Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 16
A Case Report of Cardiofaciocutaneous Syndrome with MAP2K1 Pathogenic Variant
Authors Tang Q, Gong D, Ye XM, Xu JR, Yang YC, Yan LJ, Zou L, Wen XL
Received 10 March 2023
Accepted for publication 21 August 2023
Published 8 September 2023 Volume 2023:16 Pages 817—823
DOI https://doi.org/10.2147/PGPM.S411964
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Qiong Tang, Dai Gong, Xiao-Min Ye, Jun-Ru Xu, Yi-Can Yang, Li-Juan Yan, Li Zou, Xiang-Lan Wen
Department of Children Health Care Center, Zhuzhou Hospital Affiliated to Xiangya Medical College, Central South University, Zhuzhou, People’s Republic of China
Correspondence: Xiang-Lan Wen, Department of Children Health Care Center, Zhuzhou Hospital Affiliated to Xiangya Medical College, Central South University, No. 116 of Changjiang South Road, Tianyuan District, Zhuzhou City, Hunan Province, 412007, People’s Republic of China, Tel +86 13873317331, Email [email protected]
Abstract: Craniofacial dysmorphism, cardiac abnormalities, ectodermal abnormalities, psychomotor delay, intellectual disability, and short stature are all hallmarks of the extremely rare disorder known as cardiofaciocutaneous syndrome (CFCS). Although CFCS is considered rare, approximately 300 cases have been documented in the literature. In this report, we discuss a patient diagnosed with CFCS without the typical heart malformations but with craniofacial features, skin abnormalities, intellectual disability, and short stature. Genetic testing revealed the presence of three potentially harmful variants: one in the MAP2K1 gene and two in the ATP2B3 and CDC42BPB genes, the significance of which is currently not yet found. Our findings in this case report suggest that the clinical symptoms of CFCS may be atypical, thereby expanding our understanding of the symptom spectrum of the disease. Simultaneously, the link between the clinical symptoms of the patient and the two unknown pathogenic variants has not been established. This case report supplements existing clinical reference material by providing valuable insights into the specific scenario.
Keywords: cardiofaciocutaneous syndrome, gene detection, intellectual disability, neurodevelopmental syndrome, short stature, spinocerebellar ataxia
A Letter to the Editor has been published for this article.
Introduction
Cardiofaciocutaneous syndrome (CFCS) is an autosomal-dominant genetic disease of the renin-angiotensin system (RAS) cardiomyopathy group. This syndrome refers to a group of developmental disorders caused by the germline pathogenic variant of gene encoding RAS/Mitogen-activated protein kinase (MAPK) signaling pathway protein, and the pathogenic variant of four genes related to CFCS. These are BRAF (7q34), MAP2K1 (15q22.31), MAP2K2 (19p13.3, CFCS4, OMIM#615280), and KRAS (12p12.1), which encode the RAS protein (KRAS) and/or its downstream signals serine-threonine kinase (BRAF, MEK1, MEK22). Among these, heterozygous pathogenic variants of BRAF gene are the most common, while MAP2K1, MAP2K2C2, and KRAS genes are less involved.
RAS cardiomyopathies include a wide variety of conditions, including Noonan syndrome (NS) and Costello syndrome (CS).1–3 Therefore, CFCS is similar to NS and CS while also having its own unique characteristics. The clinical phenotypes of CFCS include craniofacial features, cardiac anomalies (most commonly atrial septal defect and pulmonary artery stenosis), hair and skin abnormalities, gastrointestinal dysfunction, neurocognitive delay, epilepsy, short stature, and slow weight gain. In addition, some patients lack immunoglobulins but exhibit no symptoms.4 ATP2B3 and CDC42BPB play a key role in numerous cellular processes and physiological functions. ATP2B3 is involved in maintaining cellular calcium homeostasis, while CDC42BPB is a key regulator of actin cytoskeleton dynamics and cell migration. Both genes play critical roles in physiological processes and have been linked to diseases when their roles are disrupted.
In addition to its distinctive clinical symptoms, CFCS can be distinguished from NS and CS by genetic testing. There are few published reports on CFCS due to the MAP2K1 gene pathogenic variant in China. In this report, we describe a patient who exhibited both classic and atypical symptoms of CFCS as a result of a pathogenic variant in the MAP2K1 gene.
