")
Back to Journals » OncoTargets and Therapy » Volume 10
A blockade of PD-L1 produced antitumor and antimetastatic effects in an orthotopic mouse pancreatic cancer model via the PI3K/AkT/MTOR signaling pathway
Authors Zhao L, Li C, Liu F, Zhao Y, Liu J, Hua Y, Liu J , Huang J, Ge C
Received 17 December 2016
Accepted for publication 8 March 2017
Published 12 April 2017 Volume 2017:10 Pages 2115—2126
DOI https://doi.org/10.2147/OTT.S130481
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Carlos E Vigil
Lei Zhao,1 Cheng Li,2 Fei Liu,2 Yonghong Zhao,2 Jun Liu,2 Ye Hua,1 Jinyang Liu,1 Jiapeng Huang,1 Chunlin Ge1
1Department of Pancreatic and Biliary Surgery, The First Hospital of China Medical University, 2Department of Immunology, College of Basic Medical Sciences, China Medical University, Shenyang, Liaoning, People’s Republic of China
Background: Pancreatic cancer is one of the most aggressive and intractable malignant tumors, and most deaths from pancreatic cancer are related to metastases. It has been demonstrated in vitro that overexpression of programmed death-ligand 1 (PD-L1) correlates with a lack of phosphatase and tensin homologue (PTEN) expression in pancreatic cancer tissue. This loss of PTEN expression may aberrantly activate the phosphatidylinositol 3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) pathway, and thereby promote tumor cell survival, proliferation, and disease progression. In this study, we investigated the potential therapeutic effect of blockading PD-L1 expression on the progression of pancreatic cancer and its spontaneous liver metastases in vivo by inhibiting the PI3K/Akt/mTOR signaling pathway.
Methods: We investigated the effect of blockading PD-L1 in an orthotopic pancreatic cancer mouse model. The pancreatic tumor weights and inhibition ratios were determined after treatment with antimouse PD-L1 antibody for 5 weeks. We used immunohistochemistry methods to investigate PD-L1 expression in pancreatic cancer tissue and spontaneous liver metastasis tissue. The levels of mRNA and protein expression for various components involved in the PI3K/Akt/mTOR signaling pathway as well as for matrix metalloproteinases-2 and -9 (MMP2 and MMP9) were measured by reverse transcription polymerase chain reaction (RT-PCR) and Western blot methods, respectively.
Results: Blockading PD-L1 significantly inhibited tumor growth and decreased the levels of PD-L1 expression in tumor tissue. Furthermore, the levels of PTEN mRNA and protein expression were elevated, while the levels of phospho-Akt (p-Akt) and phospho-mTOR (p-mTOR) protein were decreased in pancreatic cancer and liver metastasis tissues after establishing a PD-L1 blockade. In addition, a PD-L1 blockade decreased the levels of MMP2 and MMP9 mRNA and protein expression in tumor tissues.
Conclusion: Our results suggest that a blockade of PD-L1 may inhibit the growth and metastasis of pancreatic cancer by modulating the PI3K/Akt/mTOR pathway.
Keywords: PD-L1 blockade, pancreatic cancer, spontaneous liver metastasis, PI3K/Akt/mTOR pathway, PTEN, MMP2, MMP9
Introduction
Pancreatic cancer is one of the most intractable and highly malignant tumors.1 Most patients with pancreatic cancer are asymptomatic until they are diagnosed at an advanced stage of their disease.2 Although great progress has been made in treating pancreatic cancer with surgery, chemotherapy, radiotherapy, and combined treatment, the 5-year survival rate among pancreatic cancer patients in the United States is ~6%.3 Moreover, pancreatic cancer is expected to become the second leading cause of cancer-related death by 2030.4 Tumor metastasis is responsible for more than 90% of pancreatic cancer mortality, and the current treatments for metastatic pancreatic cancer are only minimally effective.5
Accumulating evidence suggests that tumor cells evade host immune recognition by immune checkpoints; this lack of immune recognition results in tumor dissemination, metastasis, and disease relapse.6 The programmed death-1/programmed death-ligand 1 (PD-1/PD-L1) pathway is a normal part of the immune regulation system that signals for inhibition of T-cell activation in autoimmune diseases.7 Therefore, tumor cells can utilize the PD-1/PD-L1 pathway to silence the immune system.8 PD-1 is a member of an immunoglobulin superfamily that includes CD28 and other molecules. PD-1 is expressed on T cells, B cells, natural killer cells, monocytes, and certain subsets of dendritic cells.9 There are two PD-1 ligands: PD-L1 (B7-H1) and PD-L2 (B7-DC), of which PD-L1 is the most relevant in the tumor microenvironment.10 PD-L1 is highly expressed on the tumor-infiltrating lymphocytes found in most cancers as well as on the surface of numerous human solid tumors, such as non-small cell lung carcinoma, ovarian carcinoma, melanomas, gastrointestinal cancers, and pancreatic cancer.11 The PD-1/PD-L1 interaction inhibits T-cell receptor signaling by stimulating phosphorylation of the immunoreceptor tyrosine-based switch motif, and also induces the production of tyrosine phosphatase 2, resulting in decreased levels of T-cell proliferation and an increased proliferation of tumor-infiltrating T-regulatory cells.12 Thus, inhibiting the PD-1/PD-L1 interaction might recover T-cell activity and enhance antitumor immune responses.13 PD-L1 expression in tumor tissue is predictive of a poor prognosis for patient survival, and is correlated with a patient’s clinical response to anti-PD-L1 treatment.14 Therapies that block PD-L1 expression have produced impressive response rates in patients with different types of advanced cancers, including advanced melanoma, non-small cell lung carcinoma, and renal cell cancer.15 However, there have been no objective responses among patients with pancreatic cancer.16
An increase in PD-L1 expression is significantly correlated with the loss of phosphatase and tensin homologue (PTEN).17 The PD-1/PD-L1 interaction can reduce PTEN activity and stimulate phosphatidylinositol 3-kinase (PI3K) phosphorylation by recruiting tyrosine phosphatase 2. This series of events results in activation of the PI3K/Akt/mammalian target of rapamycin (mTOR) pathway.18 Class I PI3Ks generate lipids (PI(3,4,5)P3) that modulate the activity of a range of signaling proteins, including protein kinases (such as Akt/PKB). The regulatory subunit p85α of PI3K and Akt2 isoforms are confirmed to be overexpressed in human pancreatic adenocarcinomas.19 The PI3K/Akt/mTOR pathway plays a key role in numerous cellular processes, including metabolism, cell survival, differentiation, proliferation, motility, and angiogenesis.20 Activation of the PI3K/Akt/mTOR signaling pathway is central to the growth of many human cancers, and stimulates their metastasis.21 Matrix metalloproteinases (MMPs) play a crucial role in facilitating tumor cell migration and invasion, both of which are mediated by several signaling pathways, including the PI3K/Akt/mTOR signaling pathway.22 Moreover, MMP2 and MMP9 (gelatinase A and gelatinase B) have been implicated in the metastasis of pancreatic cancer.23
Tumor cell invasion and metastasis are the most common causes of unsuccessful pancreatic cancer treatment.24 To investigate the effect of a PD-L1 blockade on pancreatic cancer and metastasis, we established an orthotopic pancreatic cancer animal model, and then used it to analyze relationships between the levels of PD-L1 and PTEN expression in vivo. Finally, we investigated how a PD-L1 blockade and the PI3K/Akt/mTOR pathway affect pancreatic cancer progression and the occurrence of spontaneous liver metastasis. We found that blockading PD-L1 could suppress pancreatic cancer progression and the occurrence of spontaneous liver metastasis by modulating the PI3K/Akt/mTOR pathway and simultaneously downregulating MMP2 and MMP9 expression. Our study results will provide a better understanding of the mechanism by which a PD-L1 blockade affects pancreatic cancer and its metastasis.
