")
Back to Journals » Breast Cancer: Targets and Therapy » Volume 10
A combination of p53-activating APR-246 and phosphatidylserine-targeting antibody potently inhibits tumor development in hormone-dependent mutant p53-expressing breast cancer xenografts
Authors Liang Y, Mafuvadze B , Besch-Williford C, Hyder SM
Received 7 November 2017
Accepted for publication 3 January 2018
Published 22 March 2018 Volume 2018:10 Pages 53—67
DOI https://doi.org/10.2147/BCTT.S156285
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Pranela Rameshwar
Yayun Liang,1 Benford Mafuvadze,1 Cynthia Besch-Williford,2 Salman M Hyder1
1Deparment of Biomedical Sciences and Dalton Cardiovascular Research Center, Columbia, MO, USA; 2IDEXX BioResearch, Columbia, MO, USA
Background: Between 30 and 40% of human breast cancers express a defective tumor suppressor p53 gene. Wild-type p53 tumor suppressor protein promotes cell-cycle arrest and apoptosis and inhibits vascular endothelial growth factor–dependent angiogenesis, whereas mutant p53 protein (mtp53) lacks these functions, resulting in tumor cell survival and metastasis. Restoration of p53 function is therefore a promising drug-targeted strategy for combating mtp53-expressing breast cancer.
Methods: In this study, we sought to determine whether administration of APR-246, a small-molecule drug that restores p53 function, in combination with 2aG4, an antibody that targets phosphatidylserine residues on tumor blood vessels and disrupts tumor vasculature, effectively inhibits advanced hormone-dependent breast cancer tumor growth.
Results: APR-246 reduced cell viability in mtp53-expressing BT-474 and T47-D human breast cancer cells in vitro, and significantly induced apoptosis in a dose-dependent manner. However, APR-246 did not reduce cell viability in MCF-7 breast cancer cells, which express wild-type p53. We next examined APR-246’s anti-tumor effects in vivo using BT-474 and T47-D tumor xenografts established in female nude mice. Tumor-bearing mice were treated with APR-246 and/or 2aG4 and tumor volume followed over time. Tumor growth was more effectively suppressed by combination treatment than by either agent alone, and combination therapy completely eradicated some tumors. Immunohistochemistry analysis of tumor tissue sections demonstrated that combination therapy more effectively induced apoptosis and reduced cell proliferation in tumor xenografts than either agent alone. Importantly, combination therapy dramatically reduced the density of blood vessels, which serve as the major route for tumor metastasis, in tumor xenografts compared with either agent alone.
Conclusion: Based on our findings, we contend that breast tumor growth might effectively be controlled by simultaneous targeting of mtp53 protein and tumor blood vessels in mtp53-expressing cancers.
Keywords: breast cancer, blood vessel targeting agent, p53, APR-246, apoptosis, angiogenesis
Introduction
Approximately 200,000 new cases of breast cancer are detected every year in the USA and 40,000 women die of the disease annually.1 Because most deaths occur following the emergence of drug-resistant tumor cells and metastasis, it is imperative that we develop more effective treatment strategies against breast cancer. Wild-type p53 tumor suppressor protein (wtp53) promotes cell cycle arrest and apoptosis and inhibits vascular endothelial growth factor (VEGF)-dependent angiogenesis, a process essential for the rapid tumor growth that leads to metastasis and patient death.2–6 However, between 30% and 40% of all human breast cancers contain a mutated form of p53, termed mtp53.7,8 Most mutations occur in the DNA-binding domain of p53, preventing its normal regulation of the p53-dependent genes involved in apoptosis/cell cycle arrest and/or angiogenesis. Unrestrained angiogenesis results in increased blood vessel formation, facilitating rapid tumor growth and metastasis, as well as possible development of resistance to chemotherapeutic drugs.9,10 Proper functioning of wtp53 also restricts the self-renewal properties of stem cells and suppresses the epithelial-to-mesenchymal transition, which is vital for initiation of metastasis.11–13 Many wtp53 functions can be restored in p53-defective tumor cells by reactivating mtp53. These include suppression of VEGF, which, if left unfettered, promotes breast cancer progression and leads to increased mortality.14–18
APR-246 has recently been developed as a small-molecule drug that is converted to the Michael acceptor methylene quinuclidinone that binds covalently to cysteines in p53, leading to refolding and restoration of wild-type p53 function.19,20 APR-246 has entered Phase I clinical trials for the treatment of leukemia and prostate cancer.19–21 However, its ability to control the growth and metastasis of breast cancer cells is not known. Because tumor growth and survival depend on a tumor-specific vasculature,22,23 the vasculature presents a viable molecular target for antitumor therapy.24–26 Vascular-targeting agents used to selectively treat cancer swiftly disrupt the tumor vasculature by inducing ischemia and causing extensive hemorrhagic necrosis. Cell death occurs as an indirect consequence of tumor cells being deprived of their blood supply. Drugs that target the vasculature could provide an alternative means of treating tumors that are resistant to standard antiproliferative therapy.25,26 Vascular-targeting drugs that are under development fall into two main categories: those that are ligand based and agents that are small molecules. Both types of drug exhibit impressive antitumor properties in mouse breast cancer models.24 In preclinical studies, two monoclonal antibodies (3G4 and 2aG4) that target anionic phospholipids on tumor blood vessels and disrupt tumor vasculature have exhibited potent antitumor effects in several models.27–31
Using agents aimed at different tumor processes in a combination therapeutic approach may represent an innovative and effective way to treat breast cancer. Herein, we investigated the effects of APR-246, which reactivates mtp53, and 2aG4, which destroys tumor blood vessels, on xenografts derived from two different human breast cancer cell lines, BT-474 and T47-D. Both cell lines express mtp53 and estrogen and progesterone receptors; BT-474 cells also express abundant levels of Her-2-neu.32 T47-D is a breast adenocarcinoma cell line that expresses medium levels of Her-2-neu,32 and while T47-D cells do express mtp53, xenografts derived from this cell line grow more slowly than those derived from BT-474 cells.18,33 Hormone-dependent breast cancers frequently give rise to tumors that are resistant to anti-hormones such as tamoxifen.34 We found that administration of a combination of APR-246 and 2aG4 in an in vivo breast cancer tumor xenograft model inhibited tumor growth and markers of angiogenesis more effectively than APR-246 or 2aG4 alone. Our observations support the notion that a combination of these two agents is an effective form of therapy for combating hormone-dependent breast cancer.
Materials and methods
Cell lines and culture
BT-474, T47-D, and MCF-7 breast cancer cells were obtained from ATCC (Manassas, VA, USA). All cells were grown in phenol red-free DMEM/F12 medium (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich, St Louis, MO, USA). Medium for MCF-7 cells was supplemented with 10 mg/mL insulin (Thermo Fisher Scientific) for regular growth. All cells were grown in 100×20 mm tissue culture dishes and harvested with 0.05% trypsin-EDTA (Thermo Fisher Scientific).
Reagents
APR-246 (PRIMA-1MET) and PRIMA-1 were purchased from Tocris Bioscience (Bristol, UK) for in vitro studies. For in vivo experiments, APR-246 was provided by APREA AB (Solina, Sweden). The 2aG4, a mouse IgG2a monoclonal antibody that binds directly to phosphatidylserine on the tumor blood vessels, was provided by Dr Philip Thorpe from University of Texas Southwestern Medical Center (Dallas, TX, USA) and by Peregrine Pharmaceuticals, Inc. (Tustin, CA, USA). Binding of 2aG4 to anionic phospholipids relies on a 50 kD bovine plasma glycoprotein, β2-glycoprotein 1. Consequently, we mixed 2aG4 at a 1:1 ratio with β2-glycoprotein 1 to enhance binding of 2aG4 to exposed anionic phospholipids on the endothelial cell surface.30 The IgG2a mouse anti-colchicine monoclonal antibody C44 was used as a negative control for 2aG4. All other antibodies were obtained from Santa Cruz Biotechnology (Dallas, TX, USA). Antibodies for immunohistochemistry and Western blotting and their source are listed in the relevant sections below.
Activation of p53 DNA binding
The TransAM p53 Transcription Factor Assay kit (Active Motif, Carlsbad, CA, USA) was employed to analyze the conformation and activation of p53.16 Briefly, 96-well plates were coated with an oligonucleotide that contained a p53 consensus DNA-binding site. Nuclear extracts were prepared from BT-474 cells using an extraction kit (Active Motif), and 2.5 µg was incubated with the aforementioned oligonucleotide in the 96-well plates. Addition of anti-p53 antibody and a secondary antibody conjugated to horseradish peroxidase (both antibodies at dilutions of 1:1000) facilitated the detection of bound p53. A Spectra MAX 190 Microplate Reader (Molecular Devices LLC, Sunnyville, CA, USA) was used to measure the developed color at 450 nm. The background OD was determined using a 20-fold excess of free oligonucleotide. This value was subtracted from total binding to generate specific binding values. MCF-7 nuclear extract treated with H2O2 was used as a positive control.
Cell viability assay
Cell viability was measured by the sulforhodamine B assay (Sigma-Aldrich),35,36 which quantitates the protein content of surviving cells as an index of cell growth and viability. Cells (4–8×103) were seeded in 96-well plates in 100 µL culture medium and incubated overnight at 37°C in an atmosphere of 5% CO2. The medium was subsequently removed, cells were washed with DMEM/F12 medium and treated with different concentrations of APR-246 or PRIMA-1 for 24 or 48 hours in DMEM/F12 supplemented with 5% FBS. Surviving or adherent cells were fixed in situ by removing the growth medium, adding 100 µL PBS and 100 µL cold 50% trichloroacetic acid, and incubating at 4°C for 1 hour. Cells were washed with ice-cold water, dried, and stained in 50 µL 4% sulforhodamine B for 8 minutes at room temperature (RT). Unbound dye was removed by washing five times with cold 1% acetic acid. Cells were dried at RT and the bound stain solubilized in 150 µL 10 mM Tris. Sample absorbance was measured at 520 nm using a microplate reader as described above. Six wells were used for each concentration, and experiments were performed three times.
Cell apoptosis and death assay
Apoptosis and cell death in BT-474 breast cancer cells were assessed using a detection kit obtained from Biovision Research Products (Mountain View, CA, USA). BT-474 cells were grown overnight in six-well plates containing DMEM/F12 medium supplemented with 10% FBS. The medium was removed, and cells were washed and then treated for 24 hours with PBS or different concentrations of APR-246 (5, 10, or 20 µM) in 5% FBS DMEM/F12 medium. Cells were harvested with Accutase and stained with Annexin V-fluorescein isothiocyanate (FITC) and propidium iodide (PI). FITC reveals the early stages of apoptosis, while PI detects DNA fragmentation or cell death. The percentage of Annexin V-FITC–positive/PI-positive cells was ascertained by measuring the fluorescence of 10,000 cells using a FACScan flow cytometer (Becton Dickinson, San Jose, CA, USA). Experiments were performed at least twice.
