Back to Journals » OncoTargets and Therapy » Volume 13
3,6-Disubstituted 1,2,4-Triazolo[3,4-b]Thiadiazoles with Anticancer Activity Targeting Topoisomerase II Alpha
Authors Sagredou S, Dalezis P ![]() , Nikoleousakos N, Nikolaou M
, Nikoleousakos N, Nikolaou M ![]() , Voura M
, Voura M ![]() , Almpanakis K, Panayiotidis MI
, Almpanakis K, Panayiotidis MI ![]() , Sarli V
, Sarli V ![]() , Trafalis DT
, Trafalis DT
Received 21 March 2020
Accepted for publication 30 May 2020
Published 28 July 2020 Volume 2020:13 Pages 7369—7386
DOI https://doi.org/10.2147/OTT.S254856
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Leo Jen-Liang Su
Sofia Sagredou,1 Panagiotis Dalezis,1 Nikolaos Nikoleousakos,1 Michail Nikolaou,1 Maria Voura,2 Konstantinos Almpanakis,2 Mihalis I Panayiotidis,3,4 Vasiliki Sarli,2 Dimitrios T Trafalis1
1Laboratory of Pharmacology, Faculty of Medicine, National and Kapodistrian University of Athens, Athens 11527, Greece; 2Department of Chemistry, Aristotle University of Thessaloniki , Thessaloniki, 54124, Greece; 3Department of Electron Microscopy & Molecular Pathology, The Cyprus Institute of Neurology & Genetics, Nicosia 2371, Cyprus; 4The Cyprus School of Molecular Medicine, Nicosia 1683, Cyprus
Correspondence: Sofia Sagredou; Dimitrios T Trafalis Email [email protected]; [email protected]
Background: Topoisomerase IIα (topIIα) maintains the topology of DNA in order to ensure the proper functioning of numerous DNA processes. Inhibition of topIIα leads to the killing of cancer cells thus constituting such inhibitors as useful tools in cancer therapeutics. Triazolo[3,4-b]thiadiazole derivatives are known for their wide range of pharmacological activities while previous studies have documented their in vitro anticancer activity. The purpose of the current study was to investigate if these chemical compounds can act as topIIα inhibitors in cell-free and cell-based systems.
Materials and Methods: The MTT assay was performed in DLD-1, HT-29, and LoVo cancer cells so as to evaluate the antiproliferative activity of KA25, KA26, and KA39 triazolo[3,4-b]thiadiazole derivatives. The KA39 compound was tested as a potential topIIα inhibitor using the plasmid-based topoisomerase II drug screening kit. The inhibitory effect of the three derivatives on topIIα phosphorylation was studied in HT-29 and LoVo cancer cells according to Human Phospho-TOP2A/Topoisomerase II Alpha Cell-Based Phosphorylation ELISA Kit. Moreover, flow cytometry was utilized in order to explore apoptotic induction and cell cycle growth arrest, upon treatment with KA39, in DLD-1 and HT-29 cells, respectively. In silico studies were also carried out for further investigation.
Results: All three triazolo[3,4-b]thiadiazole derivatives showed an in vitro antiproliferative effect with the KA39 compound being the most potent one. Our results indicated that KA39 induced both early and late apoptosis as well as cell cycle growth arrest in S phase. In addition, the compound blocked the relaxation of supercoiled DNA while it also inhibited topIIα phosphorylation (upon treatment; P< 0.001).
Conclusion: Among the three triazolo[3,4-b]thiadiazole derivatives, KA39 was shown to be the most potent anticancer agent and catalytic inhibitor of topIIα phosphorylation as well.
Keywords: triazoles, thiadiazoles, topoisomerase IIα, catalytic cycle, Ser-1106, phosphorylation
Introduction
DNA topology is crucial for a wide range of cellular processes including cell viability, replication, transcription, as well as chromosomal segregation, recombination, and condensation/decondensation.1,2 More specifically, cells recruit a specific category of enzymes known as topoisomerases which carry out the required alterations in the topological state of DNA.3 Two types of topoisomerases, I and II, are ubiquitously expressed, both of which can regulate DNA supercoiling by i) generating transient strand breaks, ii) removing any knots and tangles, and iii) sealing the broken strands in the duplex molecule.4 The major difference between topoisomerase I and II is the generation of single and double strand breaks in the double helix, respectively.5 With regard to topoisomerase II, higher mammals express two isoforms of the enzyme, topoisomerase IIα and IIβ, which share similar amino acid sequence and catalytic activities but their cellular functions are quite distinct.6 For instance, topoisomerase IIα (topIIα) is a cell-cycle dependent enzyme (specifically at the G2/M phase) while topoisomerase IIβ’s (topIIβ) function is not in line with cell-cycle progression.7 With respect to topIIα, various posttranslational modifications (phosphorylation, ubiquitination, and SUMOylation) can modify the functionality of the enzyme in different ways.8 Among them, phosphorylation (specifically at Ser-1106) is considered to be a key mechanism capable of altering the catalytic activity of the enzyme.9,10 Topoisomerase II operates a catalytic cycle, known as a double-strand passage reaction, with two DNA duplexes (termed G- and T-segments) being formed. The enzyme binds and cleaves the G-segment which then leads to an intermediate and transient cleavage complex consisting of DNA and topoisomerase II. In the meantime, the T-segment is translocated through a “gate” provided by the cleaved G-segment. Then, topoisomerase II seals the broken strands in the duple helix of the G-segment, releases the molecule and then the “gate” closes again so that the enzyme retrieves its enzymatic activity in order to start a new catalytic cycle.5,11 The double-strand passage requires two cofactors, magnesium and adenosine triphosphate (ATP), so that the catalytic cycle can be transacted. More specifically, magnesium is required for all steps of the catalytic cycle whereas ATP is necessary for i) transportation of the T-segment through the “open gate”, and ii) conformational changes necessary for the next round of catalysis to occur.12,13
On the other hand, triazoles and thiadiazoles are heterocyclic compounds consisting of a five-membered ring where triazole contains three nitrogen atoms and thiadiazole two nitrogen and one sulfur atoms. Both classes of heterocycles are known for processing a wide spectrum of biological activities like anti-microbial, anti-inflammatory, anti-convulsant, antioxidant, anti-leishmanial, anti-viral, anti-hypertensive and anti-depressant.14–16 Of great interest is the potent anticancer activity which is attributable to reaction sites that both heterocyclic compounds carry including the toxophoric N-C-S moiety and the acidic proton at C-2 in the thiadiazole and triazole rings, respectively. Moreover, modifications at different positions of heterocyclic compounds as well as the fusion of the two core structures of triazoles and thiadiazoles rings they all constitute alternative synthetic possibilities capable of leading to novel chemical entities.17 For instance, according to current findings, fusion of 1,2,4-triazole and 1,3,4-thiadiazole rings displayed significant cytotoxic activity against several cancer cell lines implying that these newly synthesized compounds may act as potent anticancer agents.17–24 The aim of the present study was to investigate whether topIIα constitutes a potential target of triazolo[3,4-b]thiadiazole derivatives KA25, KA26, and KA39 including an evaluation of their biological activity against three human colorectal cancer cell lines and one normal. To determine the inhibitory effect of the three heterocyclic compounds, in topIIα’s activity, our study was carried out as follows: The KA39 agent, by being the most active, was tested in the DNA relaxation reaction performed by topoisomerase II whereas all three tested compounds were assayed in the inhibition of the phosphorylation of the topIIα isoform. The experimental study regarding the potential inhibition of topIIα phosphorylation was conducted in two human colorectal cancer cell lines, HT-29 and LoVo, both of which express the target protein in sufficient levels.25 Finally, other and more detailed studies were conducted about the potency of KA39 compound to induce apoptosis as well as cell cycle growth arrest.
Materials and Methods
Chemistry
The synthesis of the 1,2,4-triazolo-[3,4-b]-1,3,4-thiadiazoles was performed according to the synthetic protocol previously described by our group.17 Commercially available 2-(3,4-dimethoxyphenyl)acetic acid reacted with oxalyl chloride and esterified to yield the corresponding methyl ester, which was subsequently converted to sulfonyl chloride 2 after treatment with chlorosulfonic acid in CHCl3 (Scheme 1). Then, sulfonyl chloride 2 reacted with Me2NH to give sulfonamide 3 in excellent yield. Hydrazinolysis of this sulfonamide gave the carboxylic acid hydrazide 4, which reacted with KOH, CS2 in EtOH to give the intermediate potassium thiocarbamate that was cyclized in the presence of hydrazine hydrate to triazole 5.26 Condensation of triazole 5 with 2.5-dinitro-benzoic acid, 4-chloro-, and 4-fluoro-cinnamic acid in POCl3 produced the desired 1,2,4-triazolo[3,4-b]-1,2,4-thiadiazoles KA39, KA26, and KA25, as illustrated in Scheme 1. All reactions were carried out under an atmosphere of argon unless otherwise specified. High purity commercial reagents and solvents were purchased and used without further purification. Reactions were monitored by thin layer chromatography and using UV light as a visualizing agent and ethanolic p-anisaldehyde solution, aqueous ceric sulfate/phosphomolybdic acid, and heat as developing agents. The 1H and 13C NMR spectra were recorded at 500 and 126 MHz, and tetramethylsilane was used as an internal standard. Chemical shifts are indicated in δ values (ppm) from internal reference peaks (TMS 1H 0.00; CDCl3 1H 7.26, 13C 77.00; DMSO-d6 1H 2.50, 13C 39.51) and coupling constants (J) in Hz. Melting points (mp) are uncorrected. The LC-MS spectra were recorded on a LC20AD Shimadzu connected to Shimadzu LCMS-2010EV equipped with C18 analytical column SUPELCO Discovery (C18, 25 cmx4.6 mm, 5 μm).
 |
Scheme 1 Synthesis of 3,6-disubstituted 1,2,4-triazolo-[3,4-b]-1,3,4-thiadiazoles. |
Methyl 2-(3,4-Dimethoxyphenyl)Acetate, S1
2-(3,4-dimethoxyphenyl)acetic acid (1 g, 5.1 mmol) was dissolved in 24 mL dry DCM, a catalytic amount of dry DMF was added and the mixture was cooled at 0°C. Then, oxalyl chloride (1.32 mL, 15.31 mmol) was added dropwise and the reaction left stirring at room temperature for 0.5 hours. The addition of MeOH (20 mL) in portions at 0°C was followed and the mixture was stirred at room temperature for 10 minutes. The reaction was quenched with H2O (20 mL) and the mixture was extracted with dichloromethane (3 x 20 mL). The organic extracts were dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (eluent; hexane/ethyl acetate, 4/1), to afford S1 as a yellowish oil in 99% yield. The spectral data were accordance with those reported in the literature.27 S1: 1H NMR (500 MHz, CDCl3) δ 6.81 (s, 3H), 3.88 (s, 3H), 3.86 (s, 3H), 3.69 (s, 3H), 3.57 (s, 2H).
Methyl 2-(2-(Chlorosulfonyl)-4,5-Dimethoxyphenyl)Acetate, 2
To a solution of S1 (3.7 g, 17.64 mmol) in CHCl3 (35 mL) chlorosulfonic acid (5.28 mL, 79.4 mmol) was added dropwise at 0°C and the reaction was stirred at room temperature for 1 hour. The reaction was quenched with H2O (35 mL) and the mixture was extracted with dichloromethane (x3). The organic layers were combined, dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (eluent; hexane/ethyl acetate = 2/1), to yield compound 2 as a white solid (88% yield). 2: mp = 107–110°C; 1H NMR (500 MHz, CDCl3) δ 7.52 (s, 1H), 6.88 (s, 1H), 4.11 (s, 2H), 3.97 (s, 3H), 3.95 (s, 3H), 3.73 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 170.5, 154.1, 148.0, 134.6, 127.8, 115.3, 111.3, 56.4, 56.4, 52.4, 38.0; ESI-MS m/z for C11H13ClNaO6S [M+Na]+ calcd 331.0, found 330.85.