Material and methods
Mice and cell lines
Female C57BL/6 mice (6–8 weeks old) were purchased from Beijing Vital River Laboratory (Beijing, People’s Republic of China). The mice were housed in a pathogen-free environment that was maintained at a temperature of 25°C and a relative humidity of 45%–55%. The protocol for this study was approved by the Animal Care and Use Committee of China Medical University. All animal experiments were carried out in accordance with the guidelines established by China Medical University and were approved by the Animal Welfare and Research Ethics Committee of China Medical University. A murine pancreatic adenocarcinoma cell line was obtained from the China Infrastructure of Cell Line Resources (Beijing, People’s Republic of China). The cells were cultured in DMEM supplemented with 10% heat-inactivated fetal bovine serum. The cultured cells were maintained in a humidified 5% CO2 atmosphere at 37°C.
Experimental protocol
The C57BL/6 mice (n=10) were anesthetized with 4% chloral hydrate solution (0.1 mL/10 g), after which the abdomen of each mouse was opened by a 0.5-cm left flank incision. The pancreas was exteriorized through the incision and then slowly injected with 5×105 cells.25 Following injection, the pancreas was relocated into the abdomen, and the peritoneum and skin were closed with a 5-0 Vicryl suture. All surgical procedures were performed under sterile conditions and were well tolerated by the mice.
To ensure that pancreatic tumors had been successfully established after the intrapancreatic injection of cancer cells, serum samples were collected from the tail vein of each mouse 1 day before and 7 days after injection, and examined by an enzyme-linked immunosorbent assay for their levels of carbohydrate antigen 19-9.
Treatment protocol
Seven days after injection, the mice were randomly assigned to two separate groups. Mice in the anti-PD-L1 group were intraperitoneally injected with 0.2 mg of antimouse PD-L1 antibody (MIH5, rat IgG2a) (eBioscience; San Diego, CA, USA) three times per week for 5 weeks. Mice in the control group were intraperitoneally injected with normal saline solution.26 One week after the last injection, both groups were euthanized and dissected, and their pancreatic tumors and liver metastases were divided and examined under a dissecting microscope, and the nontumor tissue was removed from pancreatic tumor. The weights of the pancreatic tumors were measured, and growth inhibition ratios were calculated using the formula: 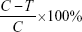 (C = mean weight of control tumors; T = mean weight of treated tumors). There is no obvious pulmonary and peritoneal metastasis in both groups.
(C = mean weight of control tumors; T = mean weight of treated tumors). There is no obvious pulmonary and peritoneal metastasis in both groups.
Immunohistochemistry
The collected pancreatic tumors and liver metastases were fixed in 4% paraformaldehyde for 24 h and then embedded in paraffin. Serial sections of 4-μm thickness were prepared for immunohistochemistry. After deparaffinization and antigen retrieval, the sections were preincubated in blocking serum, and then incubated overnight with the anti-PD-L1 antibody (Abcam; Cambridge, UK). Following incubation, the sections were washed three times in phosphate-buffered saline (PBS) and then incubated with biotin-conjugated secondary antibody for 2 h. Next, the sections were washed 3 more times in PBS and then incubated with avidin-biotinylated peroxidase complex for 30 min; they were then washed with PBS for 10 min. After being washed, 100 μL of 3, 3-diaminobenzidine substrate was placed on each tissue section, and the chromogen on each slide was allowed to develop its color. Immediately after color development, the slides were washed with ddH2O for 10 min, and then counterstained with hematoxylin. After dehydration through an ethanol series and xylene, each stained section was examined by light microscopy (Nikon; Tokyo, Japan).
Quantitative analysis of IHC
Stained tissues were defined as those showing membrane and cytoplasmic staining pattern of tumor tissue as previously described.27 PD-L1 staining intensity was graded into four groups: no staining (0), weak staining (1+), moderate staining (2+), and intense staining (3+) (Figure S1). The immunostained slide was evaluated under the microscope.
H scores, which combine components of staining intensity with the percentage of positive cells, were calculated. H scores have values ranging from 0 to 300 and are defined as:
- 1 * (percentage of cells staining at 1+ intensity)
- +2 * (percentage of cells staining at 2+ intensity)
- +3 * (percentage of cells staining at 3+ intensity)
- = H score.
All slides were scanned and graded by three independent pathologists.
Reverse transcription polymerase chain reaction (RT-PCR)
TRIzol reagent (Invitrogen; Waltham, MA, USA) was used to extract total RNA from samples of tumor tissue as previously described.28 The concentration and purity of the isolated RNA molecules were determined by spectrophotometry at wavelengths of 260, 280, and 320 nm. The ratio of A260-A320/A280-A320 was calculated to assess RNA purity, and the absorbance at 320 nm served as background absorbance. DNase I digestion was performed to remove contaminating DNA, after which, the quality of the extracted RNA was detected by 1.2% formaldehyde agarose gel electrophoresis. A 1-μg sample of RNA was reverse transcribed into cDNA using a reverse transcription kit (Takara, Japan). RT-PCR was performed to detect PTEN, PI3K, Akt, mTOR, MMP2, MMP9, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression in various tissue samples. The sequence-specific primers used for RT-PCR are shown in Table 1. Therefore, we analyzed the expression level of IFN-γ, granzyme B, and perforin in pancreatic tumor. The sequence-specific primers are shown in Table S1. RT-PCR was performed with an ABI PRISM 7700 apparatus (Applied Biosystems; Foster City, CA, USA), and the amplification program consisted of denaturation at 95°C for 30 sec followed by 40 cycles of 95°C for 5 sec and then annealing at 60°C for 40 sec. Relative levels of RNA expression were assessed using the 2−ΔΔCt method. The housekeeping gene GAPDH served as an internal control.