Western blotting
Cultured BT-474 cells were treated with 25 or 50 µM APR-246 in 100 mm culture dishes for 3 or 12 hours. After treatment, the cells were washed with cold PBS containing phosphatase inhibitors and harvested by gentle scraping with a cell lifter. Whole-cell extracts were then prepared using a nuclear extraction TransAm kit (Active Motif). In brief, cells were centrifuged at 1000 rpm for 5 minutes and pellets resuspended in complete lysis buffer containing 1 mM dithiothreitol and 1% protease inhibitor cocktail. Samples were shaken on ice for 30 minutes and then centrifuged at 14,000 rpm in an Eppendorf centrifuge at 4°C. Supernatants were transferred to microcentrifuge tubes, aliquoted, and stored at −80°C. For Western blotting, samples containing 45 µg protein were separated in a NuPAGE 10% Bis-Tris Gel (Thermo Fisher Scientific). Electrophoresis was performed at 120 V for 1.5 hours using NuPAGE MES-SDS Running Buffer (Thermo Fisher Scientific). Separated proteins were transferred to polyvinylidene difluoride membranes (Bio-Rad Laboratories, Hercules, CA, USA) at 25 V for 25 minutes. Blots were blocked at RT for 1 hour with 5% non-fat dry milk in Tris-buffered saline with 0.1% Tween 20 (TBS-T) buffer, after which they were incubated for 2 hours at RT with primary antibodies against Bax or p21 (1:200 dilution; Santa Cruz Biotechnology) or caspase-3 (1:300 dilution; R&D Systems, Minneapolis, MN, USA). After three washes with TBS-T, blots were incubated with secondary antibody for 1 hour at RT and then washed seven times with TBS-T. Immunoreactive bands were visualized using an ECL Plus detection kit (Amersham, Pharmacia Biotech, Arlington Heights, IL, USA). Membranes were stripped and re-probed using β-actin antibody (Sigma-Aldrich), which was used as a control for protein loading.
Animals
All animal studies were approved by the Animal Care and Use Committee at the University of Missouri (Columbia, MO, USA). The study adhered to the guidelines of the US Government Principles for the Utilization and Care of Vertebrate Animals Used in Testing, Research, and Training. Female athymic nude (nu/nu) mice, 5–6 weeks old and weighing 20–22 g, were purchased from Harlan Sprague Dawley, Inc., (Indianapolis, IN, USA) and housed in a laminar air-flow cabinet under specific pathogen-free conditions. All facilities were approved by the American Association for Accreditation of Laboratory Animal Care in accordance with the current regulations and standards of the United States Department of Agriculture, the Department of Health and Human Services, and the National Institutes of Health.
Treatment of nude mice bearing breast tumor xenografts with APR-246 and 2aG4
Nude mice were inoculated on the internal side of the neck with pellets containing 17-β-estradiol (E2; 1.7 mg/pellet, 60-day release) or placebo (both from Innovative Research of America, Sarasota, FL, USA) at 24–48 hours before inoculation with tumor cells. Cultured BT-474 or T47-D breast cancer cells were harvested by trypsinization and washed twice with DMEM/F12 medium. Breast cancer cells (5×106 cells) were resuspended in 0.15 mL Matrigel (BD Biosciences, Bedford, MA, USA)/DMEM/F12 medium (4:1 v/v) and injected subcutaneously into both flanks of nude mice. Every 3 days, digital calipers were used to measure tumor size. Tumor volumes were calculated by the formula (length × width × height)×π/6.17,18 Treatments commenced when the tumor volumes reached around 100–125 mm3. The treatment protocol is shown in Figure 1. Animals were assigned to four groups (control, APR-246, 2aG4, and APR-246+2aG4) with 8–10 mice/group. One group of animals received APR-246 alone (50 mg/kg/day for animals with BT-474 xenografts, 100 mg/kg/day for animals with T47-D xenografts) by intravenous (iv) injection into the tail vein. The second group received 2aG4 (100 µg/mouse/day) by intraperitoneal (ip) injection, while the third group was given both APR-246 and 2aG4 (50 mg/kg+100 µg/mouse/day for BT-474 model, 100 mg/kg+100 µg/mouse/day for T47-D model) by iv/ip injection, respectively. In both models, treatment was given every other day for a total of 16 treatments for the BT-474 model and 12 treatments for the T47-D model. In the APR-246 alone and APR-246+2aG4 treatment groups, the first treatments consisted of a double dose of APR-246. The last treatment was administered 18 hours prior to harvesting of tumors. The control group received control antibody C44 (100 µg/mouse/day, ip) and/or PBS (0.1 mL/day, iv). Animal weights were recorded twice weekly throughout the study. At the termination of the experiment, mice were sacrificed and tumors harvested. Fresh tumor tissues were immediately placed in 4% paraformaldehyde solution for immunohistochemical analysis or frozen in liquid nitrogen for future studies.
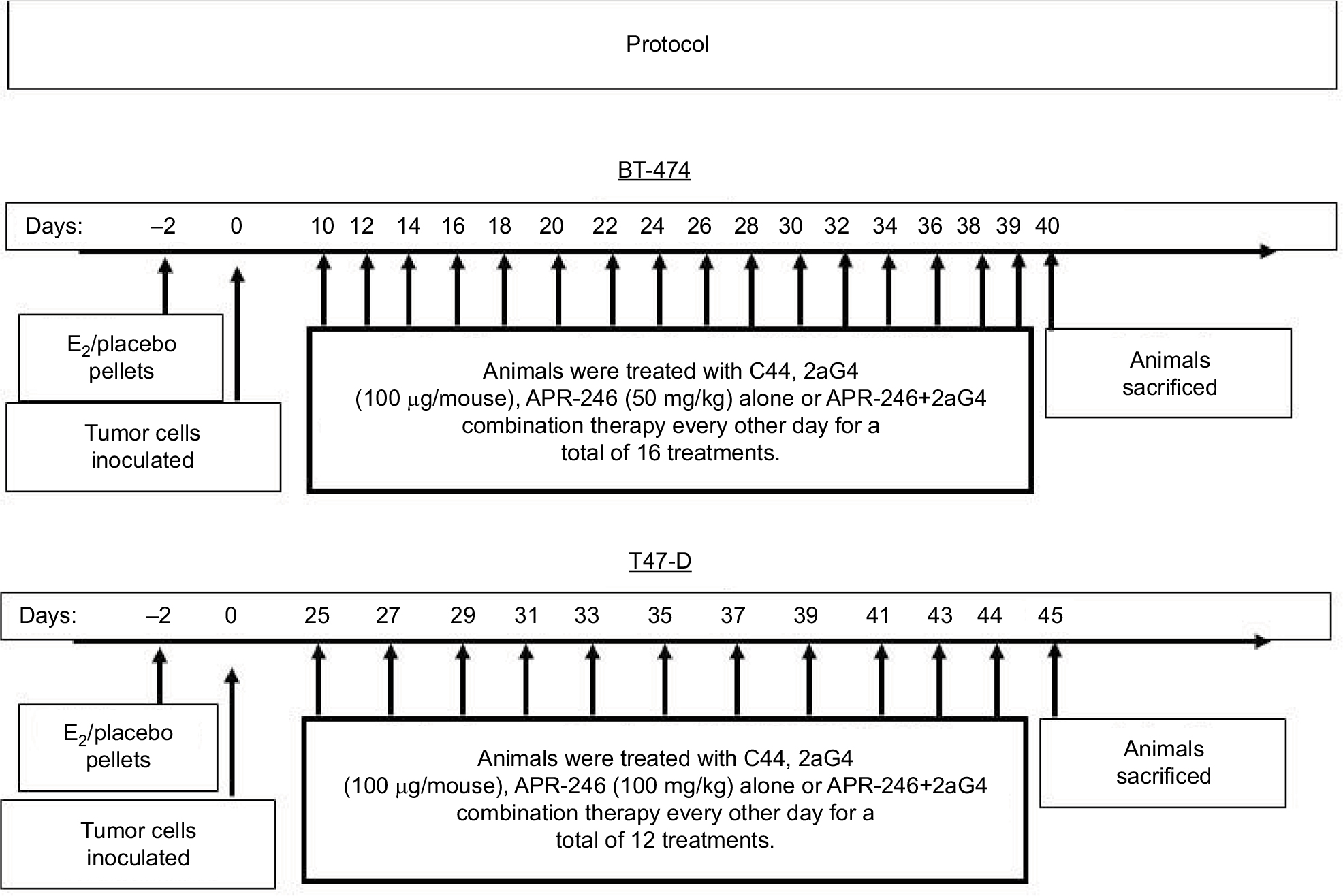 | Figure 1 Protocol for treatment of tumor xenografts in nude mice with APR-246 and 2aG4. |
Routine histochemical and immunohistochemical analysis of tumor xenograft tissues
Tumor tissue was fixed overnight in 4% paraformaldehyde prior to being infiltrated with paraffin and embedded. After mounting on ProbeOn Plus microscope slides (Fisher Scientific Inc., Pittsburgh, PA, USA), sections (5 µm) were stained with hematoxylin–eosin and examined under a light microscope for cellularity. For immunohistochemical analysis, unstained paraffin sections were dewaxed in xylene, rehydrated through graded concentrations of ethanol, rinsed in distilled water, and subjected to heat-induced epitope retrieval in 10 mM citrate buffer, pH 6.0 (Aligent, Carpenteria, CA, USA). Slides were treated with 3% H2O2 in absolute methanol to inactivate endogenous peroxidase activity, washed three times in PBS, and incubated in blocking buffer with 5% bovine serum albumin for 20 minutes. Sections were treated with the following primary antibodies for 60 minutes at RT: anti-VEGF antibody (1:100 dilution of a rabbit anti-VEGF polyclonal antibody [sc-152; Santa Cruz Biotechnology]); anti-CD31 (1:100 dilution of a rabbit anti-CD31 polyclonal antibody [Ab28364; Abcam, Cambridge, MA, USA]); or anti-Ki67 (1:300 dilution of a rabbit anti-Ki67 polyclonal antibody [RB1510-P; Thermo Fisher Scientific]). Sections were washed and sequentially incubated with a horseradish peroxidase–labeled polymer conjugated with anti-rabbit antibodies (Envision; Aligent, Santa Clara, CA, USA). Bound antibodies were visualized with 3,3′-diaminobenzidine tetrahydrochloride (0.05% with 0.015% H2O2 in PBS; Aligent). Sections were counterstained with Mayer’s hematoxylin, dehydrated, cleared, and cover-slipped for examination by light microscopy.