Methyl 2-(2-(N,N-Dimethylsulfamoyl)-4,5-Dimethoxyphenyl)Acetate, 3
To the solution of 2 (450 mg, 1.46 mmol) in THF (1.8 mL) 2.92 mmol of dimethylamine (1.47 mL, solution 2 M in THF) were added at 0°C. The reaction mixture was stirred at room temperature for 1 hour. Then, it was concentrated under reduced pressure to afford compound 3 as a pale yellow solid in 99% yield. 3: mp = 143–146°C; 1H NMR (500 MHz, CDCl3) δ 7.43 (s, 1H), 6.81 (s, 1H), 3.99 (s, 2H), 3.92 (s, 3H), 3.91 (s, 3H), 3.68 (s, 3H), 2.68 (d, 6H); 13C NMR (126 MHz, CDCl3) δ 171.5, 152.2, 147.5, 127.5, 127.3, 115.4, 113.1, 56.3, 56.1, 52.0, 37.9, 36.6; ESI-MS m/z for C13H19NNaO6S [M+Na]+ calcd 340.08, found 339.90.
2-(2-Hydrazinyl-2-Oxoethyl)-N,N-Dimethyl-4,5-Dimethoxybenzenesulfonamide,4
The mixture of 3 (300 mg, 0.95 mmol) in aqueous hydrazide 80% (0.47 mL, 7.79 mmol) was refluxed for 0.5 hour. The reaction mixture was extracted with dichloromethane (3x10 mL) and the combined organic extracts were dried over Na2SO4, filtered and concentrated under reduced pressure to afford compound 4 (yellow solid) in 66% yield. The spectral data were accordance with those reported in the literature.28 4: 1H NMR (500 MHz, CDCl3) δ 7.70 (s, 1H), 7.32 (s, 1H), 7.01 (s, 1H), 3.91 (s, 3H), 3.87 (s, 3H), 3.79 (s, 2H), 2.71 (s, 6H); 13C NMR (126 MHz, CDCl3) δ 170.6, 152.4, 147.6, 128.2, 125.9, 114.5, 112.8, 56.2, 38.4, 37.0; ESI-MS m/z for C12H19N3NaO5S [M+Na]+ calcd 340.09, found 339.85.
2-((4-Amino-5-Mercapto-4H-1,2,4-Triazol-3-Yl)Methyl)-N,N-Dimethyl-4,5-Dimethoxybenzene Sulfonamide, 5
To a flask containing compound 4 (180 mg, 0.57 mmol), abs EtOH (8.5 mL) and KOH (48 mg, 0.85 mmol) were successively added. Then, CS2 (50 μL, 0.85 mmol) was added dropwise at 0°C and the reaction mixture was allowed to warm to room temperature and stirred for 24 hours. Subsequently, diethyl ether (8.5 mL) was added and the solid product was filtered and washed with cold diethyl ether. The product S1 was dried and used in the next reaction without further purification. To a sealed vial the previous salt S1 and aqueous hydrazide 80% (2.76 mL, 45.41 mmol) were added and the mixture was refluxed for 2 hours. Acidification with 10% aq. HCl was followed and the pH was adjusted to 7. The product was formed, filtered through a Gooch funnel, washed with cold water, and dried. Compound 5 was obtained as a grey solid in 68% yield (over two steps). 5: mp = 113–116°C; 1H NMR (500 MHz, DMSO-d6) δ 13.41 (s, 1H), 7.26 (s, 1H), 7.03 (s, 1H), 5.58 (s, 2H), 4.31 (s, 2H), 3.82 (s, 3H), 3.79 (s, 3H), 2.61 (s, 6H); 13C NMR (126 MHz, DMSO-d6) δ 165.8, 151.8, 151.5, 147.1, 127.7, 126.3, 115.6, 112.7, 55.9, 36.8, 28.1 (1 carbon is missing due to overlapping); ESI-MS m/z for C13H19N5NaO4S2 [M+Na]+ calcd 396.08, found 396.00.
General Procedure for the Preparation of 1,2,4-Triazolo-[3,4-b]-1,3,4-Thiadiazoles
The equimolar mixture of 5 (0.268 mmol) and selected aromatic carboxylic acids (0.268 mmol) in phosphorous oxychloride (0.4 mL) was stirred while heated under reflux for 2 hours. The reaction mixture was cooled to room temperature and then poured into ice. Then, the pH was adjusted to 8 with solid K2CO3. The mixture was extracted with dichloromethane and the organic extracts were dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (eluent; ethyl acetate) to give the corresponding 1,2,4-triazolo-[3,4-b]-1,3,4-thiadiazoles.
(E)-2-((6-(4-Fluorostyryl)-[1,2,4]Triazolo[3,4-b][1,3,4]Thiadiazol-3-Yl)Methyl)-4,5-Dimethoxy-N,N-Dimethylbenzenesulfonamide, KA25
Yellow solid, 73% yield, mp = 210–212°C; 1H NMR (500 MHz, DMSO-d6) δ 7.70 (dd, J = 8.6, 5.6 Hz, 2H), 7.65 (d, J = 16.4 Hz, 1H), 7.58 (d, J = 16.4 Hz, 1H), 7.40–7.24 (m, 2H), 7.17 (s, 1H), 6.97 (s, 1H), 4.73 (s, 2H), 3.85 (s, 3H), 3.80 (s, 3H), 2.61 (s, 6H); 13C NMR (126 MHz, DMSO-d6) δ 166.0, 163.2, 151.9, 151.8, 147.2, 146.4, 139.6, 131.1, 130.5, 128.0, 126.0, 118.2, 116.1, 115.5, 112.7, 55.9, 36.8, 27.7 (1 carbon is missing due to overlapping); ESI-MS m/z for C22H22FN5NaO4S2 [M+Na]+ calcd 526.10, found 525.90.
(E)-2-((6-(4-Chlorostyryl)-[1,2,4]Triazolo[3,4-b][1,3,4]Thiadiazol-3-Yl)Methyl)-4,5-Dimethoxy-N,N-Dimethylbenzenesulfonamide, KA26
Yellow solid, 72% yield, mp = 229–231°C; 1H NMR (500 MHz, DMSO-d6) δ 7.84 (d, J = 8.6 Hz, 2H), 7.64 (s, 2H), 7.53 (d, J = 8.6 Hz, 2H), 7.30 (s, 1H), 7.15 (s, 1H), 4.72 (s, 2H), 3.84 (s, 3H), 3.79 (s, 3H), 2.61 (s, 6H); 13C NMR (126 MHz, DMSO-d6) δ 165.7, 151.8, 147.2, 146.3, 139.4, 134.8, 133.3, 129.8, 129.0, 127.9, 126.1, 119.0, 115.5, 112.7, 55.9, 36.8, 27.6 (2 carbons are missing due to overlapping); ESI-MS m/z for C22H22ClN5NaO4S2 [M+Na]+ calcd 542.07, found 541.85.
2-((6-(2,5-Dinitrophenyl)-[1,2,4]Triazolo[3,4-b][1,3,4]Thiadiazol-3-Yl)Methyl)-4,5-Dimethoxy-N,N-Dimethylbenzenesulfonamide, KA39
Brown solid, 29% yield, mp = 162–164°C; 1H NMR (500 MHz, DMSO-d6) δ 8.75 (s, 1H), 8.70 (d, J = 8.9 Hz, 1H), 8.50 (d, J = 9.0 Hz, 1H), 7.29 (s, 1H), 7.13 (s, 1H), 4.74 (s, 2H), 3.84 (s, 3H), 3.79 (s, 3H), 2.58 (s, 6H); 13C NMR (126 MHz, DMSO-d6) δ 161.2, 153.8, 151.9, 150.5, 149.3, 147.3, 146.3, 128.2, 127.6, 126.9, 126.1, 124.3, 115.4, 112.8, 55.9, 55.8, 36.7, 27.6 (1 carbon is missing due to overlapping); ESI-MS m/z for C20H19N7NaO8S2 [M+Na]+ calcd 572.06, found 571.75.
1H-NMR and 13C-NMR spectra as well as LC/ESI-MS analysis data for compounds S1(1), 2, 3, 4, 5, KA39, KA26, and KA25, as indicated in Scheme 1, are presented as Supplementary Material, in Figures S1–S15, respectively.
Drug Preparation
Three novel triazolo[3,4-b]thiadiazole derivatives, KA25, KA26, and KA39 (Figure 1), were synthesized according to a previously described experimental procedure.17,26–28 Stock solutions were prepared by dissolving the required amount of the substances in DMSO solvent in order to reach the desired concentration of 15 mM. The final volume of DMSO did not exceed 1% of the culture medium.
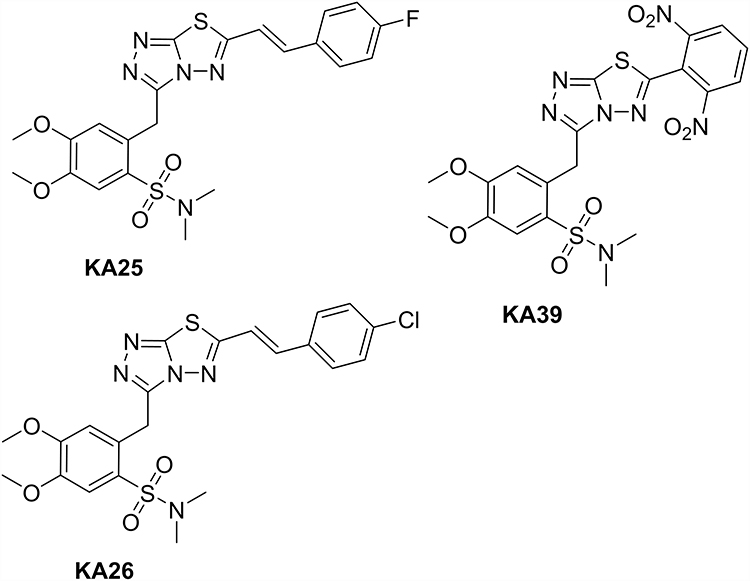 |
Figure 1 Chemical structures of KA25 (2-((6-(4-fluorostyryl)-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazol-3-yl)methyl)-4,5-dimethoxy-N,N-dimethylbenzenesulfonamide), KA26 (2-((6-(4-chlorostyryl)-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazol-3-yl)methyl)-4,5-dimethoxy-N,N-dimethylbenzene sulfonamide), and KA39 (2-((6-(2,5-dinitrophenyl)-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazol-3-yl)methyl)-4,5-dimethoxy-N,N-dimethylbenzenesulfonamide),triazolo[3,4-b]thiadiazole derivatives. |
Cell Lines and Culture Conditions
Our study was conducted in four human cell lines, of which three were of colorectal cancer origin (DLD-1, HT-29, and LoVo) and one from normal lung (MRC5). All cell lines were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA) and cultured in different culture mediums according to the supplier instructions. All growth mediums were supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. All cancer cell lines were cultured as monolayers and maintained at 37°C in a humidified 5% CO2 atmosphere.
