Back to Journals » OncoTargets and Therapy » Volume 14
A Combination of RNA-Seq Analysis and Use of TCGA Database for Determining the Molecular Mechanism and Identifying Potential Drugs for GJB1 in Ovarian Cancer
Authors Yang J, Fan Y, Xie B, Yang D ![]()
Received 27 January 2021
Accepted for publication 25 March 2021
Published 14 April 2021 Volume 2021:14 Pages 2623—2633
DOI https://doi.org/10.2147/OTT.S303589
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Federico Perche
Jie Yang,1 Yaqin Fan,1 Beibei Xie,2 Dan Yang1,2
1Obstetrics and Gynecology, Yuncheng County People’s Hospital, Heze, People’s Republic of China; 2Faculty of Health, Yantai Nanshan University, Yantai, People’s Republic of China
Correspondence: Dan Yang
Obstetrics and Gynecology, Yuncheng County People’s Hospital, No. 184 Tangta Road, Yuncheng County, Heze, Shandong, 274700, People’s Republic of China
Tel +86 18660036376
Email [email protected]
Background: There has been increasing evidence for the vital role played by gap junction protein beta-1 (GJB1) in ovarian cancer (OC) and for the possibility of this protein serving as a therapeutic target. However, the detailed mechanism of GJB1 in OC has not yet been clearly determined. The current study aimed to establish the molecular mechanisms of the involvement of GJB1 in OC and to further predict potential drugs targeting this protein.
Methods: To better understand the molecular mechanisms of the involvement of GJB1 in OC, RNA-Seq transcriptome sequencing was performed. Then, we carried out an RNA-Seq analysis to determine the genes differentially co-expressed with GJB1. Subsequently, we carried out bioinformation methods to study the upstream regulatory transcriptional factor (TF) of GJB1. Further, the binding of FOXA1 and GJB1 promoter was tested using ChIP-qPCR. Moreover, we performed pathway enrichment to identify the downstream regulatory mechanisms of GJB1. Furthermore, potential drugs targeting GJB1 were screened using AutoDock 4.2.
Results: We constructed the transcriptional factor FOXA1 regulatory network based on the AnimalTFDB, JASPAR, RNA-Seq, TCGA cohort and ChIP-qPCR to study the upstream regulation of GJB1. In addition, two key pathways for the involvement of GJB1 in OC—namely the “ECM–receptor interaction” and “focal adhesion” KEGG pathways—were identified. Furthermore, ZINC000005552022 was found in a screening to be a potentially promising drug targeting GJB1.
Conclusion: Our study results suggested that the transcriptional factor FOXA1 regulates the involvement of GJB1 in OC through ECM–receptor interaction and focal adhesion KEGG pathways, and that ZINC000005552022 may have promising potential as a drug targeting GJB1; this finding might be used to help accelerate drug development and improve the outcomes for patients with OC.
Keywords: transcriptional factor, ChIP-qPCR, KEGG, virtual screening, omics
Introduction
Although ovarian cancer (OC) diagnostic criteria and clinical therapeutic strategies have evolved in recent decades, this disease is the leading cause of death among women with gynecological malignancies, and it is characterized by high recurrence and mortality.1 The high mortality rate is due to most cases of OC being identified at advanced stages, that is stages III or IV, and due to its increased risk of recurrence even after any initial response to traditional platinum-based therapy.2 Thus, there is an urgent need to identify new molecular targets and develop more effective therapies against ovarian cancer.
Connexins (Cxs) normally assemble into gap junctions (GJs), which directly exchange cytoplasmic substances with molecular weights of < 1 kDa, including ions, second messengers and metabolites, between adjacent cells. The gap junction protein beta-1 (GJB1) gene encodes specifically the transmembrane channel protein connexin 32 (Cx32), which belonged to Cxs family.3 GJB1 might play an essential role in OC and hence might also be a therapeutic target for this disease.4–6 Previous work has shown the abnormal distribution of Cx32 promoting the progress of cervical cancer and hepatocellular carcinoma. Further, upregulated GJB1 in OC cell line A2780 could contribute to resistance to cisplatin.7 In addition, Lai et al reported that GJB1 suppressed extrinsic apoptosis in OC via the NF‑κB signaling pathway.8 Further, Zhao et al suggested the ability of GJB1 to induce an anti-apoptotic effect via epidermal growth factor receptor (EGFR) upregulation.5 Moreover, Zhang et al showed that GJB1 might be involved in cisplatin resistance by modulating the expression of drug efflux transporters and activating the EGFR‑Akt anti‑apoptosis axis.9 These studies suggested the potential mechanisms of the involvement of GJB1 in OC to include multiple pathways. However, the key signaling pathway for this mechanism is still not clear. There is, therefore, a crucial need to systematically investigate the underlying mode of action of GJB1 in OC.
In the present study, we carried out a bioinformatics analysis based on an RNA-Seq analysis and the large-scale multi-omics data in The Cancer Genome Atlas (TCGA) to investigate the detailed molecular mechanism of the involvement of GJB1 in the OC, and we found potential drugs for targeting GJB1. Our study has provided new insight into OC at the molecular level. In the study, we investigated the therapeutic potential of medicines to overcome OC. The results might lead to improvements in the outcomes for OC patients.
Materials and Methods
Cell Culture
As previously described,5 HeLa-GJB1 (purchased from Shanghai Genechem Co., Ltd.) with a tetracycline-inducible promoter was found to be capable of constitutively expressing GJB1, when treated with 1 μg of doxycycline (Sangon Biotech) for 48 h. OC cell lines A2780 and SKOV3 were purchased from the cell library of the Chinese Academy of Sciences. Cells were cultured in DMEM with 10% fetal bovine serum at 37 °C, 5% CO2, in a humidified incubator.