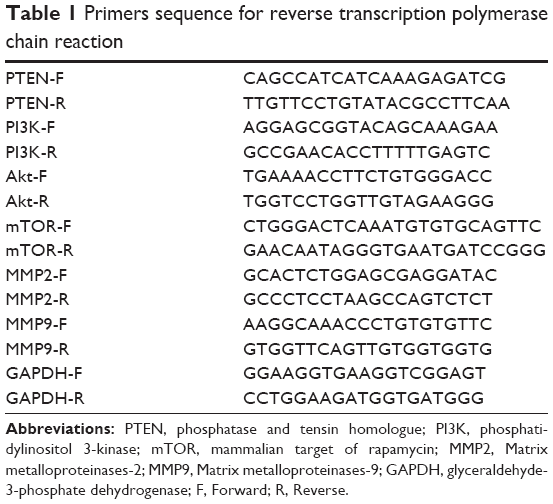 | Table 1 Primers sequence for reverse transcription polymerase chain reaction |
Western blot analysis
Ice-cold RIPA lysis buffer containing 1 mM phenylmethylsulfonyl fluoride was used to extract the total proteins from samples of tumor tissue. The extraction of phosphorylated proteins required inclusion of phosphatase inhibitors in the extraction buffer. Samples of tumor tissue (~50 mg) were homogenized in 500 μL of RIPA lysis buffer and then incubated on ice for 20 min, after which the samples were centrifuged at 14,000 rpm (4°C) for 30 min. The supernatant fractions containing soluble proteins were collected and stored at −80°C. Protein concentrations were determined using a bicinchoninic acid protein assay kit (Pierce; Rochford, IL, USA), according to the manufacturer’s instructions. Equal amounts of total protein (30 μg) were fractionated by 12% polyacrylamide gel electrophoresis, and the separated protein bands were transferred onto a PVDF membrane (BioRad; Hercules, CA, USA) by the wet transfer method. The membrane was then blocked with 5% skimmed dry milk in TBST (50 mM Tris-HCl, 150 mM NaCl, 0.1% Tween 20, pH 7.4) for 2 h at room temperature. It was then washed 3 times with washing buffer (TBS with 0.1% Tween 20, pH 7.6) for 10 min each time. After being washed, the membrane was incubated with the primary antibodies (rabbit anti-PTEN, rabbit anti-PI3K, rabbit anti-phospho-PI3K [Tyr458], rabbit anti-Akt, rabbit anti-phospho-Akt [Ser473], rabbit anti-mTOR, and rabbit anti-phospho-mTOR [Ser2448] [Cell Signaling Technology; Danvers, MA, USA]) and also mouse anti-MMP2, mouse anti-MMP9, and mouse anti-GAPDH (Santa Cruz Biotechnology; Santa Cruz, CA, USA) at dilutions of 1:1,000 in TBST at 4°C overnight.29 The following day, the membrane was washed three times with TBST (5 min per wash) and then incubated with goat antimouse or goat antirabbit IgG (Santa Cruz Biotechnologies) at a dilution of 1:5,000 in TBST at room temperature for 2 h. Next, the membrane was washed three more times with TBST, and ECL Plus substrate (Thermo Fisher Scientific; Waltham, MA, USA) was applied to the membrane in a darkroom. The staining images were captured with a Chemiluminescence Imaging System (Tanon Science & Technology; Shanghai, People’s Republic of China), and the gray scale of each band was analyzed using ImageJ software. Protein expression was normalized to that of GAPDH.
Statistical analysis
All data were analyzed using GraphPad Prism version 6.01 software (GraphPad Prism; San Diego, CA, USA). Differences between two groups were evaluated using Student’s t-test. All data were from three separate experiments and analyzed in a blinded manner, and values were expressed as the mean ± standard deviation. A P-value <0.05 was considered statistically significant.
Results
Effects of anti-PD-L1 antibody on the growth of pancreatic cancer and its spontaneous liver metastasis in vivo
We established an orthotopic mouse model of pancreatic adenocarcinoma for use in determining the therapeutic effect of blocking the PD-1/PD-L1 pathway on pancreatic cancer and its metastasis in vivo. The mean carbohydrate antigen 19-9 serum level increased from 13.898±2.272 U/mL at 1 day before injection of the pancreatic cancer cells to 64.787±8.052 U/mL at 7 days after injection (Figure S2; P<0.01). After treatment with anti-PD-L1 antibody for 5 weeks, all experimental mice exhibited a gradual weight loss and reduced activity; however, the average weight of the mice in the two groups was not significantly different (Figure 1A; P>0.05). After sacrificing the mice, we found that all the mice in both groups had pancreatic tumors. The mean weights of the pancreatic tumors in the control group and in the anti-PD-L1 group were 0.236±0.050 g and 0.126±0.041 g, respectively (Figure 1B; P<0.01). The anti-PD-L1 treatment inhibited pancreatic tumor growth by 46.6%. Liver metastases were found in 5 of 5 mice in the control group and 2 of 5 mice in the anti-PD-L1 group.
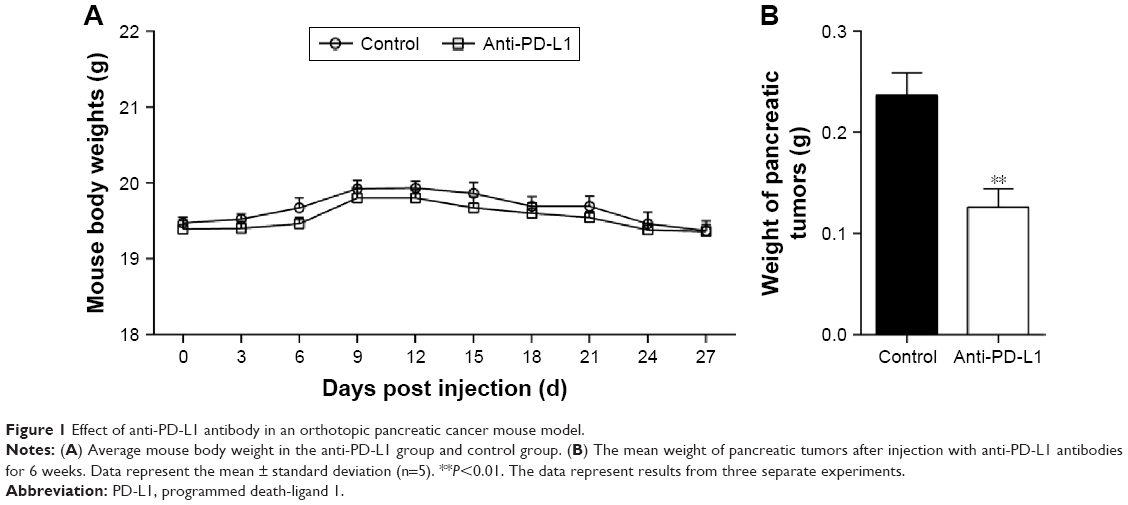 | Figure 1 Effect of anti-PD-L1 antibody in an orthotopic pancreatic cancer mouse model. |
Anti-PD-L1 antibody reduced PD-L1 expression in pancreatic cancer tissue and metastases
We used immunohistochemistry methods to examine PD-L1 expression in pancreatic cancer tissue and spontaneous liver metastases. All immunostained tumor sections were examined by bright-field microscopy, and images were photographed with a microscope camera (Nikon; Tokyo, Japan) at a magnification of ×400. We found that the PD-L1 expression was significantly reduced in pancreatic cancer tissue and liver metastasis tissue when compared with its expression in the control group (Figure 2 and Table S2; P<0.0001 and P<0.0001), and mainly expressed on pancreatic carcinoma cells. RT-PCR demonstrated that the expression level of IFN-γ, granzyme B, and perforin was significantly higher in the anti-PD-L1 group than in the control (Figure S3; IFN-γ: P<0.01, granzyme B: P<0.01, and perforin: P<0.01, respectively).
