Back to Journals » Infection and Drug Resistance » Volume 14
(2Z)-3-Hydroxy-3-(4-R-Phenyl)-Prop-2-Enedithioic Acids as New Antituberculosis Compounds
Authors Pretelín-Castillo G, Silva Miranda M, Espitia C, Chávez-Santos RM, Suárez-Castro A, Chacón-García L, Aguayo-Ortiz R, Martinez R ![]()
Received 30 July 2021
Accepted for publication 28 September 2021
Published 20 October 2021 Volume 2021:14 Pages 4323—4332
DOI https://doi.org/10.2147/IDR.S328132
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Héctor Mora-Montes
Gustavo Pretelín-Castillo,1 Mayra Silva Miranda,2,3 Clara Espitia,2,3 Rosa María Chávez-Santos,1 Abel Suárez-Castro,4 Luis Chacón-García,4 Rodrigo Aguayo-Ortiz,5 Roberto Martinez1
1Instituto de Química, Universidad Nacional Autónoma de México, Circuito Exterior, Ciudad Universitaria, Cd. México, 04510, México; 2Catedrática CONACYT adscrita al Instituto de Investigaciones Biomédicas, Universidad Nacional Autónoma de México, Ciudad Universitaria, Cd. México, 04510, México; 3Instituto de Investigaciones Biomédicas, Universidad Nacional Autónoma de México, Departamento de Inmunología, Ciudad Universitaria, Cd. México, 04510, México; 4Instituto de Investigaciones Químico-Biológicas, Universidad Michoacana de San Nicolás de Hidalgo, Edificio B-1, Ciudad Universitaria, Morelia Michoacán, 58030, México; 5Departamento de Farmacia, Facultad de Química, Universidad Nacional Autónoma de México, Circuito Exterior, Ciudad Universitaria, Cd. México, 04510, México
Correspondence: Roberto Martinez
Instituto de Química, Universidad Nacional Autónoma de México, Circuito Exterior, Ciudad Universitaria, Cd. México, 04510, México
Tel +52 55 56224441
Fax +52 55 56162217
Email [email protected]
Background: Tuberculosis is an infectious disease caused by the bacillus Mycobacterium tuberculosis. Compounds including a sulfur-containing scaffold have been shown to be key scaffolds in various antituberculosis agents. Interestingly, the 3-hydroxy-3-phenyl-prop-2-enedithioic acids 11a-j have, to the best of our knowledge, not been previously described as antituberculosis agents.
Purpose: In the present study, we investigated the role of substituents attached to the phenyl ring of a 3-hydroxy-3-phenyl-prop-2-enedithioic acid scaffold (compounds 11a–j) in inhibiting the growth of M. tuberculosis strain H37Rv.
Methods: (Z)-3-hydroxy-3-(4-R-phenyl)-prop-2-enedithioic acids 11b–j, with R groups including various electron-donating or electron-withdrawing groups, were designed by structurally modifying the lead compound 11a. The syntheses of 11a–j involved each one-step procedure starting from the corresponding substituted acetophenone. Compounds 11a–j were tested against M. tuberculosis strain H37Rv to evaluate their bacterial growth inhibitory activities. ADMET profiles were predicted by employing three different methods. In addition, molecular docking studies were carried out, based on the molecular similarities of the synthesized compounds with ethionamide ( 5), on the active site of the M. tuberculosis H37Rv (3R)-hydroxyacyl-ACP (HadAB) dehydratase heterodimer.
Results: The antituberculosis activities of compounds 11a–j could be explained in terms of the presence of electron-donating or electron-withdrawing substituents on the aromatic ring of the substituted 3-hydroxy-3-phenyl)-prop-2-enedithioic acid core. The activity and selectivity index (SI) value of (Z)-3-hydroxy-3-(4-nitrophenyl)-prop-2-enedithioic acid 11e suggested that this compound could be used for the design of novel antituberculosis agents. Most of the synthesized molecules showed an acceptable ADME profile and a low probability of being toxic. Docking studies of 11d and 11e showed them forming hydrogen bonds with the ACys61 residue of the HadAB enzyme.
Conclusion: Our results suggested that the antituberculosis compound 11e could be used for the design of novel antituberculosis agents.
Keywords: synthesis, 3-hydroxy-3-(4-R-phenyl)-prop-2-enedithioic acids, antituberculosis agents, docking, theoretical ADME
Graphical Abstract:

Introduction
Mycobacterium tuberculosis is a pathogenic bacterium that is well known to be the causative agent of tuberculosis. The World Health Organization (WHO) has pinpointed this pathogen as one of the two leading causes worldwide of higher mortality resulting from an infectious agent. The increase in the number of cases of tuberculosis, the presence of M. tuberculosis strains showing resistance to first-line drugs, and the adverse effects or long treatment durations of current tuberculosis medications have increased concerns about the ability to find a safe and effective treatment for this disease but at the same time have led researchers in medicinal chemistry to search for new compounds that are more effective against this disease.1 Most drugs launched in the last decade were derived by modifying known drugs or lead structures, and structural modification has been shown to be an effective approach for understanding the mechanism of drug action and for designing better drugs.2,3
A sulfur-containing scaffold, for example, is a key structural feature present in most compounds displaying antituberculosis activity (Figure 1). Several drugs targeting various diseases contain this scaffold, including the anticoagulant agent rivaroxaban 1,4 the prostate cancer drug enzalutamide 2,5 and disulfiram 3, which is used for the treatment of alcohol abuse.6 Compounds containing a sulfur-containing scaffold and displaying antituberculosis activity, however, have not been reported on much, but with examples that have been reported including thioacetazone 4, ethionamide 5, and prothionamide 6 as prodrugs.7 In the present study, we investigated the role of substituents attached to the phenyl ring of a 3-hydroxy-3-phenyl-prop-2-enedithioic acid scaffold (to form compounds 11a–j) in inhibiting the growth of M. tuberculosis strain H37Rv. Compounds 11a-j have, to the best of our knowledge, not been previously described as antituberculosis agents—but some of them were found in the current work to display antituberculosis activity.