Terminal dUTP-mediated nick end labeling (TUNEL) assay
A TUNEL staining kit (Roche Applied Science, Madison, WI, USA) was used to detect fragmented DNA of apoptotic tumor cells. Briefly, dewaxed paraffin sections of tumors were rehydrated through graded ethanol solutions with a final immersion in water. Sections were heated for 1 minute in a microwave in 0.1 M citrate buffer, pH 6.0 and cooled before being incubated in 3% H2O2 for 5 minutes. After being rinsed in PBS, they were then incubated for 5 minutes with a peptide-based block solution (Background Buster; Innovex Biosciences, Richmond, CA, USA). After rinsing, a reaction medium containing equilibration buffer, nucleotide mix, and terminal deoxynucleotidyl transferase was added. Sections were then incubated in a dark humidified chamber for 1 hour at 37°C. The reaction was terminated by immersing sections in three washes of PBS. Sections were then incubated with an anti-fluorescein antibody conjugated to horseradish peroxidase for 30 minutes at 37°C, rinsed with PBS, and incubated with 3,3′-diaminobenzidine tetrahydrochloride (0.016% with 0.05% H2O2 in PBS). After rinsing, sections were dehydrated in graded ethanol solution and xylene and cover-slipped. Images were captured with a DP-70 digital camera (Olympus America, Inc., Center Valley, PA, USA) mounted on a Zeiss light microscope. DP Manager software was utilized for subsequent analysis. Apoptotic index of tumor sections was calculated by analyzing 3000 cells from each tumor in four to five fields from eight to 10 sections, examining only one section from each tumor. Necrotic areas were avoided to reduce error in the results.
Quantification of VEGF and tumor blood vessel immunohistochemical staining
VEGF expression in immunohistochemically stained sections of tumor tissue was quantitated by measuring the immunolabeled pixels in standardized digital images, photographed at 20× magnification, using the Fovea Pro 3.0 imaging program (Reindeer Graphics, Asheville, NC, USA). The distribution of VEGF was determined on every cell in each tumor image, of which 8–12 were analyzed per treatment group from three to five tumors. Data are reported as the mean number of labeled pixels per group. Quantitation of blood vessels was achieved by photographing CD31-labeled tissue sections from three to four tumors per treatment group at 20× magnification. The total number of vessels in these digital images was counted in 8–15 fields per treatment group, each field representing ~0.39 mm2. Vessel density was calculated as vessel number per field.
Statistical analysis
Differences among groups were tested using one-way analysis of variance (ANOVA). The assumption of the ANOVA was examined, and if necessary, a non-parametric measure based on ranks was used. If normality failed, Kruskal–Wallis one-way ANOVA on ranks was used in place of regular ANOVA. In cases where a significant effect was shown by ANOVA (F-ratio, P<0.05), the Student–Newman–Keuls multirange test was employed to compare the means of the individual groups. SigmaPlot software version 12.5 was used for statistical analysis. Data are reported as mean ± standard error of the mean. For all comparisons, P<0.05 was considered significant.
Results
APR-246 selectively reduces the viability of mtp53-expressing breast cancer cells
Treatment of mtp53-expressing BT-474 and T47-D cells with PRIMA-1 or APR-246 dose dependently reduced cell viability. Half-maximal effects occurred in response to concentrations between 5 and 40 µM, depending on the length of exposure (24 or 48 hours) of cells to the two compounds (Figure 2; Table 1). APR-246 more potently reduced the viability of both cell lines than PRIMA-1. In contrast, neither APR-246 nor PRIMA-1 affected the viability of MCF-7 breast cancer cells (which express wtp53 protein), unless they were exposed to toxic levels of the compound. These findings indicate that high concentrations of APR-246 or PRIMA-1 may exert a p53-independent effect on viability in cells that express wtp53.
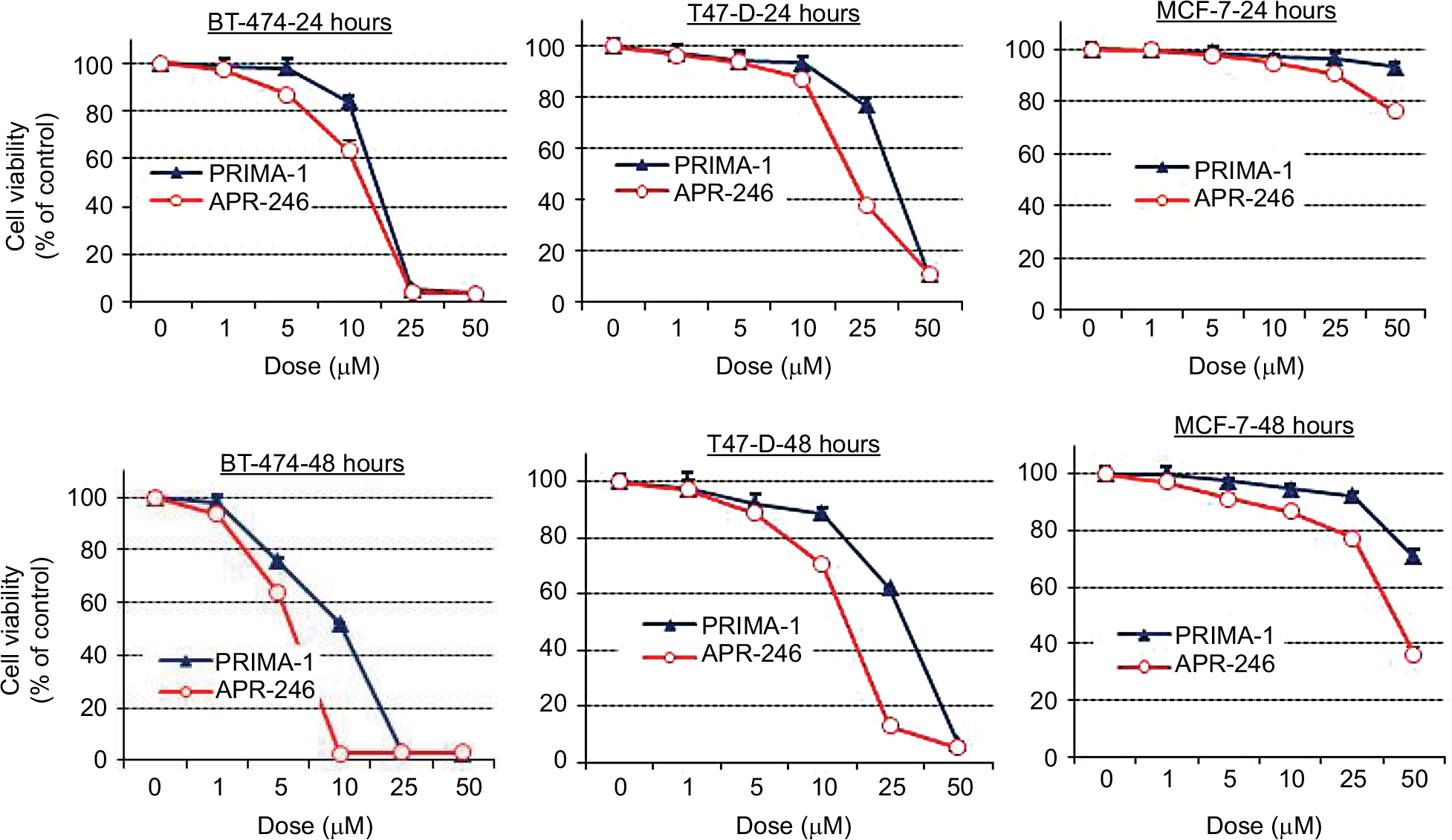 | Figure 2 APR-246 and PRIMA-1 reduce cell viability of mtp53-expressing breast cancer cells. Notes: Cells (4–8×103) were grown in DMEM/F12 medium with 5% FBS and incubated with vehicle (O) or increasing concentrations of APR-246 or PRIMA-1 for 24 or 48 hours. Cell viability was measured using SRB assays. Data are shown as the mean±SEM from six different determinations. Abbreviations: FBS, fetal bovine serum; mtp53, mutated form of p53; SEM, standard error of the mean; SRB, sulforhodamine B. |
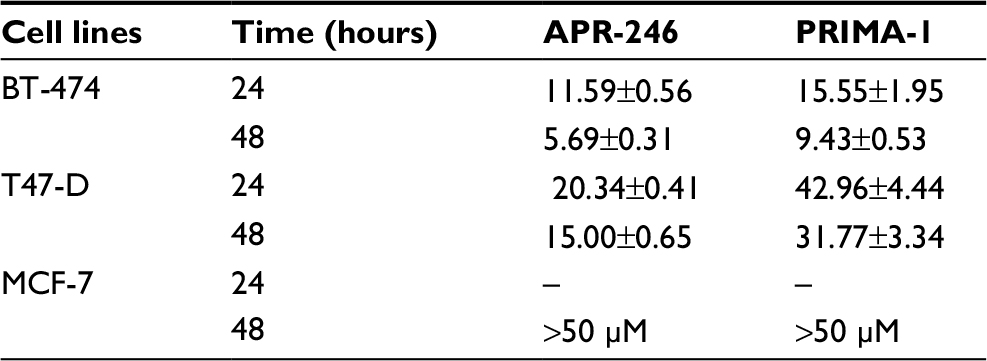 | Table 1 IC50s (in mM) of APR-246 and PRIMA-1 |
APR-246 converts mtp53 protein into a wtp53 DNA-binding form in BT-474 breast cancer cells
As BT-474 cells were more responsive to APR-246, we used this mtp53-expressing cell line to determine whether APR-246 converts mtp53 protein into a form that can bind DNA. APR-246 significantly increased DNA binding in BT-474 cells compared with that observed with untreated BT-474 cells (Figure 3), indicating that APR-246 converted the low DNA-binding form of mtp53 in BT-474 cells into a form that bound DNA.