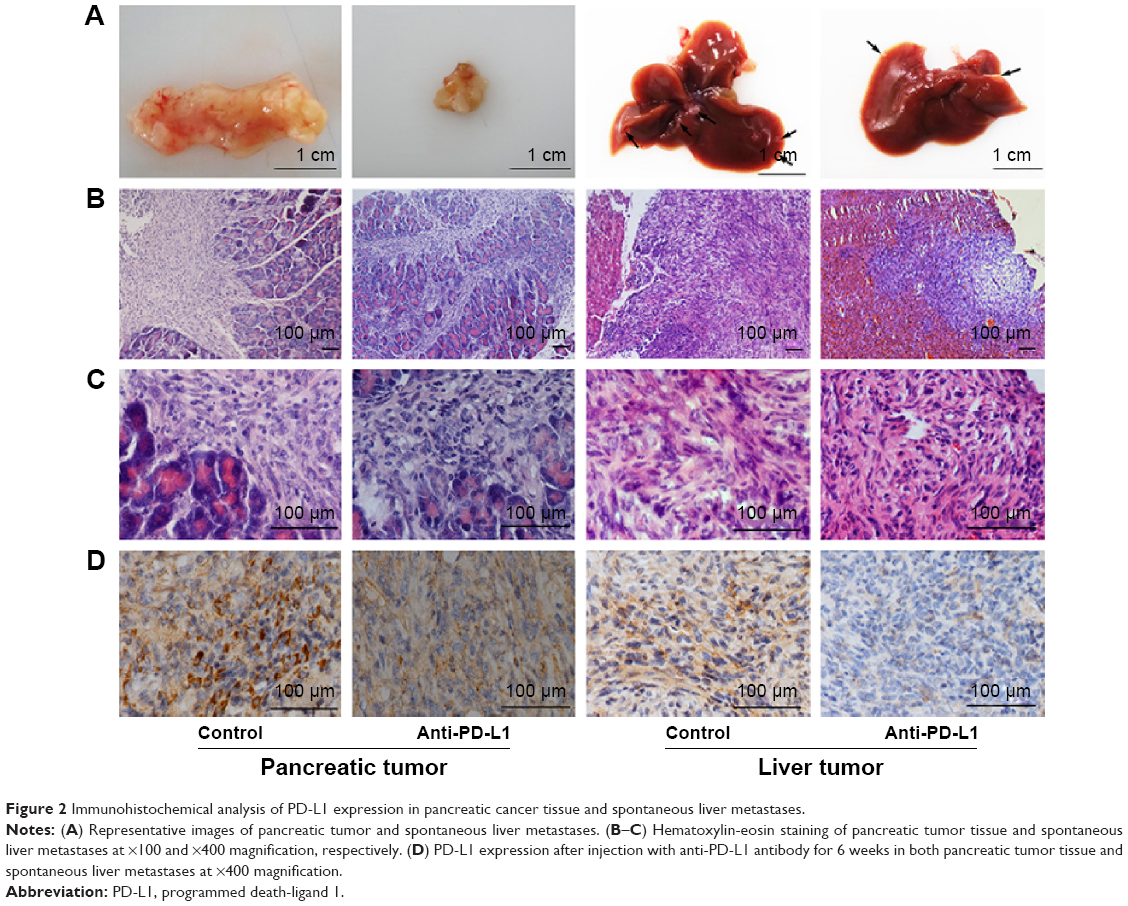 | Figure 2 Immunohistochemical analysis of PD-L1 expression in pancreatic cancer tissue and spontaneous liver metastases. |
Anti-PD-L1 antibody influenced the expression of components involved in the PI3K/Akt/mTOR signaling pathway
To determine how a PD-L1 blockade affects the PI3K/Akt/mTOR signaling pathway, we examined the levels of PTEN, PI3K, Akt, and mTOR mRNA expression in pancreatic cancer tissue and spontaneous liver metastases after treatment with the anti-PD-L1 antibody (Figure 3). We found that the levels of PTEN mRNA expression in pancreatic cancer tissue and liver metastases were significantly higher in the anti-PD-L1 group than in the control (pancreatic cancer tissue: Figure 3A; P<0.01) (liver metastasis tissue: Figure 3B; P<0.01). Similar levels of PI3K, Akt, and mTOR expression were found in pancreatic cancer tissue (Figure 3A; P>0.05) and liver metastasis tissue (Figure 3B; P>0.05) from the control group and the anti-PD-L1 group. We next performed Western blot studies to examine the levels of PTEN, PI3K, p-PI3K, Akt, p-Akt, mTOR, and p-mTOR protein expression in the tumors (Figure 4) after treatment with anti-PD-L1 antibody. The results showed that treatment with anti-PD-L1 antibody inhibited the PD-1/PD-L1 pathway and resulted in increased levels of PTEN protein expression in pancreatic cancer tissue (Figure 4A; P<0.01) and liver metastasis tissue (Figure 4B; P<0.01). We also found significant decreases in p-Akt and p-mTOR protein expression in pancreatic cancer tissue (Figure 4A; P<0.01) and liver metastasis tissue (Figure 4B; P<0.01) in the anti-PD-L1 group compared with the control. However, both groups did not show any significant differences in their respective levels of PI3K, p-PI3K, Akt, and mTOR protein expression in pancreatic cancer tissue or spontaneous liver metastasis tissue after treatment (Figure 4; P>0.05).