In vitro Anticancer Activity
The in vitro anticancer activity of the three disubstituted triazolo[3,4-b]thiadiazole derivatives (KA25, KA26, and KA39) against DLD-1, HT-29, and LoVo cancer cells was evaluated using the [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] MTT assay, a well-known quantitative colorimetric method, which associates cell viability with color formation (absorbance). Moreover, in order to gain a better insight into the tumor specificity of KA25, KA26, and KA39, we explored their cytostatic and cytotoxic properties on healthy lung (MRC5) cells as well. Etoposide (a known topIIα poison) was included in our experiments in order to evaluate the in vitro anti-tumor activity of the newly synthesized triazolo[3,4-b]thiadiazole derivatives by comparing them to etoposide’s therapeutic effectiveness. Briefly, cells were seeded into a 96-well plate at a density of 8×103 cells per well and maintained for 72 hours at 37°C in humidified 5% CO2. After 24 hours of cell growth, cells were treated with the tested compounds in concentrations of 1–100 μΜ. Following 48 hours of drug exposure, culture medium was discarded and replaced with 100 μL of fresh medium. To determine cell viability, 50 μL of MTT (5 mg/mL) were added in each well and cells were incubated for 3 hours. Incubation time was followed by discard of supernatant and addition of 100 μL of DMSO so the formazan crystals could be solubilized. The absorbance of the converted dye was recorded at a wavelength of 540 nm on an ELISA reader (Versamax, Orleans, USA).29, 30 The MTT assay provides the required absorption values for determining three dose-response parameters, GI50, TGI, and IC50, using the linear progression method. According to the National Cancer Institute (NCI), GI50 and TGI are the drug concentrations which signify the cytostatic effect of a tested compound and induce 50% and 100% of cell growth inhibition, respectively; IC50 is the drug concentration which implies the cytotoxic effect of a tested compound and leads to a 50% decrease of cell viability.31,32 The three parameters are determined using the mean of cell survival in all nine absorbance measurements including control 24h (Ct24), control 72h (Ct72) and the seven drug concentrations (Tt72). The percentage of growth inhibition was calculated as NCI indicates: [(Tt72x)-(Ct24)/(Ct72)-(Ct24)]×100 for concentrations for which Tt72x>Ct24 and [(Tt72x)-(Ct24)/Ct24]×100 for concentrations for which Tt72x<Ct24; GI50 was calculated from [(Tt72x)-(Ct24)/(Ct72)-(Ct24)]×100=50, TGI from [(Tt72x)-(Ct24)/(Ct72)-(Ct24)]×100=0, and IC50 from [(Tt72x)-(Ct24)/Ct24]×100=50. All the experiments were carried out in triplicate.
Flow Cytometric Analysis of Apoptosis
Apoptosis was conducted by the FITC Annexin V Apoptosis Detection Kit with 7-AAD (Biolegend, San Diego, California, USA). DLD-1 cancer cells were seeded (1.0×106 cells/well) in a 6-well plate and maintained for 24 hours at 37°C in humidified 5% CO2. After 24 hours of cell growth, culture medium was replaced with fresh medium and cells were exposed to KA39 (IC50=9 μM) and etoposide (IC50=27 μΜ) as reference drug compound, for 48 and 72 hours, while unexposed cells served as controls. For apoptosis assay, cells were washed with ice-cold PBS (pH 7.4) and detached enzymatically with standard trypsinization. All required centrifugations were carried out in 1500 rpm for 5 minutes, including culture medium discard and two washing steps with 2 mL of cold cell staining buffer (BioLegend, San Diego, CA, USA). Subsequently, pellets were re-suspended with 150 μL of Annexin V Binding Buffer and 100 μL of cell suspension were transferred. Afterwards, cells were stained with 5 μL of FITC Annexin V and 5 μL of 7-AAD Viability Staining Solution and then cells were incubated for 15 minutes at room temperature in the dark. Cell staining was followed by the addition of 400 μL of Annexin V Binding Buffer and 400 μL of cell staining buffer before analyzing on the flow cytometer (CyFlow®, SL, Partec, GmbH, Germany). For each sample, 1×104 events were acquired and analysis was carried out in triplicate. Flow cytometric analysis was performed using the Partec FloMax software.
Flow Cytometric Analysis of Cell Cycle Growth Arrest
Cell cycle progression was analyzed by the CyStain PI Absolute T reagent kit (Sysmex, Partec, GmbH, Germany). HT-29 cancer cells were seeded in a 6-well plate and maintained for 24 hours at 37°C in humidified 5% CO2. After 24 hours of cell growth, culture medium was replaced with fresh medium; untreated cells represented controls whilst cells were treated with KA39 at TGI and IC50 concentrations of 15.9 and 19.5 μΜ, respectively. Treated cells were allowed to grow for 48 and 24 hours, respectively. In order to compare cell cycle progression under treatment with KA39, HT-29 cancer cells were treated with etoposide at a TGI concentration of 69 μΜ for 48 hours and IC50 concentration for 24 hours. An MTT assay was performed in order to determine the required IC50 concentration of etoposide in HT-29 cancer cells. To analyze cell cycle, cells were washed with ice-cold PBS (pH 7.4) and detached enzymatically with standard trypsinization. As previously described, all centrifugations were carried out at 1500 rpm for 5 minutes and cells were washed each time with 2 mL of cold cell staining buffer (BioLegend, San Diego, CA, USA). Subsequently, pellets were resuspended with 150 μL of nuclei extraction buffer and cells were incubated for 15 minutes in the dark with gentle shaking. Following DNA extraction, DNA contents were stained with 2 mL of cell staining solution (containing staining buffer, PI and RNAse) and incubated at least for 30–60 minutes while they were not exposed to light before analyzing on the flow cytometer (CyFlow®, SL, Partec, GmbH, Germany). For each sample, 1×104 events were acquired and analysis was carried out in triplicate. Flow cytometric analysis was performed using Partec FloMax software.
DNA Topoisomerase II Assay
The inhibition of the catalytic activity of topoisomerase II was estimated by the plasmid-based topoisomerase II drug screening kit (TopoGen, Buena Vista, CO, USA). The inhibitory effect of KA39 against topoisomerase II was tested in a supercoiled DNA relaxation assay in a range of concentrations (0.1, 1.0, 7.5, and 20 μΜ). According to manufacturer’s recommendations, each reaction included 150 ng of pHOT1 supercoiled DNA, 4 μL complete buffer [0.5 M Tris-HCl, pH (8.0), 150 M NaCl, 100 mM Mg2Cl, 5 mM Dithiothreitol, 300 μg BSA/mL and 20 mM ATP], nuclease free water whose volume was variable depending on the case and 6 units of topoisomerase II. Concerning the tested compounds, 2 μL were added to the mixture before reaction was started by the addition of topoisomerase II. All reactions were incubated at 37°C for 30 minutes and terminated by adding 0.1 volume of 10% SDS. To digest DNA/topoisomerase II complexes, 50 μg/mL of proteinase K were added and samples were further incubated at 37°C for 15 minutes. Afterwards, 0.1 volume of loading buffer was added and samples were cleaned up by adding chloroform/isoamyl alcohol (24:1, v/v). After a brief vortex mixing, samples were spun in a microcentrifuge for 5 seconds. The upper aqueous phase of DNA samples as well as 2 μL of each marker (supercoiled and linear DNA) were loaded onto 0.8% agarose gel in TBE buffer and run at 50 V for 120 minutes (electrophoresis system, BioRad, USA). As a non-ethidium bromide gel, the agarose gel was stained with 0.5 μg/mL of ethidium bromide and destained with water prior to the photo documentation of DNA electrophoresis. The non-ethidium bromide gel was photodocumented in Geldoc It2 imager UVP (Cambridge, UK).
DNA Topoisomerase IIα Phosphorylation
TopIIα phosphorylation (Ser1106) was determined by the LSBioTM Human Phospho-TOP2A/Topoisomerase II Alpha Cell-Based Phosphorylation ELISA Kit (LSBio, LifeSpan Biosciences, Seattle, USA). The inhibitory impact of the three disubstituted triazolo[3,4-b]thiadiazole derivatives (KA25, KA26 and KA39) in the phosphorylation of topIIα was tested in two colorectal cancer cell lines, LoVo and HT-29. In order to determine the required TGI concentrations of KA25 and KA26 derivatives in both cancer cell lines, further MTT assays were performed, as previously described. According to manufacturer’s instructions, cells were seeded onto a 96 well plate at a density of 15–20×103 cells per well and maintained for 24 hours at 37°C in humidified 5% CO2. After 24 hours of cell culture, cells were treated with KA39, KA25, and KA26 at TGI and IC50 concentrations (μΜ) for 6 and 24 hours. Cells were washed with 1×TBS and fixed with 4% fixing solution for 20 minutes. Afterwards, cells were washed three times with 1× Wash Buffer and incubated with 1× Quenching Buffer for 1 hour. Then, cells were washed three times with 1× Wash Buffer and incubated with 1× Blocking Buffer for 1 hour. Once cells were washed three times with 1× Wash Buffer, 50 μL of 1× primary antibodies (Anti-TOP2A (Phospho-Ser1106), Anti-TOP2A and GAPDH antibody) were added and cells were incubated for 16 hours at 4°C. Incubation with primary antibodies was followed by three washing steps with 1× Wash Buffer. Then, 50 μL of 1× secondary antibodies were added (HRP-Conjugated with Anti-Rabbit IgG antibody or HRP-Conjugated with Anti-Mouse IgG antibody) to the corresponding wells and cells were incubated for 1.5 hours. After washing the cells three times with 1× Wash Buffer, 50 μL of 1× Ready to Use Substrate were added and cells were incubated in the dark for 30 minutes with gentle shaking. The reaction was terminated by the addition of 50 μL of 1× Stop Solution and the absorption was immediately measured at 450 nm on an ELISA reader (Versamax, Orleans, USA). After reading the absorbance at 450 nm, the plate was washed twice with 200 μL of 1× Wash Buffer and twice with 200 μL of 1× TBS. After letting the plate dry for 5 minutes, cells were stained with 50 μL of crystal violet and incubated for 30 minutes at room temperature. Subsequently, the plate was well-washed with continually running and distilled water as well, and wells were allowed to dry for 30 minutes. Then, 100 μL of SDS solution were added to each well and the plate was incubated at room temperature for 1 hour. Finally, the absorbance was measured at 595 nm on an ELISA reader (Versamax, Orleans, USA). The experiment was carried out in triplicate.
Computational Studies for Determining Direct Effects on Topoisomerase IIα
Molecular docking was performed into the ATP-binding pocket of the human topIIα ATPase in an attempt to explore the possible binding modes of the synthesized compounds KA25, KA26, and KA39 with the ATPase domain while etoposide was also tested. More specifically, the alpha isoform of human topoisomerase II structure, containing ANP as the co-crystallized ligand (PDB ID: 1ZXM) was selected and retrieved from the RCSB Protein Data Bank (www.rcsb.org).33 The co-crystallized ligand and non-bonded heteroatoms were removed, the polar hydrogens were added and the non-polar were merged through AutoDockTools (1.5.6 package). In addition, Gasteiger’s charges were added and the structure was saved in the pdbqt format. The structures of the ligands were generated and energy minimized. AutoDock vina program was used for the docking study following the standard procedure.34 The three-dimensional grid with 20×20×20 points and a spacing of 1 Å was created and centered at the area surrounding the co-crystallized ligand AMP-PNP (X=36.071; Y=−0.286; Z=38.215). The exhaustiveness value was set to 100. Figures for best scoring docking complexes were generated by PyMOL viewer and Discovery Studio visualizer.
Statistical Analysis
Student’s t-test was used to compare the level of significance between the experimental groups. Differences with a P-value less than 0.05 were considered significant. Microsoft Excel (Microsoft Hellas, Athens, Greece).
Results
In vitro Anticancer Activity
In order to determine the in vitro anti-proliferative activity of the three novel triazolo[3,4-b]thiadiazole derivatives (KA25, KA26, and KA39), the MTT assay was conducted in three human colorectal cancer cell lines (DLD-1, HT-29, and LoVo) while etoposide was utilized as a reference drug compound. Briefly, KA39 showed the most potent anticancer activity against all cancer cell lines, demonstrating significant cytostatic and cytotoxic effects (P<0.001). Moreover, KA25 displayed higher cytostatic activity than KA26 derivative (P<0.01), while etoposide induced cytostatic activity against all three colorectal cancer cell lines and cytotoxic activity against DLD-1 cancer cells only. Overall, it is suggested that KA39 inhibits cell proliferation and induces cytotoxicity to a greater extent than KA25, KA26, and etoposide (Tables 1 and 2). In addition, the selectivity of all three derivatives was assessed in a lung normal cell line as well. As Tables 1 and 2 demonstrate, the three triazolo[3,4-b]thiadiazole derivatives induced significant cytostatic and cytotoxic activities against all cancer cell lines while they exhibited rather low activity in non-tumorigenic (MRC5) cells. Based on our results, GI50, TGI, and IC50 values of KA25, KA26, and KA39 compounds were 3–5 times higher than those in MRC5 cells, indicating significant selectivity of the tested triazolo[3,4-b]thiadiazole derivatives.
