations in each tissue. Additionally, Multi-marker Analysis of Genomic Annotation (MAGMA) was utilized for validation. Subsequently, we performed Mendelian randomization (SMR), colocalization analysis, and independence analysis of candidate genes, followed by bioinformatics analysis to explore their biological characteristics. Consequently, this study aims to systematically identify and validate novel susceptibility genes for COPD through an integrated analytical framework, providing novel research perspectives on the genetic mechanisms of COPD and laying a research foundation for its prevention and treatment.
Method
The workflow diagram of susceptibility gene screening in this study is shown in Figure 1.
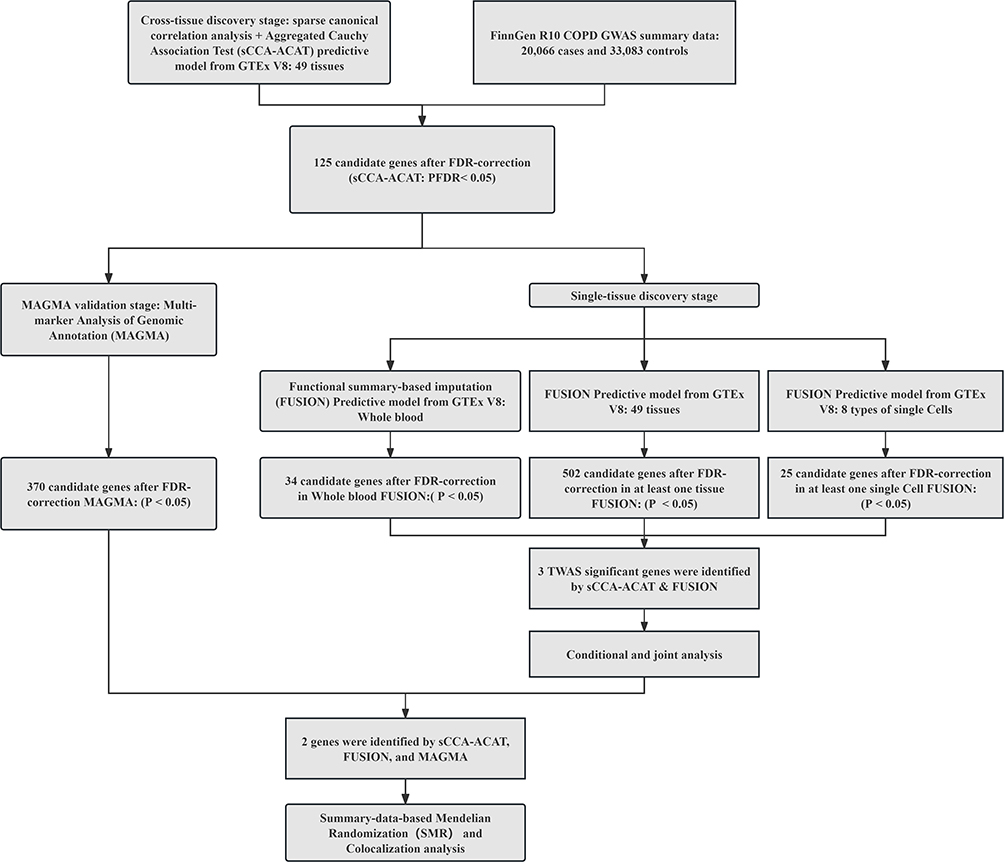 |
Figure 1 The workflow diagram of susceptibility gene screening in this study. Abbreviations: GWAS, genome-wide association; GTEx, Genotype-Tissues Expression Project; TWAS, transcriptome-wide association studies; FUSION, functional summary-based imputation; MAGMA, multi-marker Analysis of GenoMic Annotation. |
GWAS Data Source for COPD
GWAS data for COPD was retrieved from the Finn-Gen R10 dataset (https://www.finngen.fi/en). The dataset comprised 20066 COPD cases and 338303 controls of European descent. All GWAS summary data used were derived from the original studies and public databases, which had undergone corresponding ethical review and obtained informed consent from participants, with the data presented in anonymized format.
eQTL Files Source
Whole blood gene expression data was obtained from FUSION’s official website (https://gusevlab.org/projects/fusion/) and Gusev’s study, while the analysis of the SMR and the eQTL files were accessible upon the whole blood eQTLGen database.14 48 in different groups of rich gene expression data were obtained from the GTEx V8 dataset, comprising 838 samples collected post-mortem from donors (https://ftp.ebi.ac.uk/pub/data-bases/spot/eQTL/imported/GTEx_V8).18 Sample sizes for different tissues varied, ranging from 73 samples for the kidney cortex to 706 samples for skeletal muscle. The weight files for single cells came from Mike Thompson’s work.19
Cross-Tissue TWAS Analyses
We employed sparse canonical correlation analysis (sCCA) integrated with aggregated Cauchy association test (ACAT) for cross-tissue TWAS analysis. When eQTL data of genetic predictor expression had a small sample size, the results obtained by SCCA-ACAT were characterized by a higher sensitivity and lower false positive rate.20 We adopted sCCA weights built on GTEx v8 for analysis, combining the results of individual tissue TWAS tests with the results of sCCA-TWAS tests utilizing ACAT.
Single-Organization Multilevel TWAS Analysis
We initially performed TWAS analysis of whole blood, tissues, and cells from patients with COPD. In whole blood and tissues, this study utilized the FUSION tool to integrate GWAS data for COPD with eQTL data from 48 tissues in YFS (Blood) and GTEx V8 for TWAS to investigate the association of each gene with the disease. Firstly, the LD between the prediction models and the SNPs at each site of the GWAS was estimated using a European sample of 1000 genomes. FUSION then integrated several predictive models (BLUP, BSLMM, LASSO, Elastic Net, and top 1) to assess the overall impact of SNPs on gene expression weights. The model manifests the highest predictive performance for determining gene weights.21 Subsequently, we combined the genetic effects of COPD (COPD GWAS Z-score) with these genetic weights to perform TWAS for COPD. Similarly, we employed the FUSION tool to integrate COPD GWAS data with single-cell gene expression data constructed by the CONTENT method for TWAS in cells.19 Candidate genes that met the criterion of FDR < 0.05 were included in subsequent studies.