Experimental Design
The workflow of this study is shown in Figure 1. To better understand the molecular mechanisms of GJB1 in OC, RNA-Seq transcriptome sequencing was performed on HeLa-GJB1 and control (Cntrl) groups. Then, we carried out an RNA-Seq analysis to determine the genes differentially co-expressed with GJB1. Subsequently, we investigated the upstream regulatory transcriptional factor (TF) of GJB1 by carrying out various analyses, including those involving AnimalTFDB,10 JASPAR,11 genes positively co-expressed with GJB1 determined using RNA-Seq, and upregulated DEGs in TCGA. We also performed a pathway enrichment analysis to identify the mechanisms controlling the downstream regulation of GJB1. Furthermore, potential drugs targeting GJB1 were screened using AutoDock 4.2.
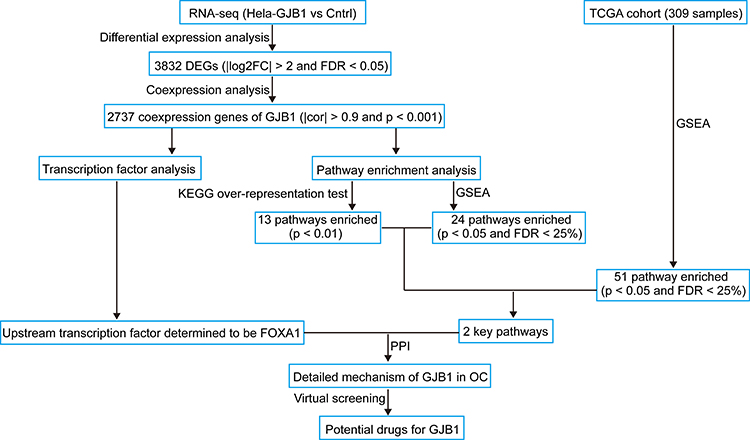 |
Figure 1 Flow diagram of the current study: data collection, preprocessing, analysis, and validation. Abbreviations: GJB1, gap junction protein beta 1; RNA-Seq, RNA-Sequencing; DEGs, differentially expressed genes; OC, ovarian cancer; PPI, protein-protein interaction; cor, correlation; Cntrl, control; GSEA, gene set enrichment analysis. |
RNA Isolation and Sequencing
Cells were collected and pooled for RNA-Seq analysis, with each treatment run in triplicate. Total RNAs of the cell lines from the control groups and HeLa-GJB1 groups were extracted using an RNeasyMini Kit (Qiagen), and RNA quality was assessed by using NanoDrop (Thermo Fisher Scientific, Waltham, MA) and Qubit (Invitrogen). High-quality RNA samples (28 S/18 S = 2.0−2.2, RIN > 9.0) were used to construct libraries following a standardized procedure at the Beijing Genome Institute on a BGISEQ-500 platform, with three replicates per treatment group.
TCGA Data Collection
HTSeq-Counts of gene expression data (3 normal samples and 306 tumor samples) and corresponding OC clinical data were collected from The Cancer Genome Atlas (TCGA) database (https://www.cancer.gov/tcga; accessed on February 4, 2020). In the present study, we used TCGA data based on the TCGA publication guidelines.
Identification of Differentially Expressed Genes
The R package edgeR12 was used to normalize and analyze genes differentially expressed between HeLa-GJB1 groups (n = 3) and the Cntrl groups (n = 3) significantly. Further, the analysis was also performed on the TCGA cohort (306 OC samples and 3 normal samples). A gene was considered differentially expressed to a statistically significant extent if its |log2 fold change| ≥ 2 and FDR < 0.05. Then, DEGs were identified based on the above threshold for RNA-Seq and the TCGA cohort. Furthermore, we performed an analysis of the co-expression of RNA-Seq-derived DEGs with GJB1 using a Pearson correlation value cutoff > 0.9 and p < 0.001.
Prediction of the Transcription Factor Binding Site in the GJB1 Promoter
To study the upstream transcription factor (TF) regulation, we downloaded the sequence of the GJB1 promoter from UCSC (http://www.genome.ucsc.edu/). Then, we carried out analyses to predict the transcription factor binding site (TFBS) for GJB1 based on the AnimalTFDB10 and JASPAR databases.11 As we previously5 showed GJB1 to be highly expressed in OC cells, we carried out various analyses, including those of the genes positively co-expressed with GJB1 and upregulated genes in TCGA to further screen for the TF. We also investigated Chiq-seq data in the Cistrome database to further identify the binding of the TF to the GJB1 promoter.13
Chromatin Immunoprecipitation (ChIP)-qPCR
Chromatin immunoprecipitation (ChIP) assays were performed according to the manufacturer’s instructions for the Chromatin Immunoprecipitation Kit (17–10,086, Merck) and according to the instructions in a previous study.14 In brief, cells were fixed with 1% formaldehyde to cross-link protein and chromatin, and then sonicated to produce 200–1000-bp DNA fragments (ultrasound 10 s, stop 10 s, repeated seven times). The lysate was precleared with protein A/G agarose and incubated with specific antibodies, namely anti-FOXA1 (ab23738, Abcam) or IgG (ab171870, Abcam), at 4 °C overnight. IgG antibody was used as a control. Then, the protein/DNA complexes were eluted and reverse cross-linking of the protein/DNA complexes was carried out according to manufacturer’s instructions in order to free the DNA. The free DNA was purified and analyzed using real-time quantitative PCR (qPCR, denaturation at 95°C for 20 sec, followed by 40 cycles of treatment at 95°C for 10 sec, 60°C for 20 sec and 70°C for 10 sec.). The primer sequences against the GJB1 promoter were as follows: forward 5ʹ‑GTCTTCAGGTAAGCCGTCAGT‑3; reverse 5ʹ‑AATTGCTTTCTACCCTCAAGGC‑3ʹ at the location shown in Figure S1.