Case Description
A two-year six-month-old boy was diagnosed with “slow growth in height and weight and developmental delay for more than two years”. The child presented with a neurodevelopmental delay, an inability to sit independently, and no verbal communication; the child had a height of 78.6 cm (−3.7 SD) and weight of 8.34 kg (−3.9 SD) at the time of first visit.
The child was born to a then 35-year-old G7P3 mom at the gestation age of 33 weeks and 4 days by a cesarian section due to premature rupture of membranes. The neonate measured 45 cm in length and weighed 2.35 kg. During pregnancy, fetal measurements showed that both the head and the abdomen circumferences were larger than normal for the gestational age, but no complications were seen in the neonatal period. The child was initially breastfed and introduced to complementary foods at six months of age.
During the initial visit, the child had difficulty in consuming food and preferred semi-solid and liquid foods, although the child’s intake was on par with that of other children of the same age. Both parents were healthy, and the mother did not suffer from any of the conditions that put her at risk during pregnancy. Physical examination indicated significant growth retardation compared to children of the same age, with the child’s height and weight measuring −3SD to −4SD. However, the head circumference (47 cm) and abdominal circumference (47.5 cm) fell within the normal range, which contrasted with the child’s height and weight.
There were significant delays in cognitive, language, and motor development (Figure 1). The child was unable to sit or stand unassisted and did not smile until he was 2 years old. He responded to his name without using words, and his tongue stuck out of his mouth frequently. He showed abnormal irritability, distress, and low muscle tension in all four limbs, as evidenced by his resistance to the prone position and the flexion of his two hips into a head-low-hip-high position while in the prone position, as part of the neurological examination.
 |
Figure 1 Severe neurodevelopmental delay observed in the child. |
There were abnormalities in the child’s craniofacial and skin features (Figure 2), including a slightly wide forehead, a short face, sparse eyebrows, narrow coloboma of eyelids, and a hypertrophy and depressed bridge of the nose. The child had a long philtrum, with a wide mouth, low ear position, and rough skin. The skin of the auricles, the front of the ears, and on the extended surface of the limbs felt grainy like sand (Figure 3).
 |
Figure 2 Craniofacial features of the child. |
 |
Figure 3 Skin of the auricles and the front of the ears observed in the child. |
Results from the physical examination included severe neurodevelopmental delay, stereotypical behavior of regularly touching the tongue with the hands, skin that was rough to the touch like grainy sand, and an abnormal prone position. Growth retardation was also observed but did not correlate with appetite (Figure 4).
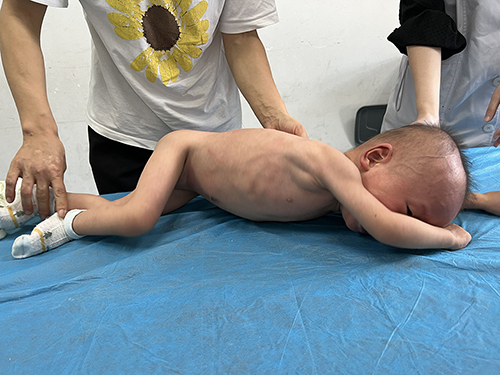 |
Figure 4 Inconsistent head circumference, abdominal circumference, height and weight, and abnormal posture observed in the child. |
The Gesell developmental evaluation of the child was as follows: Adaptability: 31 points, severe developmental delay; Gross motor: 13 points, very severe developmental delay; Fine motor: 33 points, severe developmental delay; Language: 21 points, very severe developmental delay; Individual-social: 18 points, very severe developmental delay.
Peabody Motor Development Scale assessment was as follows: Gross Motor Quotient (GMQ) 45 points, <1%; Fine Motor Quotient (FMQ) 52 points, <1%; Total Motor Quotient (TMQ) 44 points, <1%.
The child’s parents took him to the hospital when he was 11 months old, where he underwent several tests including thyroid function, complete set of toxoplasmosis, rubella cytomegalovirus, herpes simplex, and HIV (TORCH) antibodies, fasting blood glucose, electrolytes, liver function, renal function, myocardial enzymes, blood ammonia, and phenylalanine. The results of other tests were normal, while the TORCH test revealed a positive result for IgG against herpes simplex and rubella virus.