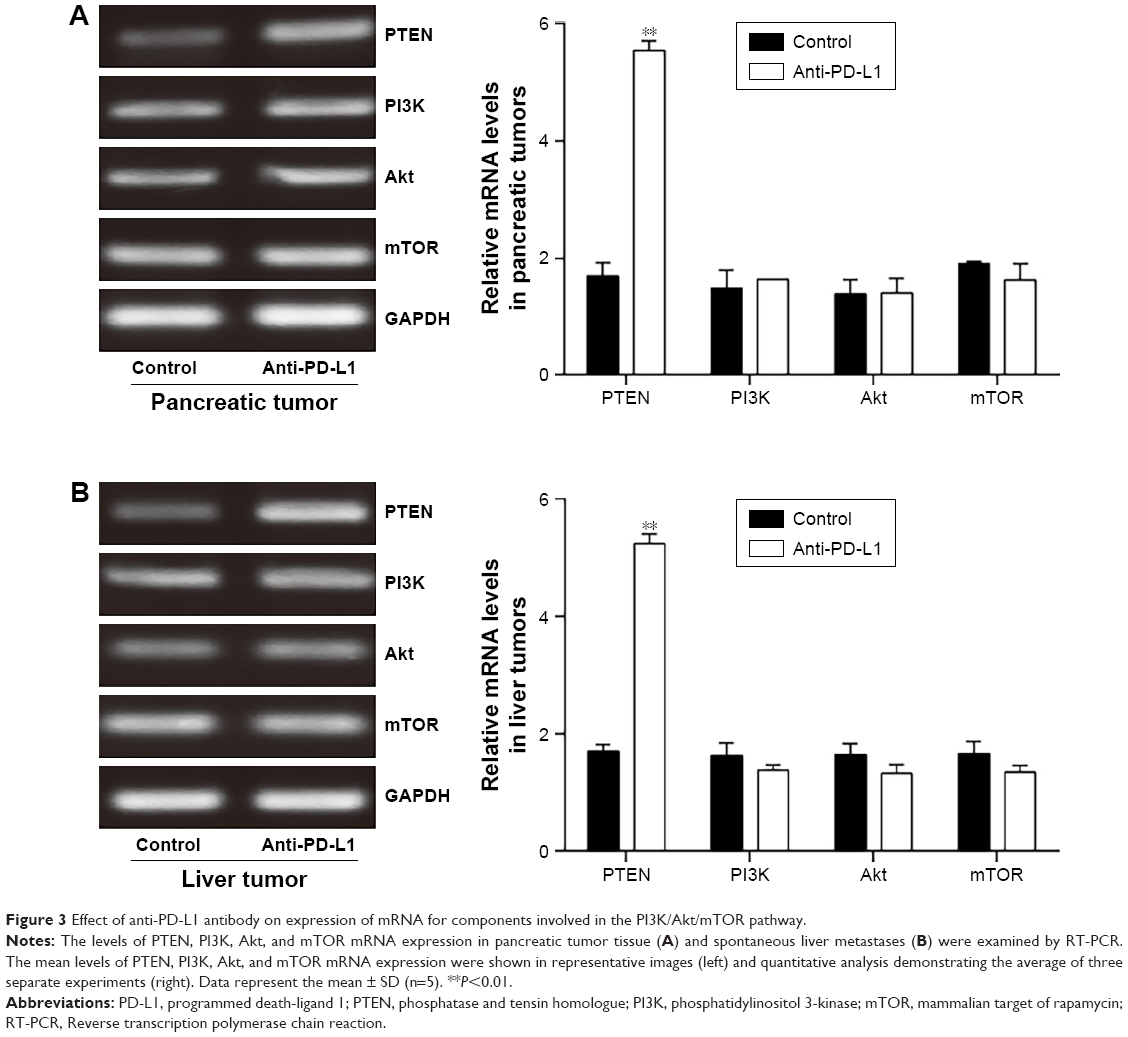 | Figure 3 Effect of anti-PD-L1 antibody on expression of mRNA for components involved in the PI3K/Akt/mTOR pathway. |
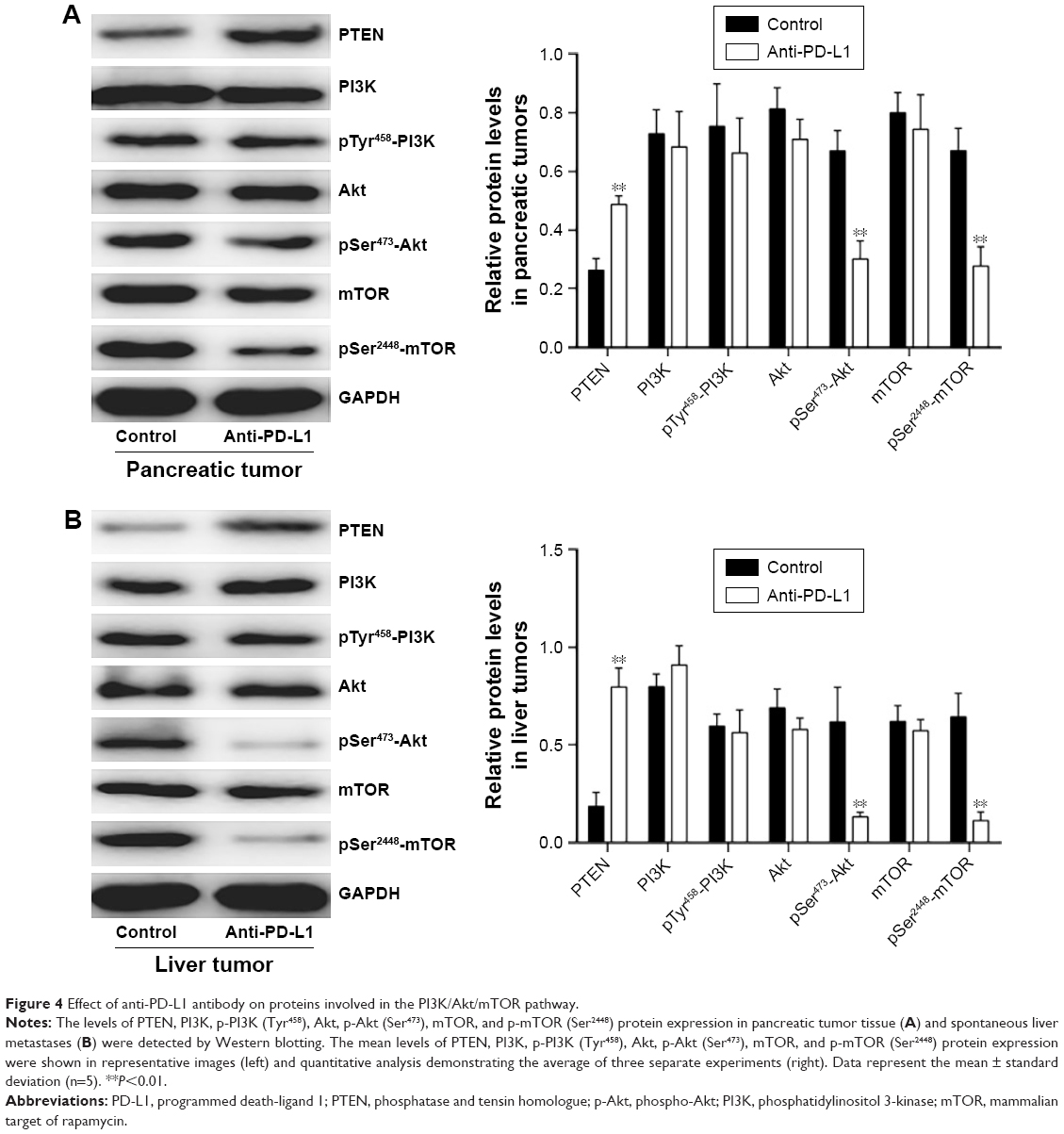 | Figure 4 Effect of anti-PD-L1 antibody on proteins involved in the PI3K/Akt/mTOR pathway. |
Anti-PD-L1 antibody reduced MMP2 and MMP9 expression in pancreatic cancer tissue and spontaneous liver metastasis tissue
It is well known that MMP2 and MMP9 are involved in the systemic dissemination of tumors.30 Furthermore, high levels of MMP2 and MMP9 have been found in pancreatic cancer tissue.31 In this study, we performed RT-PCR analyses (Figures 5A and B) and Western blot studies (Figures 5C and D) to measure MMP2 and MMP9 expression in samples of pancreatic cancer tissue and spontaneous liver metastasis tissue. We found that the levels of MMP2 and MMP9 mRNA expression were significantly decreased in both pancreatic cancer tissue (Figure 5A; P<0.05 and P<0.01, respectively) and liver metastasis tissue (Figure 5B; P<0.01) after treatment with anti-PD-L1 antibody. Similar decreases were seen in the levels of MMP2 and MMP9 protein expression in pancreatic cancer tissue (Figure 5C; P<0.05 and P<0.01, respectively) and liver metastasis tissue (Figure 5D; P<0.01).