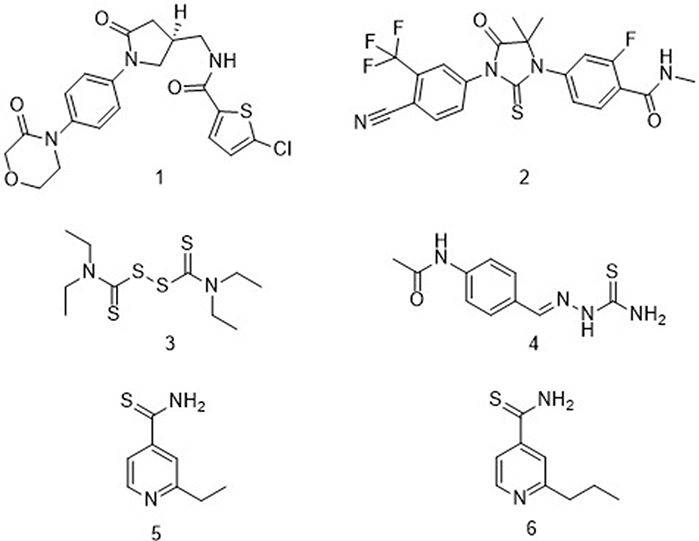 |
Figure 1 Examples of compounds that both contain a thio scaffold and exhibit biological activity. |
Materials and Methods
Melting points were measured for the compounds in open capillaries using a Mel-Temp apparatus. 1H-NMR spectra were recorded using a 300 MHz Jeol Eclipse spectrometer with the samples dissolved in deuterated chloroform (CDCl3) solutions containing tetramethylsilane (TMS) as an internal standard (δ = 0 ppm), and 13C-NMR spectra were recorded at 75 MHz using the same instrument. Regarding these NMR spectra, the chemical shift (δ) values are reported in parts per million (ppm), and the peak shapes are indicated as s for singlet, d for doublet, t for triplet, q for quartet, m for multiplet, and bs for broad signal. The coupling constant (J) values are reported in hertz (Hz). IR spectra were obtained using a Magna-IR spectrometer. Mass spectra were recorded using a Jeol AccuTOF Direct Analysis in Real Time (DART) spectrometer with time-of-flight detection for low- and high-resolution measurements. Flash column chromatography was carried out using silica gel 60 (230–400 mesh ASTM) from Macherey–Nagel GmbH & Co. Reactions were monitored using thin-layer chromatography (TLC). The TLC plates were visualized using a dual short-wavelength/long-wavelength UV lamp or by staining them with an ethanol solution of potassium permanganate, vanillin, or p-anisaldehyde. The reagents 12a–j, sodium hydride, and carbon disulfide were used as obtained from Sigma-Aldrich, St. Louis, MO, USA, as commercial raw materials.
Chemistry
General Procedure for Preparing (Z)-3-Hydroxy-3-(4-R-Phenyl)-Prop-2-Enedithioic Acids 11a–j
Ten samples of ice-bath-cooled NaH (12.0 mmol) suspensions in pre-dried THF (5.0 mL) were prepared, and to each sample was added, dropwise, a THF solution (2.0 mL) of a different acetophenone 12a–j (4 mmol). Each resulting mixture was stirred for 30 minutes, and then to each mixture was added, dropwise, a THF solution (1.0 mL) of CS2 (6 mmol)). Each resulting mixture was stirred for over 12–15 hours at room temperature. In each case, after the starting material was consumed (as monitored using TLC), the crude reaction product was poured into a water–ice suspension and extracted three times with ethyl acetate. The resulting aqueous phase was acidified with 2 N sulfuric acid to a pH of 2 and then extracted three times with ethyl acetate, while the resulting organic layer was dried with Na2SO4. Removal of the solvent under reduced pressure gave an amorphous solid that was crystallized from an appropriate solvent.8 Spectroscopic data collected from each of the resulting compounds 11a-j fully supported their expected structures (see Spectral Data in the Supplementary Information).9,10
Antitubercular Activity Evaluation
Preparations of Stock Solutions
DMSO (Sigma-Aldrich, St. Louis, MO, USA) stock solutions of the synthesized compounds at a concentration of 10 mg/mL were prepared for carrying out in each case the resazurin microtiter assay (REMA). Each compound was diluted in a Middlebrook 7H9 broth medium (Difco, Sparks, MD, USA). Stock solutions for each of the reference compounds at 64 μg/mL were prepared and filtered employing a 0.22 μm-diameter pore membrane (Millipore, Darmstadt, Germany). The prepared stock solutions were stored at −20°C prior to their evaluation.
Cell Culture
The Vero cell line (African green monkey kidney) from American Type Culture Collection (ATCC) was employed to carry out the cytotoxicity assays. RPMI 1640 medium (Gibco, Grand Island, NE, USA) supplemented with 10% fetal bovine serum (FBS) (Gibco) and nonessential amino acids (Gibco) was used to culture these cells.