Conditional and Joint Analysis
FUSION enables us to identify multiple related features within a site and to determine which of them are conditionally independent. Therefore, we performed conditional and joint (COJO) analysis (post-processing module in FUSION) to recognize independent genetic signals.22 COJO analysis ensures a comprehensive understanding of the genetic structure of trait variation by considering LD between markers.23 Consequently, genes representing independent associations are deemed jointly significant, while those no longer exhibiting significance are considered marginally significant.
Gene Analysis
For gene analysis, we used MAGMA software with default parameters (version 1.08) to aggregate SNP level association statistics into gene scores, thereby quantifying the degree to which each gene is associated with the phenotype.24,25 The details on parameter setting and synthetic method argumentation can be referred to in the original MAGMA documentation.26
Summary-Data-Based Mendelian Randomization and Bayesian Colocalization
Summary-data-based Mendelian randomization (SMR) analysis based on aggregated data was further performed as a complementary method to verify the causal relationship between genes and COPD.27 SMR and HEIDI tests were conducted using SMR software (SMR v1.3.1). We then performed a Bayesian colocalization analysis using the “coloc” R package to determine if there were overlaps between GWAS and eQTL signals at causal variant sites.28,29 This analysis highlights the posterior probability (PP) of five relationships, denoted as PPH0 to PPH4, which quantify support for all hypotheses. Among them, PPH0 means unassociated with any trait; PPH1 connects to gene expression but not COPD risk; PPH2 is related to COPD risk but not gene expression; PPH3 links to both COPD risk and gene expression, reflecting different causal variations; PPH4 associates with COPD risk and gene expression, sharing a common causal variation. Due to the limited power of colocalization analysis, we set criterion of the analysis to genes with PPH3 + PPH4 ≥ 0.8.29
Animals and Cigarette Smoke Exposure
We used the ARRIVE checklist when writing our report.30 Eight mice each for the normal control group and the COPD group were housed in SPF environments and randomly assigned to cages of four mice each. All operations have been approved by the Laboratory Animal Ethics and Welfare Committee of Shandong University of Traditional Chinese Medicine (SDUTCM20241028001). Moreover, they strictly adhere to the relevant regulations of the “Guidelines for the Care and Use of Laboratory Animals” issued by the National Institutes of Health (NIH) and the “Implementation Measures for Laboratory Animal Management” of China. Sixteen 7-week-old male mice were obtained from Jinan Pengyue Laboratory Animal Company (SCXK (Lu) 20220006). Mice in the smoke group were exposed to 10 cigarettes a day, twice a day, five days a week for six weeks. Euthanasia was performed using a commercial animal euthanasia chamber (Model SMQ-II, Shanghai Shangli Laboratory Equipment Co., Ltd). This entire procedure, including the use of pre-anesthesia and controlled CO2 flow, was conducted in strict accordance with the American Veterinary Medical Association (AVMA) Guidelines for the Euthanasia of Animals. After mouse lung tissue was extracted, mRNA expression levels of IL-1β, TNF-α, DNAJA4 and IREB2 were detected by RNA extraction kit, reverse transcription reagent and amplification reagent (GeneCopoeia).
Cell Culture and Treatment
Beas-2b cells were obtained from Quicell Biotechnology. Beas-2b cells were cultured in DMEM (Corning), supplemented with 10% foetal bovine serum (Cytiva) and grown in room air at 37°C with 5% CO2. The cigarette smoke was drawn into a glass syringe containing 10 mL DMEM. And then DMEM was filtered and titrated to pH 7.35–7.45. This liquid was regarded as 100% concentration of CSE. When the cell confluence reached 70%, 3% CSE was added to simulate the COPD model for 24 h. DNAJA4 plasmid (GenePharma) and IREB2 siRNA (GeneChem) were transferred into Beas-2b cells by transfection agent lipo2000 for 24 h. After cell collection, mRNA expression of IL-1β and TNF-α in cells was detected by RT-PCR. The expression of IL-1β and TNF-α was detected by Elisa kit (Solarbio).
Single-Cell RNA Sequencing Data Processing and Analysis
To further investigate the differential expression of identified susceptibility genes between COPD and control groups, single-cell RNA sequencing data (GSE173896) from Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/) was processed and analyzed utilizing the “Seurat” v5 R package.31,32 Low-quality cells were filtered out by excluding cells expressing fewer than 200 genes and genes that expressed in less than three cells. Additionally, the percentage of mitochondrial genes in the cells was calculated, and only cells that expressed between 300 and 4500 genes and had mitochondrial reads less than 10% were considered for further analysis. Then, data normalization was performed using the LogNormalize function, and highly variable genes were identified with the FindVariableFeatures function. The dataset was scaled using ScaleData, followed by principal component analysis (PCA) for dimensionality reduction. Cell clustering was conducted utilizing the FindNeighbors and FindClusters functions (dim = 21, resolution = 0.5), while the batch correction was implemented via the “Harmony” R package.33 Finally, we employed the “SingleR” R package for cell type annotations. The FindAllMarkers function was utilized to locate the identified susceptibility genes among the annotated cell types, with the expression level compared between COPD and control groups.
The single-cell transcriptome data used in this study were sourced from public databases. The original research was ethically approved, and the data were anonymized.
Gene MANIA Analysis
The GeneMANIA platform (https://genemania.org/) integrates multiple genetic interactions, pathways, and co-expression datasets of target genes, as well as other gene-function relationships, to enhance understanding of the underlying biological functions of these target genes.34,35
Statistical Analysis
Data were presented as the mean ± standard deviation (SD). An unpaired Student’s t-test was employed to analyze the statistical difference between the two groups. The charts were generated using GraphPad Prism 8 software, and statistical analyses were conducted with SPSS 23.0 software. P-value less than 0.05 was considered statistically significant.
Result
TWAS Analyses in Cross-Tissue
Through sCCA-ACAT analysis, a total of 1842 genes were identified as significant (p < 0.05), with 125 remaining significant after FDR correction (Table S1).