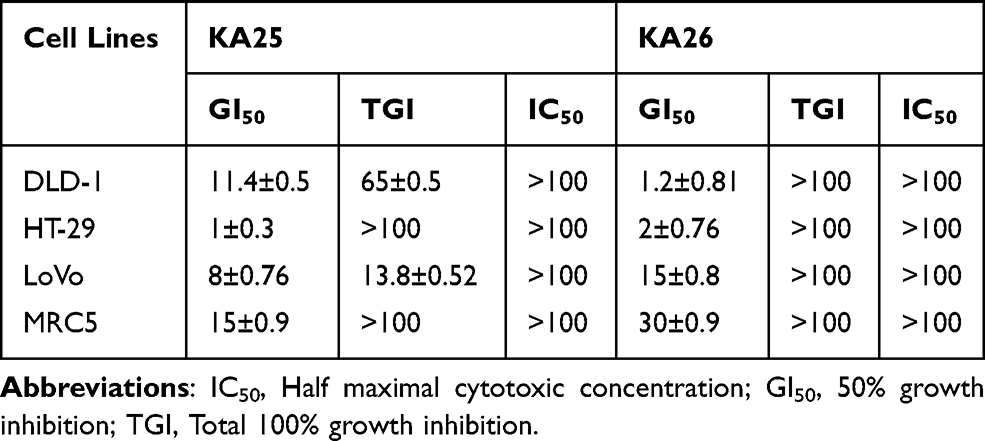 |
Table 1 In vitro Antiproliferative Activity of KA25 and KA26 Against Four Human Cell Lines (Three Colorectal Cancer Cell Lines and One Normal) |
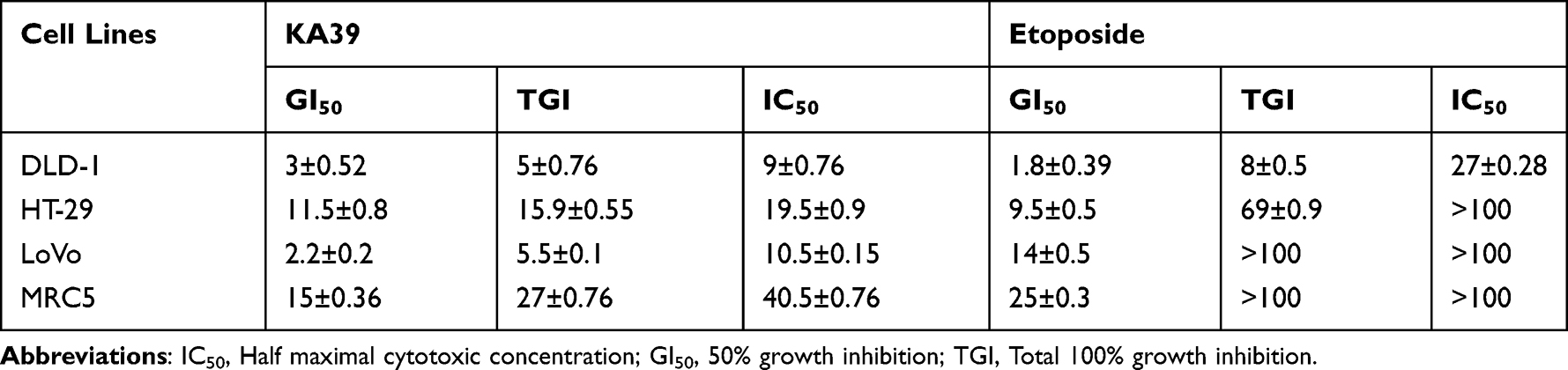 |
Table 2 In vitro Antiproliferative Activity of KA39 and Etoposide Against Four Human Cell Lines (Three Colorectal Cancer Cell Lines and One Normal) |
Apoptotic Analysis
DLD-1 cancer cells were treated with KA39 at IC50 concentration (μM), for 48 and 72 hours, while its apoptotic potency was compared to that of etoposide, also utilized at IC50 concentration (μM) for 48 and 72 hours. Untreated DLD-1 cells (control) were also cultured for 48 and 72 hours as well. As flow cytometric analysis indicated, treatment with ΚΑ39 at IC50 concentration (μM) for 48 hours displayed high rates of early (17.35%) and late apoptosis (13.12%) compared to the corresponding untreated cells (P<0.001) (Figure 2). On the other hand, exposure to KA39 (at the same concentration for 72 hours) led to a significant decrease of the percentage of viable cells and consequently to an increase of early (20.68%) and late (20.96%) apoptosis (Figure 2). As expected, treatment with KA39, for 72 hours, contributed to an enhanced late apoptosis (20.96%) in contrast to treatment for 48 hours (13.12%; P<0.01) (Figure 2). On the other hand, cell exposure to etoposide at IC50 concentration (μM), for 48 hours, remarkably decreased cell viability, thereby yielding high rates of early (51.12%) and late (18.84%) apoptosis (P<0.001). Treatment with etoposide at the same concentration, for 72 hours, significantly increased the rates of late apoptosis (30.48%) with a concomitant decrease of those of early apoptosis (42.01%; P<0.001). As Table 2 shows, the IC50 value of etoposide in DLD-1 cancer cells is 3-fold higher compared to that of KA39 (Figure 2).
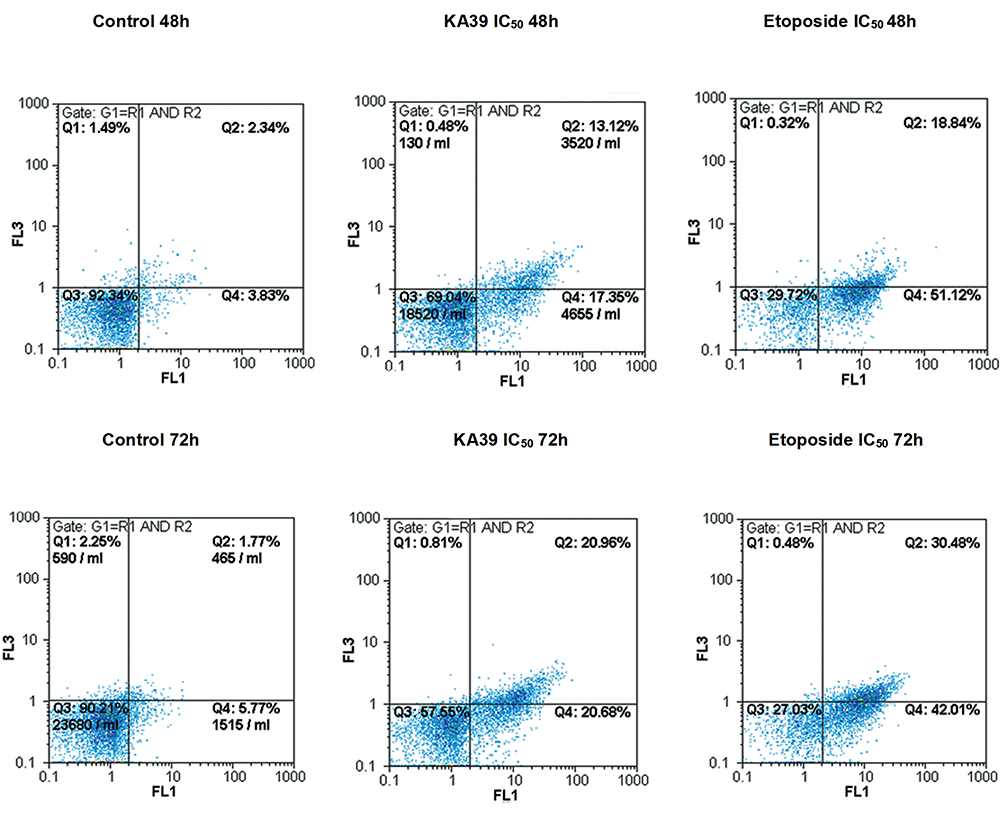 |
Figure 2 Apoptotic induction in DLD-1 cancer cells, upon treatment with KA39 and etoposide, at IC50 concentration (μΜ), for 48 and 72 hours, compared with those of untreated cells (controls). The percentages of viable, early-apoptotic, late-apoptotic, and necrotic cells are demonstrated. |
Cell Cycle Analysis
HT-29 cancer cells were treated with KA39 at TGI and IC50 concentrations (μΜ), for 48 and 24 hours, respectively. In order to determine the effect of KA39 on cell cycle distribution, untreated HT-29 cells were also cultured for 48 and 24 hours, thereby acting as controls. In addition, etoposide was also utilized under the same experimental conditions. Based on flow cytometric analysis, the highest percentage of untreated HT-29 cells, at 24 hours, were distributed in the G1 phase (65.32%) whereas treatment with KA39, at IC50 concentration (μΜ) for 24 hours, induced significant cell cycle growth arrest indicated by cellular distribution at the S phase (21.52%; P<0.001). At the same time, treatment with etoposide, at IC50 concentration for 24 hours, led to cell cycle growth arrest at the G2/M phase, a 2-fold increase when compared to untreated cells (at G2/M; P<0.001) (Table 3, Figure 3). On the other hand, untreated HT-29 cells, at 48 hours, were shown to be distributed at the G1 phase (64.68%) while exposure to KA39, at TGI concentration (μΜ) for 48 hours, displayed a relative increase in the G2/M phase (9.48%) (P<0.001). Upon treatment with etoposide, at TGI concentration (μΜ) for 48 hours, the highest percentage of HT-29 cells was recorded in SubG1 phase (55.80%) with a concomitant decrease in G1 phase (P<0.001) (Table 3, Figure 3). Nevertheless, the percentages of cell cycle distribution in the S phase were quite similar in HT-29 cancer cells, treated and untreated for 48 hours (Table 3). All treatment conditions induced a decrease in G1 phase with a significant increase (P<0.001) in SubG1 probably due to apoptotic induction by KA39 and etoposide in HT-29 cancer cells. Overall, treatment with KA39 led to inhibition of cell proliferation as well as cell cycle growth arrest, in S phase, while exposure for a greater time interval enhanced the percentage of growth arrested cells in the G2/M phase (P<0.001).