Pathway Enrichment Analysis Based on RNA-Seq and a TCGA Cohort
A KEGG over-representation test was performed by using the R package clusterProfiler,15 at a cutoff of p < 0.01, with the co-expression of GJB1 from RNA-Seq results. Simultaneously, gene set enrichment analysis (GSEA) was performed16 to explore the potential molecular mechanisms of the HeLa-GJB1 groups (n = 3) and Cntrl groups (n = 3) based on the expression profile of all of the protein-coding genes with a false discovery rate (FDR) cutoff of < 0.25 and p < 0.05. Furthermore, GSEA was also used to examine the enrichment pathways of the normal samples (n = 3) and OV samples (n = 306). Finally, the critical pathways of GJB1 involved in OC were identified based on the pathway enrichment results.
Protein–Protein Interaction Analysis
A protein–protein interaction (PPI) network was constructed using the GJB1 and co-expressed genes involved in crucial pathways based on the STRING database (medium confidence: 0.4)17 and visualized using Cytoscape 3.7.1.18 We performed a shortest-pathway analysis by using the Python package NetworkX (http://networkx.github.io) to find possible connections between GJB1 and the key signaling pathways.
Identifying a Potential Drug to Target GJB1
We obtained the 3D structure of GJB1 from SWISS-MODEL (https://swissmodel.expasy.org/). Then, Binding Site Detect in AutoDock 4.219 was employed to find the binding site of GJB1. Subsequently, we used a library of 50,353 drug-like chemical compounds obtained from the ZINC15 database.20 Finally, virtual screening and molecular docking were performed using AutoDock 4.2 to find a potential GJB1-targeting drug.
Statistical Analysis
Statistical analysis was performed by using R 3.6.3. A normal distribution test and homogeneity of variance test were performed before carrying out statistical analysis. A value of p < 0.05 was considered statistically significant.
Results
DEGs and Genes Co-Expressed with GJB1 Based on RNA-Seq Analysis
To investigate the mechanism of the involvement of GJB1 in ovarian cancer, the genes differentially expressed between HeLa-GJB1 and Cntrl groups were analyzed, according to the count values of all detected genes. A volcano plot of the DEGs is shown in Figure 2A. A total of 3832 genes differentially expressed between the HeLa-GJB1 and Cntrl groups (1979 upregulated, indicated by the red dots in the figure, and 1853 downregulated, indicated by the green dots; Figure 2A and Table S1) were identified at a cutoff of |log2Fold Change| > 2 and FDR < 0.05. A heat map of the top 50 DEGs was produced to visualize their expression levels in the different samples (Figure 2B). Then, we carried out a co-expression analysis of GJB1 with the all 3832 DEGs by performing RNA-Seq. We obtained 2737 genes co-expressed with GJB1 (1400 positively co-expressed genes and 1337 negatively co-expressed genes; Table S2), with the 10 genes most strongly co-expressed with GJB1 shown in Figure 2C.
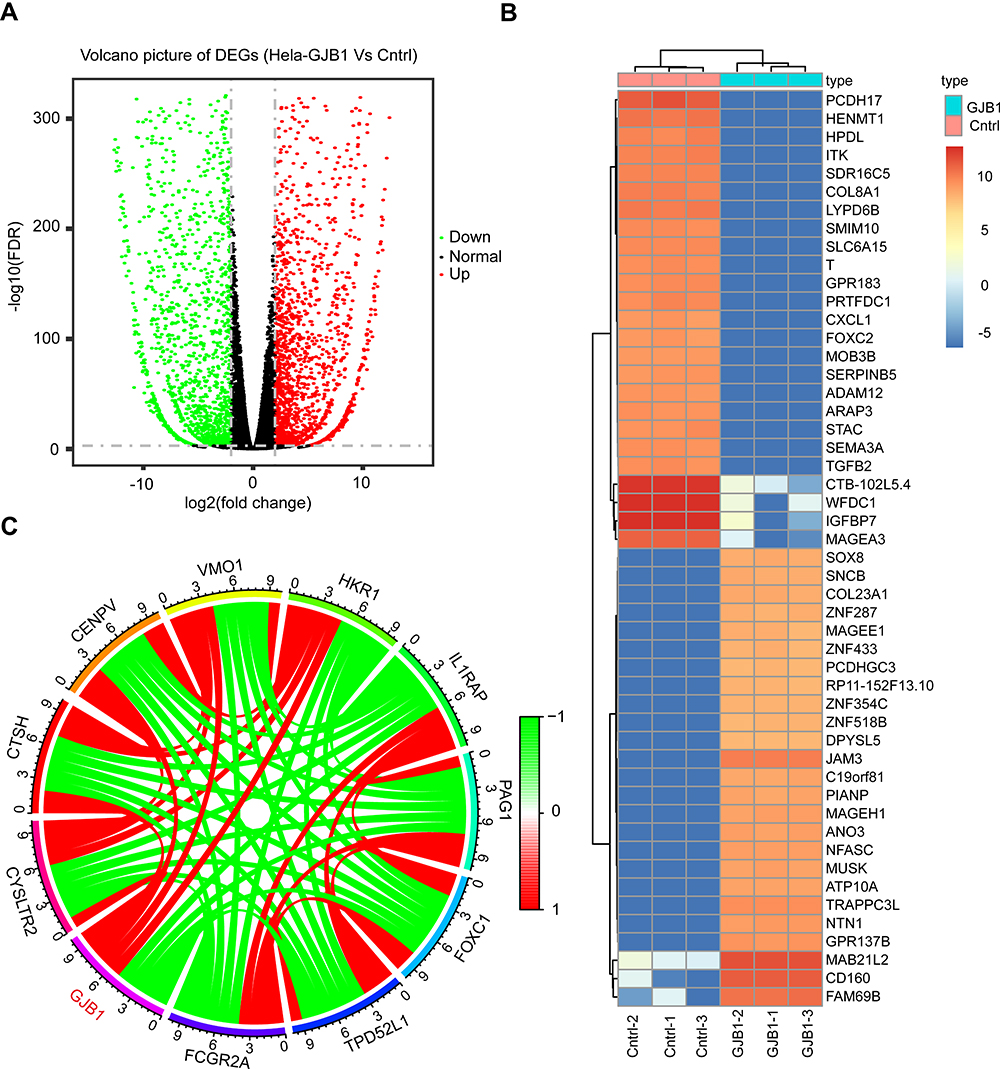 |