Both the adrenal and abdominal color Doppler ultrasounds came back normal. X-rays of both hip joints showed slightly larger collo-diaphyseal angles of the femur, and the echocardiography was normal (patent foramen ovale at birth and closed at 6 months of age). A head MRI showed patchy and slightly longer T2 signals in the white matter of both parietal lobes as well as slightly increased extra-cerebral space in both frontotemporal regions. The electroencephalogram (EEG) showed a slightly slower background rhythm, and there were no epileptic waves detected throughout the amplitude.
Due to the multi-system abnormality, we performed whole exome sequencing, and the results showed one pathogenic variant of MAP2K1 (Figure 5), specifically chr15: 66436843-66436843, exon3, NM 002755.4c.389A>G:p.Y130C, missense variant. Two pathogenic variants with unclear significance were also found in the gene detection. These were: ATP2B3 chrX:153541460-153541460, exon4, NM_0010013 44.3: c.310G>A: p.E104K, missense_vari, hemizygote and CDC42BPB chr14: 102963 077-102963077, exon20, NM_006035.4: c.2805A>C: p.K935N, missense_vari, heterozygosis; the mutation types of these pathogenic variants are missense mutation, mutation rate: 8.38054372967718e-06 and 3.32336e-05, respectively, and the mutation rating is VUS, indicating that the clinical significance is unknown. The MAP2K1 gene validation test results of the parents were all normal.
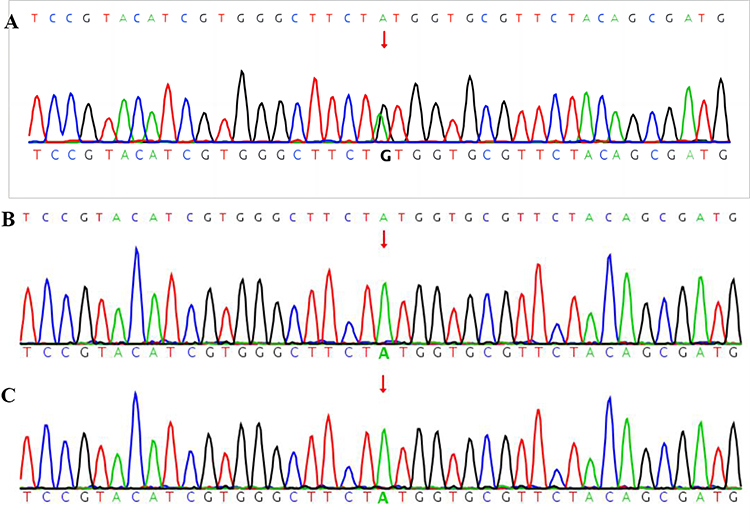 |
Figure 5 The SNV and InDel maps of genes in the child and parents. (A) Child; (B) Father; (C) Mother. Note: Red arrows represent the change of the base. |
Discussion
CFCS was first described in 1986 by Reynolds et al and Baraitser & Patton as an autosomal dominant syndrome. It remains an extremely rare syndrome,5 and approximately 300 cases have been documented so far. The diagnosis of CFCS cannot be made solely based on clinical manifestations, as it belongs to the “RAS cardiomyopathy group”, which is part of a group of developmental disorders with similar clinical symptoms. However, CFCS often has more serious medical complications and developmental delay/intellectual disability,6 and its effect on the skin also is discernible,7 but this is not adequate for diagnosis; genetic testing must be performed on all suspected patients.
Current research shows that four gene pathogenic variants are associated with CFCS, namely BRAF (7q34), MAP 2K1 (15q22.1-q22.33), MAP 2K2 (19p13.3), and KRAS (12p12.1), which encode the RAS protein (KRAS) and/or its downstream signal serine-threonine kinase (BRAF, MEK1, and MEK23). Among them, the heterozygous pathogenic variant of the BRAF gene (75%) is the most common, while MAP2K1, MAP2P2 (25%), and KRAS (<2%) genes may be less involved;8 novel pathogenic variants have been reported in most of the affected individuals to date.