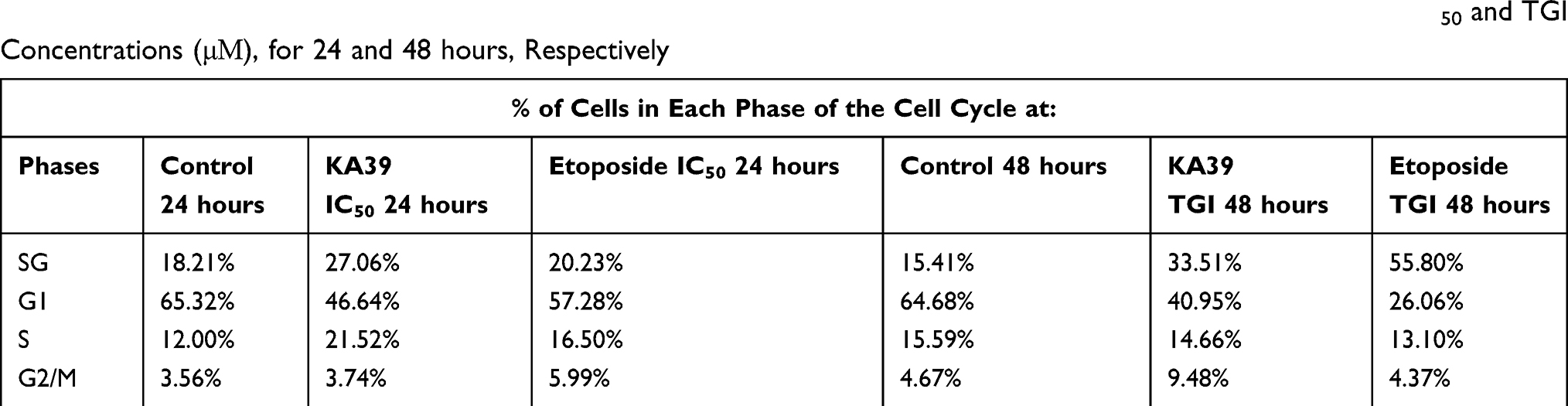 |
Table 3 Cell Cycle Distribution in Untreated (Control) and Treated HT-29 Cancer Cells with KA39 and Etoposide, at IC50 and TGI Concentrations (μΜ), for 24 and 48 hours, Respectively |
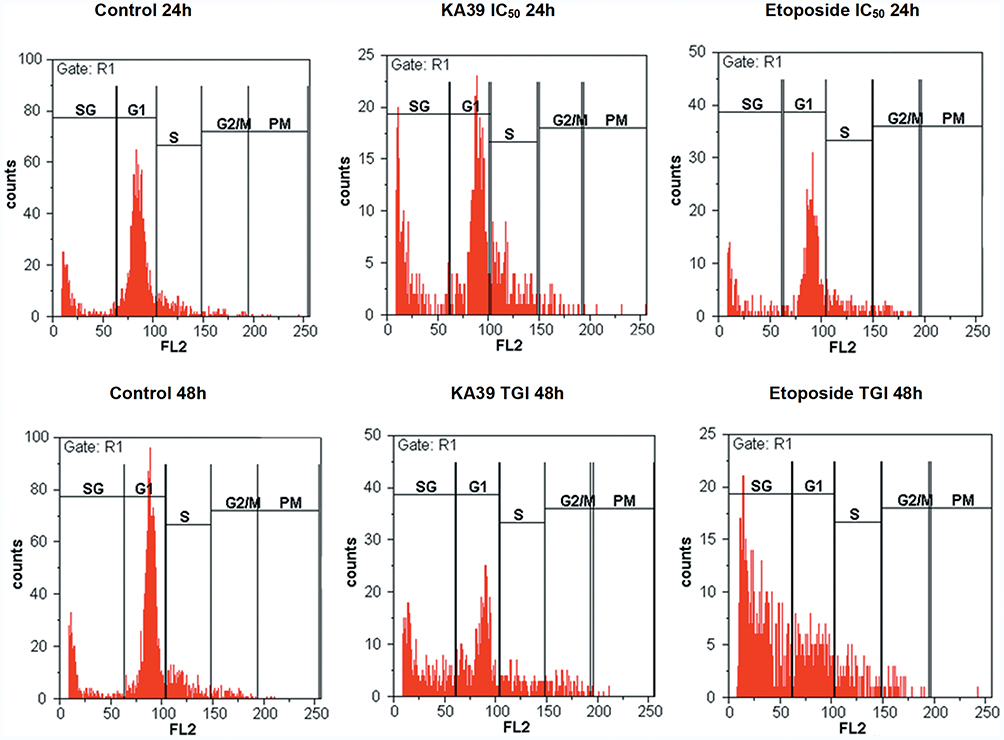 |
Figure 3 Cell cycle growth arrest in HT-29 cancer cells induced by KA39 and etoposide. HT-29 cells were arrested in S- and G2/M phase after being treated at IC50 concentration (μΜ), for 24 hours, respectively. Treatment with both compounds at TGI concentration (μΜ), for 48 hours, significantly increased SubG1 phase. |
DNA Topoisomerase II Assay
To investigate whether KA39 derivative inhibits the catalytic activity of topoisomerase II, the DNA decatenation assay was performed by using plasmid DNA as a substrate while VP-16 was used as a reference compound. According to the data in Figure 4, in the absence of any drug, topoisomerase II catalyzes the reaction cycle through which normal circular relaxed DNA products are formed, known as topoisomers (lane 3). During DNA electrophoresis, KA39 induced the formation of supercoiled DNA in all concentrations used (eg, 0.1, 1.0, 7.5, and 20 μΜ) in contrast to VP16-induced linear DNA (lanes 4–8). The KA39-induced supercoiled DNA reaction product indicates that KA39 acts as a topoisomerase II inhibitor, either by blocking the DNA binding activity of the enzyme or by affecting the ATPase action and consequently preventing any necessary conformational transitions of topoisomerase II.
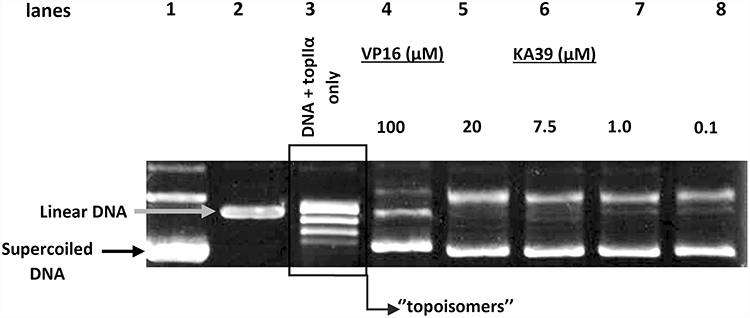 |
Figure 4 Supercoiled and linear DNA are reactions products, induced by KA39 and VP-16, respectively. The samples were loaded onto the agarose gel as follows: lane 1 represents pHOT1 supercoiled DNA (marker), lane 2 linear pHOT1 DNA (marker), lane 3 relaxed circular DNA (topoisomers), lane 4 VP16-induced linear DNA, and lanes 5–8 KA39-induced supercoiled DNA. All topoisomerase II reaction products are resolved on a non EB gel. |
Phosphorylation of DNA Topoisomerase IIα
The inhibitory impact of KA25, KA26, and KA39 in phosphorylating topIIα was conducted in two human colorectal cancer cell lines, LoVo and HT-29. According to our data, the phosphorylation of topIIα was significantly inhibited by KA39, at TGI and IC50 concentrations of 5.5 μΜ and 10.5 μΜ respectively, in LoVo cells when treated for 24 hours (P<0.001). Anti-topIIα normalization indicated that topIIα phosphorylation was decreased upon treatment with KA39, at IC50 concentration, after exposure for 24 hours (Figure 5A). Regarding GAPDH normalization, results were in line with those of anti-topIIα normalization, that is greater inhibition of topIIα phosphorylation by KA39 at IC50 concentration, for 24 hours (Figure 5B). By contrast, treatment with the same compound, at TGI and IC50 concentrations for 6 hours, enhanced the phosphorylation status of topIIα. More specifically, according to anti-topIIα normalization, a considerable increase of topIIα phosphorylation was demonstrated, at TGI concentration, whilst no significant alteration was observed, at IC50 concentration, when compared to the untreated cells (Figure 5A). Furthermore, GAPDH normalization indicated a remarkable increase in topIIα phosphorylation for both treatment concentrations, at 6 hours (Figure 5B).
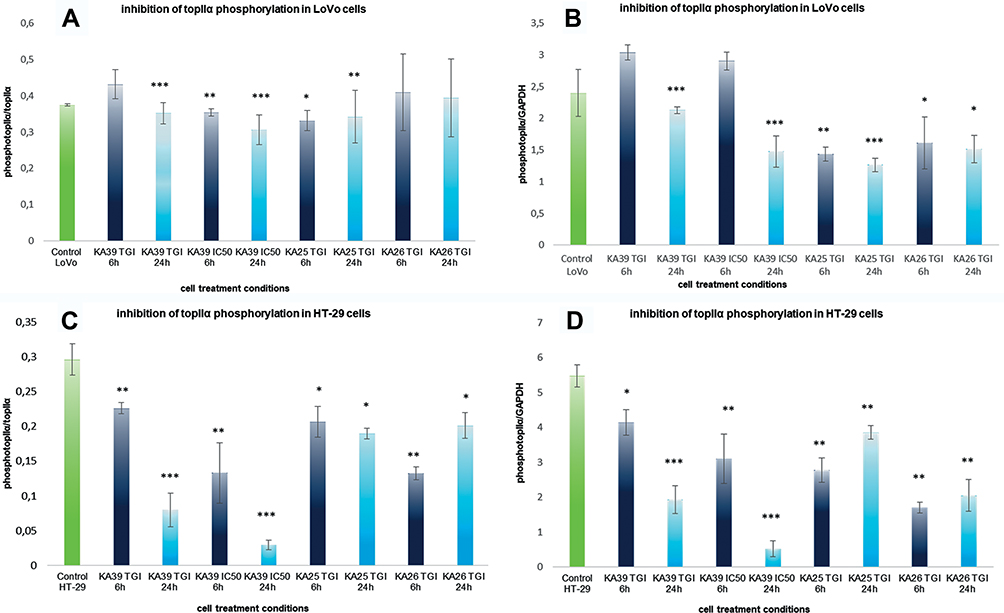 |
Figure 5 Inhibition of topIIα phosphorylation in LoVo and HT-29 cancer cell lines, treated with KA25, KA26, and KA39, at TGI and IC50 concentrations, for 6 and 24 hours. (A) The diagram shows inhibition of topIIα phosphorylation, according to anti-topIIα normalization, in LoVo cancer cells. (Β) The diagram illustrates inhibition of topIIα phosphorylation, according to GAPDH normalization, in LoVo cancer cells. (C) The diagram demonstrates inhibition of topIIα phosphorylation, according to anti-topIIα normalization, in HT-29 cancer cells. (D) The diagram displays inhibition of topIIα phosphorylation, according to GAPDH normalization, in HT-29 cancer cells. Anti-topIIα normalization is expressed by the ratio phosphotopIIα/topIIα whereas GAPDH normalization by the rate of phosphotopIIα/GAPDH. * P<0.05, ** P<0.01, *** P<0.001. |
As Figure 5A shows, ΚΑ25 inhibited the phosphorylation of topIIα in LoVo cancer cells treated at TGI=13.8 μΜ, for 24 hours (P<0.001). According to GAPDH and anti topIIα normalization, topIIα phosphorylation was sufficiently reduced after cells being exposed to KA25, for 24 hours (Figure 5A and B). However, treatment with KA25 at the same drug concentration, for 6 hours induced less significant inhibition of topIIα phosphorylation (P<0.01). Particularly, the proportion of phosphorylated and total topIIα is decreased under treatment with KA25, for 6 hours, while greater inhibition was shown through GAPDH normalization (Figure 5A and B). Moreover, KA26-induced inhibition of topIIα phosphorylation was not statistically significant since treatment at TGI=100 μΜ, for 6 and 24 hours, did not affect the phosphorylation status of topIIα. More specifically, the ratio of phosphorylated and total topIIα remained similar with that of untreated cells although GAPDH normalization demonstrated quantitative alterations in the phosphorylated form of topIIα (Figure 5B).
Regarding HT-29 cells, the inhibition in the phosphorylation of topIIα was significantly induced by KA39 in all treatment conditions. More precisely, the inhibition of topIIα phosphorylation was statistically significant upon treatment with KA39 at TGI and IC50 concentrations of 15.9 and 19.5 μΜ respectively, for 24 hours (P<0.001). Concerning TGI, the ratio of phosphorylated with total topIIα was considerably decreased in all treated cells at 6 and 24 hours (Figure 5C). In addition, GAPDH normalization also confirmed the inhibitory impact of KA39 at TGI concentration over 6 and 24 hour treatment periods (Figure 5D). Nevertheless, the most outstanding inhibitory effect of KA39 was displayed at IC50 concentration when cells were treated for 24 hours (P<0.001). Based on both ways of normalization, the phosphorylated form of topIIα was highly reduced compared to the untreated cells. However, inhibition of topIIα phosphorylation was observed upon treatment with KA39, at IC50 concentration for 6 hours, which was not shown to be statistically significant (Figure 5C and D). In general, the greatest inhibition of topIIα phosphorylation was induced by KA39 at IC50 concentration under all treatment times (P<0.001). On the other hand, KA25 and KA26 did not show statistically significant inhibition in topIIα phosphorylation though anti-topIIα and GAPDH normalizations indicated that both compounds contributed to a reduction of the phosphorylated form of topIIα at TGI concentrations of 120 μΜ (for KA25) and 200 μΜ (for KA26) over 6 and 24 hour treatment periods (Figure 5C and D). According to our data, the maximum inhibitory effects of KA39, KA25, and KA26 on topIIα phosphorylation, at either TGI or IC50 concentrations over 6 or 24 hour treatment periods, in LoVo cells were at 60%, 47%, and 18%, respectively, whereas they were at 76%, 33%, and 50% in HT-29 cells for the same compounds, respectively.