TWAS Analysis of Blood, Tissues, and Cells
In whole blood TWAS analysis, 503 genes were identified with p < 0.05, of which 34 remained significant after FDR correction (FDR < 0.05) (Table S2). Tissue TWAS analysis revealed 8480 genes with p < 0.05, with 501 remaining significant post-FDR correction (FDR < 0.05) (Table S3). Single-cell TWAS analysis identified 2206 genes with p < 0.05, with 26 remaining significant after FDR correction (FDR < 0.05) (Table S4).
COJO Analysis
The intersection of cross-tissue and single-tissue screening yielded 13 candidate genes (Table 1), followed by COJO analysis in their respective tissues to eliminate false positives caused by LDS (Table S5). In Adipose_Subcutaneous and Nerve_Tibial, the TWAS signal of IREB2 significantly decreased when conditioned on the predicted expression of PSMA4 (Figure S1A and F). In Brain_Frontal_Cortex_BA9, the TWAS signal of DNAJA4 decreased significantly when conditioned on the predicted expression of PSMA4 (Figure S1B). Similarly, in Colon_Sigmoid, Gastroesophageal_Junction, Muscle_Skeletal, Pancreas, and Whole_Blood, modulating the predicted expression of BAG5 led to a significant reduction in the TWAS signal of XRCC3 (Figure S1C, D, E, H and I). In Ovary, the TWAS signal of BAG5 decreased significantly when conditioned on the predicted expression of XRCC3 (Figure S1G). In Whole_Blood, the TWAS signal of ORMDL3 significantly decreased when conditioned on the predicted expression of GSDMB (Figure S1J).
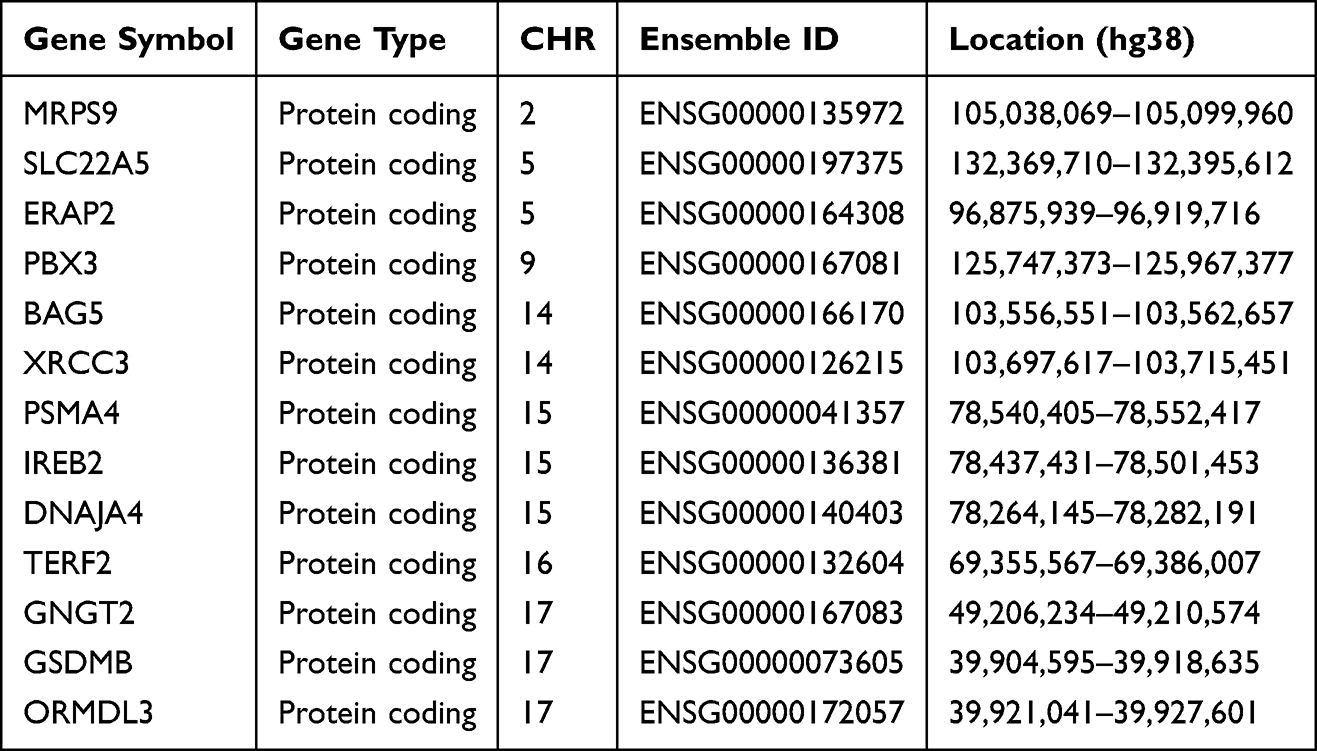 |
Table 1 Candidate Genes for COPD Risk Factors Obtained by Cross-Tissue sCCA-ACAT Analysis Overlapping with Single-Tissue Fusion Analysis |
Gene Analysis of MAGMA
MAGMA analysis identified 369 significant genes associated with COPD (FDR < 0.05) (Table S6). To enhance the robustness of our findings, we conducted cross-tissue analyses of sCCA-ACAT and single-tissue multi-level FUSION results, alongside MAGMA analysis. The results mutually verified each other and manifested two promising candidate genes (IREB2 and DNAJA4) (Figure 2 and Table S7). Their relevance is illustrated in Figure 3A.