Cytotoxicity Assay
A total of 10,000 Vero cells were placed in a 96-well plate and then incubated in an RPMI medium (100 μL) for 24 h. Then, the plate was washed and fresh medium containing the synthesized compounds at different concentrations was added to it. Each compound was incubated under a 5% CO2 atmosphere for 48 h at 37°C. A 10 μL MTT (Sigma-Aldrich) solution (5 mg/mL in sterile PBS) was added to each well, and the resulting mixtures were incubated for another 4 h. The medium was then removed, and a volume of 100 μL of DMSO was used to solubilize the formazan (Sigma-Aldrich, St. Louis, MO, USA). The formazan absorbance (ABS) at a wavelength of 570 nm was measured, and cytotoxicity was calculated as %toxicity = (1 − (ABS experimental/ABS control)) × 100. Controls were cells without treatment but following the same procedures as described above.11
M. tuberculosis Inocula Preparation
A 7H9–glycerol–10% ADC–0.01% tyloxapol medium was used to cultivate M. tuberculosis strains H37Rv, H37Ra (virulent and non-virulent reference strains, respectively) and 209 (clinical isolated strain, rifampicin resistant) at 37°C until an OD600nm of 0.4 was reached.
Antimicrobial Susceptibility Test Using the Resazurin-Based Microtiter Plate Assay (REMA)
The REMA procedure used in the current work was developed based on a microplate alamar Blue assay (MABA) used in a previous work for determining antimycobacterial activities of various compounds.12 In brief, the outer wells of a 96-well plate were each filled with 200 μL of sterile PBS to prevent dehydration from occurring during the long incubation period (8 days). Here, rifampicin (RIF) (Sigma-Aldrich, St. Louis, MO, USA) was included in each plate as a reference drug (16-0.001 μg/mL serial two-fold dilutions), DMSO, DMSO+Mtb, medium, media+Mtb, and compound alone were added as control to validate the plate. The compounds were evaluated at various concentrations, ranging from 0.98 μg/mL to 250 μg/mL and in triplicate. After 6-day incubations of the plates, a volume of 30 µL of 0.01% (weight/volume) resazurin (Sigma-Aldrich, St. Louis, MO, USA) instead of MABA was added to each well, and the plates were incubated for additional 2 days. Visual inspection was done to determine the color of each well, with blue interpreted as a lack of cells growth and pink as growth of cells. The MIC value for each experiment was defined as the lowest concentration of the compound in those wells where the blue color was observed.
Selectivity Index (SI)
This index was calculated by comparing the IC50 values obtained in Vero cells with the bacteria MIC100 determined by performing the REMA.
Minimal Bactericidal Concentration (MBC)
Following the methodology described in a previous study, some of the compounds were selected to determine their respective MBCs. Here, MBC was defined as the minimum extract concentration that did not cause a color shift in cultures reincubated in fresh medium. Briefly, 5 μL volumes of REMA duplicate bacteria suspensions were transferred to a new microplate containing 195 μL of fresh culture medium in each well. Subsequently, the microplates were incubated as described previously for the REMA.13
Physicochemical Properties and ADMET Prediction
The ADME-Tox filtering tool FAF-Drugs4 was used to evaluate the physicochemical properties of the synthesized compounds.14 The ADMET profiles were predicted by employing the consensus of the results of vNN-ADMET,15 admetSAR 2.0 (unrestricted applicability domain),16 and pkCSM.17
In silico Molecular Docking Studies
Validation of the Docking Procedure
The complex of 4RLU with 2ʹ,4,4ʹ-trihydroxychalcone (HCC) was selected for docking studies and done so due to 4RLU having been identified as the protein receptor for HadAB.33 To validate the docking process, the co-crystallized ligand (HCC) was re-docked into the active site, and an RMSD value of 3.0 Å between the cocrystallized structure and the redocked structure was obtained.
Ligand Preparation
The structures of compounds 11a-j were each modeled in their two tautomeric forms (keto and enol) as 2D structures by using the software ChemBio Draw Ultra 12.0 and were converted into 3D structures in MDL format.18 Their protonated states were then modeled using the online tool Chemicalize.19 The geometries of the compounds and co-crystalized ligand were calculated at the density functional B3LYP level using the 6‐31G (d, p) basis set in the Gaussian 16 software package.20 Finally, using Autodock Tools, the ligands were prepared by including in them polar hydrogens and Gasteiger charges as well as rotatable (ie, single) bonds that were assigned by default, and a PDBQT file was generated.21
Receptor Preparation
The X-ray coordinates of the HadAB receptor were retrieved from the Protein Data Bank (PDB code:4RLU).22 Molecular water was removed from the crystallographic structure, and the final preparation and minimization of the receptor structure was carried out by deploying the Dock Prep module of Chimera software23 using the AMBER-ff14SB force field. Lastly, Kollman charges were added to the obtained structure by using Autodock Tools, and a PDBQT file was generated.