Figure 2 Determination of genes differentially expressed between HeLa-GJB1 and control (Cntrl) groups. (A) Volcano plot of all unique genes from the genes differentially expressed between HeLa-GJB1 and Cntrl groups. Vertical and horizontal lines indicate the significance thresholds of |log2 Fold Change| > 2 and false discovery rate (FDR) < 0.05, respectively. (Blue and red dots indicate significantly downregulated and significantly upregulated genes, respectively; while black dots indicate genes with nonsignificant changes in expression levels). (B) Heat map of the top 50 DEGs (Red indicate upregulated and Blue indicate downregulated). (C) Circos plot of the top 10 genes co-expressed with GJB1 (Red indicate positive co-expression and Green indicate negative co-expression). Abbreviation: GJB1, gap junction protein beta 1. |
Constructing the TF Upstream Regulatory Network of GJB1 in OC
To understanding the upstream regulation mechanism of GJB1 in OC, we performed a bioinformatics analysis to find the TF that could regulate the GJB1 gene. We downloaded the GJB1 promoter from the UCSC genome website (~2K, Table S3). Then, we used the AnimalTFDB and JASPAR databases to predict which TF could bind to the promoter of GJB1. Here, 252 and 486 TFs were obtained from the AnimalTFDB (p < 0.05; Table S4) and JASPAR (score > 6; Table S5), databases, respectively. As we previously showed GJB1 to be upregulated in OC cells, we speculated that its TF should be highly expressed in OC and that this level of expression should be positively correlated with the level of expression of GJB1. By carrying out various analyses, including those involving AnimalTFDB, JASPAR, genes positively co-expressed with GJB1 using RNA-Seq, and upregulated DEGs in TCGA (Table S6), we suggested that FOXA1 might regulate the GJB1 gene in OC (Figure 3A). The FOXA1 binding site on the promoter of GJB1 is shown in Figure 3B. We also explored the ChIP-seq data in the Cistrome database to identify the relationship between FOXA1 and GJB1. Based on this analysis of the ChIP-seq data, we found that FOXA1 could bind to the GJB1 promoter (Figure S1). Furthermore, we performed ChIP-qPCR in cells of the OC cell lines A2780 or SKOV3 to further determine whether FOXA1 directly binds to the promoter region of GJB1. As shown in Figure 3C and D, FOXA1 could directly bind to the GJB1 promoter.
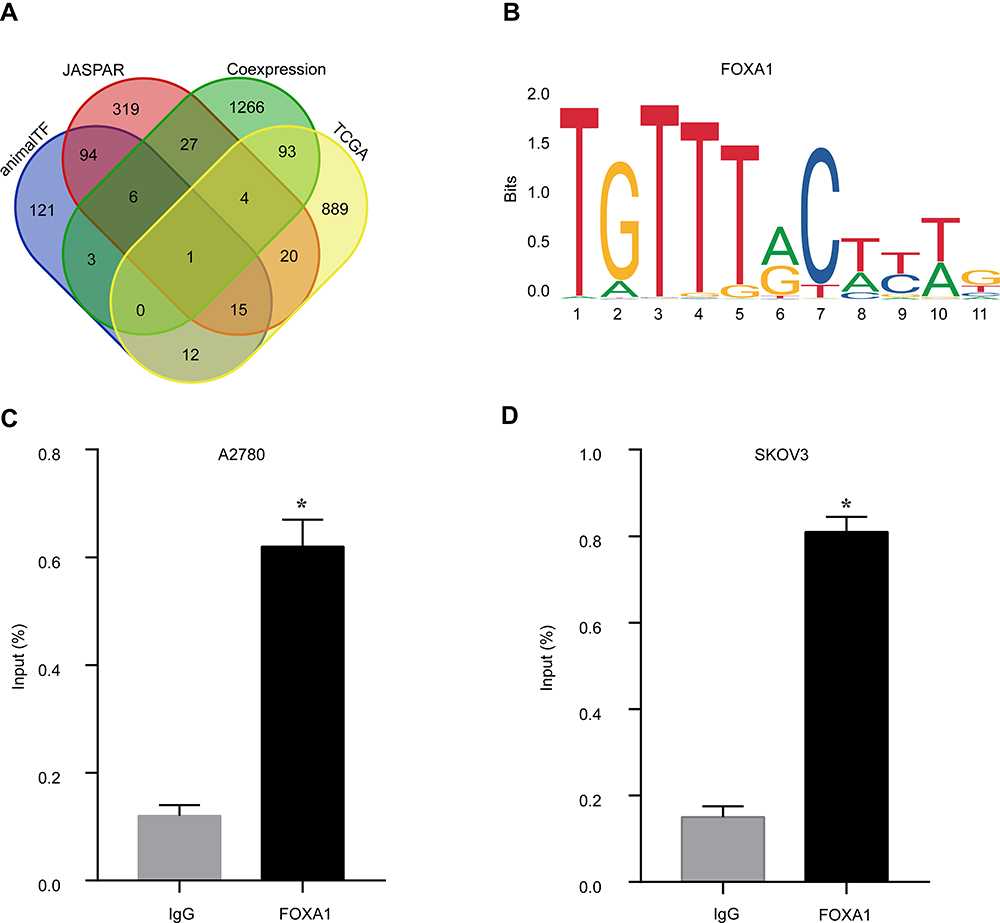 |
Figure 3 Constructing the TF upstream regulatory network of GJB1 in ovarian cancer (OC). (A) Venn diagram of the results of the analyses of AnimalTFDB, JASPAR, genes positively co-expressed with GJB1 using RNA-Seq, and upregulated differentially expressed genes (DEGs) in TCGA. (B) Sequence logo display of the binding site of the FOXA1 transcription factor (TF) on the GJB1 promoter. (C and D) Chromatin immunoprecipitation was performed using antibodies against FOXA1 or using a control IgG in (C) A2780 and (D) SKOV3 cells. Here, DNA immunoprecipitated by the IgG or the anti-FOXA1 antibody was in each case observed to be enriched compared with the input chromatin. Data are expressed each as mean ± SD (n = 3). *p < 0.05 compared with the IgG group (unpaired t-test). Abbreviation: GJB1, gap junction protein beta 1. |
Identification of the Downstream Regulation Mechanism of GJB1