Computational Studies of Direct Effects on Topoisomerase IIα
As previously described, KA25, KA26, KA39, and etoposide were all docked, using the AutoDock vina software, with only the lowest energy conformations being considered for analysis. According to the results, all three compounds showed comparable binding scores and were found to occupy the active site in a manner similar to that of etoposide, as illustrated in Figures 6 and 7. The binding affinity scores and the interacting residues are shown in Table 4.
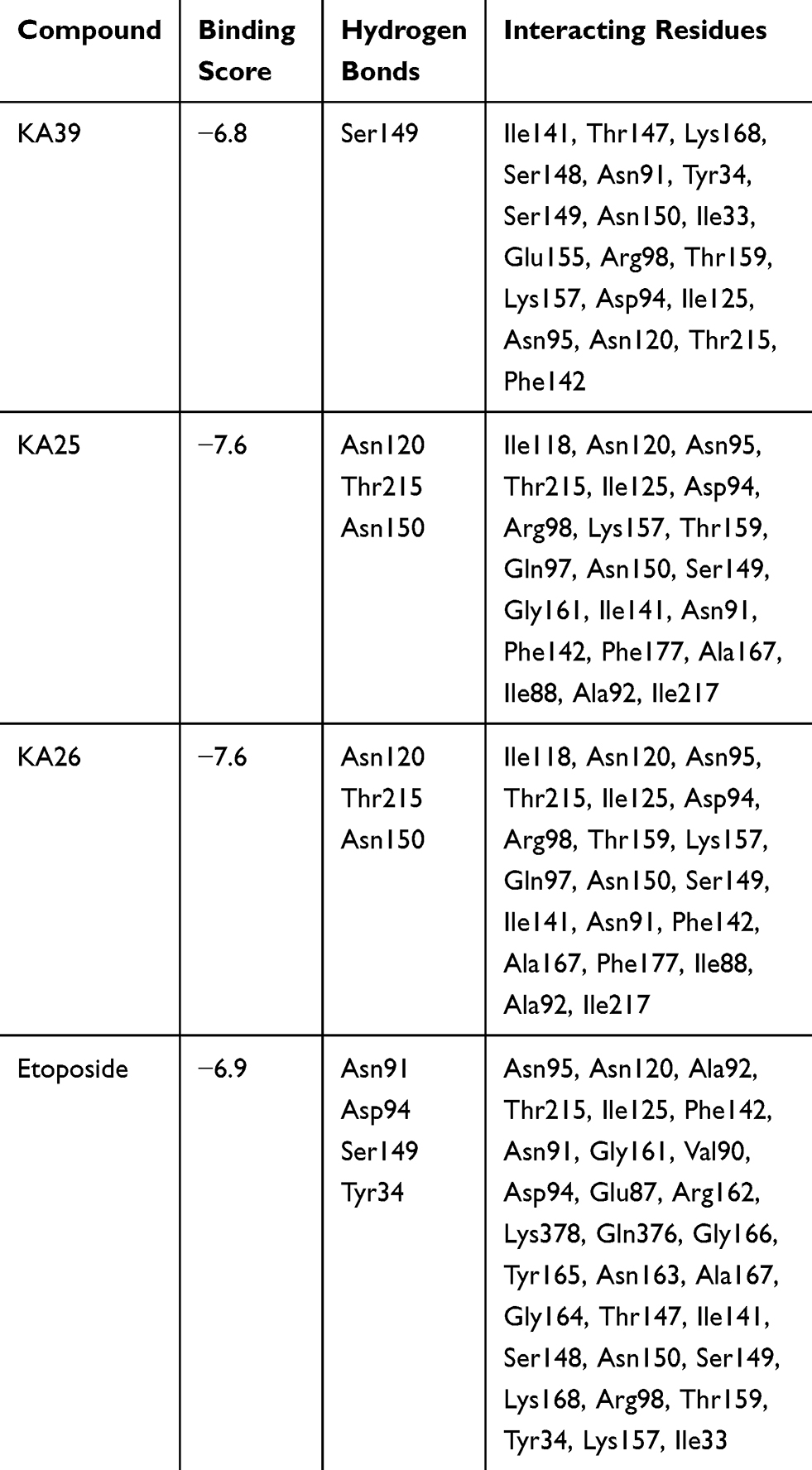 |
Table 4 Docking Interactions Between Human topIIα ATPase and KA39, KA25, KA26, and Etoposide |
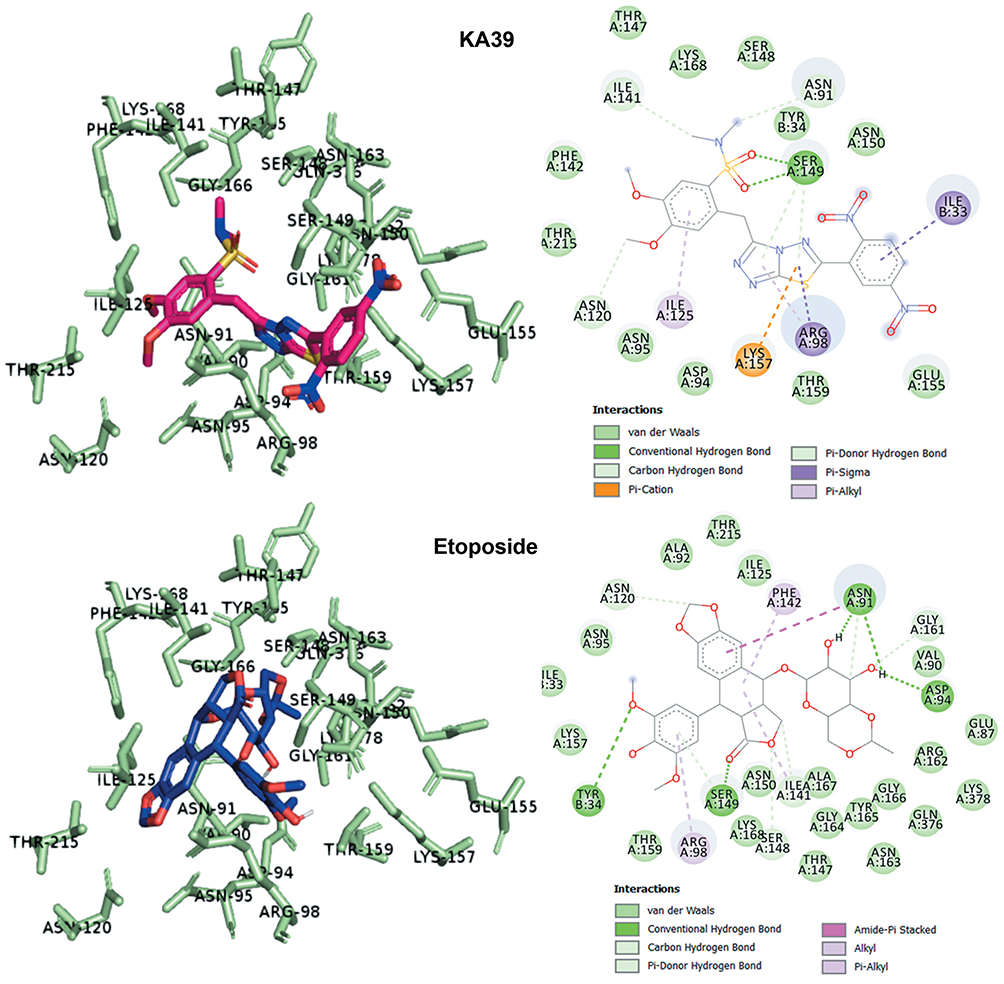 |
Figure 6 Predicted docking modes and 2D interactions of KA39 (magenta) and etoposide (blue) in the ATP-binding domain of human topIIα (PDB: 1ZXM). |
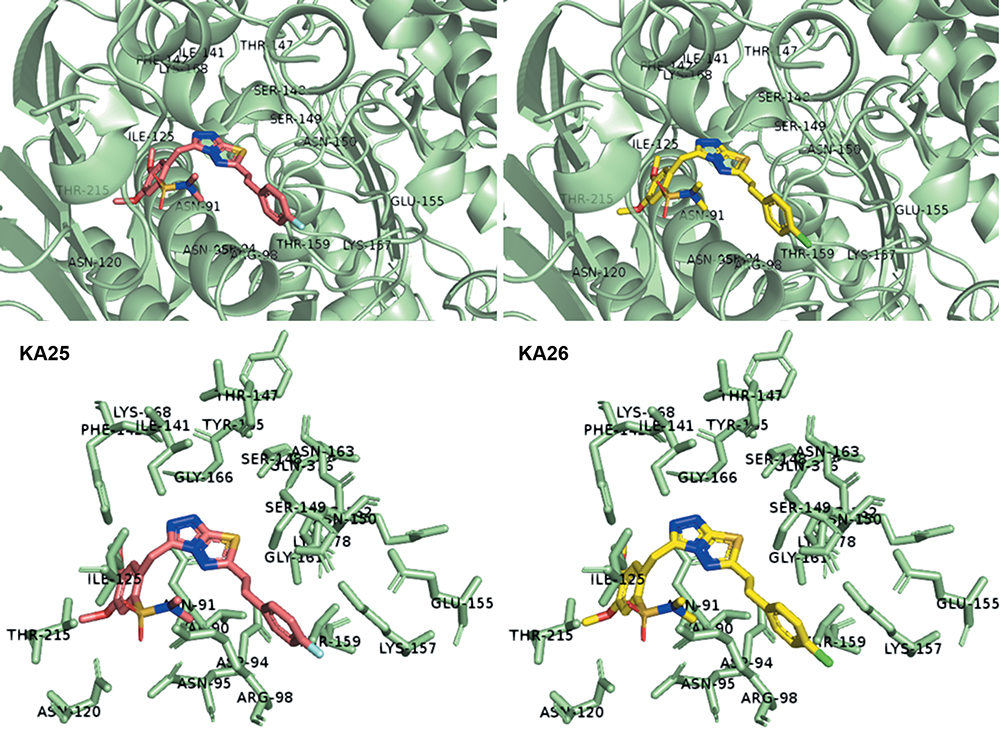 |
Figure 7 3D interactions between KA25 (pink sticks) and KA26 (yellow sticks) with the ATP-binding site of human topIIα. |
Discussion
TopIIα is a versatile molecule whose functionality may be affected by its cellular abundance, binding interactions, changes in DNA topology, and post-translational modifications.35 With respect to posttranslational modifications, phosphorylation seems to play a leading role in the catalytic activity of the enzyme with numerous proteins being involved including casein kinase IIβ, protein kinase C (PKC), extracellular signal-related kinase 2 (ERK2), p38γ, and members of the polo-like kinase (Plk) family. Casein kinase IIβ, PKC, and ERK2 augment the catalytic activity of topIIα via its phosphorylation, while p38γ ensures the activity as well as the stability of the enzyme by phosphorylating Ser-1542.36 In addition, members of the Plk family like Plk1, Plk2, and Plk3 activate topIIα through its phosphorylation in specific serine and threonine residues, for example Plk1 phosphorylates Ser-1337 and Ser-1524, whereas Plk3 phosphorylates Thr-1342.37,38
Due to the fact that most of the phosphorylation sites of topIIα are located into the C-terminal domain without influencing its enzymatic activity, but only being responsible for its nuclear localization, deletion of this region or mutations in such phosphorylation sites like Ser-1376 and Ser-1524 do not affect the activation of topIIα.10,39–42 Phosphorylation sites like Ser-29 and Ser-1106 are located in the ATP-binding domain within the N-terminal region and catalytic core of topIIα, respectively.43 Prior studies showed that the phosphorylation at Ser-1106 activates the decatenation function of topIIα, suggesting that the phosphorylation site Ser-1106 plays a major role in the catalytic activity of topIIα while mutation of Ser-1106 to alanine leads to decreased activity of topIIα.9 Later studies revealed two kinases, casein kinase I (CKI) and II (CKII), which catalyze the phosphorylation at Ser-1106. However, further investigation demonstrated that the isoenzymes of CKI superfamily, CKIδ/ε, play a more essential role in the phosphorylation at this site than CKII. Grovaz et al43 showed that CKIδ/ε kinases are specifically involved in the phosphorylation at Ser-1106, enhancing the in vivo activity of topIIα and as a result leading to increased topIIα-DNA cleavage. On the other hand, inhibition or downregulation of CKIδ/ε decreases the phosphorylation at Ser-1106, implying that a lack of CKIδ/ε kinase activity or absence of phosphorylation at Ser-1106 suppresses the function of topIIα.43 Experimental studies using cell lines strongly support a linkage of p53 and levels of topIIα expression. Given that the wild-type p53, as a tumor suppressor protein, regulates the transcriptional activity of several genes including topIIα, a mutated p53 may cause unregulated expression of the genes involved in the regulation of topIIα. Wang et al44 demonstrated that the reduction of both mRNA and protein levels arise through p53-induced inhibition of topIIα gene transcription. More precisely, wild-type p53 suppresses the activity of human topIIα gene promoter while loss of the p53 transcriptional regulator stimulates the expression of topIIα 5–10 times higher in tumor cells. Therefore, mutated p53 leads to overexpression of topIIα which in turn contributes to accelerated cell proliferation, chromosomal rearrangements, abortive cell-cycle arrest and higher sensitivity to topIIα inhibitors.44,45 In our study, the inhibition of topIIα phosphorylation was conducted in LoVo and HT-29 cancer cells which were treated with KA25, KA26, and KA39 at TGI and IC50 concentrations, for 6 and 24 hours. Of utmost significance was the KA39-induced inhibition of topIIα phosphorylation in both cancer cell lines treated, at TGI and IC50 concentrations, for 24 hours. Nonetheless, the greatest inhibition was observed in HT-29 cells whose topIIα phosphorylation was significantly reduced. However, the two colorectal cancer cell lines do not express the same p53 status as LoVo cancer cells express the wild-type p53 whilst HT-29 cancer cells express the mutated p53 (p.R273H) (Table 5). In addition, LoVo cells express mutated Plk2 (p.S636N) leading to decreased topIIα phosphorylation.48 Consequently, the combinatory effect of wild type p53 and mutated Plk2 can reduce the expression levels of total and phosphorylated topIIα, in LoVo cancer cells. On the other hand, in HT-29 cancer cells, the loss of functional p53 results in overexpression of topIIα which, in turn, may enhance the KA39-induced inhibition of topIIα phosphorylation. In reference to the crystal violet stained cells, no alterations in topIIα expression were observed either in transcriptional or translational level (data not shown).