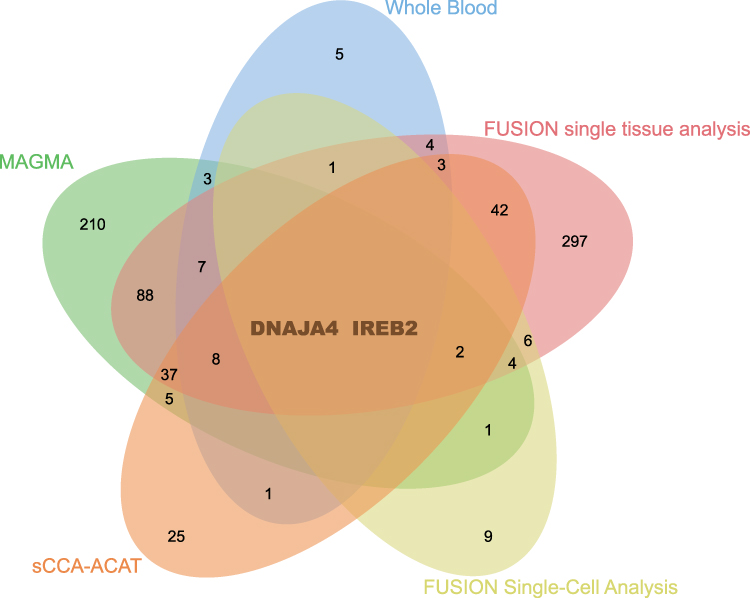 |
Figure 2 A Venn diagram to illustrate the number of significant genes identified by each method. Notes: MAGMA identified 369 significant genes related to COPD, while FUSION identified 34, 501, and 26 significant genes in blood, tissues and single cells, respectively. Additionally, sCCA-ACAT cross-tissue analysis identified 125 genes, of which two were common genes. |
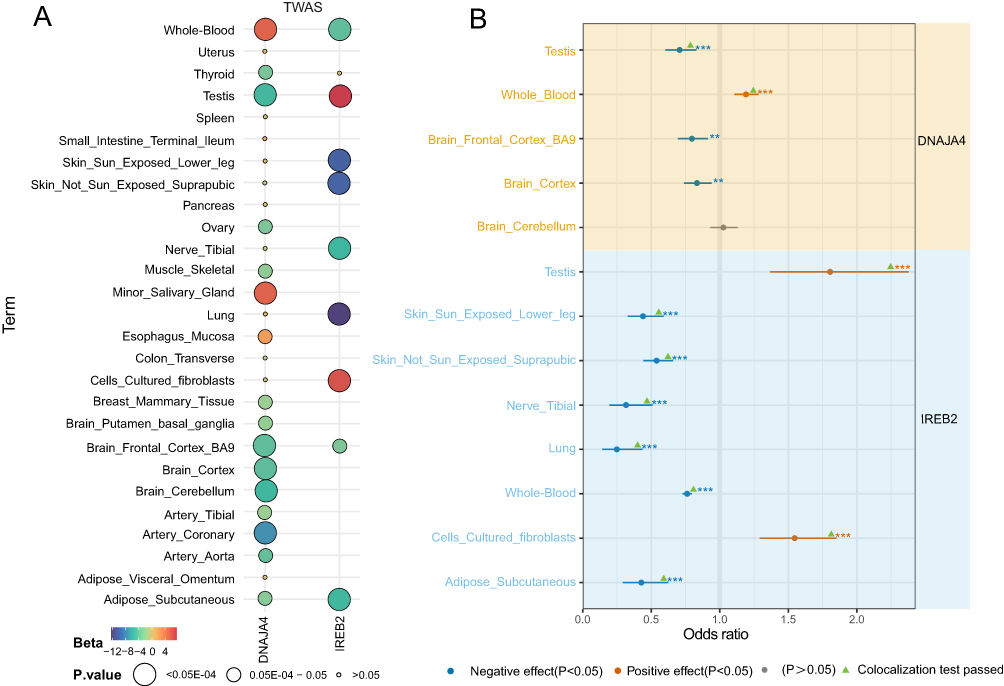 |
Figure 3 Visualization of candidate gene tissue correlation and SMR analysis results. Notes: (A) Visualization of the correlation results of the candidate genes in the tissue. (B) SMR results confirming the causal relationship between the two candidate genes and COPD. Each point represents a tissue-gene pair. Blue dots: negative effect (p < 0.05); orange dots: positive effect (p < 0.05); gray dots: non-significant association (p > 0.05). **p<0.01 and ***p < 0.001. Green triangles indicate colocalization test passed. |
SMR and Colocalization Results
The IREB2 gene, located at 15q25.1, exhibits significant expression in whole blood, tissues, and cells, confirmed by subsequent colocalization analyses in whole blood, testes, and cerebellum (Table S8). The odds ratio (OR) (95% confidence interval (CI)) for whole blood in SMR analysis is 1.19 (1.10, 1.28), while the OR for testis is 0.70 (0.60, 0.82) (Figure 3B and Table S8). The DNAJA4 gene, also located at 15q25.1, shows significant expression in whole blood, tissues, and cells. In subsequent co-localization analyses, whole blood, subcutaneous fat, lung, tibial nerve, testis, cultured fibroblasts, suprapubic skin not exposed to sunlight, and skin of lower limbs exposed to sunlight all passed the co-localization test (Table S8). In SMR analysis, DNAJA4 expression in testis and cultured fibroblasts is positively correlated with COPD, while being negatively correlated with other tissues (Figure 3B and Table S8).
DNAJA4 and IREB2 are Associated with an Inflammatory State Caused by Smoke Exposure
In order to further validate the DNAJA4 and IREB2 obtained in the preliminary screening, we applied the smoke-induced COPD mouse model and cell model for experimental verification. In smoke-exposed mice, we observed significantly elevated mRNA expression of inflammatory factors and decreased expression of DNAJA4 and IREB2 (Figure 4A and B). We further verified the role of these two genes in COPD in cell experiments. When DNAJA4 was overexpressed, the expression level of inflammatory factors induced by smoke decreased significantly (Figure 4C and D). When IREB2 was silenced, the inflammatory state caused by smoke was aggravated (Figure 4C and D).