Docking Calculations
Rigid receptor molecular docking was carried out in Autodock4 using the Lamarckian genetic algorithm.24 We used grid maps with 70 × 70 × 70 points in the active site of the receptor with the coordinates x=0.272, y=22.253, z=−30.833, and a grid-point spacing of 0.375 Å. AD4.dat parameters were applied to all the ligands. The parameters used were 10 runs, a population size of 100, and a run-termination criterion of a maximum of 27,000 generations or a maximum of 250,000 energy evaluations. The visualization and analysis of the nonbonded interactions as hydrogen bonds of the best poses were carried out using Discovery Studio Visualizer software.25
Results and Discussion
Chemistry
The desired 3-hydroxy-3-(4-R-phenyl)-prop-2-enedithioic acids 11a–j (Scheme 1) were prepared with good yields by carrying out respective coupling reactions between carbon disulfide and the substituted acetophenones 12a–j in the presence of sodium hydride in dry tetrahydrofuran.8 The spectroscopic data of the 11a–j products were fully consistent with their expected structures. Note that the 1H-NMR and 13C-NMR spectra of these products showed the signals characteristic for their enol and not keto forms.9,10
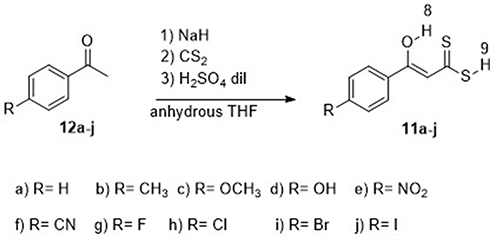 |
Scheme 1 Synthetic route to compounds 11a–j. |
Antitubercular Activity Evaluation
Compounds 11a–j were evaluated as M. tuberculosis strain H37Rv growth inhibitors in 250 μg/mL–0.98 μg/mL serial two-fold dilutions (Table 1). These activities were compared with those of RIF, which is usually indicated for the treatment of Mycobacterium infections, including tuberculosis (TB).11–13 First, we synthesized compound 11a (R=H) and measured its antituberculosis minimal inhibition concentration (MIC) to be 62.5 μg/mL. Then, to examine the influence of electron-donating and electron-withdrawing groups on the antituberculosis activity, we synthesized compounds 11b (R=CH3), 11c (R=OCH3), and 11d (R=OH). Surprisingly, 11b did not inhibit the growth of M. tuberculosis cells (MIC=250 μg/mL). The compound resulting from replacing the 4-CH3 substituent with the 4-OCH3 group (4c) exhibited inhibition levels similar to those of 11a (R=H) but with a lower SI [SI (11a)= 3.46 vs SI (11c)= 1.60].
Compound 11d, generated by incorporating an -OH group at position 4 of the phenyl ring of compound 11a, showed an inhibitory activity four times that of 11a and a toxicity level lower than that of 11a; and compound 11e, generated by incorporating an -NO2 group at position 4 of the phenyl ring of compound 11a, showed an even greater inhibitory activity – eight times that of 11a – and was observed to be much less toxic [SI (11a)= 3.46 vs SI (11e)= 32.69)]. Because the reduction in the aromatic nitro group in vivo produces hydroxylamine and nitrosamine,26 both highly reactive and carcinogenic species, this group was replaced with an isosteric group, namely –CN, which produces fewer toxic compounds than to nitro compounds.27 Unexpectedly, when the 4-NO2 substituent was replaced with 4-CN (11f), the new compound exhibited inhibition levels similar to those of 11a (R=H) but with a lower SI [SI (11a)=3.45 vs SI (11f)=0.80)]. We also investigated the effects of halogen substituents on the antituberculosis activities by synthesizing compounds containing fluorine, chloro, bromo, and iodine groups at the 4 position of the phenyl ring. Compound 11g, having a fluorine at position 4 of the phenyl group, exhibited an antituberculosis activity essentially the same as that of 11a (MIC=62.50 μg/mL) but with lower toxicity [SI (11a)= 3.46 vs SI (11g)=7.09]. The inhibitory activity of compound 11h, generated by the incorporation of a chlorine substituent at position 4 of the phenyl ring, was four times that of 11a, and the compound was essentially equally toxic. In contrast, when 4-H was replaced with 4-Br, the generated compound 11i exhibited inhibition levels similar to those of 11a (R=H), and with a lower SI [SI (11a)=3.46 vs SI (11i)=1.60]. And compound 11j, having an iodine substituent, showed no inhibitory activity.
All told, none of the compounds with halogen substituents showed an antituberculosis activity as good as that of 11e. Based on our results, derivatization of the phenyl ring with substituents such as -OH and especially -NO2 offered a suitable approach for optimizing the antituberculosis activities of the 3-hydroxy-3-(4-R-phenyl)-prop-2-enedithioic acids. In both cases, the presence of hydrogen bonds apparently influenced the antituberculosis activity. The presence of a nitro group has been proposed to increase the number of hydrogen bond interactions with the target receptor.28 In vitro activity against M. tuberculosis strains that are resistant to a single TB drug (eg, isoniazid or RIF) has been used as a key criterion to determine the reliability of a compound to be considered as an anti-TB drug.29 Therefore, we also evaluated compound 11e with non-virulent M. tuberculosis H37Ra bacteria, which are more susceptible to lower concentrations of drugs than are virulent strains. Specifically, compound 11e activities against M. tuberculosis H37Ra and 209 (an RIF-resistant strain) were evaluated using 250 μg/mL–0.98 μg/mL serial two-fold dilutions (Table 2). Table 2 also lists the MIC100 values of RIF. Compound 11e was found to be active against M. tuberculosis strains H37Ra and 209.