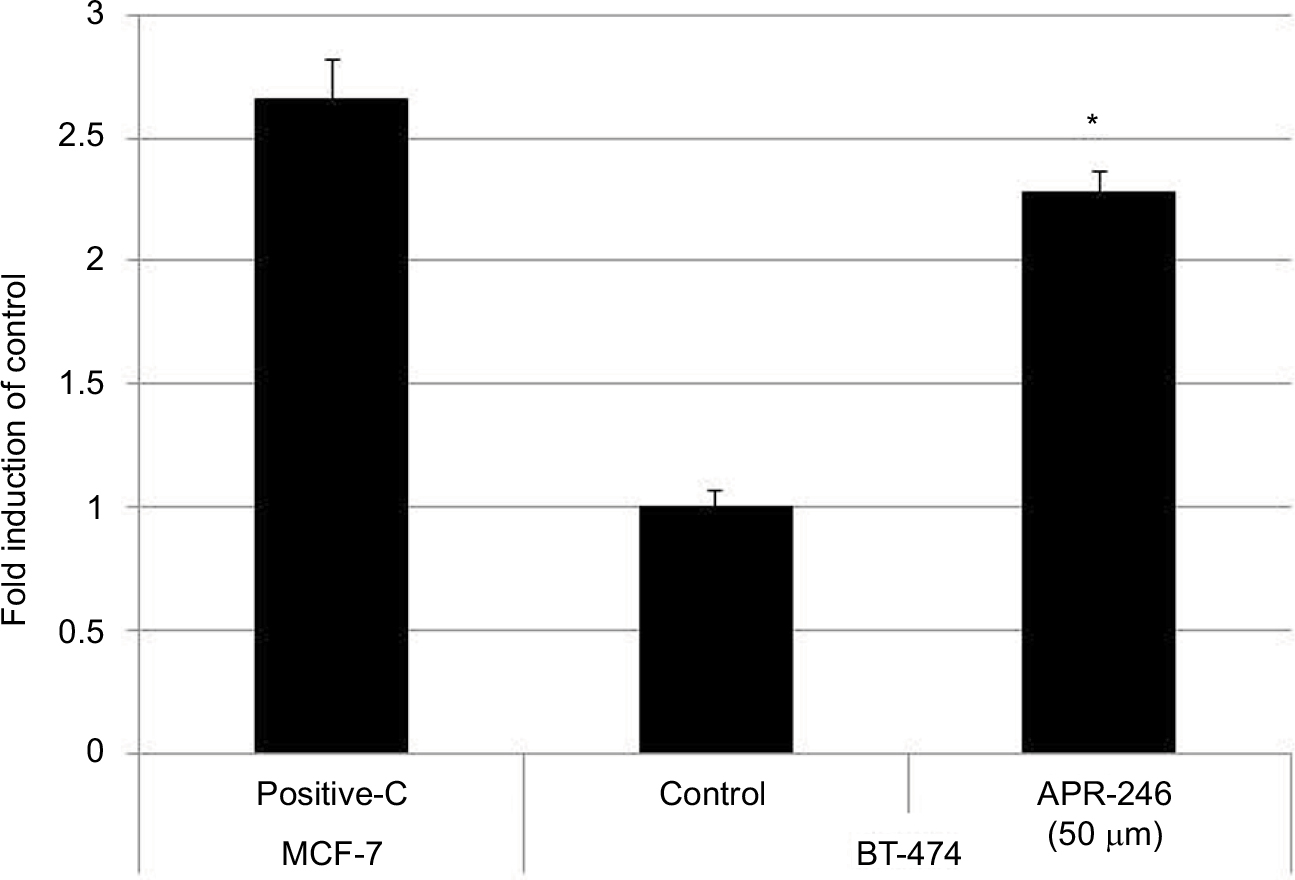 | Figure 3 APR-246 increases DNA binding of mtp53 in BT-474 breast cancer cells. Notes: Cells were grown overnight in DMEM/F12 medium supplemented with 5% FBS. Cells were then washed once with PBS and treated with 50 μM APR-246 for 1 hour, prior to harvest by scraping and subsequent preparation of nuclear extracts. TransAM assays were performed using nuclear extracts (2.5 μg), with each sample being analyzed in triplicate. Activation of DNA-binding ability was compared with the vehicle-treated group (control), whose value was set at 1. Nuclear extract of wtp53-expressing MCF-7 cells (provided with the TransAM kit) was used as a positive control (Positive-C). Data are shown as the mean±SEM from three different determinations. *Significantly different from untreated controls (P<0.05; ANOVA). Abbreviations: ANOVA, analysis of variance; FBS, fetal bovine serum; mtp53, mutated form of p53; SEM, standard error of the mean. |
APR-246 induces apoptosis and cell death in BT-474 breast cancer cells
We next conducted studies to ascertain whether APR-246 causes apoptosis in BT-474 cells, which express mtp53. Flow cytometry analysis showed that in BT-474 cells exposed for 24 hours to APR-246, apoptosis and cell death were significantly induced in a dose-dependent manner (Figure 4A). We then treated cells for either 3 or 12 hours with 25 or 50 µM APR-246, and performed Western blot analysis of whole-cell extracts to study the expression of mitochondria-dependent components of the apoptotic pathway. APR-246 treatment elevated Bax and p21 protein expression and caused cleavage of caspase-3 (Figure 4B). Taken together, these studies suggest that APR-246 induces apoptosis in BT-474 cells by activating a signaling pathway that is mitochondria dependent.
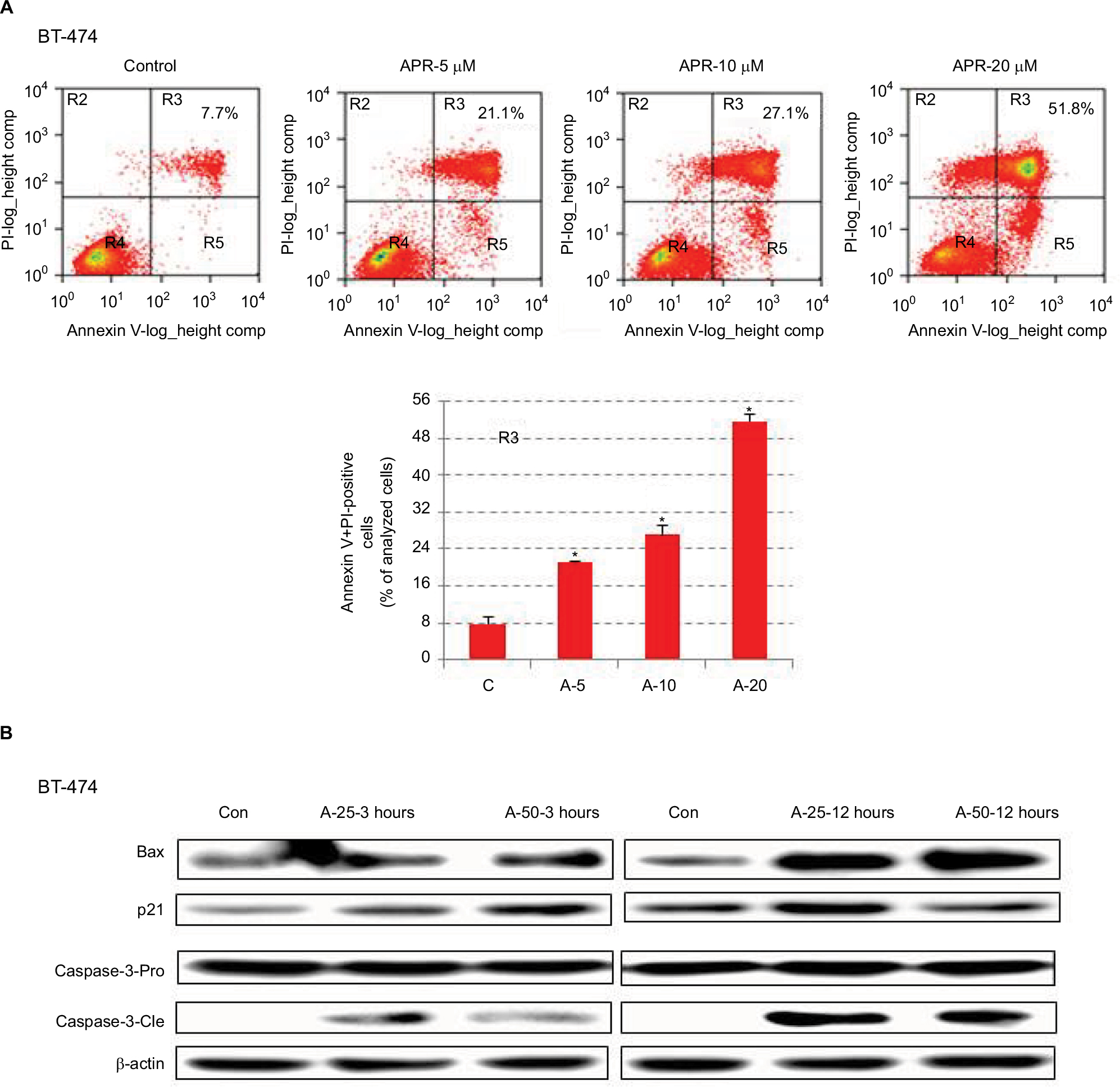 | Figure 4 APR-246 induces apoptosis in BT-474 breast cancer cells. Notes: (A) Cells were grown for 24 hours in DMEM/F12 medium containing 5% FBS and then treated with 0 (vehicle control), 5, 10, or 20 µM APR-246 (A-5, A-10, A-20) for 24 hours. Cells were stained with Annexin V-FITC (BioVision) and PI, and the percentage of Annexin V+PI-positive cells (quadrant R3) was quantified by FACScan flow cytometry. Representative data are shown; inset values represent the percentage of Annexin V+PI-positive cells in the example shown. Bar graph presents the mean±SEM from three determinations. *Significantly different from control (C) (P<0.05; ANOVA). (B) Cells were treated with 0 (vehicle control; Con), 25, or 50 μM APR-246 for 3 or 12 hours. After cell harvest, whole-cell lysates (45 μg protein) were analyzed by Western blot for Bax and p21 protein expression and caspase-3 cleavage (caspase-3-Cle) from the caspase-3-Pro band. β-actin was used as a loading control. Abbreviations: ANOVA, analysis of variance; Con, control; FBS, fetal bovine serum; FITC, fluorescein isothiocyanate; PI, propidium iodide; SEM, standard error of the mean. |
A combination of APR-246 and the phosphatidylserine-targeting antibody 2aG4 has additive effects with respect to inhibiting the growth of BT-474 and T47-D tumor xenografts in nude mice
BT-474 tumor xenografts were grown in nude mice. When the tumor volumes were ~100–125 mm3, we began treatment with APR-246 and/or 2aG4 monoclonal antibody, which targets phosphatidylserine residues on tumor vasculature (Figure 5A). We then monitored tumor volume for the next few weeks. In animals administered the control IgG C44 and PBS, tumors continued to grow. However, by the time treatment with 2aG4 or APR-246 ended, tumor growth (based on mean tumor volume) was significantly inhibited with both agents, by ~30% and 50%, respectively (Figure 5A, left panel). Importantly, a combination of APR-246 and 2aG4 had an additive effect on both inhibition of tumor growth (~65%) and tumor eradication (Figure 5A, right top panel) over the course of the study. Animals maintained their weight throughout the study (Figure 5A, right bottom panel), indicating that the treatment protocol was not toxic. Figure 5B shows examples of tumors in situ at the end of treatment (four left-most panels) and an example of complete tumor regression (right-most panel). Thus, treatment with APR-246 alone or in combination with 2aG4 effectively limited the growth of BT-474–derived xenografts, and completely eradicated some tumors.