Currently, there are few studies on the genotype-phenotype of CFCS, but studies indicate that 50% of patients diagnosed with CFCS with the BRAF gene pathogenic variant and 37% of patients with the MAP2K1 gene pathogenic variant have pulmonary artery stenosis. The MAP2K1 gene pathogenic variant may be related to premature birth, ventricular septal defect, thoracic dysmorphia, urogenital system abnormality, and dermatological abnormality. Hypertrophic cardiomyopathy, atrial septal defect, moderate and severe intellectual disability, and severe feeding difficulties may be more common in patients with the BRAF gene pathogenic variant.9
In this case, the genetic test result revealed one pathogenic variation of MAP2K1 (Figure 5, A: Child; B: Father; C: Mother), specifically chr15: 66436843-66436843, exon3, NM 002755.4c.389A>G:p.Y130C, missense variant, which is related to the autosomal dominant disease of CFCS type 3; the main manifestations of CFCS3 type 3 are short stature, hypotonus, curly hair, wide mouth, scoliosis, heat intolerance, epileptic seizure, comprehensive developmental delay, short neck, slow growth, keratosis follicularis, nystagmus, hypertrophic cardiomyopathy, pectus excavatum, hyperhidrosis, pulmonary artery stenosis, atrial septal defect, ventricular septal defect, cervical web, decreased bone density, and others. The symptoms that this child presented with were short stature, hypotonus, generalized developmental delay, wide mouth, and hyperkeratosis of skin and hair follicles. Using symptom comparison and gene test results, we confirmed the diagnosis of CFCS in our patient.
We found two unknown pathogenic variants in this patient, which were ATP2B3 chrX:153541460–153541460, exon4, NM_0010013 44.3: c.310G>A: p.E104K, missense_vari, hemizygote and CDC42BPB (Allele frequency 0.00003323) chr14:102963 077–102963077, exon 20, NM_006035.4: c.2805A>C: p.K935N, missense_vari, heterozygosis. These two pathogenic variants lead to X-linked spinocerebellar ataxia and neurodevelopmental syndrome, but their pathogenicity remains unclear. However, a diagnosis was not considered due to the significant differences in age of onset, progression, and main symptoms between the patient and these two diseases.10,11
The patient in this case report, however, also had some inconsistencies with the diagnosis of CFCS, for example, when the child presented to the hospital, he did not have a heart-related phenotype. We discovered that this patient shared the same site of the pathogenic variant as a patient with CFCS caused by a MAP2K1 pathogenic variant reported by Wang et al in 2020,12 but the patient in their case was younger. The two patients had similar clinical symptoms, such as no feeding difficulties, but there was significant growth retardation. In previous literature reports, the cardiac phenotype of CFCS was primarily pulmonary artery stenosis, atrial septal defect, and hypertrophic cardiomyopathy,13 and the case reported by Wang et al also had pulmonary artery stenosis; but, the child described in our case report has no cardiac abnormality, and it is not known whether or not he will develop hypertrophic cardiomyopathy in the future.
Studies show that between 40 and 50% of children with CFCS experience epileptic symptoms,14 with most of these children having their first seizure in infancy or as young children. Unlike the case described by Wang et al, in which an EEG was abnormal despite the absence of an epileptic seizure, the EEG of the patient in our case suggested a critical state in this case, and it was clear that the child was not developing normally. The child was unable to stretch his body in the prone position and bend his hip joint because he could not lie in that position. This position is inexplicable based on the X-ray findings of the hip and pelvis. The child’s stereotypical behavior of touching his tongue with his hand was indicative of his low cognitive ability and poor prognosis in terms of his intelligence.
Conclusion
The evaluation of this case provides a profound and significant comprehension of the clinical phenotype connected to the MAP2K1 gene pathogenic variant in CFCS. Although the patient did not exhibit any signs of congenital heart disease during the evaluation, continued monitoring is necessary to detect the emergence of cardiac abnormalities such as hypertrophic cardiomyopathy, in the future. Establishing the precise genetic diagnosis based on the clinical presentation of the patient allows for targeted and appropriate medical management, better prognosis, and genetic counseling for the parents; this is especially important given the uncertain nature of the prognosis for children with neurological abnormalities such as cognitive developmental delays and behavioral issues.
Data Sharing Statement
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Ethics Approval and Consent to Participate
This study was conducted with approval from the Ethics Committee of Zhuzhou Hospital Affiliated to Xiangya Medical College. This study was conducted in accordance with the declaration of Helsinki. Written informed consent was obtained from patient guardians.