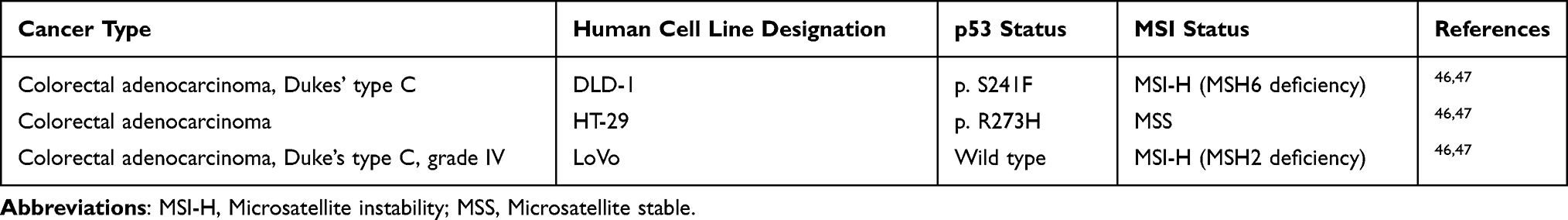 |
Table 5 Description of Histotypes and Special Characteristics of the Three Human Colorectal Cancer Cell Lines Involved in the Investigation of the Biological Activity of Triazolo[3,4-b]thiadiazole Derivatives KA25, KA26, and KA39 |
The catalytic activity of topIIα can be inhibited either by targeting the strand DNA passage reaction or preventing the required ATP hydrolysis. The double-strand DNA passage reaction can be blocked in several steps including DNA binding to topIIα, formation of a topIIα-DNA cleavage complex and DNA religation. TopIIα blockers, like etoposide, trap topIIα cleavage complexes, constituting DNA religation as impossible, whereas DNA intercalators prevent topIIα from binding to DNA through alterations in DNA structure.49–51 With reference to ATP cofactor, closing and opening of the topIIα clamp are coupled with ATP binding and hydrolysis. Inhibition of ATP hydrolysis does not allow the ATPase domain to be reopened, thus trapping topological DNA complexes inside the enzyme.52 As Figure 4 shows, KA39-induced supercoiled DNA and VP16-induced linear DNA underline the different pharmacological mechanisms through which KA39 and VP16 inhibit topIIα activity. According to the data from the DNA decatenation assay, the formation of KA39-induced supercoiled DNA indicates that this compound interferes with the catalytic activity of topIIα either by blocking the access of topIIα to its plasmid substrate or by inhibiting the ATPase action of the enzyme.
So far, we demonstrated that all three derivatives inhibit topIIα phosphorylation which is critical for topIIα activity. Furthermore, we showed that KA39 specifically inhibits topIIα catalytic activity, thus inducing the formation of supercoiled DNA which may be due to the inhibition of critical steps of the catalytic cycle. Nonetheless, it is unclear whether the triazolo[3,4-b]thiadiazole derivatives affect topIIα in a direct or indirect manner. In silico studies revealed that all three compounds may act directly through binding to the ATPase domain of topIIα (Figures 6 and 7). According to the results, the binding scores of KA25, KA26, KA39, and etoposide (used as a positive control) were quite similar (Table 4). For both KA25 and KA26 compounds, they share the same hydrogen bonds, whereas the interacting residues are pretty similar, indicating that the two derivatives interact with the ATPase domain in the same way. In contrast, KA39 interacts with the ATPase domain in a very different way since the hydrogen bonds and interacting residues are unlike those of KA25 and KA26. It is noteworthy that KA39 develops hydrogen bonds with the ATPase domain only through one residue, Ser149, indicating a highly specific way of interaction (Table 4). Comparing with the three triazolo[3,4-b]thiadiazole derivatives, etoposide interacts with the ATPase domain through different hydrogen bonds and interacting residues, sharing only one common residue with KA39 at Ser149. With respect to the most potent anticancer agent, KA39 derivative, it is possible that it alters the ATPase action of topIIα, leading to the formation of supercoiled DNA as DNA decatenation assay demonstrated.
Being a cell-cycle dependent enzyme, the expression of topIIα varies during the different phases of cell-cycle. More specifically, the expression of topIIα is at its lowest levels during the G1 phase while it starts to increase before the S phase, remains stable during most of the S and raises again in the late S phase. The expression of topIIα peaks in the late G2 phase due to its necessity in cellular events like chromatin condensation, chromosome assembly, and segregation of sister chromatids. Once mitosis has been completed, topIIα is rapidly decreased.53,54 The phosphorylation of topIIα is increased in a cell-cycle dependent manner as well, with most of topIIα being phosphorylated in the G2 phase.53 Our data from cell-cycle analysis suggest that the KA39 derivative constitutes a potent topIIα inhibitor since 21.52% of HT-29 cancer cells were arrested in the S phase upon treatment at IC50 concentration for 24 hours. Increased percentages in the SubG1 phase were also recorded in all treatment conditions, signifying the apoptotic activity of our compound (Table 3). However, the apoptotic potency of KA39 was confirmed by flow cytometry analysis in DLD-1 cells where apoptosis was at its highest levels at 72 hours (Figure 2).
Furthermore, Tables 1 and 2 indicate that KA39 exhibits the greatest cytostatic and cytotoxic effect in comparison with those of KA25 and KA26. According to the IC50 values, of great interest is the sensitivity of the MMR-deficient cells, DLD-1 and LoVo, after exposure to KA39 when compared to the HT29-MMR proficient cell line (Table 5). It is very likely that a correlation between mismatch repair system (MMR) and drug sensitivity to topIIα inhibitors may exist. In fact, later studies have shown enhanced drug sensitivity in MSI-H colorectal cancer cell lines that have been treated with etoposide, a known topIIα inhibitor that interrupts the catalytic cycle by targeting to the topIIα cleavage complex.25
Conclusion
DNA topoisomerases, known for resolving topological issues and relaxing supercoiled DNA, are introduced as potent chemotherapeutic drug targets attracting more and more attention in the area of drug discovery.6,55 In our study, we successfully demonstrated that all three 3.6-disubstituted-1,2,4-triazolo[3,4-b]thiadiazole derivatives exhibited in vitro anticancer activity and induced the inhibition of topIIα phosphorylation. Nevertheless, among the three triazolo[3,4-b]thiadiazole derivatives, the most potent one was shown to be KA39 in terms of an in vitro anti-proliferative and inhibitory activity in topIIα phosphorylation. The inhibition of topIIα phosphorylation was specifically shown to occur at Ser-1106, which is closely related to the decatenation activity of topIIα. In addition to the above, cell cycle arrest in the S phase also highlighted KA39’s action as a topIIα inhibitor while the increased percentages in SubG1 phase indicated its apoptotic potency. Overall, the triazolo[3,4-b]thiadiazole derivatives constitute a chemical class with potent anticancer properties which enable the advancement of new avenues in the development of topIIα inhibitors.
Acknowledgments
This work was supported by the GALENICA S.A. and ENERGON BIOTECHNOLOGIES S.A. and granted by the Hellenic Society of Medical Oncology (HESMO) and Athena Institute of Biomedical Sciences, Greece.
Disclosure
The tested investigational compounds were covered in the international patent application WO2018/011414 (PCT/EP2017/067908). The authors declare no conflicts of interest in this work.
References
1. Nitiss JL, Soans E, Rogojina A, Seth A, Mishina M. Topoisomerase assays. In: Enna S, editor. Current Protocols in Pharmacology. New York: Wiley; 2012:1–27.
2. Wilstermann AM, Osheroff N. Stabilization of eukaryotic topoisomerase II-DNA cleavage complexes. Curr Top Med Chem. 2003;3(3):1349–1364. doi:10.2174/1568026033452519
3. Bettotti P, Visone V, Lunelli L, Perugino G, Ciaramella M, Valenti A. Structure and properties of DNA molecules over the full range of biologically relevant supercoiling states. Sci Rep. 2018;8(1):6163. doi:10.1038/s41598-018-24499-5
4. Wang J. Cellular roles of DNA topoisomerases: a molecular perspective. Nat Rev Mol Cell Biol. 2002;3(6):430–440. doi:10.1038/nrm831
5. Wendorff T, Schmidt B, Heslop P, Austin C, Berger J. The structure of DNA-bound human topoisomerase II alpha: conformational mechanisms for coordinating inter-subunit interactions with DNA cleavage. J Mol Biol. 2012;424(3–4):109–124. doi:10.1016/j.jmb.2012.07.014
6. Champoux J. DNA topoisomerases: structure, function, and mechanism. Annu Rev Biochem. 2001;70(1):369–413. doi:10.1146/annurev.biochem.70.1.369
7. Zhao W, Jiang GBC, Li Y, et al. The dual topoisomerase inhibitor A35 preferentially and specially targets topoisomerase 2α by enhancing pre-strand and post-strand cleavage and inhibiting DNA relegation. Oncotarget. 2015;6(35):37871–37894. doi:10.18632/oncotarget.5680
8. Watt PM, Hickson ID. Structure and function of type II DNA topoisomerases. Biochem J. 1994;303(Pt 3):681–695. doi:10.1042/bj3030681
9. Chikamori K, Grabowski DR, Kinter M, et al. Phosphorylation of serine 1106 in the catalytic domain of topoisomerase II alpha regulates enzymatic activity and drug sensitivity. J Biol Chem. 2003;278(15):12696–12702. doi:10.1074/jbc.M300837200
10. Luo K, Yuan J, Chen J, Lou Z. Topoisomerase IIαlpha controls the decatenation checkpoint. Nat Cell Biol. 2009;11(2):204–210. doi:10.1038/ncb1828
11. Wang JC. Moving one DNA double helix through another by a type II DNA topoisomerase: the story of a simple molecular machine. Q Rev Biophys. 1998;31(2):107–144. doi:10.1017/S0033583598003424
12. Osheroff N. Eukaryotic topoisomerase II. Characterization of enzyme turnover. J Biol Chem. 1986;261(21):9944–9950.
13. Baird CL, Harkins TT, Morris SK, Lindsley JE. Topoisomerase II drives DNA transport by hydrolyzing one ATP. Proc Natl Acad Sci U S A. 1999;96(24):13685–13690. doi:10.1073/pnas.96.24.13685
14. Kharb R, Sharma P, Yar M. Pharmacological significance of triazole scaffold. J Enzyme Inhib Med Chem. 2010;26(1):1–21. doi:10.3109/14756360903524304
15. Siddiqui N, Ahujaa P, Ahsana W, Pandeyab SN, Alama MS. Thiadiazoles: progress report on biological activities. J Chem Pharm Res. 2009;1(1):19–30.