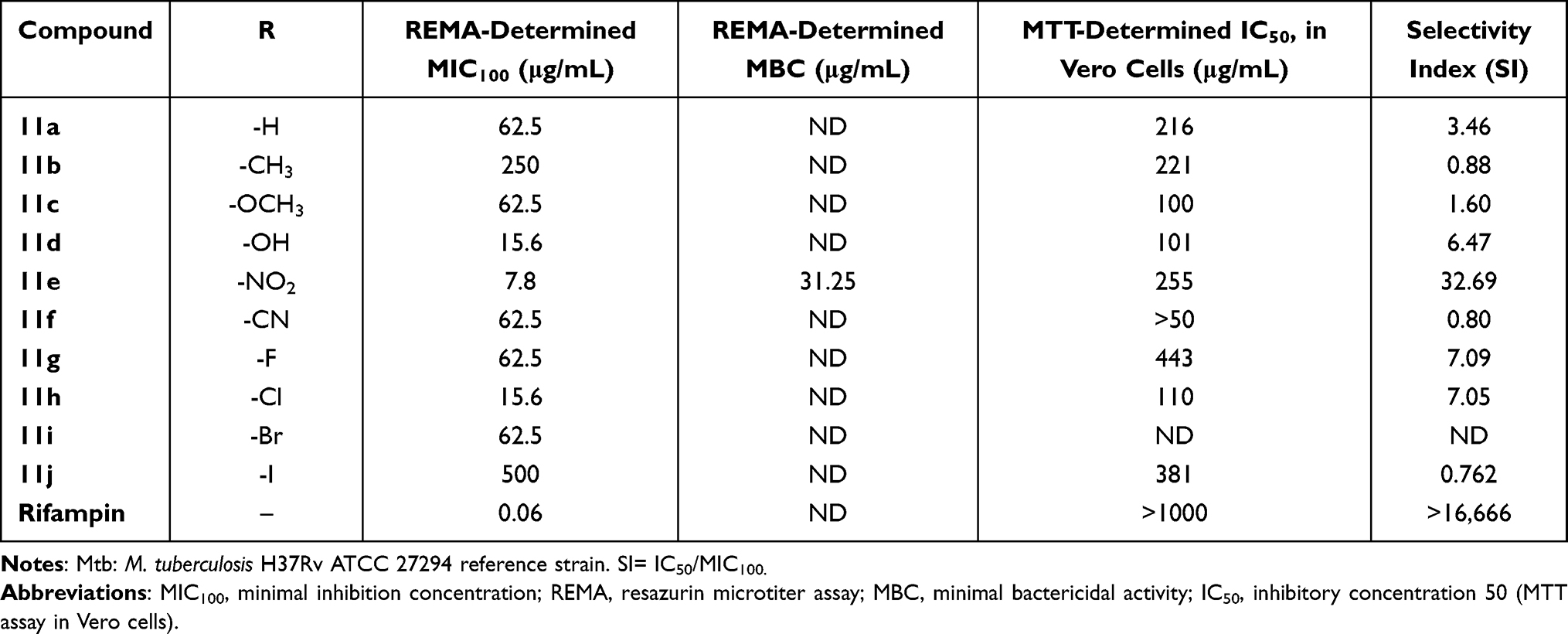 |
Table 1 Anti-M. tuberculosis Effects and Cytotoxicity Levels of Compounds 11a–j |
 |
Table 2 REMA-Determined MIC100 Values of 11e Against Virulent, Non-Virulent and RIF-Resistant M. tuberculosis Strains |
ADMET Predictions
Table 3 shows the determined physicochemical properties of the ten synthesized compounds. Assessment of these molecules using the Lipinski,30 Veber,31 and Egan32 rules suggested that they each had a high probability of exhibiting good oral bioavailability. An absorption, distribution, metabolism, excretion, and toxicity (ADMET) consensus analysis indicated that none of the compounds could inhibit P-glycoprotein and that only compounds 11b and 11d could be pumped out of the cell through this multidrug-resistant protein (Table 3).
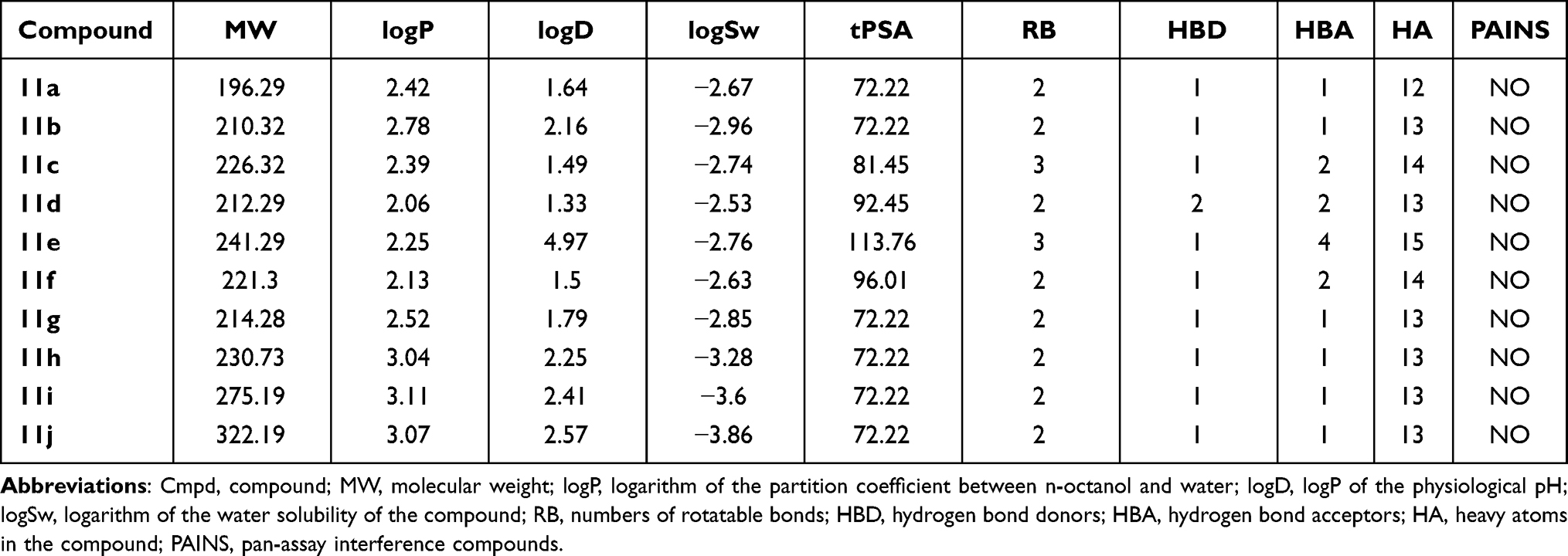 |
Table 3 Calculated Physicochemical Properties of Compounds 11a-j |
The prediction also suggested that all the compounds could lower CYP1A2 activity, and that only 11c, 11h, 11i, and 11j could inhibit CYP2C19, while 11b and 11h would be metabolized by CYP2C9 (Table 3). The compounds were concluded to have a low probability of exerting mutagenicity, cardiotoxicity, mitochondrial toxicity, and cytotoxicity, but have a high risk of damaging the liver and the skin (see Table 4). Therefore, it will apparently be necessary to modify the structure at the hit-to-lead stage to reduce its hepatotoxicity.