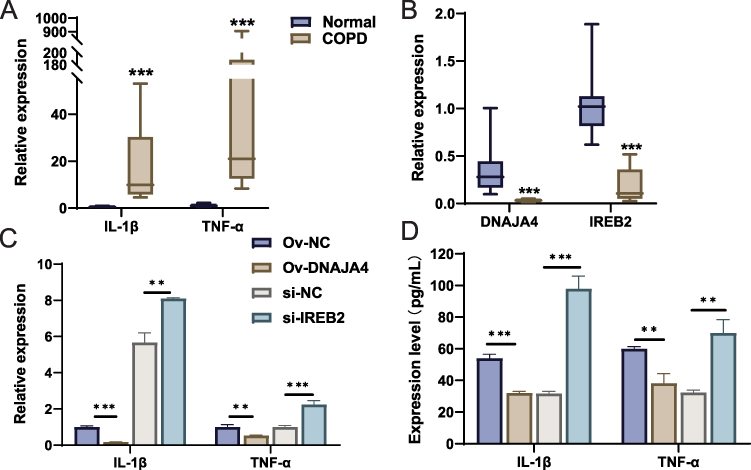 |
Figure 4 Inflammatory status and expression of DNAJA4 and IREB2 in lung tissue of smoke-exposed mice and smoke-exposed epithelial cells. Notes: (A) mRNA expression of IL-1β, TNF-α, and (B) DNAJA4, IREB2 in lung tissue of smoke-exposed mice and normal mice. (C) mRNA expression of IL-1β, TNF-α in Ov-NC, Ov-DNAJA4, si-NC, si-IREB2 cells. (D) Expression of IL-1β, TNF-α in Ov-NC, Ov-DNAJA4, si-NC, si-IREB2 cells. **p < 0.01 and ***p < 0.001. |
Cell Type-Specific Expression in Lung Tissue of COPD Patients
To explore cell type-specific enrichment of the two genes in the lung tissue of COPD patients, we performed single-cell expression analysis using RNA sequencing data from GEO.The cells were classified into 11 clusters and further divided into nine cell types: T cells, NK cells, endothelial cells, neutrophils, fibroblasts, mast cells, dendritic cells (DCs), type 2 alveolar cells (T2 alveolar cells), macrophages, club epithelial cells, smooth muscle cells, B cells, ciliated epithelial cells, and type 1 alveolar cells (T1 alveolar cells) (Figure 5A). Both genes exhibit specific cell type enrichment in the lung tissue of COPD patients (Log2FC > 0.5 and p < 0.05) (Figure 5B–D).
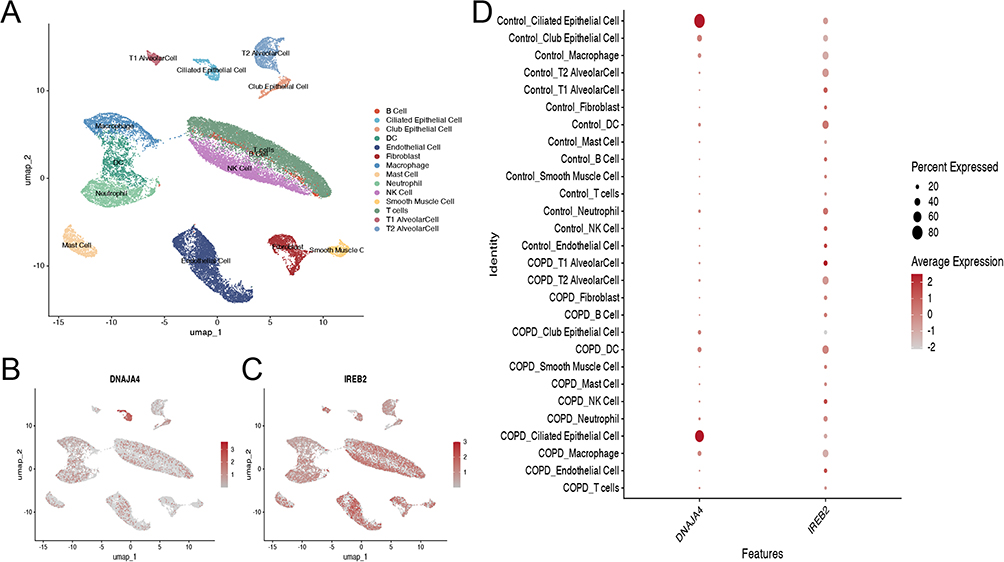 |
Figure 5 Single-cell type expression in lung tissue for DNAJA4 and IREB2. Notes: (A) A total of nine cell types were identified. (B) DNAJA4 and (C) IREB2 show the expression level in each cluster. (D) DNAJA4 and IREB2 expression in different cell clusters of COPD and control samples. |
Gene MANIA Analysis
Figure 6A depicts the potential interacting gene network constructed with IREB2 being the core. The top-ranked functional pathways enriched in IREB2-related networks include hydrogenase activity, tricarboxylic acid cycle metabolism and carbohydrate metabolism (Table S9). The gene interaction network centered around DNAJA4 is shown in Figure 6B. The highlighted functional pathways associated with DNAJA4 include protein folding, post-translational modification, and chaperone-mediated protein folding (Table S10).
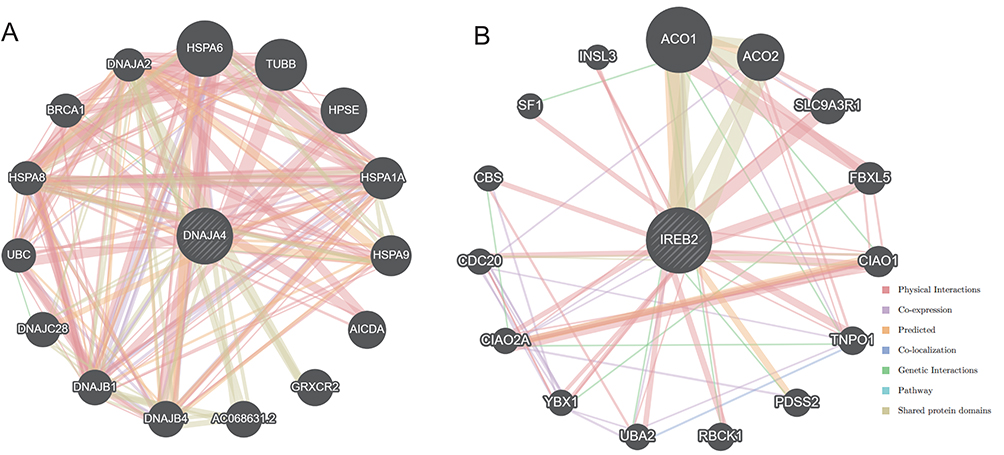 |
Figure 6 GeneMania gene network. Notes: (A) DNAJA4 as the core, and (B) IREB2 as the core. |
Discussion
Using COPD GWAS and GTEx V8 eQTL data, we systematically assessed the relationship between genetic susceptibility, gene expression, and COPD risk. Our analysis identified two significant COPD susceptibility genes, IREB2 and DNAJA4, through cross-tissue and single-tissue TWAS analysis, reinforced by MAGMA. Further confirmation was achieved through SMR and colocalization analyses. Additionally, single-cell omics analysis demonstrated distinct expression patterns of these genes across various cell types in lung tissue. Finally, gene-function relationship analysis provided deeper insights into the roles of these susceptibility genes in COPD.