To investigate the involvement of the region downstream of the GJB1 gene in OC, we performed a pathway enrichment analysis. Specifically, we performed a KEGG over-representation test based on genes differentially expressed according to an RNA-Seq analysis; this test revealed an enrichment of 13 KEGG pathways associated with GJB1 (Figure 4A). Pathway analysis was also performed by carrying out GSEA, which targets the expression of all of the genes. The GSEA analysis produced a total of 2 pathways for the HeLa-GJB1 groups and 22 pathways (Table S7) for the Cntrl groups according to the cutoff of FDR < 25% and p < 0.05. To make the analysis more accurate, we also employed GSEA in the TCGA cohort. This analysis yielded 34 pathways for the OC groups and 20 for the normal groups (Table S8). Based on the KEGG over-representation test and GSEA results (Figure 4B), two overlapping pathways—namely, the “ECM receptor interaction” and “focal adhesion” KEGG pathways—were identified. To investigate the interaction of GJB1 with these two key KEGG pathways, we searched for the genes (involved in the two overlapping pathways) in the STRING database, and this search yielded a combined score of > 0.4. The resulting PPI network comprised 35 nodes and 239 edges. From the PPI network and shortest path analysis, we found that GJB1 could only directly interact with EGFR (Figure 4C), a receptor belonging to the focal adhesion KEGG pathway. Then, we showed the first-stage nodes (nodes directly connected) of EGFR, to attempt to help understand the interactions between GJB1 and the two key pathways.
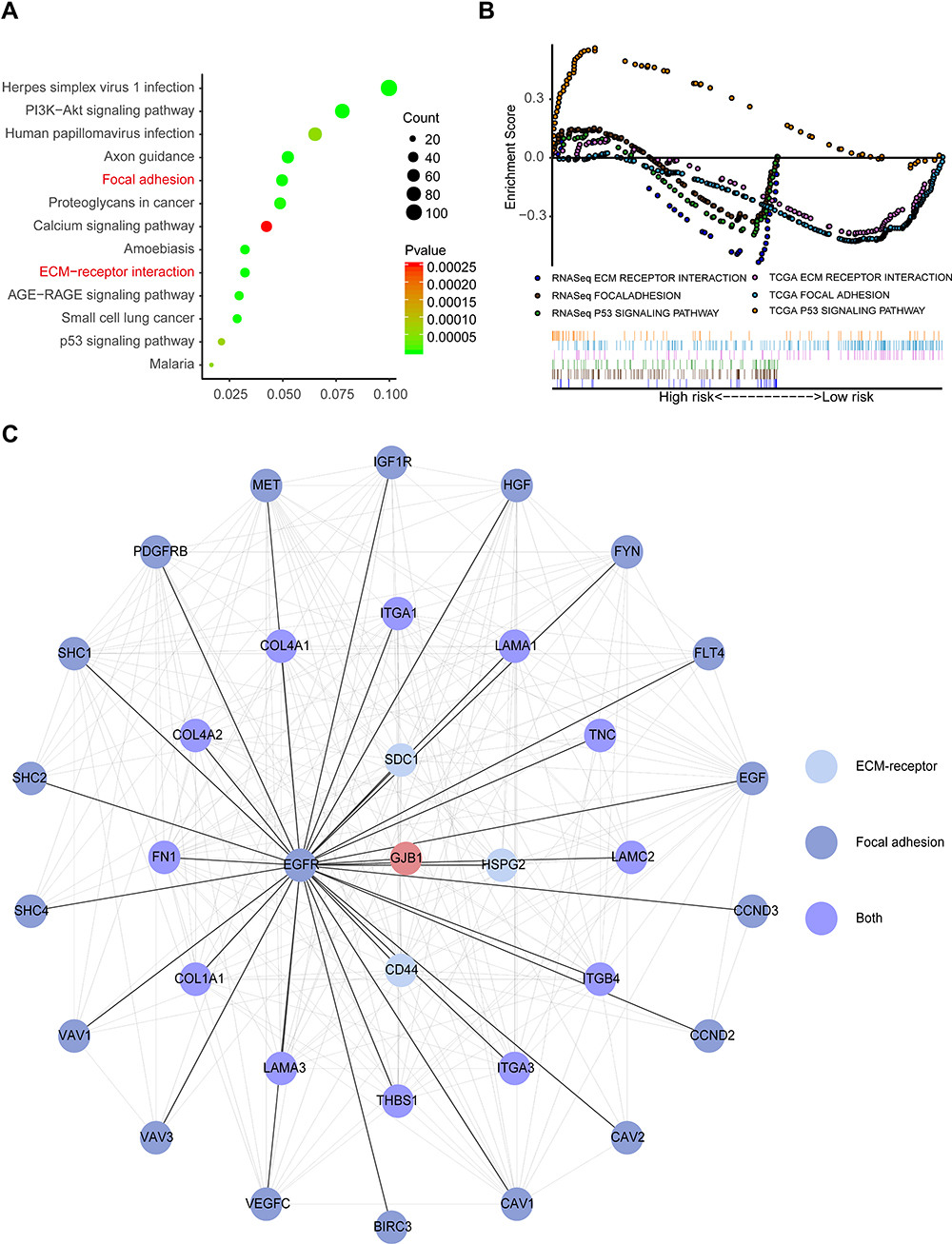 |
Figure 4 Downstream regulation mechanism of GJB1 in OC. (A) KEGG over-representation test based on genes differentially expressed according to an RNA-sequencing (RNA-Seq) analysis achieved using clusterProfiler (p < 0.01). (B) The results of gene set enrichment analysis (GSEA) of RNA-Seq and a TCGA cohort (p < 0.05 and FDR < 25%, False discovery rate (FDR). (C) Protein–protein interaction network was constructed based on the String database. Abbreviation: GJB1, gap junction protein beta 1. |
Identifying Potential Drugs Targeting GJB1
Virtual screening was performed using AutoDock 4.2 to identify potential GJB1-targeting drugs. We built the 3D protein structure of GJB1 based on the SWISS-MODEL database (Figure S2). Its active site was found using AutoDock 4.2 and is shown in Figure 5A. ZINC000005552022, whose 3D structure is shown in Figure 5B, was the top hit of a structure-based virtual screening process based on glide scores (Table S9). The 3D structure of ZINC000005552022 docked into GJB1 is shown in Figure 5C, and their interaction with each other was indicated according to this docking to include one H-bond and one π-π interaction as well as hydrophobic interactions.
 |
Figure 5 The potential drugs based on the three-dimensional (3D) structure of GJB1. (A) The active site of GJB1 was found using AutoDock 4.2 2019–1. (B) The structure of ZINC000005552022 based on ZINC15 database. (C) AutoDock-derived structure of ZINC000005552022 bound to GJB1. The yellow dashed line represents a hydrogen bond, and the blue one a π-π interaction. Abbreviation: GJB1, gap junction protein beta 1. |
Discussion