Consent for Publication
The patient guardians signed a document of informed consent for publication of the case details included publication of the images.
Acknowledgments
We would like to acknowledge the hard and dedicated work of all the staff that implemented the intervention and evaluation components of the study.
Funding
No external funding was received to conduct this study.
Disclosure
The authors declare that they have no competing interests in this work.
References
1. Pierpont ME, Magoulas PL, Adi S, et al. Cardio-facio-cutaneous syndrome: clinical features, diagnosis, and management guidelines. Pediatrics. 2014;134(4):e1149–62. doi:10.1542/peds.2013-3189
2. Rodriguez-Viciana P, Tetsu O, Tidyman WE, et al. Germline pathogenic variants in genes within the MAPK pathway cause cardio-facio-cutaneous syndrome. Science. 2006;311(5765):1287–1290. doi:10.1126/science.1124642
3. Serra G, Felice S, Antona V, et al. Cardio-facio-cutaneous syndrome and gastrointestinal defects: report on a newborn with 19p13.3 deletion including the MAP 2 K2 gene. Ital J Pediatr. 2022;48(1):65. doi:10.1186/s13052-022-01241-6
4. Leoni C, Tedesco M, Onesimo R, Giorgio V, Rigante D, Zampino G. Immunoglobulin deficiency associated with a MAP2K1-related pathogenic variant causing cardio-facio-cutaneous syndrome. Immunol Lett. 2020;227:79–80. doi:10.1016/j.imlet.2020.08.009
5. Jurcă MC, Iuhas OA, Puiu M, et al. Cardiofaciocutaneous syndrome - a longitudinal study of a case over 33 years: case report and review of the literature. Rom J Morphol Embryol. 2021;62(2):563–568. doi:10.47162/RJME.62.2.23
6. Noonan JA. Noonan syndrome and related disorders: alterations in growth and puberty. Rev Endocr Metab Disord. 2006;7(4):251–255. doi:10.1007/s11154-006-9021-1
7. Siegel DH, McKenzie J, Frieden IJ, Rauen KA. Dermatological findings in 61 pathogenic variant-positive individuals with cardiofaciocutaneous syndrome. Br J Dermatol. 2011;164(3):521–529. doi:10.1111/j.1365-2133.2010.10122.x
8. Ciara E, Pelc M, Jurkiewicz D, et al. Is diagnosing cardio-facio-cutaneous (CFC) syndrome still a challenge? Delineation of the phenotype in 15 Polish patients with proven pathogenic variants, including novel pathogenic variants in the BRAF1 gene. Eur J Med Genet. 2015;58(1):14–20. doi:10.1016/j.ejmg.2014.11.002
9. Allanson JE, Annerén G, Aoki Y, et al. Cardio-facio-cutaneous syndrome: does genotype predict phenotype? Am J Med Genet C Semin Med Genet. 2011;157(2):129–135. doi:10.1002/ajmg.c.30295
10. Feyma T, Ramsey K, Huentelman MJ, et al; C4RCD Research Group. Dystonia in ATP2B3-associated X-linked spinocerebellar ataxia. Mov Disord. 2016;31(11):1752–1753. doi:10.1002/mds.26800
11. Chilton I, Okur V, Vitiello G, et al. De novo heterozygous missense and loss-of-function variants in CDC42BPB are associated with a neurodevelopmental phenotype. Am J Med Genet A. 2020;182(5):962–973. doi:10.1002/ajmg.a.61505
12. Wang QM, Chen PL, Peng Q. 心-面-皮肤综合征MAP2K1基因新发突变一例 [A case of a new pathogenic variant of MAP2K1 gene in heart-face-skin syndrome]. Chin J Med Genet. 2020;37(8):567–569. Chinese. doi:10.1136/jmedgenet-2019-106698
13. Rauen KA. Cardiofaciocutaneous syndrome [EB/OL]. In: GeneReviews®. Seattle (WA): University of Washinton, Seattle; 2007:1993–2018.
14. Yoon G, Rosenberg J, Blaser S, Rauen KA. Neurological complications of cardio-facio-cutaneous syndrome. Dev Med Child Neurol. 2007;49(12):894–899. doi:10.1111/j.1469-8749.2007.00894.x
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.