With the advancement of GWAS, numerous studies have identified susceptibility genes and single nucleotide polymorphisms (SNPs) associated with COPD. Lin et al identified nine novel susceptibility sites in the Chinese population using GWAS and polygenic risk scores.36 Sun et al employed single-cell sequencing and Mendelian randomization to analyze four immune cell-associated marker genes causally linked to COPD.37 A comprehensive study compiled 129 non-overlapping COPD risk sites and 60 candidate genes from GWAS literature, identifying 12 new candidate genes on a genome-wide scale.38 Additionally, using TWAS applied to lung eQTL data from GTEx, 23 genes were replicated. Variations in results can be attributed to differences in sample sizes, sources, and the algorithms utilized in these studies. Thus, a combination of multiple approaches is essential to pinpoint potential genetic factors contributing to COPD susceptibility. Building on single-layer TWAS (whole blood, tissue, and cell), this study integrated cross-tissue TWAS analysis, updated the GTEx version, and enhanced the statistical power to identify complex shape-related genes while addressing missing associations at a single level. Notably, this study is the first to identify two genes, DNAJA4 and IREB2, associated with COPD risk through cross-tissue and single-layer TWAS analysis, validated by SMR and co-localization, with DNAJA4 being previously unreported.
DNAJA4 (DNAJ Heat Shock Protein Family Member A4) is part of the HSP40/DNAJ family and is widely expressed in various tissues, located in the cytosol and membranes.39 It initiates chaperone binding activity and unfolded protein binding, participating in biological processes such as cholesterol biosynthesis and keratinocyte migration.40,41 Diseases associated with DNAJA4 include melanoma and nasopharyngeal cancer.42 In this study, we identified, for the first time, a significant association between decreased DNAJA4 expression and an increased risk of COPD. Multiple studies and our results have linked the risk of COPD to members of the heat shock protein family. Specifically, clinical studies have shown that SNPs in HSPs correlate with a reduced risk of COPD in patients.43 Laboratory studies have found that the loss of HSP27 after epithelial-mesenchymal transformation in COPD is one of the causes of downregulation of STAT3 signaling.44 This suggests that the causal relationship between DNAJA4 and COPD may be related to its regulatory role in molecular chaperone-mediated protein folding.
IREB2 (Iron-binding regulatory protein 2) is another COPD susceptibility gene identified through GWAS. IREB2 plays a crucial role in maintaining iron balance and works with cytoplasmic iron regulatory protein (IRP) to regulate the expression of cellular iron metabolism factors.45 One study found that a specific pattern of iron deficiency, accompanied by nitrosation and oxidative stress, is linked to reduced antioxidant capacity in COPD patients.46 During acute exacerbations, the body releases numerous inflammatory mediators that can disrupt lung iron homeostasis, resulting in compensatory changes in IREB2 expression.47 Iron deficiency can lead to anemia and impaired oxygen transport, negatively affecting cell function. Conversely, excess iron promotes the formation of reactive oxygen species and lipid peroxidation, leading to cytotoxicity.48 Given IREB2’s critical role in regulating iron metabolism and its strong association with COPD risk, we hypothesize that the link between IREB2 expression and COPD risk may stem from its involvement in iron metabolism regulation.
DNAJA4 and IREB2 identification provides potential new targets and biomarkers for precision intervention of COPD. From the perspective of therapeutic targets, the molecular chaperone network associated with DNAJA4 and the iron metabolism pathway regulated by IREB2 are both well-established biological processes with pharmacological potential. The development of small-molecule modulators targeting these pathways may provide novel strategies to correct protein homeostasis imbalance and oxidative damage in COPD. Furthermore, the differential expression of these biomarkers in specific cell types of lung tissue suggests their potential value as disease subtypes or therapeutic efficacy predictors. In the future, their association with disease progression can be further validated through preclinical models, and the feasibility and safety of targeted interventions can be evaluated.
To summarize, this study identified two genes (DNAJA4 and IREB2) associated with COPD susceptibility and speculated on their distributions and potential functions based on public data. However, our research exhibits certain limitations. Firstly, our analysis relied on the FinnGen cohort of European ancestry, which may limit the generalizability of our findings to other ethnic populations; therefore, these results require validation in diverse racial groups. Additionally, experimental studies would benefit from validating our hypothesized pathophysiological mechanisms. Nevertheless, this research offers new insights into the underlying mechanisms of COPD and provides a foundational basis for its prevention and treatment.
Conclusion
In summary, TWAS analysis of blood, tissues, and cells identified two susceptibility genes, DNAJA4 and IREB2, whose down-regulation is associated with an increased risk of COPD. Subsequent in vitro and in vivo experiments further verified the crucial regulatory functions of DNAJA4 and IREB2 in the inflammatory responses of COPD induced by smoke. Additionally, we utilized bioinformatics to map the expression distribution of these genes in specific lung cell types and speculate on their biological functions, offering new insights into the genetics of the disease.
Data Sharing Statement
The data that support the findings of this study are publicly available. This sources have been supplemented in the methods section of the article.
Ethics Approval and Consent to Participate and for Publication
This study consists of two components: (1) The animal experiment segment was approved by the Laboratory Animal Ethics and Welfare Committee of Shandong University of Traditional Chinese Medicine (No.: SDUTCM20241028001). Furthermore, it is necessary to strictly adhere to the guidelines promulgated by the National Institutes of Health (NIH) and the relevant provisions of China’s “Implementation Measures for Laboratory Animal Management”. (2) The analysis segment grounded in public databases (encompassing GWAS summary data and single-cell data) employed publicly accessible anonymized data. In line with Article 32, items 1 and 2 of the “Ethical Review Measures for Life Science and Medical Research Involving Human Subjects” (2023), this secondary analysis of publicly anonymized data is exempted from repeated approval by the Institutional Review Board (IRB). The entire research process complies with the principles of the Declaration of Helsinki.