About 60% of OC patients are diagnosed at an advanced clinical stage21 and have a high risk of recurrence despite any initial response to traditional platinum-based therapy.2 These issues ultimately lead to the poor prognosis for OC patients, with a five-year survival rate of only 25%–30%.22,23 Therefore, it is critical to identify new molecular targets and develop more effective anti-cancer therapies. There has been increasing evidence of the important role played by GJB1 in OC and increasing evidence that it might be a therapeutic target,24,25 but the key signaling pathway of GJB1 involved in OC is not understood. In the present study, we carried out a systematic bioinformatics analysis to investigate the detailed molecular mechanism of the involvement of GJB1 in OC and searched for potential GJB1-targeting drugs. These efforts were made with the aim of providing new information and supporting data to improve the outcomes for OC patients.
Recently, many studies have demonstrated GJB1 to be upregulated in OC cells.4–6,8 However, the mechanism underlying the upregulation of GJB1 in OC remains poorly understood. In the present study, we showed the upregulated expression of GJB1 to coincide with a coordinated increase in the transcript levels of transcription factors. Therefore, we performed an upstream regulator analysis to find potential relevant transcription factors. Since GJB1 was found to be upregulated in OC, we presumed that its transcription factors would be positively co-expressed with GJB1 and would also be upregulated in OC. By carrying out various analyses, including those involving AnimalTFDB and JASPAR databases, we identified upregulation of the transcription factor FOXA1 as corresponding to the upregulation of GJB1 in OC. We also found, by performing ChIP-qPCR, that FOXA1 could bind to the GJB1 promoter. FOXA1 was previously ascribed oncogenic roles in breast cancer26 and prostate cancer.27,28 Moreover, FOXA1 was shown to be related to the epithelial-to-mesenchymal transition in colorectal cancer.29 The previous studies were in line with our laboratory results showing a tumor-promoting effect of GJB1 in OC. By taking these results together, we found that FOXA1 was related to the high GJB1 expression levels in OC.
Though most of the studies found GJB1 playing an essential role in OC, the molecular mechanism of GJB1 has remained controversial. To clarify this issue, we performed a systematic bioinformatics analysis to identify the key pathways based on RNA-Seq and a TCGA cohort. First, we employed pathways enrichment by two methods (KEGG over-representation test and GSEA) according to an RNA-Seq analysis. Secondly, to make the results more accurate, we also performed GSEA based on the TCGA cohort. Then, the ECM–receptor interaction and focal adhesion KEGG pathways were identified as key pathways of the involvement of GJB1 in OC. Previous studies found the ECM–receptor interaction KEGG pathway playing a key role in OC and affecting the survival of OC patients.30 Moreover, several reports showed that the focal adhesion KEGG pathway could influence cell proliferation, migration, and invasion in OC.31–33 Taken together, we provided evidence for the involvement of GJB1 in OC through ECM–receptor interaction and focal adhesion KEGG pathways.