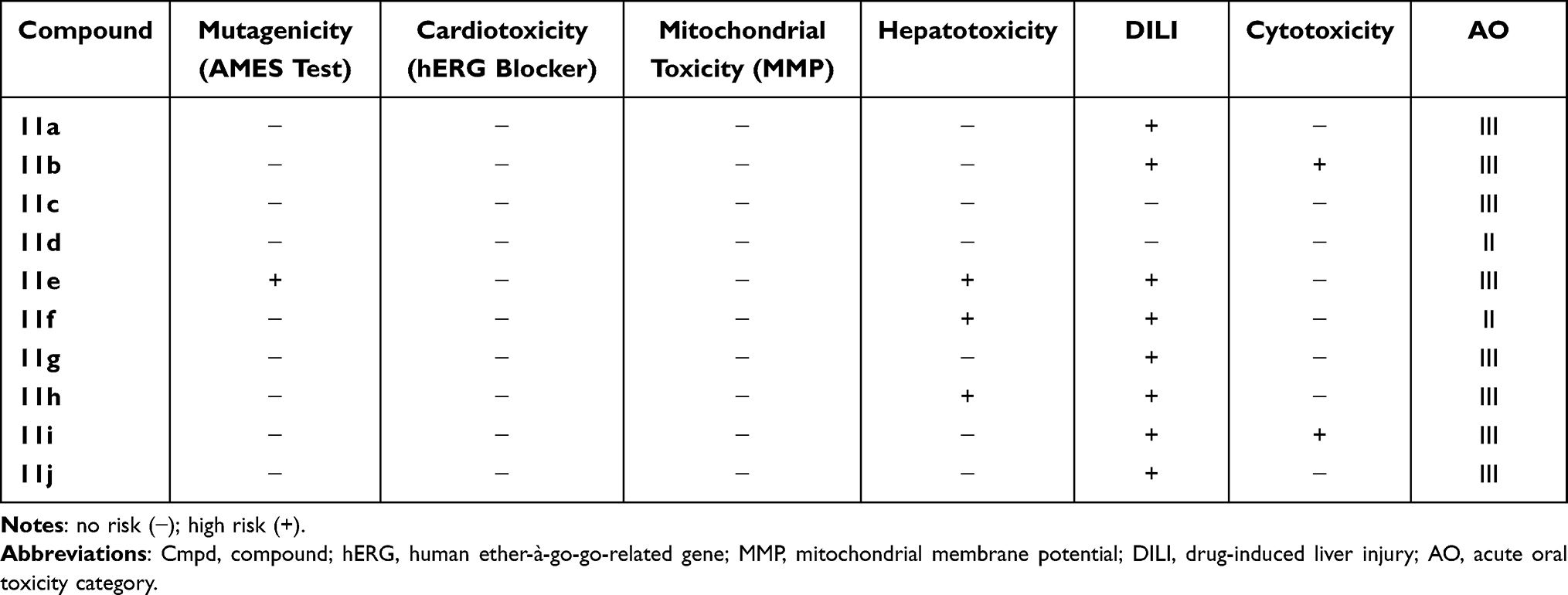 |
Table 4 Predicted Toxicities of the Synthesized Compounds 11a-j |
Docking Studies
To investigate the possible mode or modes of interaction of compounds 11a-j with M. tuberculosis, molecular docking studies were carried out, based on the molecular similarity of the synthesized compounds with ethionamide (5), on the active site of the (3R)-hydroxyacyl-ACP (HadAB) dehydratase heterodimer (PDB: 4RLU).33 It is important to note that each of compounds 11a-j can be found in a tautomeric ketone or enol form. Although the enol form of each of them was found according to 1H-NMR observations to be predominant in organic media (see above), the relative amounts of the two forms of each compound may differ when the compound is placed in aqueous media or when interacting with a receptor biomolecule. Therefore, we decided to carry out in silico studies of the interactions of both the keto and enol forms of each compound with the HadAB enzyme. The predicted free energy and affinity constant values are listed in Table 5.