Acknowledgments
The authors would like to thank Jiachen Gao, for assistance with language editing.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work has no fund support.
Disclosure
The authors declare that they have no conflicts of interest.
References
1. Stolz D, Mkorombindo T, Schumann DM, et al. Towards the elimination of chronic obstructive pulmonary disease: a Lancet Commission. Lancet Lond Engl. 2022;400(10356):921. doi:10.1016/S0140-6736(22)01273-9
2. GBD Chronic Respiratory Disease Collaborators. Prevalence and attributable health burden of chronic respiratory diseases, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Respir Med. 2020;8(6):585–13. doi:10.1016/S2213-2600(20)30105-3
3. Chen S, Kuhn M, Prettner K, et al. The global economic burden of chronic obstructive pulmonary disease for 204 countries and territories in 2020–50: a health-augmented macroeconomic modelling study. Lancet Glob Health. 2023;11(8):e1183. doi:10.1016/S2214-109X(23)00217-6
4. Qi Y, Yan Y, Tang D, et al. Inflammatory and immune mechanisms in COPD: current status and therapeutic prospects. J Inflamm Res. 2024;17:6603–6618. doi:10.2147/JIR.S478568
5. Yang IA, Jenkins CR, Salvi SS. Chronic obstructive pulmonary disease in never-smokers: risk factors, pathogenesis, and implications for prevention and treatment. Lancet Respir Med. 2022;10(5):497–511. doi:10.1016/S2213-2600(21)00506-3
6. Løkke A, Lange P, Scharling H, Fabricius P, Vestbo J. Developing COPD: a 25 year follow up study of the general population. Thorax. 2006;61(11):935–939. doi:10.1136/thx.2006.062802
7. Hooper R, Burney P, Vollmer WM, et al. Risk factors for COPD spirometrically defined from the lower limit of normal in the BOLD project. Eur Respir J. 2012;39(6):1343–1353. doi:10.1183/09031936.00002711
8. Ingebrigtsen T, Thomsen SF, Vestbo J, et al. Genetic influences on Chronic Obstructive Pulmonary Disease - a twin study. Respir Med. 2010;104(12):1890–1895. doi:10.1016/j.rmed.2010.05.004
9. Zhou JJ, Cho MH, Castaldi PJ, Hersh CP, Silverman EK, Laird NM. Heritability of chronic obstructive pulmonary disease and related phenotypes in smokers. Am J Respir Crit Care Med. 2013;188(8):941–947. doi:10.1164/rccm.201302-0263OC
10. Silverman EK. Genetics of COPD. Annu Rev Physiol. 2020;82:413–431. doi:10.1146/annurev-physiol-021317-121224
11. Werder RB, Zhou X, Cho MH, Wilson AA. Breathing new life into the study of COPD with genes identified from genome-wide association studies. Eur Respir Rev Off J Eur Respir Soc. 2024;33(172):240019. doi:10.1183/16000617.0019-2024
12. Shrine N, Izquierdo AG, Chen J, et al. Multi-ancestry genome-wide association analyses improve resolution of genes and pathways influencing lung function and chronic obstructive pulmonary disease risk. Nat Genet. 2023;55(3):410–422. doi:10.1038/s41588-023-01314-0
13. Cho MH, Boutaoui N, Klanderman BJ, et al. Variants in FAM13A are associated with chronic obstructive pulmonary disease. Nat Genet. 2010;42(3):200–202. doi:10.1038/ng.535
14. Gamazon ER, Wheeler HE, Shah KP, et al. A gene-based association method for mapping traits using reference transcriptome data. Nat Genet. 2015;47(9):1091–1098. doi:10.1038/ng.3367
15. Ni J, Wang P, Yin KJ, et al. Novel insight into the aetiology of rheumatoid arthritis gained by a cross-tissue transcriptome-wide association study. RMD Open. 2022;8(2):e002529. doi:10.1136/rmdopen-2022-002529
16. Gu SC, Welton T, Sun Q, Wu YC, Tan EK, Zhou ZD. The integration of genome-wide and transcriptome-wide association studies in neurodegenerative diseases: opportunities, challenges, and current methodological innovations. Brief Bioinform. 2025;26(4):bbaf350. doi:10.1093/bib/bbaf350
17. Hu SY, Jiang F, Song HM, et al. Synovial transcriptome-wide association study implicates novel genes underlying rheumatoid arthritis risk. Rheumatol Oxf Engl. 2025;64(5):2515–2524. doi:10.1093/rheumatology/keae654
18. GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;45(6):580–585. doi:10.1038/ng.2653
19. Thompson M, Gordon MG, Lu A, et al. Multi-context genetic modeling of transcriptional regulation resolves novel disease loci. Nat Commun. 2022;13(1):5704. doi:10.1038/s41467-022-33212-0
20. Feng H, Mancuso N, Gusev A, et al. Leveraging expression from multiple tissues using sparse canonical correlation analysis and aggregate tests improves the power of transcriptome-wide association studies. PLoS Genet. 2021;17(4):e1008973. doi:10.1371/journal.pgen.1008973
21. Li SJ, Shi JJ, Mao CY, et al. Identifying causal genes for migraine by integrating the proteome and transcriptome. J Headache Pain. 2023;24(1):111. doi:10.1186/s10194-023-01649-3
22. Gusev A, Ko A, Shi H, et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat Genet. 2016;48(3):245–252. doi:10.1038/ng.3506
23. Liao C, Laporte AD, Spiegelman D, et al. Transcriptome-wide association study of attention deficit hyperactivity disorder identifies associated genes and phenotypes. Nat Commun. 2019;10(1):4450. doi:10.1038/s41467-019-12450-9
24. de Leeuw CA, Neale BM, Heskes T, Posthuma D. The statistical properties of gene-set analysis. Nat Rev Genet. 2016;17(6):353–364. doi:10.1038/nrg.2016.29