There used to be a debate about whether GJB1 plays a role in OC with or without EGFR. A previous report showed that GJB1 could inhibit apoptosis without EGFR.6 However, GJB1 was proposed by Xie et al to induce resistance to afatinib via a transcriptional increase in EGFR expression.14 Further, GJB1 was reported in previous studies to exert anti-apoptotic and pro-tumor effects via EGFR.3,5 Additionally, previous research suggested that GJB1 could interact with EGFR to promote resistance to cisplatin in cells of the OC cell line A2780.9 Moreover, based on the STRING database,34 GJB1 and EGFR were co-expression (score 0.064) and co-mentioned in Pubmed Abstracts (score 0.591). In the present study, we hypothesized that GJB1 in general interacts with EGFR in OC. Based on our co-expression analysis resulting from RNA-Seq, GJB1 was found to be positively co-expressed with EGFR (cor > 0.9 and p < 0.001). Further, based on the results of our PPI and shortest path analysis (Figure 4C), we concluded that GJB1 indeed in general interacts with EGFR in OC. In previous study,3 GJB1 in OC cells was shown to contribute to tumor growth and to have an anti-apoptotic effect. Then, based on these previous results combined with our current pathway enrichment results (Figure 4A and B), we speculated that GJB1 in general interacts with EGFR, and has an anti-apoptotic effect through the ECM–receptor interaction and focal adhesion KEGG pathways in OC. Because previous studies indicated the participation of the ECM–receptor interaction and focal adhesion KEGG pathways in the process of apoptosis,35,36 we carried out a systemic investigation of the detailed mechanism of the involvement of GJB1 in OC, the results of which we expect to provide a basis for subsequent corresponding drug research and clinical research.
In contrast to other related studies,5,14,37 we not only identified the potential mechanism for the involvement of GJB1 in OC, but also found potential drugs targeting GJB1. We sought out the potential drugs targeting GJB1 by carrying out a virtual screening. This technique and the corresponding results we obtained might expand the possible therapeutic strategies for achieving OC treatments—and in this way accelerate the development of a drug or drugs to improve the outcomes for OC patients.
Conclusions
In summary, we carried out a bioinformatics analysis to construct a TF network for GJB1, the results of which expanded our understanding of the upstream regulation mechanism for GJB1 in OC and might provide therapeutic targets to improve OC prognoses. Moreover, we uncovered an interaction of GJB1 with EGFR, and showed GJB1 to be involved in OC through the ECM–receptor interaction and focal adhesion KEGG pathways by performing co-expression analysis and pathway enrichment. Furthermore, we found ZINC000005552022 to be a potential GJB1-targeting drug by carrying out virtual screening based on the structure of GJB1, and this finding might accelerate drug development to improve the outcomes for OC patients.
Data Sharing Statement
Publicly available datasets were analyzed in this study. The data can be found at TCGA, https://portal.gdc.cancer.gov; ZINC15, http://zinc15.docking.org/; AnimalTFDB, http://bioinfo.life.hust.edu.cn/AnimalTFDB/#!/; and JASPAR, http://jaspar.genereg.net/.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors declare that they have no competing interests.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7–34. doi:10.3322/caac.21551
2. Yue H, Wang J, Chen R, Hou X, Li J, Lu X. Gene signature characteristic of elevated stromal infiltration and activation is associated with increased risk of hematogenous and lymphatic metastasis in serous ovarian cancer. BMC Cancer. 2019;19(1):1266. doi:10.1186/s12885-019-6470-y
3. Xiang Y, Wang Q, Guo Y, et al. Cx32 exerts anti-apoptotic and pro-tumor effects via the epidermal growth factor receptor pathway in hepatocellular carcinoma. J Exp Clin Cancer Res. 2019;38(1):145. doi:10.1186/s13046-019-1142-y
4. Tanaka T, Yamasaki H, Mesnil M. Induction of a bystander effect in HeLa cells by using a bigenic vector carrying viral thymidine kinase and connexin32 genes. Mol Carcinog. 2001;30(3):176–180. doi:10.1002/mc.1026
5. Zhao Y, Lai Y, Ge H, et al. Non-junctional Cx32 mediates anti-apoptotic and pro-tumor effects via epidermal growth factor receptor in human cervical cancer cells. Cell Death Dis. 2017;8(5):e2773. doi:10.1038/cddis.2017.183
6. Lai Y, Tao L, Zhao Y, et al. Cx32 inhibits TNFalpha-induced extrinsic apoptosis with and without EGFR suppression. Oncol Rep. 2017;38(5):2885–2892. doi:10.3892/or.2017.5950
7. Wu W, Fan L, Bao Z, et al. The cytoplasmic translocation of Cx32 mediates cisplatin resistance in ovarian cancer cells. Biochem Biophys Res Commun. 2017;487(2):292–299. doi:10.1016/j.bbrc.2017.04.053
8. Lai Y, Fan L, Zhao Y, et al. Cx32 suppresses extrinsic apoptosis in human cervical cancer cells via the NFkappaB signalling pathway. Int J Oncol. 2017;51(4):1159–1168. doi:10.3892/ijo.2017.4106
9. Zhang Y, Tao L, Fan LX, et al. Cx32 mediates cisplatin resistance in human ovarian cancer cells by affecting drug efflux transporter expression and activating the EGFRAkt pathway. Mol Med Rep. 2019;19(3):2287–2296. doi:10.3892/mmr.2019.9876