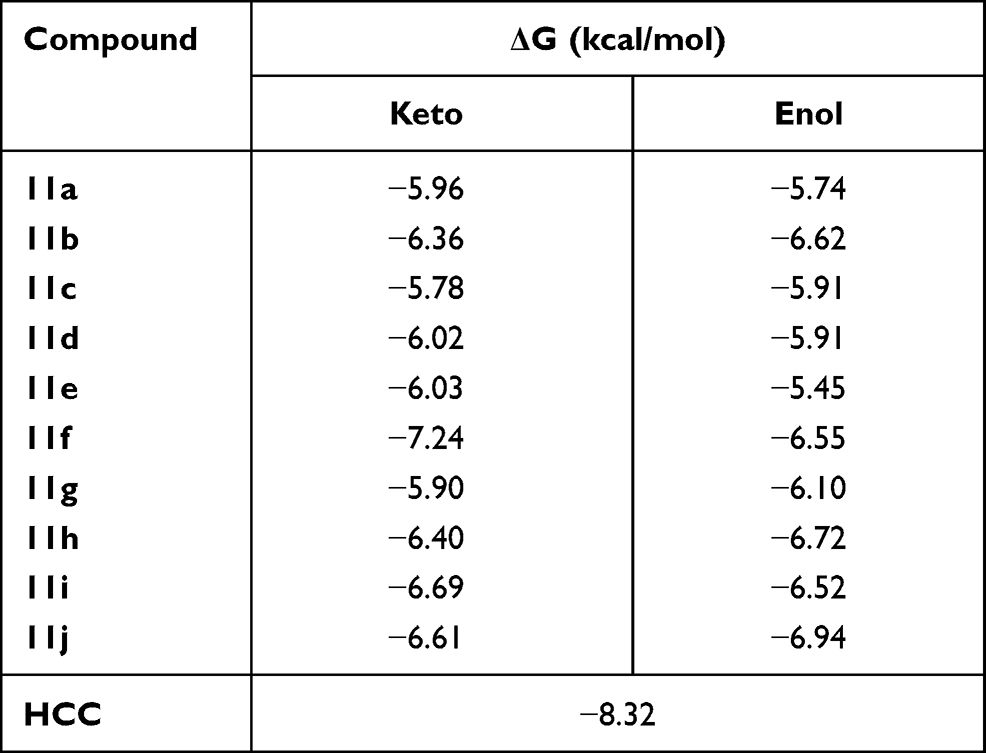 |
Table 5 Calculated Binding Free Energy (ΔG) from Molecular Docking Results of the Two Tautomeric Forms (Keto and Enol) of 11a-j Compounds Within HadAB Active Site |
Molecular Dockings of Compounds 11a-j
The above-described docking protocol was implemented to analyze the binding interactions between each of the compounds 11a-j and HadAB. To compare these results with that for a leading compound known to interact with the same receptor, an anchoring study involving the co-crystallized enzyme complex and the 2ʹ,4,4ʹ-trihydroxychalcone (HCC) inhibitor was carried out under the same conditions as those used with 11a-j. Here, the free energy of the binding of HCC was found to be −8.32 kcal/mol, within about 2 kcal/mol of the values obtained for the tested compounds. All compounds were analyzed in their best pose with the lowest binding free energy with the nonbonding interactions at the active site. While 11f, 11h, 11i, 11j, and 11b in both tautomeric forms showed better-than-predicted free energy values, only 11e in its keto and enol forms and 11d in its enol form showed nonbonding interactions with ACys61 (Figure 2). Similar free energy values were predicted for the ketonic tautomers of 11d and 11e, but quite different ones for their enolic forms, with 11d predicted to have the lowest binding energy. This difference may have been due to their making different numbers of hydrogen bonds with the enzyme according to the docking studies—with 11d predicted to make four hydrogen bonds with the enzyme, specifically two with ACys61 and at the same time two with AGln86, and the 11e enolic form predicted to only make two hydrogen bonds, one with ACys61 and the other with BAsp36. Moreover, each of the two tautomeric forms of 11e was predicted to make hydrogen bond interactions with ACys61. Thus, the molecular docking studies indicated that the experimentally measured antituberculosis activity of 11d could be explained by its forming hydrogen bonds with HadAB.
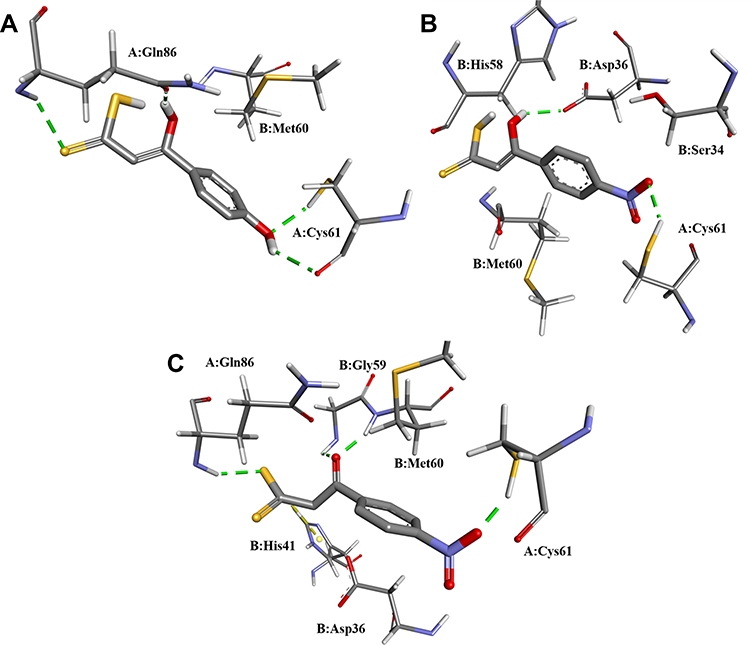 |
Figure 2 Nonbonding interactions between the active site of HadAB and (A) the 11d enolic tautomer, (B) 11e enolic tautomer, and (C) 11e ketonic tautomer. |
Conclusions
The present results indicated that derivatizing the phenyl group of 3-hydroxy-3-phenyl-prop-2-enedithioic acids with electron-donating and electron-withdrawing groups modified its antituberculosis activity. We investigated the role of electron-donating groups in the antituberculosis activity of this type of acid by synthesizing compounds 11b–d. Our results showed that the 4-hydroxy derivative of 11a (ie, 11d) was the most active of these compounds. The data presented here were found to be inconclusive regarding the relationship between the electronegativity of the halogen substituent on the phenyl group and the inhibition of the growth of M. tuberculosis cells, as well as regarding the relationship between the size of the halogen substituent and this inhibition (compounds 11g–j).