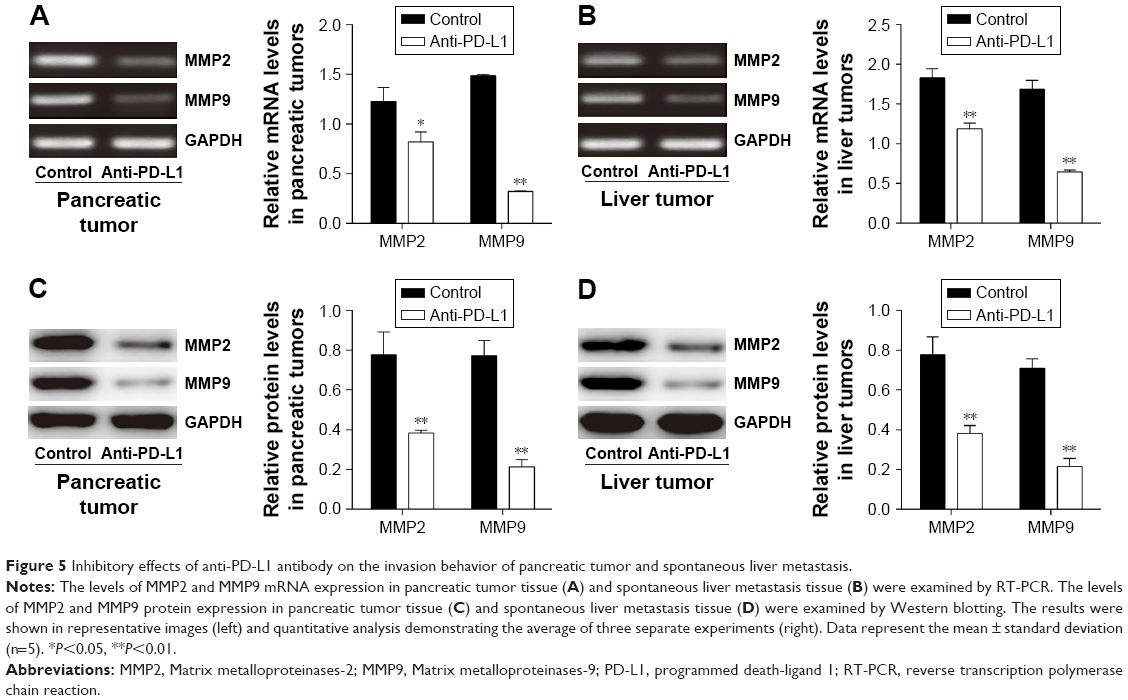 | Figure 5 Inhibitory effects of anti-PD-L1 antibody on the invasion behavior of pancreatic tumor and spontaneous liver metastasis. |
Discussion
Pancreatic cancer remains one of the most lethal human malignancies1 and is difficult to treat owing to its high incidence of metastasis to distant organs.2 Tumor cells can employ various mechanisms within the tumor microenvironment to evade the host’s immune surveillance system.32 Interactions between immune-checkpoint receptor PD-1 and its ligands (PD-L1 and PD-L2) on tumor cells can seriously impair the ability of T cells to target tumor cells.33 The PD-1/PD-L1 pathway has emerged as a promising target for immune modulation of cancer. Furthermore, such modulation may be achievable by inducing antitumor immune responses by the patient’s own immune system.32 Nomi et al34 demonstrated the antitumor effects of a PD-L1 blockade in a mouse subcutaneous pancreatic cancer model. However, that study lacked an orthotopic environment for pancreatic tumor formation, which is essential for analyses of metastasis and response to treatment.
Our pancreatic cancer model was established by a local injection of murine pancreatic cancer cells into the pancreas, and was more similar to the actual in vivo environment in which pancreatic tumors grow and metastasize. The model we established had a 0% mortality rate. Furthermore, the orthotopic model is very important to our study because cancer immunity is highly regulated by organ-specific leukocyte recruitment.26 After treatment with anti-PD-L1 antibody for 5 weeks, the growth rates of the pancreatic tumors were inhibited by 46.6%, suggesting that the PD-L1 blockade had a significant effect on tumor growth. The occurrence of spontaneous liver metastasis was significantly decreased in the anti-PD-L1 group compared with the control group.
Several studies have confirmed that overexpression of PD-L1 in tumor cells is predictive of a poor prognosis.34–37 Blockade PD-L1 could enhance the infiltration of tumor-infiltrating lymphocyte, especially CD8+T cells, which might directly lead to the antitumor effect.34 We found that in the anti-PD-L1 group, PD-L1 expression was significantly decreased in pancreatic cancer tissue and spontaneous liver metastases. The expression level of several effectors, including IFN-γ, granzyme B, and perforin, was significantly higher than that in the control, which demonstrates that the majority of CD8+T cells infiltrated into the pancreatic tumor in the model. Parsa et al17 reported that glioma cells with genetic deletions or mutations of PTEN had higher levels of PD-L1 protein expression and PI3K/Akt/mTOR pathway activity. Moreover, an in vitro study performed by Zhang et al38 demonstrated that PD-L1 expression is regulated by PTEN via the PI3K/Akt/mTOR pathway, and that a loss of PTEN induced tumor cell proliferation and invasion. These findings indicate that PTEN is a major regulator of downstream activation of the PI3K/Akt/mTOR pathway.39
The PI3K/Akt/mTOR signaling pathway is an evolutionarily conserved pathway that participates in a variety of physiological processes, including embryonic development, cell mitogenesis, and inflammatory reactions.40 The aberrant activation of this pathway, especially when measured by the loss of PTEN expression, leads to mutations in the PI3K enzyme, and thereby promotes carcinogenesis and tumor angiogenesis.41 PI3K phosphorylates phosphatidylinositol 3, 4, 5-triphosphate and then transmits signals from the cell surface into the cytoplasm to activate Akt. Akt modulates cell growth and protein synthesis via the phosphorylation of mTOR.42 To determine whether a PD-L1 blockade could effectively suppress pancreatic tumor invasion and metastasis in vivo, we examined the expression of various components involved in the PI3K/Akt/mTOR signaling pathway. These studies revealed that the levels of PTEN expression in pancreatic cancer and spontaneous liver metastasis tissue were significantly higher in the anti-PD-L1 treated group than in the control group, which further demonstrated that PD-L1 expression in pancreatic cancer is correlated with PTEN loss in vivo. There were no significant differences in the levels of PI3K, Akt, and mTOR mRNA and protein expression in the anti-PD-L1 treated group when compared with the control group. However, the levels of p-Akt and p-mTOR protein expression in the anti-PD-L1 group were significantly lower than those in the control group. These results indicated that the PD-L1 blockade had inhibited the PI3K/Akt/mTOR signaling pathway by causing an upregulation of PTEN expression and inhibiting Akt and mTOR phosphorylation.