10. Hu H, Miao YR, Jia LH, Yu QY, Zhang Q, Guo AY. AnimalTFDB 3.0: a comprehensive resource for annotation and prediction of animal transcription factors. Nucleic Acids Res. 2019;47(D1):D33–D38. doi:10.1093/nar/gky822
11. Fornes O, Castro-Mondragon JA, Khan A, et al. JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2020;48(D1):D87–D92. doi:10.1093/nar/gkz1001
12. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–140. doi:10.1093/bioinformatics/btp616
13. Li S, Wan C, Zheng R, et al. Cistrome-GO: a web server for functional enrichment analysis of transcription factor ChIP-seq peaks. Nucleic Acids Res. 2019;47(W1):W206–W211. doi:10.1093/nar/gkz332
14. Xie J, Wang X, Ge H, et al. Cx32 mediates norepinephrine-promoted EGFR-TKI resistance in a gap junction-independent manner in non-small-cell lung cancer. J Cell Physiol. 2019;234(12):23146–23159. doi:10.1002/jcp.28881
15. Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–287. doi:10.1089/omi.2011.0118
16. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–15550. doi:10.1073/pnas.0506580102
17. Szklarczyk D, Morris JH, Cook H, et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017;45(D1):D362–D368. doi:10.1093/nar/gkw937
18. Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. doi:10.1101/gr.1239303
19. Morris GM, Huey R, Lindstrom W, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem. 2009;30(16):2785–2791. doi:10.1002/jcc.21256
20. Irwin JJ, Sterling T, Mysinger MM, Bolstad ES, Coleman RG. ZINC: a free tool to discover chemistry for biology. J Chem Inf Model. 2012;52(7):1757–1768. doi:10.1021/ci3001277
21. Torre LA, Trabert B, DeSantis CE, et al. Ovarian cancer statistics, 2018. CA Cancer J Clin. 2018;68(4):284–296. doi:10.3322/caac.21456
22. Cortes-Guiral D, Elias D, Cascales-Campos PA, et al. Second-look surgery plus hyperthermic intraperitoneal chemotherapy for patients with colorectal cancer at high risk of peritoneal carcinomatosis: does it really save lives? World J Gastroenterol. 2017;23(3):377–381. doi:10.3748/wjg.v23.i3.377
23. Damia G, Broggini M. Platinum resistance in ovarian cancer: role of DNA repair. Cancers. 2019;11(1):119. doi:10.3390/cancers11010119
24. Januchowski R, Swierczewska M, Sterzynska K, Wojtowicz K, Nowicki M, Zabel M. Increased expression of several collagen genes is associated with drug resistance in ovarian cancer cell lines. J Cancer. 2016;7(10):1295–1310. doi:10.7150/jca.15371
25. Leask A. A centralized communication network: recent insights into the role of the cancer associated fibroblast in the development of drug resistance in tumors. Semin Cell Dev Biol. 2020;101(5):111–114. doi:10.1016/j.semcdb.2019.10.016
26. Robinson JL, Macarthur S, Ross-Innes CS, et al. Androgen receptor driven transcription in molecular apocrine breast cancer is mediated by FoxA1. EMBO J. 2011;30(15):3019–3027. doi:10.1038/emboj.2011.216
27. Robinson JL, Hickey TE, Warren AY, et al. Elevated levels of FOXA1 facilitate androgen receptor chromatin binding resulting in a CRPC-like phenotype. Oncogene. 2014;33(50):5666–5674. doi:10.1038/onc.2013.508
28. Pomerantz MM, Li F, Takeda DY, et al. The androgen receptor cistrome is extensively reprogrammed in human prostate tumorigenesis. Nat Genet. 2015;47(11):1346–1351. doi:10.1038/ng.3419
29. Matsuda Y, Miura K, Yamane J, et al. SERPINI1 regulates epithelial-mesenchymal transition in an orthotopic implantation model of colorectal cancer. Cancer Sci. 2016;107(5):619–628. doi:10.1111/cas.12909
30. Tassi RA, Gambino A, Ardighieri L, et al. FXYD5 (Dysadherin) upregulation predicts shorter survival and reveals platinum resistance in high-grade serous ovarian cancer patients. Br J Cancer. 2019;121(7):584–592. doi:10.1038/s41416-019-0553-z
31. Zhang M, Zhao L. CKAP2 promotes ovarian cancer proliferation and tumorigenesis through the FAK-ERK pathway. DNA Cell Biol. 2017;36(11):983–990. doi:10.1089/dna.2017.3876
32. Sima LE, Yakubov B, Zhang S, et al. Small molecules target the interaction between tissue transglutaminase and fibronectin. Mol Cancer Ther. 2019;18(6):1057–1068. doi:10.1158/1535-7163.MCT-18-1148
33. Zeng B, Zhou M, Wu H, Xiong Z. SPP1 promotes ovarian cancer progression via Integrin beta1/FAK/AKT signaling pathway. Onco Targets Ther. 2018;11:1333–1343. doi:10.2147/OTT.S154215
34. Szklarczyk D, Gable AL, Lyon D, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47(D1):D607–D613. doi:10.1093/nar/gky1131
35. Agrawal R, Garg A, Malgulwar PB, Sharma V, Sarkar C, Kulshreshtha R. p53 and miR-210 regulated NeuroD2, a neuronal basic helix-loop-helix transcription factor, is downregulated in glioblastoma patients and functions as a tumor suppressor under hypoxic microenvironment. Int J Cancer. 2018;142(9):1817–1828. doi:10.1002/ijc.31209
36. Nourmohammadi S, Aung TN, Cui J, et al. Effect of compound kushen injection, a natural compound mixture, and its identified chemical components on migration and invasion of colon, brain, and breast cancer cell lines. Front Oncol. 2019;9:314. doi:10.3389/fonc.2019.00314
37. Aasen T, Sansano I, Montero MA, et al. Insight into the role and regulation of gap junction genes in lung cancer and identification of nuclear Cx43 as a putative biomarker of poor prognosis. Cancers. 2019;11(3):320. doi:10.3390/cancers11030320
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.