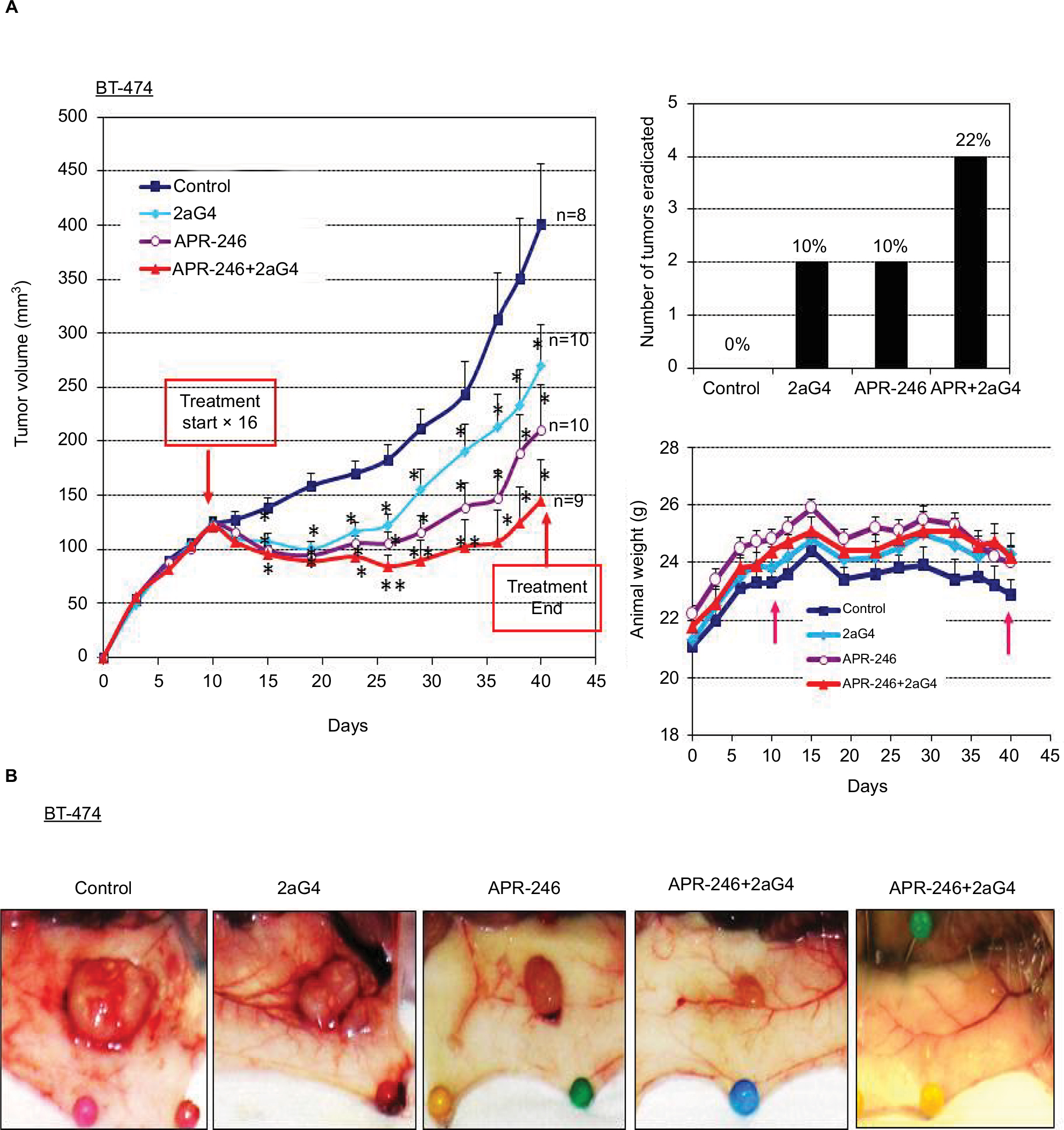 | Figure 5 APR-246 and 2aG4 exert additive inhibitory effects on BT-474 tumor xenograft growth in nude mice. Notes: Nude mice (n=8–10/group) were inoculated with an estrogen pellet 48 hours prior to injection of 5×106 BT-474 cells into both flanks. Tumors were measured every 3 days with a digital caliper to determine volume. (A) Left panel, growth curves for tumors, with points representing mean tumor volumes±SEM in each group of mice. *Significantly different from the control group; **significantly different from the control and 2aG4 groups (P<0.05; ANOVA). Top right panel, number of tumors completely eradicated at the end of the experiment. The number of tumors eradicated was converted to a percentage of the tumor total and presented above each bar in the graph. Bottom right panel, animal weight during treatment period. Arrows indicate treatment start and end points. Values represent mean±SEM. (B) Representative images of tumor size from each group at the end of the experiment. Abbreviations: ANOVA, analysis of variance; SEM, standard error of the mean. |
In a parallel study, we established T47-D tumor xenografts in nude mice and initiated treatment once the tumor volumes reached ~100 mm3 (Figure 6A). For T47-D xenografts, we used an increased dose of APR-246 (100 mg/kg/day) in both the single and combination treatment groups, because T47-D cells were relatively less sensitive to APR-246 than were BT-474 cells (Figure 2; Table 1). As with the BT-474 xenografts, T47-D tumors continued to grow in controls; however, tumor volumes were significantly reduced in all three treatment groups compared with the control group (Figure 6A). Mean tumor volumes were reduced compared with controls by 23%, 32%, and 70% following treatment with 2aG4, APR-246, or a combination of APR-246 and 2aG4, respectively (Figure 6A, left panel). Whereas no tumor eradication was observed in response to either single agent by the end of the study, combination APR-246 and 2aG4 treatment led to the destruction of 44% of tumors present (Figure 6A, top right panel). No toxicity, based on animal weight during the study, was observed (Figure 6A, lower right panel). Figure 6B shows examples of tumors in situ at the end of treatment, and illustrates two examples of complete tumor eradication following treatment with a combination of APR-246 and 2aG4 (right-most two panels).
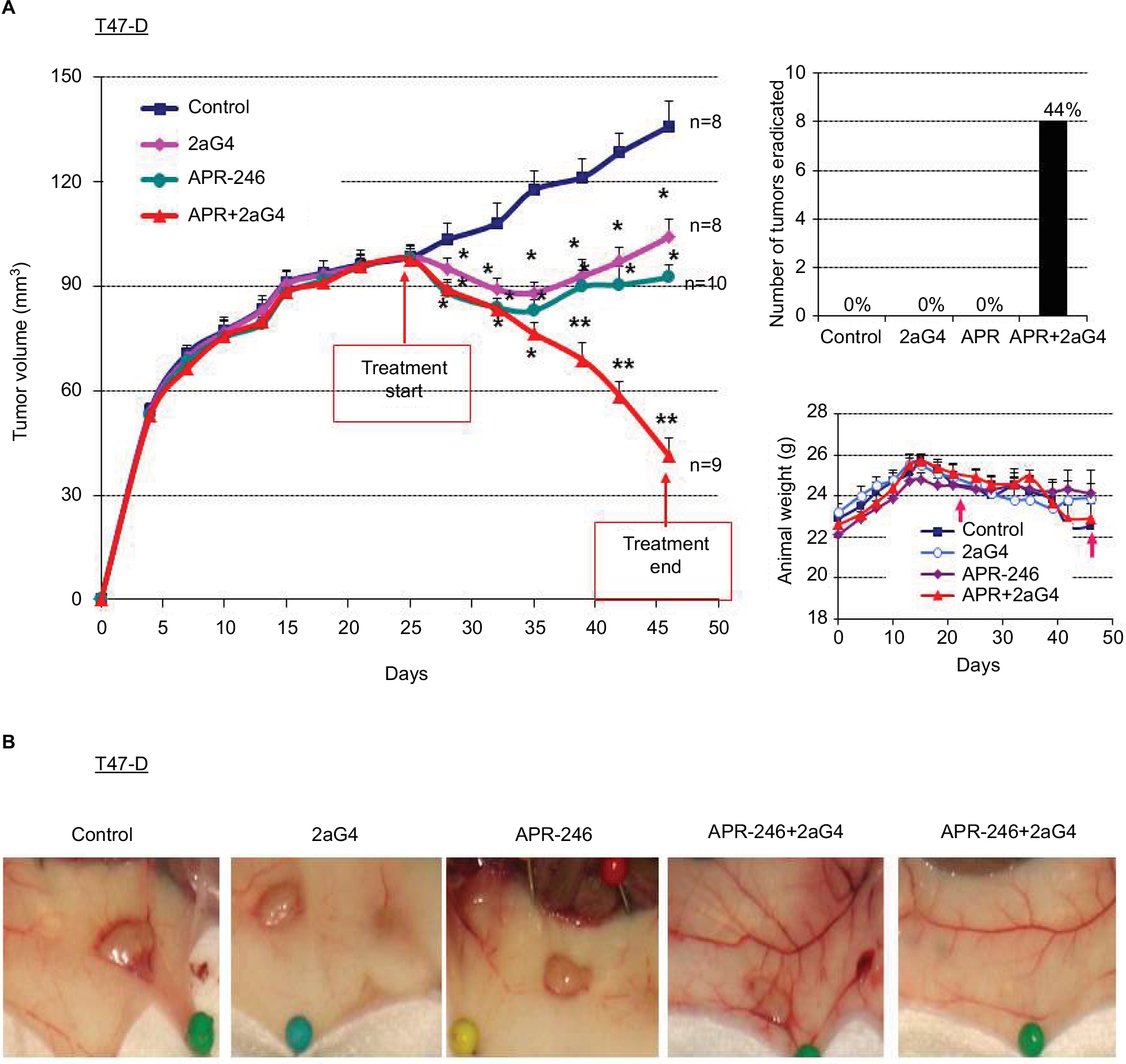 | Figure 6 APR-246 and 2aG4 exert additive inhibitory effects on T47-D tumor xenograft growth in nude mice. Notes: Nude mice (n=8–10/group) were inoculated with an estrogen pellet 48 hours prior to injection of 5×106 T47-D cells into both flanks. Tumors were measured every 3 days with a digital caliper to determine volume. (A) Left panel, growth curve for tumors, with points representing mean tumor volumes±SEM in each group of mice. *Significantly different from the control group; **significantly different from the control, APR-246, and 2aG4 groups (P<0.05; ANOVA). Top right panel, number of tumors eradicated at the end of the experiment. The number of tumors eradicated was converted to a percentage of the tumor total and presented above each bar in the graph. Right bottom panel, animal weight during the treatment period. Arrows indicate treatment start and end points. Values represent mean±SEM. (B) Representative images of tumor size from each group at the end of the experiment. Abbreviations: ANOVA, analysis of variance; SEM, standard error of the mean. |
APR-246 and 2aG4 combination treatment reduces cell proliferation and induces apoptosis in BT-474 and T47-D tumor xenografts in nude mice
We measured cell proliferation in tumor sections obtained from the treated tumors shown in Figures 5A and 6A using Ki67 as the proliferation marker. Both APR-246 and 2aG4 alone significantly reduced cell proliferation compared with controls in T47-D cells; however, in BT-474 cells, only APR-246 alone significantly reduced proliferation (Figure 7). When used in combination, the two agents significantly reduced proliferation in both BT-474 and T47-D cells compared with the effects of either agent alone (Figure 7).