16. Gomha S, Abdel-aziz H, Badrey M, Abdulla M. efficient synthesis of some new 1,3,4‐thiadiazoles and 1,2,4‐triazoles linked to pyrazolylcoumarin ring system as potent 5α‐reductase inhibitors. J Heterocycl Chem. 2019;56(4):1275–1282. doi:10.1002/jhet.3487
17. Charitos G, Trafalis D, Dalezis P, et al. Synthesis and anticancer activity of novel 3,6-disubstituted 1,2,4-triazolo-[3,4-b]-1,3,4-thiadiazole derivatives. Arab J Chem. 2016;12(8):4784–4794. doi:10.1016/j.arabjc.2016.09.015
18. Chowrasia D, Karthikeyan C, Choure L, Sahabjada GM, Arshad M, Trivedi P. Synthesis, characterization and anticancer activity of some fluorinated 3,6-diaryl-[1,2,4] triazolo[3,4-b][1,3,4]thiadiazoles. Arab J Chem. 2017;10:S2424–S2428. doi:10.1016/j.arabjc.2013.08.026
19. Gomha S, Riyadh S. Synthesis under microwave irradiation of [1,2,4]triazolo[3,4-b] [1,3,4]thiadiazoles and other diazoles bearing indole moieties and their antimicrobial evaluation. Molecules. 2011;16(10):8244–8256. doi:10.3390/molecules16108244
20. Gomha S, Abdel-aziz H. Synthesis and antitumor activity of 1,3,4-thiadiazole derivatives bearing coumarine ring. Heterocycles. 2015;91(3):583–592. doi:10.3987/COM-14-13146
21. Gomha S, Riyadh S, Mahmmoud E, Elaasser M. Synthesis and anticancer activity of arylazothiazoles and 1,3,4-thiadiazoles using chitosan-grafted-poly(4-vinylpyridine) as a novel copolymer basic catalyst. Chem Heterocycl Compd. 2015;51(11–12):1030–1038. doi:10.1007/s10593-016-1815-9
22. Gomha S, Badrey M, Edrees M. Heterocyclisation of 2,5-diacetyl-3,4-disubstituted-thieno[2,3-b]thiophene bis-thiosemicarbazones leading to bis-thiazoles and bis-1,3,4-thiadiazoles as anti-breast cancer agents. J Chem Res. 2016;40(2):120–125. doi:10.3184/174751916X14537182696214
23. Gomha S, Abdel-aziz H, Khalil K. Synthesis and SAR study of the novel thiadiazole–imidazole derivatives as a new anticancer agents. Chem Pharm Bull (Tokyo). 2016;64(9):1356–1363. doi:10.1248/cpb.c16-00344
24. Gomha S, Abdelhamid A, Kandil O, Kandeel S, Abdelrehem N. Synthesis and molecular docking of some novel thiazoles and thiadiazoles incorporating pyranochromene moiety as potent anticancer agents. Mini Rev Med Chem. 2018;18(19):1670–1682. doi:10.2174/1389557518666180424113819
25. Jacob S, Aguado M, Fallik D, Praz F. The role of the DNA mismatch repair system in the cytotoxicity of the topoisomerase inhibitors camptothecin and etoposide to human colorectal cancer cells. Cancer Res. 2001;61(17):6555–6562.
26. Reid JR, Heindel ND. Improved syntheses of 5-substituted-4-amino-3-mercapto-(4H)-1,2,4-triazoles. J Heterocycl Chem. 1976;13(4):925–926. doi:10.1002/jhet.5570130450
27. Chang DJ, An H, Kim KS, et al. Design, synthesis, and biological evaluation of novel deguelin-based heat shock protein 90 (HSP90) inhibitors targeting proliferation and angiogenesis. J Med Chem. 2012;55(24):10863–10884. doi:10.1021/jm301488q
28. Ezabadi IR, Camoutsis C, Zoumpoulakis P, et al. Sulfonamide-1,2,4-triazole derivatives as antifungal and antibacterial agents: synthesis, biological evaluation, lipophilicity, and conformational studies. Bioorg Med Chem. 2008;16(3):1150–1161. doi:10.1016/j.bmc.2007.10.082
29. Trafalis DT, Camoutsis C, Papageorgiou A. Research on the anti-tumour effect of steroid lactam alkylator (NSC-294859) in comparison with conventional chemotherapeutics in malignant melanoma. Melanoma Res. 2005;15(4):273–281. doi:10.1097/00008390-200508000-00007
30. Trafalis DT, Camoutsis C, Dalezis P, et al. Antitumour effect of a- and d- lactam androgen nitrogen mustards on non-small cell lung carcinoma. J BUON. 2004;9(3):275–282.
31. Elkamhawy A, Park J, Cho N, Sim T, Pae A, Roh E. Discovery of a broad spectrum antiproliferative agent with selectivity for DDR1 kinase: cell line-based assay, kinase panel, molecular docking, and toxicity studies. J Enzyme Inhib Med Chem. 2015;31(1):158–166. doi:10.3109/14756366.2015.1004057
32. Paull KD, Shoemaker RH, Hodes L, et al. Display and analysis of patterns of differential activity of drugs against human tumor cell lines: development of mean graph and COMPARE algorithm. J Natl Cancer Inst. 1989;81(14):1088–1092. doi:10.1093/jnci/81.14.1088
33. Wei H, Ruthenburg AJ, Bechis SK, Verdine GL. Nucleotide-dependent domain movement in the ATPase domain of a human type IIA DNA topoisomerase. J Biol Chem. 2005;280(44):37041–37047. doi:10.1074/jbc.M506520200
34. Trott O, Olson AJ. AutoDock vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461. doi:10.1002/jcc.21334
35. Lee J, Berger J. Cell cycle-dependent control and roles of DNA topoisomerase II. Genes. 2019;10(11):859. doi:10.3390/genes10110859
36. Qi X, Hou S, Lepp A, et al. Phosphorylation and stabilization of topoisomerase IIα protein by p38γ mitogen-activated protein kinase sensitize breast cancer cells to its poisons. J Biol Chem. 2011;286(41):35883–35890. doi:10.1074/jbc.M111.229260
37. Li H, Wang Y, Liu X. Plk1-dependent phosphorylation regulates functions of DNA topoisomerase II alpha in cell cycle progression. J Biol Chem. 2008;283(10):6209–6221. doi:10.1074/jbc.M709007200
38. Iida M, Matsuda M, Komatani H. Plk3 phosphorylates topoisomerase I Ialpha at Thr(1342), a site that is not recognized by Plk1. Biochem J. 2008;411(1):27–32. doi:10.1042/BJ20071394
39. Ishida R, Iwai M, Marsh KL, et al. Threonine 1342 in human topoisomerase IIαlpha is phosphorylated throughout the cell cycle. J Biol Chem. 1996;271(47):30077–30082. doi:10.1074/jbc.271.47.30077
40. Daum JR, Gorbsky GJ. Casein kinase II catalyzes a mitotic phosphorylation on threonine 1342 of human DNA topoisomerase IIαlpha, which is recognized by the 3F3/2 phosphoepitope antibody. J Biol Chem. 1998;273(46):30622–30629. doi:10.1074/jbc.273.46.30622
41. Escargueil AE, Plisov SY, Filhol O, Cochet C, Larsen AK. Mitotic phosphorylation of DNA topoisomerase II alpha by protein kinase CK2 creates the MPM-2 phosphoepitope on Ser-1469. J Biol Chem. 2000;275(44):34710–34718. doi:10.1074/jbc.M005179200
42. Redwood C, Davies SL, Wells NJ, Fry AM, Hickson ID. Casein kinase II stabilizes the activity of human topoisomerase IIα in a phosphorylation-independent manner. J Biol Chem. 1998;273(6):3635–3642. doi:10.1074/jbc.273.6.3635
43. Grozav A, Chikamori K, Kozuki T, et al. Casein kinase I δ/ε phosphorylates topoisomerase IIα at serine-1106 and modulates DNA cleavage activity. Nucleic Acids Res. 2008;37(2):382–392. doi:10.1093/nar/gkn934
44. Wang Q, Zambetti G, Suttle D. Inhibition of DNA topoisomerase II alpha gene expression by the p53 tumor suppressor. Mol Cell Biol. 1997;17(1):389–397. doi:10.1128/MCB.17.1.389
45. Liu D, Huang C, Kameyama K, et al. Topoisomerase IIα gene expression is regulated by thep53 tumor suppressor gene in nonsmall cell lung carcinoma patients. Cancer. 2002;94(8):2239–2247. doi:10.1002/cncr.10450
46. Berg K, Eide P, Eilertsen I, et al. Multi-omics of 34 colorectal cancer cell lines - a resource for biomedical studies. Mol Cancer. 2017;16(1):116. doi:10.1186/s12943-017-0691-y
47. Ahmed D, Eide P, Eilertsen I, et al. Epigenetic and genetic features of 24 colon cancer cell lines. Oncogenesis. 2013;2(9):e71. doi:10.1038/oncsis.2013.35
48. Broad Institute Cancer Cell Line Encyclopedia (CCLE). Portals broad institute org; 2020. Available from: https://portals.broadinstitute.org/ccle/page?cell_line=LOVO_LARGE_INTESTINE.
49. Fortune JM, Osheroff N. Topoisomerase II as a target for anticancer drugs: when enzymes stop being nice. Prog Nucleic Acid Res Mol Biol. 2000;64:221–253.
50. Pommier Y, Schwartz RE, Kohn KW, Zwelling LA. Formation and rejoining of deoxyribonucleic acid double-strand breaks induced in isolated cell nuclei by antineoplastic intercalating agents. Biochemistry. 1984;23(14):3194–3201. doi:10.1021/bi00309a013
51. Tewey KM, Chen GL, Nelson EM, Liu LF. Intercalative antitumor drugs interfere with the breakage-reunion reaction of mammalian DNA topoisomerase II. J Biol Chem. 1984;259(14):9182–9187.
52. Pommier Y, Leo E, Zhang H, Marchand C, Topoisomerases DNA. Their poisoning by anticancer and antibacterial drugs. Chem Biol. 2010;17(5):421–433. doi:10.1016/j.chembiol.2010.04.012
53. Larsen A, Skladanowski A, Bojanowski K. The roles of DNA topoisomerase II during the cell cycle. In: Meijer L, Guidet S, Vogel L, editors. Progression in Cell Cycle Research. Boston: Springer; 1996:229–239.
54. Heck MMS, Hittelman WN, Earnshaw WC. Differential expression of DNA topoisomerases I and II during the eukaryotic cell cycle. Proc Natl Acad Sci U S A. 1988;85(4):1086–1090. doi:10.1073/pnas.85.4.1086
55. Bush NG, Evans-Roberts K, Maxwell A. DNA topoisomerases. EcoSal Plus. 2015;6(2). doi:10.1128/ecosalplus.ESP-0010-2014
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.