Activation of the PI3K/Akt/mTOR signaling pathway is associated with a poor clinical prognosis.39 MMP2 and MMP9 are believed to be important facilitators of pancreatic cancer cell invasion and migration.43 Findings in recent studies suggest that MMP2 helps to regulate the inflammatory response, and MMP9 affects the angiogenesis process.31 Our results showed that the levels of MMP2 and MMP9 mRNA and protein expression were significantly decreased in both pancreatic cancer tissue and spontaneous liver metastases after treatment. These findings indicate that a PD-L1 blockade inhibits the PI3K/Akt/mTOR signaling pathway and thereby downregulates MMP2 and MMP9 expression and activity.
Conclusion
In this study, we showed that blockading PD-L1 prevented the loss of PTEN and affected the PI3K/Akt/mTOR signaling pathway in vivo. These mechanisms may contribute to the antitumor and antimetastasis effects of a PD-L1 blockade in cases of pancreatic cancer. We also demonstrated that a PD-L1 blockade could inhibit the progression of pancreatic cancer and its spontaneous liver metastasis by downregulating MMP2 and MMP9 expression and activity. The results of this in vivo study suggest that PD-L1 is a critical regulator of pancreatic tumor growth, invasion, and metastasis.
Acknowledgments
The study was supported by the Science and Technology Planning Project of Liaoning Province, People’s Republic of China (Grant number F15-199-1-48) and Educational Commission of Liaoning Province, People’s Republic of China (Grant number L2014294). We would like to thank the native English-speaking experts from BioMed Proofreading LLC (Cleveland, OH, USA) for editing this manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Kamisawa T, Wood LD, Itoi T, Takaori K. Pancreatic cancer. Lancet. 2016;388(10039):73–85. | ||
Amundadottir LT. Pancreatic Cancer Genetics. Int J Biol Sci. 2016;12(3):314–325. | ||
Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9–29. | ||
Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74(11):2913–2921. | ||
Keleg S, Buchler P, Ludwig R, Buchler MW, Friess H. Invasion and metastasis in pancreatic cancer. Mol Cancer. 2003;2:14. | ||
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264. | ||
Freeman GJ, Long AJ, Iwai Y, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192(7):1027–1034. | ||
Pedoeem A, Azoulay-Alfaguter I, Strazza M, Silverman GJ, Mor A. Programmed death-1 pathway in cancer and autoimmunity. Clin Immunol. 2014;153(1):145–152. | ||
Bally AP, Austin JW, Boss JM. Genetic and epigenetic regulation of PD-1 expression. J Immunol. 2016;196(6):2431–2437. | ||
Okazaki T, Chikuma S, Iwai Y, Fagarasan S, Honjo T. A rheostat for immune responses: the unique properties of PD-1 and their advantages for clinical application. Nat Immunol. 2013;14(12):1212–1218. | ||
Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev. 2010;236:219–242. | ||
Okazaki T, Maeda A, Nishimura H, Kurosaki T, Honjo T. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc Natl Acad Sci U S A. 2001;98(24):13866–13871. | ||
Swaika A, Hammond WA, Joseph RW. Current state of anti-PD-L1 and anti-PD-1 agents in cancer therapy. Mol Immunol. 2015;67(2 Pt A):4–17. | ||
Patel SP, Kurzrock R. PD-L1 Expression as a predictive biomarker in cancer immunotherapy. Mol Cancer Ther. 2015;14(4):847–856. | ||
Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455–2465. | ||
Sunshine J, Taube JM. PD-1/PD-L1 inhibitors. Curr Opin Pharmacol. 2015;23:32–38. | ||
Parsa AT, Waldron JS, Panner A, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med. 2007;13(1):84–88. | ||
Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med. 2012;209(6):1201–1217. | ||
Baer R, Cintas C, Therville N, Guillermet-Guibert J. Implication of PI3K/Akt pathway in pancreatic cancer: When PI3K isoforms matter? Adv Biol Regul. 2015;59:19–35. | ||
Mayer IA, Arteaga CL. The PI3K/AKT Pathway as a Target for Cancer Treatment. Ann Rev Med. 2016;67:11–28. | ||
Yothaisong S, Dokduang H, Techasen A, et al. Increased activation of PI3K/AKT signaling pathway is associated with cholangiocarcinoma metastasis and PI3K/mTOR inhibition presents a possible therapeutic strategy. Tumour Biol. 2013;34(6):3637–3648. | ||
Adya R, Tan BK, Punn A, Chen J, Randeva HS. Visfatin induces human endothelial VEGF and MMP-2/9 production via MAPK and PI3K/Akt signalling pathways: novel insights into visfatin-induced angiogenesis. Cardiovasc Res. 2008;78(2):356–365. | ||