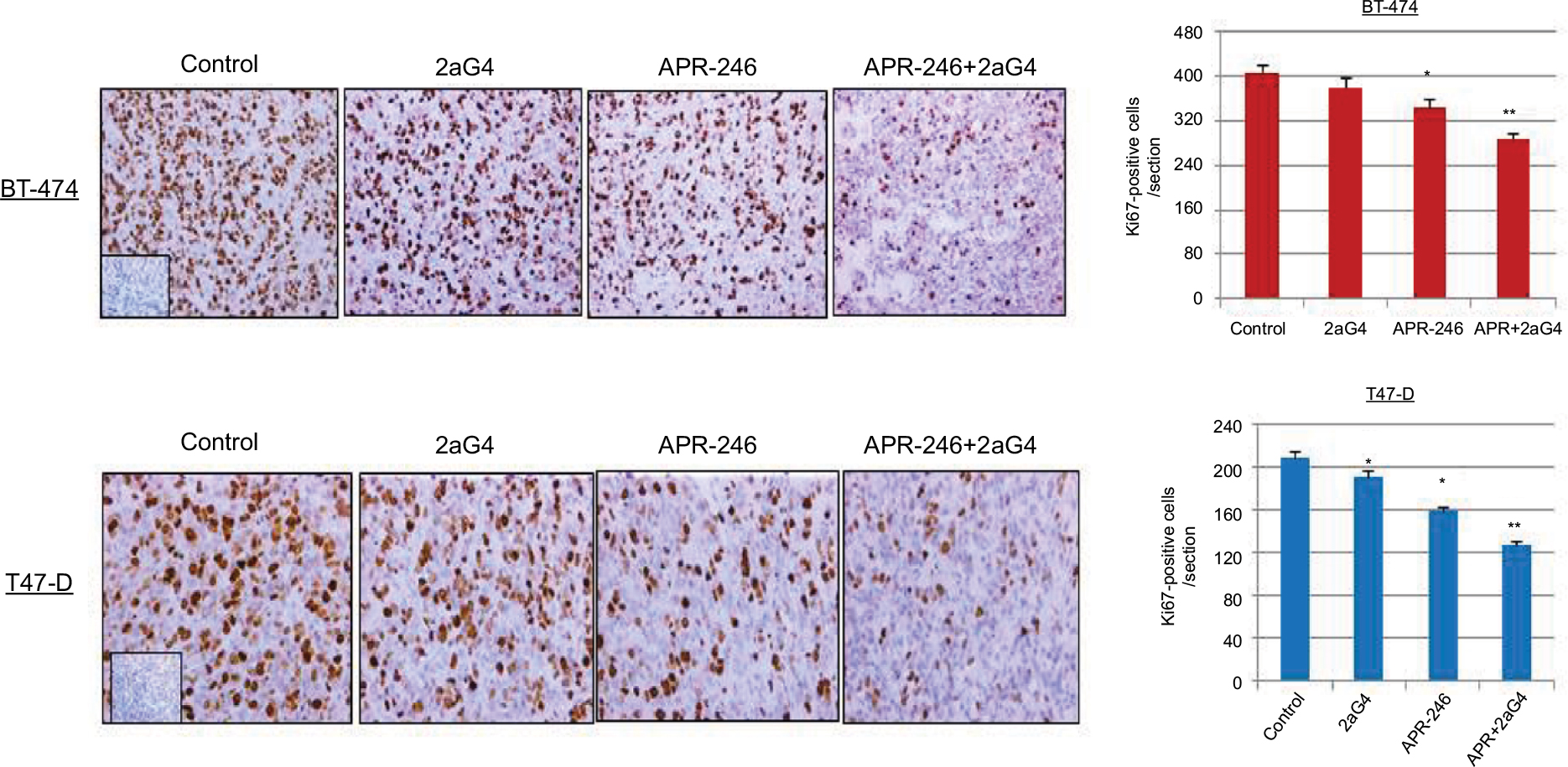 | Figure 7 APR-246 and 2aG4 combination treatment reduces the expression of Ki67 proliferation markers in breast tumor xenografts in nude mice. Notes: At the termination of the experiments tumor tissue sections were immunostained for Ki67 and the signal was quantified. Left, representative images of immunohistochemical staining. Right, bar graph represents quantitated Ki67 signal shown as the mean±SEM. *Significantly different from the control group; **significantly different from APR-246 and 2aG4 groups (P<0.05; ANOVA). Inserts represent negative controls. Abbreviations: ANOVA, analysis of variance; SEM, standard error of the mean. |
We next measured apoptosis in tumor sections obtained at the end of the experiments shown in Figures 5A and 6A using terminal dUTP-mediated nick end labeling (TUNEL) assays. In BT-474 tumors, 2aG4, APR-246, and a combination of the two significantly induced apoptosis compared with controls. Both APR-246 alone and a combination of APR-246+2aG4 more potently induced apoptosis compared with controls and animals given only 2aG4 (Figure 8). In T47-D–derived tumors, induction of apoptosis was significantly higher with all three treatments (2aG4, APR-246, or a combination of the two), as shown in Figure 8.
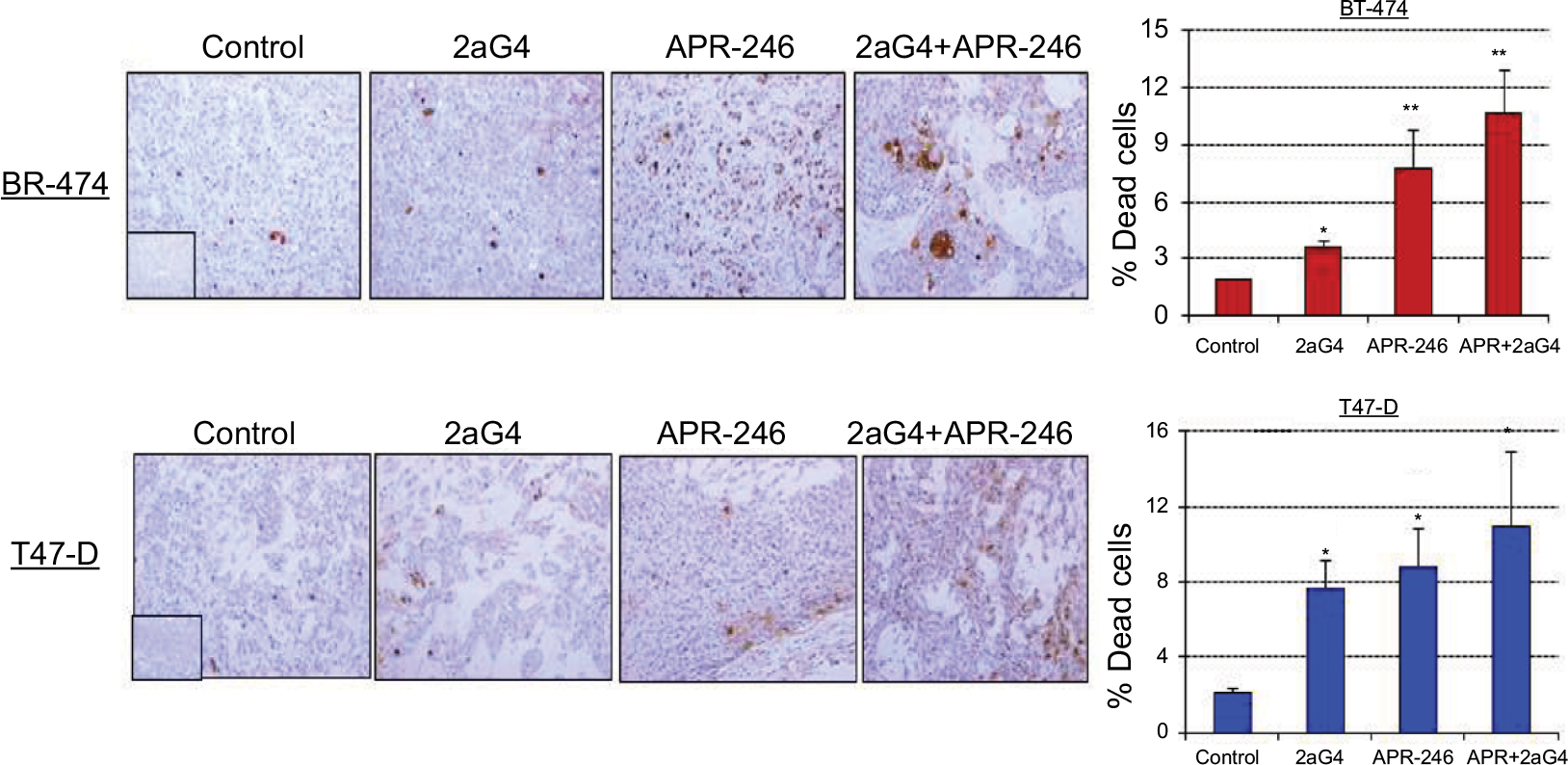 | Figure 8 APR-246 and 2aG4 combination treatment induces apoptosis in breast tumor xenografts in nude mice. Notes: At the termination of the experiments tumor tissue sections were subjected to TUNEL assays and the signal was quantified. Left, representative images of TUNEL staining. Right, bar graph represents quantitated TUNEL signal shown as the mean±SEM. Inserts represent negative controls. *Significantly different from the control group; **significantly different from the control and 2aG4 groups (P<0.05; ANOVA). Abbreviations: ANOVA, analysis of variance; SEM, standard error of the mean; TUNEL, terminal dUTP-mediated nick end labeling. |
APR-246 and 2aG4 combination treatment reduced markers of angiogenesis in BT-474 and T47-D tumor xenografts in nude mice
Lastly, we measured VEGF expression and tumor blood vessel density in sections of tumor harvested at the end of the experiments shown in Figures 5A and 6A. Tumor blood vessel density was determined immunohistochemically using the blood vessel marker CD31. Compared with controls, VEGF expression was significantly elevated in 2aG4-treated BT-474–derived tumors. However, combination therapy significantly and drastically reduced VEGF expression in both BT-474– and T47-D–derived tumors (Figure 9A). Interestingly, CD31 expression was significantly reduced in both types of tumor by all treatments, with APR-246 and 2aG4 combination therapy being especially effective (Figure 9B).