25. de Leeuw CA, Stringer S, Dekkers IA, Heskes T, Posthuma D. Conditional and interaction gene-set analysis reveals novel functional pathways for blood pressure. Nat Commun. 2018;9(1):3768. doi:10.1038/s41467-018-06022-6
26. de Leeuw CA, Mooij JM, Heskes T, Posthuma D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput Biol. 2015;11(4):e1004219. doi:10.1371/journal.pcbi.1004219
27. Wu Y, Zeng J, Zhang F, et al. Integrative analysis of omics summary data reveals putative mechanisms underlying complex traits. Nat Commun. 2018;9(1):918. doi:10.1038/s41467-018-03371-0
28. Zhu M, Fan J, Zhang C, et al. A cross-tissue transcriptome-wide association study identifies novel susceptibility genes for lung cancer in Chinese populations. Hum Mol Genet. 2021;30(17):1666–1676. doi:10.1093/hmg/ddab119
29. Giambartolomei C, Vukcevic D, Schadt EE, et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 2014;10(5):e1004383. doi:10.1371/journal.pgen.1004383
30. Percie du Sert N, Ahluwalia A, Alam S, et al. Reporting animal research: explanation and elaboration for the ARRIVE guidelines 2.0. PLoS Biol. 2020;18(7):e3000411. doi:10.1371/journal.pbio.3000411
31. Adams TS, Schupp JC, Poli S, et al. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci Adv. 2020;6(28):eaba1983. doi:10.1126/sciadv.aba1983
32. Hao Y, Stuart T, Kowalski MH, et al. Dictionary learning for integrative, multimodal and scalable single-cell analysis. Nat Biotechnol. 2024;42(2):293–304. doi:10.1038/s41587-023-01767-y
33. Korsunsky I, Millard N, Fan J, et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods. 2019;16(12):1289–1296. doi:10.1038/s41592-019-0619-0
34. Mostafavi S, Ray D, Warde-Farley D, Grouios C, Morris Q. GeneMANIA: a real-time multiple association network integration algorithm for predicting gene function. Genome Biol. 2008;9(Suppl 1):S4. doi:10.1186/gb-2008-9-s1-s4
35. Warde-Farley D, Donaldson SL, Comes O, et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010;38(Web Server issue):W214. doi:10.1093/nar/gkq537
36. Lin WD, Liao WL, Chen WC, Liu TY, Chen YC, Tsai FJ. Genome-wide association study identifies novel susceptible loci and evaluation of polygenic risk score for chronic obstructive pulmonary disease in a Taiwanese population. BMC Genomics. 2024;25(1):607. doi:10.1186/s12864-024-10526-5
37. Sun G, Zhou Y, Han X, et al. Potential marker genes for chronic obstructive pulmonary disease revealed based on single-cell sequencing and Mendelian randomization analysis. Aging. 2024;16(10):8922–8943. doi:10.18632/aging.205849
38. Lamontagne M, Bérubé JC, Obeidat M, et al. Leveraging lung tissue transcriptome to uncover candidate causal genes in COPD genetic associations. Hum Mol Genet. 2018;27(10):1819–1829. doi:10.1093/hmg/ddy091
39. Abdul KM, Terada K, Gotoh T, Hafizur RM, Mori M. Characterization and functional analysis of a heart-enriched DnaJ/ Hsp40 homolog dj4/DjA4. Cell Stress Chaperones. 2002;7(2):156–166.
40. Liu RJ, Niu XL, Yuan JP, Chen HD, Gao XH, Qi RQ. DnaJA4 is involved in responses to hyperthermia by regulating the expression of F-actin in HaCaT cells. Chin Med J. 2020;134(4):456–462. doi:10.1097/CM9.0000000000001064
41. Hodge G, Roscioli E, Jersmann H, et al. Steroid resistance in COPD is associated with impaired molecular chaperone Hsp90 expression by pro-inflammatory lymphocytes. Respir Res. 2016;17(1):135. doi:10.1186/s12931-016-0450-4
42. Zhang Q, Feng P, Zhu XH, et al. DNAJA4 suppresses epithelial-mesenchymal transition and metastasis in nasopharyngeal carcinoma via PSMD2-mediated MYH9 degradation. Cell Death Dis. 2023;14(10):697. doi:10.1038/s41419-023-06225-w
43. Ambrocio-Ortiz E, Pérez-Rubio G, Ramírez-Venegas A, et al. Protective role of genetic variants in HSP90 genes-complex in COPD secondary to biomass-burning smoke exposure and non-severe COPD forms in tobacco smoking subjects. Curr Issues Mol Biol. 2021;43(2):887–899. doi:10.3390/cimb43020063
44. Desai P, Yang J, Tian B, et al. Mixed-effects model of epithelial-mesenchymal transition reveals rewiring of signaling networks. Cell Signal. 2015;27(7):1413–1425. doi:10.1016/j.cellsig.2015.03.024
45. Xia H, Wu Y, Zhao J, et al. N6-Methyladenosine-modified circSAV1 triggers ferroptosis in COPD through recruiting YTHDF1 to facilitate the translation of IREB2. Cell Death Differ. 2023;30(5):1293. doi:10.1038/s41418-023-01138-9
46. Pérez-Peiró M, Alvarado Miranda M, Martín-Ontiyuelo C, Rodríguez-Chiaradía DA, Barreiro E. Nitrosative and Oxidative Stress, Reduced Antioxidant Capacity, and Fiber Type Switch in Iron-Deficient COPD Patients: analysis of Muscle and Systemic Compartments. Nutrients. 2023;15(6):1454. doi:10.3390/nu15061454
47. Yoshida M, Minagawa S, Araya J, et al. Involvement of cigarette smoke-induced epithelial cell ferroptosis in COPD pathogenesis. Nat Commun. 2019;10(1):3145. doi:10.1038/s41467-019-10991-7
48. Pantopoulos K, Porwal SK, Tartakoff A, Devireddy L. Mechanisms of mammalian iron homeostasis. Biochemistry. 2012;51(29):5705–5724. doi:10.1021/bi300752r
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.