Bloomston M, Zervos EE, Rosemurgy AS 2nd. Matrix metalloproteinases and their role in pancreatic cancer: a review of preclinical studies and clinical trials. Ann Surg Oncol. 2002;9(7):668–674. | ||
Hartwig W, Werner J, Jager D, Debus J, Buchler MW. Improvement of surgical results for pancreatic cancer. Lancet Oncol. 2013;14(11):e476–e485. | ||
Zou Y, Li J, Chen Z, et al. miR-29c suppresses pancreatic cancer liver metastasis in an orthotopic implantation model in nude mice and affects survival in pancreatic cancer patients. Carcinogenesis. 2015;36(6):676–684. | ||
Okudaira K, Hokari R, Tsuzuki Y, et al. Blockade of B7-H1 or B7-DC induces an anti-tumor effect in a mouse pancreatic cancer model. Int J Oncol. 2009;35(4):741–749. | ||
Smith J, Robida MD, Acosta K, et al. Quantitative and qualitative characterization of Two PD-L1 clones: SP263 and E1L3N. Diagn Pathol. 2016;11(1):44. | ||
Cao Y, Wang Q, Du Y, et al. l-arginine and docetaxel synergistically enhance anti-tumor immunity by modifying the immune status of tumor-bearing mice. Int Immunopharmacol. 2016;35:7–14. | ||
He L, Wu Y, Lin L, et al. Hispidulin, a small flavonoid molecule, suppresses the angiogenesis and growth of human pancreatic cancer by targeting vascular endothelial growth factor receptor 2-mediated PI3K/Akt/mTOR signaling pathway. Cancer Sci. 2011;102(1):219–225. | ||
Vargova V, Pytliak M, Mechirova V. Matrix metalloproteinases. EXS. 2012;103:1–33. | ||
Jakubowska K, Pryczynicz A, Januszewska J, et al. Expressions of matrix metalloproteinases 2, 7, and 9 in carcinogenesis of pancreatic ductal adenocarcinoma. Dis Markers. 2016;2016:9895721. | ||
Postow MA, Callahan MK, Wolchok JD. Immune checkpoint blockade in cancer therapy. J Clin Oncol. 2015;33(17):1974–1982. | ||
Riley JL. PD-1 signaling in primary T cells. Immunol Rev. 2009;229(1):114–125. | ||
Nomi T, Sho M, Akahori T, et al. Clinical significance and therapeutic potential of the programmed death-1 ligand/programmed death-1 pathway in human pancreatic cancer. Clin Cancer Res. 2007;13(7):2151–2157. | ||
Muenst S, Schaerli AR, Gao F, et al. Expression of programmed death ligand 1 (PD-L1) is associated with poor prognosis in human breast cancer. Breast Cancer Res Treat. 2014;146(1):15–24. | ||
Ohigashi Y, Sho M, Yamada Y, et al. Clinical significance of programmed death-1 ligand-1 and programmed death-1 ligand-2 expression in human esophageal cancer. Clin Cancer Res. 2005;11(8):2947–2953. | ||
Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016;8(328):328rv4. | ||
Zhang Y, Zhang J, Xu K, et al. PTEN/PI3K/mTOR/B7-H1 signaling pathway regulates cell progression and immuno-resistance in pancreatic cancer. Hepatogastroenterology. 2013;60(127):1766–1772. | ||
Ocana A, Vera-Badillo F, Al-Mubarak M, et al. Activation of the PI3K/mTOR/AKT pathway and survival in solid tumors: systematic review and meta-analysis. PLoS One. 2014;9(4):e95219. | ||
Polivka J Jr, Janku F. Molecular targets for cancer therapy in the PI3K/AKT/mTOR pathway. Pharmacol Ther. 2014;142(2):164–175. | ||
Burris HA 3rd. Overcoming acquired resistance to anticancer therapy: focus on the PI3K/AKT/mTOR pathway. Cancer Chemother Pharmacol. 2013;71(4):829–842. | ||
Akinleye A, Avvaru P, Furqan M, Song Y, Liu D. Phosphatidylinositol 3-kinase (PI3K) inhibitors as cancer therapeutics. J Hematol Oncol. 2013;6(1):88. | ||
Maatta M, Soini Y, Liakka A, Autio-Harmainen H. Differential expression of matrix metalloproteinase (MMP)-2, MMP-9, and membrane type 1-MMP in hepatocellular and pancreatic adenocarcinoma: implications for tumor progression and clinical prognosis. Clin Cancer Res. 2000;6(7):2726–2734. |
Supplementary materials
 | Figure S1 Immunohistochemical score of PD-L1 expression in pancreatic tumor and spontaneous liver metastases. |
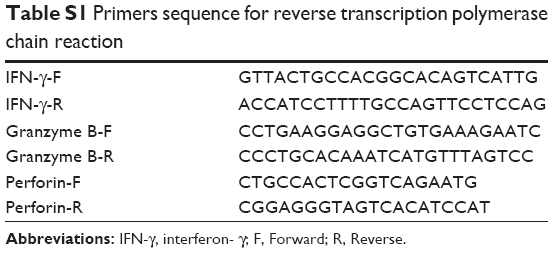 | Table S1 Primers sequence for reverse transcription polymerase chain reaction |
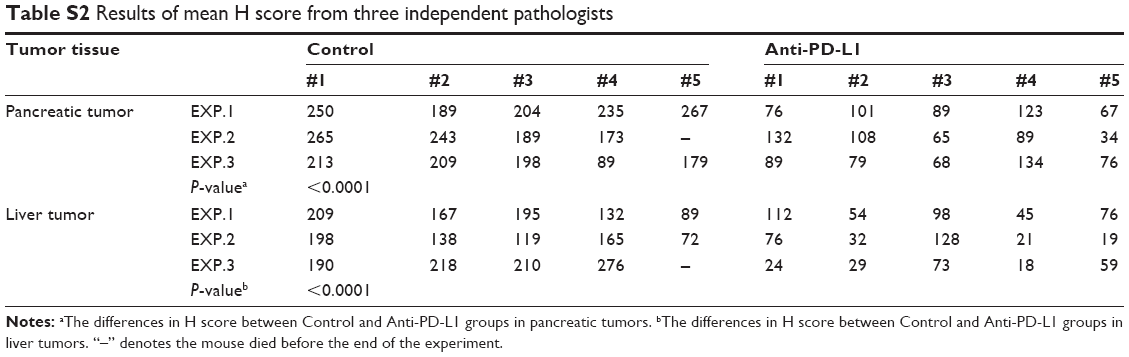 | Table S2 Results of mean H score from three independent pathologists |
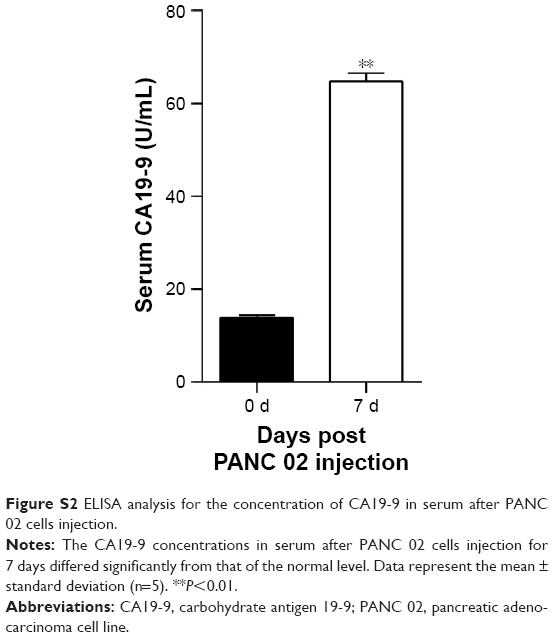 | Figure S2 ELISA analysis for the concentration of CA19-9 in serum after PANC 02 cells injection. |
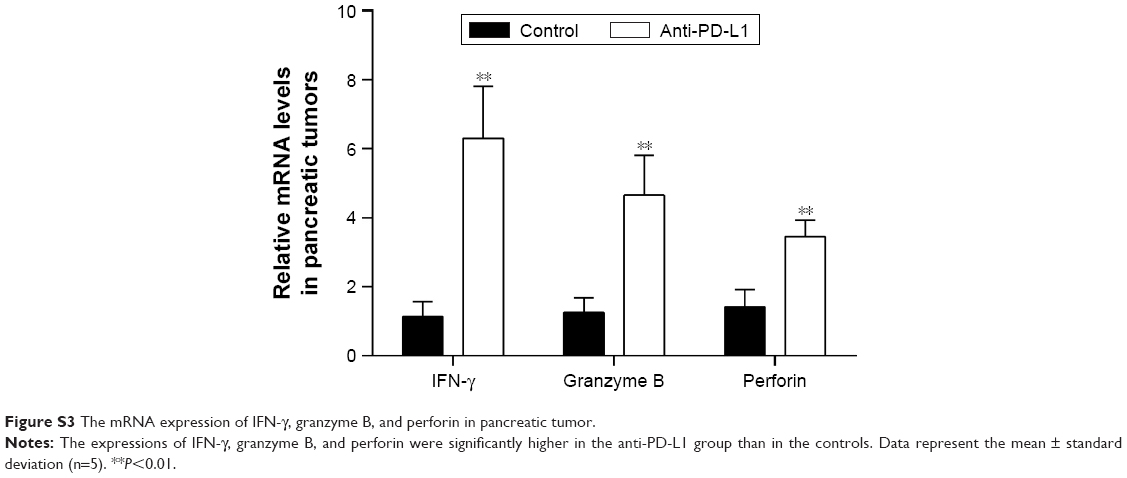 | Figure S3 The mRNA expression of IFN-γ, granzyme B, and perforin in pancreatic tumor. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.