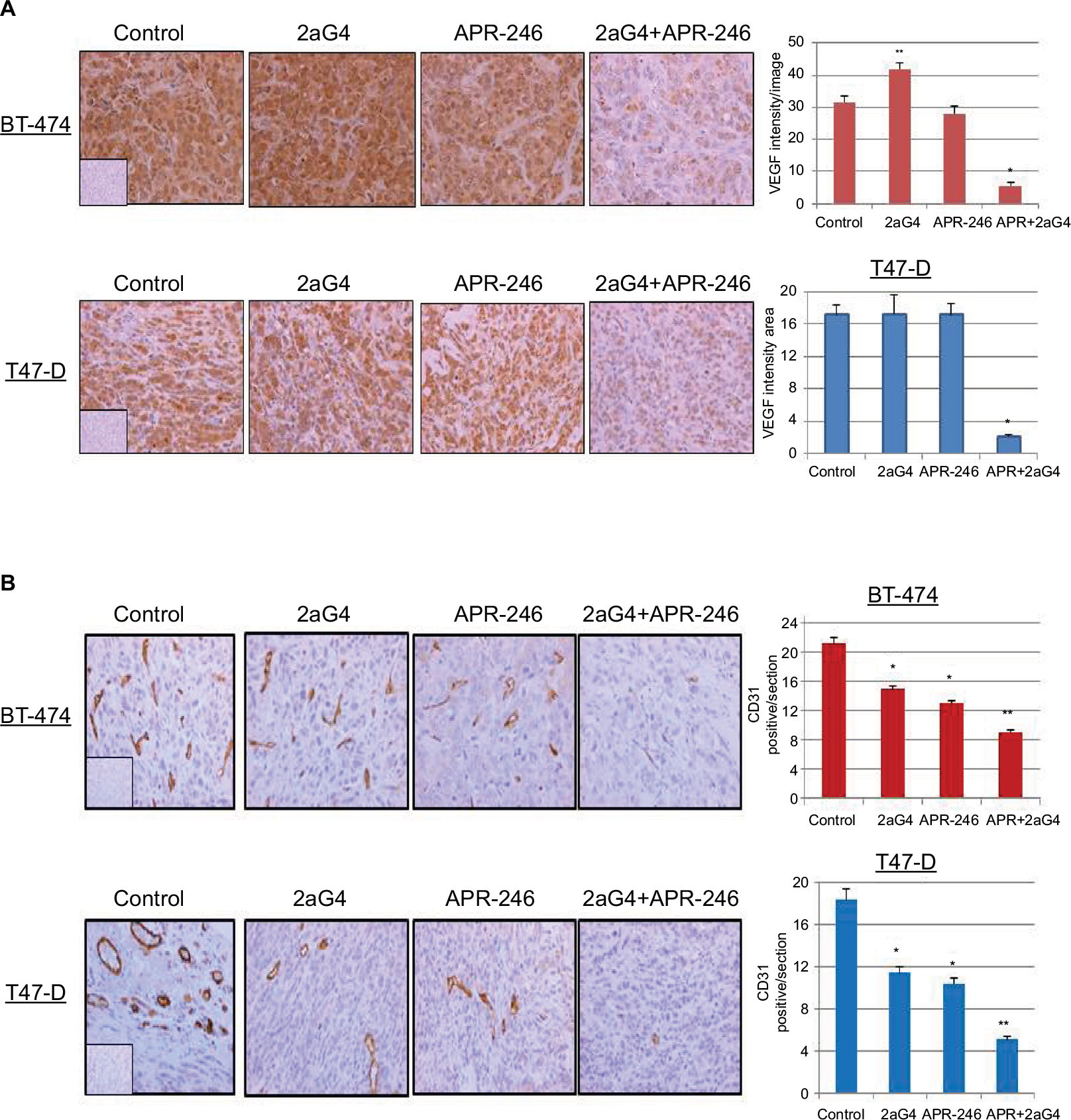 | Figure 9 APR-246 and 2aG4 combination treatment reduces angiogenesis markers in breast tumor xenografts in nude mice. Notes: At the termination of the experiments tumor tissue sections were immunostained for (A) VEGF and (B) CD31 and the signal was quantified. Left, representative images of immunostaining. Right, bar graph represents quantitated (A) VEGF signal or (B) blood vessel density shown as the mean±SEM. CD31 staining was used to count the blood vessels and determine blood vessel density. Inserts represent negative controls. *Significantly different from the control group; **significantly different from the control, 2aG4, and APR-246 groups (P<0.05; ANOVA). Abbreviations: ANOVA, analysis of variance; SEM, standard error of the mean; VEGF, vascular endothelial growth factor. |
Discussion
Tumors derived from BT-474 and T47-D breast cancer cells, both of which express mtp53 as well as estrogen and progesterone receptors, were grown in nude mice prior to treatment with APR-246, an mtp53 activator, and 2aG4, an antibody that binds phosphatidylserine residues present on the surface of tumor endothelial cells. None of the treatment regimens elicited any major toxic effects in experimental animals. We found that treatment of tumor-bearing animals with a combination of APR-246 and 2aG4 reduced the proliferation of cancer cells, induced their apoptosis, and inhibited tumor growth more effectively than either agent alone. Furthermore, combination treatment caused eradication of a number of tumor xenografts. Complete tumor eradication did not occur in BT-474–derived xenografts as frequently as in the T47-D model, suggesting that multiple mechanisms might be responsible for the antitumor effects elicited by combination therapy, or that an effective dose of one or both of the agents was not achieved in the BT-474 cells as a consequence of incomplete cellular uptake. Efficiency of drug delivery to tumor cells and blood vessels may also play a role in this phenomenon.
APR-246 significantly increased the levels of p53 with the wild-type conformation in mtp53-expressing BT-474 cells in vitro, and significantly increased TUNEL staining in tumor xenograft tissues. As wtp53 induces the expression of genes related to apoptosis, we propose that switching the conformation of mtp53 to wtp53 is an important molecular mechanism through which APR-246 exerts its antitumor effects. Western blot analysis yielded further support for this type of mechanism, demonstrating that APR-246 increased Bax and p21 expression in BT-474 cells in vitro. Immunohistochemical data also demonstrated that combination treatment inhibited VEGF expression in both BT-474 and T47-D tumor xenograft models. This observation concurs with our previously published findings in progesterone-dependent tumor prevention models, in which APR-246 diminished VEGF levels, both in vitro and in vivo.16–18 By virtue of its ability to inhibit VEGF expression, which is key to the survival of tumor cells, it is clear that APR-246 has important antiangiogenic properties. APR-246 probably stimulates the destruction of endothelial cells indirectly, by reducing the levels of VEGF produced by both endothelial and epithelial cells.37,38 However, because our analysis was conducted in tumors at the endpoint of the experiment, collected 18 hours after the last treatment, we cannot totally dismiss the possibility that blood vessels were lost through endothelial cell destruction at earlier phases of tumor inhibition. The delay in collection of tumor tissues might also account for our inability to detect a significant response with respect to reduced VEGF in response to treatment of tumor cells with a single agent, possibly allowing VEGF levels to rebound. However, expression of VEGF was diminished in the combination treatment group. This was likely due to increased effectiveness of combined APR-246 and 2aG4 treatment on inhibiting the expression of VEGF by tumor epithelial cells, which were probably the source of much of the locally derived growth factor. Future studies involving analysis of VEGF from tumors collected immediately after the cessation of treatment should help us understand the inhibitory process more fully.
Mechanistically, a combination of APR-246 and 2aG4 might cause increased antitumor activity by affecting separate signal transduction pathways. Our data indicate that in tumor epithelial cells, APR-246 activates mtp53 and induces the expression of genes related to apoptosis, suggesting that it may exert its antitumor effects by instigating cell death via the mitochondrial-dependent apoptotic pathway.14,17 By binding to phosphatidylserine on the surface of tumor endothelial cells, 2aG4 mediates antibody-dependent cellular cytotoxicity and effectively prevents tumor blood vessels from functioning.28,29 Recently, it has also been proposed that 2aG4 may influence the immune system to eradicate tumors.39,40 In addition, because APR-246 promotes tumor epithelial cell apoptosis, it probably causes the generation of reactive oxygen species, which may then enhance phosphatidylserine exposure on the surface of tumor endothelial cells. Such an action of APR-246 would augment the capacity of 2aG4 to disrupt normal blood vessel function in tumor tissue. Hence, because APR-246 may also induce phosphatidylserine exposure on tumor epithelial cells, its use in combination with 2aG4 may be synergistically effective, since the antibody would not only neutralize tumor epithelial cells but also disrupt blood vessels. The latter concept remains to be tested.
High levels of Her-2-neu are expressed by BT-474 cells. Her-2-neu is overexpressed in 15%–30% of human breast cancers and correlates with both a poor prognosis and limited response to hormonal and chemotherapeutic treatment.41–46 Several strategies have, therefore, been proposed for treating Her-2-neu–positive tumors.47 Our experimental findings provide evidence that such tumors might succumb to treatment with either 2aG4 or APR-246 alone, though we believe that a combination of the two would be most effective. Administration of APR-246, followed by 2aG4, could be especially effective against anti-hormone resistant tumors that express both Her-2-neu and mtp53. In a sequential treatment regimen involving the two agents, APR-246 would induce apoptosis of tumor cells while paving the way for subsequent 2aG4 action by exposing phosphatidylserine residues on tumor endothelial and epithelial cells. While this idea remains to be tested, findings with tamoxifen-resistant BT-474 cells suggest that it may be a viable way to treat this type of tumor.
In summary, ~50% of breast cancers express mtp53, which is not only a prognostic indicator but also causes resistance to chemotherapeutic drugs. Our study provides a strong rationale for using a therapy regimen that combines APR-246, which targets mtp53, with 2aG4, which disrupts tumor blood vessels, to treat mtp53-expressing breast cancers. Such treatment could be effective against hormone-dependent breast tumors, which is the most common type of breast cancer in women worldwide.1 We contend that our findings support the further study of APR-246 and 2aG4 as possible therapeutic options to combat breast cancer in humans. We further contend that combination therapy involving APR-246 and 2aG4 might be effective against other types of cancer.
Acknowledgments
This research was initially conducted with Dr Philip Thorpe from the University of Texas Southwestern Medical Center (Dallas, TX, USA), who sadly passed away recently. We would like to thank him for providing antibodies, including 2aG4, used in our research. We would also like to thank Peregrine Pharmaceuticals Inc., (Tustin, CA, USA) for providing additional 2aG4 and acknowledge APREA AB (Solna, Sweden) for providing partial funding and APR-246 for our in vivo studies. Additional financial support was provided by a peer-reviewed faculty research grant from the College of Veterinary Medicine, University of Missouri. SMH is the Zalk Missouri Professor of Tumor Angiogenesis.
Disclosure
The authors report no conflicts of interest in this work.
References
DeSantis C, Ma J, Bryan L, Jemal A. Breast cancer statistics, 2013. CA Cancer J Clin. 2014;64:52–62. | ||
Garritano S, Inga A, Gemignani F, Landi S. More targets, more pathways and more clues for mutant p53. Oncogenesis. 2013;2:e54. | ||
Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. | ||
Turner N, Moretti E, Siclari O, et al. Targeting triple negative breast cancer: is p53 the answer? Cancer Treat Rev. 2013;39:541–550. | ||
Shapira I, Lee A, Vora R, Budman DR. p53 mutations in triple negative breast cancer upregulate endosomal recycling of epidermal growth factor receptor (EGFR) increasing its oncogenic potency. Crit Rev Oncol Hematol. 2013;88:284–292. | ||
Bassett EA, Wang W, Rastinejad F, El-Deiry WS. Structural and functional basis for therapeutic modulation of p53 signaling. Clin Cancer Res. 2008;14:6376–6386. | ||
Lacroix M, Toillon RA, Leclercq G. p53 and breast cancer, an update. Endocr Relat Cancer. 2006;13:293–325. | ||
Kumar S, Walia V, Ray M, Elble RC. p53 in breast cancer: mutation and counter measures. Front Biosci. 2007;12:4168–4178. | ||
Rahko E, Blanco G, Soini Y, Bloigu R, Jukkola A. A mutant TP53 gene status is associated with a poor prognosis and anthracycline- resistance in breast cancer patients. Eur J Cancer. 2003;39:447–453. | ||
Muller M, Wilder S, Bannasch D, et al. p53 activates the CD95 (APO-1/Fas) gene in response to DNA damage by anticancer drugs. J Exp Med. 1998;188:2033–2045. | ||
Wang W, Rastinejad F, EL-Deiry WS. Restoring p53-dependent tumor suppression. Cancer Biol Ther. 2003;2:S55–S63. | ||
Stoklosa T, Golab J. Prospects for p53-based cancer therapy. Acta Biochimica Polonica. 2005;52:321–328. | ||
Willis AC, Chen X. The promise and obstacle of p53 as a cancer therapeutic agent. Curr Mol Med. 2002;2:329–345. | ||
Foster BA, Coffey HA, Morin MJ, Rastinejad F. Pharmacological rescue of mutant p53 conformation and function. Science. 1999;286:2507–2510. | ||
Bykov VJ, Issaeva N, Shilov A, et al. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat Med. 2002;8:282–888. | ||
Liang Y, Wu J, Stancel GM, Hyder SM. p53-dependent inhibition of progestin-induced VEGF expression in human breast cancer cells. J Steroid Biochem Mol Biol. 2005;93:173–182. | ||
Liang Y, Besch-Williford C, Benakanakere I, Hyder SM. Re-activation of the p53 pathway inhibits in vivo and in vitro growth of hormone- dependent human breast cancer cells. Int J Oncol. 2007;31:777–784. | ||
Liang Y, Besch-Williford C, Hyder SM. PRIMA-1 inhibits growth of breast cancer cells by re-activating mutant p53 protein. Int J Oncol. 2009;35:1015–1023. | ||
Bykov VJ, Wiman KG. Mutant p53 reactivation by small molecules makes its way to the clinic. FEBS Lett. 2014;588:2622–2627. | ||
Bykov VJ, Zhang Q, Zhang M, Ceder S, Abrahmsen L, Wiman KG. Targeting of mutant p53 and the cellular redox balance by APR-246 as a strategy for efficient cancer therapy. Front Oncol. 2016;6:21. | ||
Deneberg S, Cherif H, Lazarevic V, et al. An open-label phase I dose- finding study of APR-246 in hematological malignancies. Blood Cancer J. 2016;6:e447. | ||
Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med. 1995;1:27–31. | ||
Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nat Rev Cancer. 2003;3:401–410. | ||
Thorpe PE. Vascular targeting agents as cancer therapeutics. Clin Cancer Res. 2004;10:415–427. | ||
Burrows FJ, Thorpe PE. Vascular targeting agents as cancer therapeutics. Clin Cancer Res. 2004;10:415–27. | ||
Denekamp J. Angiogenesis, neovascular proliferation and vascular pathophysiology as targets for cancer therapy. Br J Radiol. 1993;66:181–196. | ||
Ran S, Downes A, Thorpe P. Increased exposure of anionic phospholipids on the surface of tumor blood vessels. Cancer Res. 2002;62:6132–6140. | ||
Ran S, He J, Huang X, Soares M, Scothorn D, Thorpe PE. Antitumor effects of a monoclonal antibody that binds anionic phospholipids on the surface of tumor blood vessels in mice. Clin Cancer Res. 2005;15:1551–1562. | ||
Luster TA, He J, Huang X, et al. Plasma protein beta-2-glycoprotein 1 mediates interaction between the anti- tumor monoclonal antibody 3G4 and anionic phospholipids on endothelial cells. J Biol Chem. 2006;281:29863–29871. | ||
Huang X, Bennett M, Thorpe PE. A monoclonal antibody that binds anionic phospholipids on tumor blood vessels enhances the antitumor effect of docetaxel on human breast tumor in mice. Cancer Res. 2005;65:4408–4416. | ||
He J, Luster TA, Thorpe PE. Radiation-enhanced vascular targeting of human lung cancers in mice with a monoclonal antibody that binds anionic phospholipids. Clin Cancer Res. 2007;13:5211–5218. | ||
Emde A, Mahlknecht G, Maslak K, et al. Simultaneous inhibition of estrogen receptor and the HER2 pathway in breast cancer: effects of HER2 abundance. Transl Oncol. 2011;4:293–300. | ||
Carroll CE, Liang Y, Benakanakere I, Besch-Williford C, Hyder SM. The anticancer agent YC-1 suppresses progestin-stimulated VEGF in breast cancer cells and arrests breast tumor development. Int J Oncol. 2013;42:179–187. | ||
Ali S, Rasool M, Chaoudhry H, et al. Molecular mechanisms and mode of tamoxifen resistance in breast cancer. Bioinformation. 2016;12:135–139. | ||
Rubinstein LV, Shoemaker RH, Paull KD, et al. Comparison of in vitro anticancer-drug-screening data generated with a tetrazolium assay versus a protein assay against a diverse panel of human tumor cell lines. J Natl Cancer Inst. 1990;82:1113–1118. | ||
Skehan P, Storeng R, Scudiero D, et al. New colorimetric cytotoxicity assay for anti-cancer-drug screening. J Natl Cancer Inst. 1990;82:1107–1112. | ||
Breen EC. VEGF in biological control. J Cell Biochem. 2007;102:1358–1367. | ||
Liang Y, Brekken RA, Hyder SM. Vascular endothelial growth factor induces proliferation of breast cancer cells and inhibits the anti- proliferative activity of anti- hormones. Endocrinology. 2006;13:905–919. | ||
Yin Y, Huang X, Lynn KD, Thorpe PE. Phosphatidylserine-targeting antibody induces M1 macrophage polarization and promotes myeloid-derived suppressor cell differentiation. Cancer Immunol Res. 2013;1:256–268. | ||
Cheng X, Li L, Thorpe PE, Yopp AC, Brekken RA, Huang X. Antibody-mediated blockade of phosphatidylserine enhances the antitumor effect of sorafenib in hepatocellular carcinomas xenografts. Ann Surg Oncol. 2016;23(Suppl 5):583–591. | ||
Harris LN, Liotcheva V, Broadwater G, et al. Comparison of methods of measuring HER-2 in metastatic breast cancer patients treated with high-dose chemotherapy. J Clin Oncol. 2001;19:1698–1706. | ||
Badowska-Kozakiewicz AM, Sobol M, Patera J, Kozłowski W. Immunohistochemical evaluation of human epidermal growth factor receptor 2 and estrogen and progesterone receptors in invasive breast cancer in women. Arch Med Sci. 2013;9:466–471. | ||
Slamon DJ, Godolphin W, Jones LA, et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science. 1989;244:707–712. | ||
Paik S, Hazan R, Fisher ER, et al. Pathologic findings from the national surgical adjuvant breast and bowel project: prognostic significance of erbB-2 protein overexpression in primary breast cancer. Clin Oncol. 1990;8:103–112. | ||
Toikkanen S, Helin H, Isola J, Joensuu H. Prognostic significance of HER-2 oncoprotein expression in breast cancer: a 30-yr follow-up. J Clin Oncol. 1992;10:1044–1048. | ||
Isola J, Holli K, Oksa H, Teramoto Y, Kallioniemi OP. Elevated erbB2 oncoprotein levels in preoperative and follow-up serum samples define an aggressive disease course in patients with breast cancer. Cancer. 1994;73:652–658. | ||
Savellano MD, Pogue BW, Hoopes PJ, Vitetta ES, Paulsen KD. Multiepitope HER-2 targeting enhances photoimmunotherapy of HER-2-Overexpressing cancer cells with pyropheophorbide-a immunoconjugates. Cancer Res. 2005;65:6371–6379. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.