The role of the electron-withdrawing groups in the antituberculosis activity was investigated by synthesizing and measuring the activity levels of compounds 11e and 11f; in these experiments, compound 11e (R=NO2) was more active and showed a remarkable selectivity index (SI=32.69). All of the tested molecules were also shown to not be pan-assay interference compounds (PAINS), so the antiproliferative activities reported in this study could be due to a target-specific interaction. In our docking studies, 11d and 11e made hydrogen bond interactions with ACys61 of the HadAB enzyme, leading us to think that HadAB should be considered as a possible therapeutic target for these compounds and suggesting that the development of new compounds based on these structures should be pursued. Accepted criteria for developing new drugs against infectious diseases such as tuberculosis include an SI higher than 10, an MIC lower than 10 μM, and activity against a mono-resistant strain.29 Therefore, our results suggested that the antituberculosis compound 11e might be useful for the design of novel antituberculosis agents. Further optimization of this hit compound is expected to provide information about the structural requirements for achieving high antitubercular activity. Overall, this study represents an important attempt towards the development of novel tuberculosis treatments.
Acknowledgments
We thank R. Patiño, A. Peña, E. Huerta, B. Quiroz, L. Velasco, J. Pérez, and E. Segura Salinas for technical support. During the experimental phase of the study, Gustavo Pretelin Castillo was a CONACyT Graduate Scholarship holder (308250).
Funding
Financial support from the DGAPA (project PAPIIT 204619), CONACyT (project A1-S-16584) and NUATEI-IIB-UNAM is gratefully acknowledged.
Disclosure
The authors declare no conflicts of interest, financial or otherwise.
References
1. World Health Organization [homepage on the internet]. Geneva; 1948. Available from: https://www.who.int/en/news-room/fact-sheets/detail/tuberculosis.
2. Teague SJ. Learning lessons from drugs that have recently entered the market. Drug Discov Today. 2011;16(9,10):398–411. doi:10.1016/j.drudis.2011.03.003
3. Flick AC, Ding HX, Leverett CA, et al. Synthetic approaches to the 2014 new drugs. Bioorg Med Chem. 2016;24(9):1937–1980. doi:10.1016/j.bmc.2016.03.004
4. Mueck W, Schwers S, Stampfuss J. Rivaroxaban and other novel oral anticoagulants: pharmacokinetics in healthy subjects, specific patient populations and relevance of coagulation monitoring. Thromb J. 2013;11(10):1–17. doi:10.1186/1477-9560-11-10
5. Bassetto M, Ferla S, Pertusati F, et al. Design and synthesis of novel bicalutamide and enzalutamide derivatives as antiproliferative agents for the treatment of prostate cancer. Eur J Med Chem. 2016;118:230–243. doi:10.1016/j.ejmech.2016.04.052
6. Pal A, Pattanayak RD, Sagar R. Tracing the journey of disulfiram: from an unintended discovery to a treatment option for alcoholism. J Mental Health Hum Behav. 2015;20(1):41–43. doi:10.4103/0971-8990.164826
7. Laborde J, Deraeve C, Bernardes-Génisson V. Update of antitubercular prodrugs from a molecular perspective: mechanisms of action, bioactivation pathways, and associated resistance. ChemMedChem. 2017;12(20):1657–1676. doi:10.1002/cmdc.201700424
8. Konreddy AK, Toyama M, Ito W, Bal C, Baba M, Sharon A. Synthesis and anti-HCV activity of 4-hydroxyamino α-pyranone carboxamide analogues. ACS Med Chem Lett. 2014;5(3):259–263. doi:10.1021/ml400432f
9. Garcia-Orozco I, Lopez-Cortes JG, Ortega-Alfaro MC, Toscano RA, Penieres-Carrillo G, Alvarez-Toledano C. Synthesis, characterization, and tautomerism of four novel copper(I) complexes from 3-hydroxy-3-(p-R-phenyl)-2-propenedithioic acids. Inorg Chem. 2004;43(26):8572–8576. doi:10.1021/ic0488132
10. Larsson FCV, Lawesson SO. Preparation and alkylation of substituted β-hydroxydithiocinnamic acids. Tetrahedron. 1972;28(21):5341–5347. doi:10.1016/S0040-4020(01)93857-8
11. Mossman T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65(1–2):55–63. doi:10.1016/0022-1759(83)90303-4
12. Collins LA, Franzblau SG. Microplate alamar blue assay versus BACTEC 460 system for high-throughput screening of compounds against Mycobacterium tuberculosis and Mycobacterium avium. Antimicrob Agents Chemother. 1997;41(5):1004–1009. doi:10.1128/AAC.41.5.1004
13. Molina SGM, Ramos GMC, Vargas VJ, Mata CBD, Becerril MP, Said FS. Activity of organic extracts from Flourensia cernua DC against strains of Mycobacterium tuberculosis. Arch Med Res. 2006;37(1):45–49. doi:10.1016/j.arcmed.2005.04.010
14. Lagorce D, Bouslama L, Becot J, Miteva MA, Villoutreix BO. FAF-Drugs4: free ADME-tox filtering computations for chemical biology and early stages drug discovery. Bioinformatics. 2017;33(22):3658–3660. doi:10.1093/bioinformatics/btx491
15. Schyman P, Liu R, Desai V, Wallqvist A. vNN web server for ADMET predictions. Front Pharmacol. 2017;8:889. doi:10.3389/fphar.2017.00889
16. Yang H, Lou C, Sun L, et al. admetSAR 2.0: web-service for prediction and optimization of chemical ADMET properties. Bioinformatics. 2019;35(6):1067–1069. doi:10.1093/bioinformatics/bty707
17. Pires DEV, Blundell TL, Ascher DB. pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J Med Chem. 2015;58(9):4066–4072. doi:10.1021/acs.jmedchem.5b00104
18. Cousins KR. Computer review of ChemDraw Ultra 12.0. J Am Chem Soc. 2011;133(21):8388. doi:10.1021/ja204075s
19. Swain M. Chemicalize.org. J Chem Inf Mod. 2012;52(2):613–615. doi:10.1021/ci300046g
20. Frisch MJ, Trucks GW, Schlegel HB, et al. Gaussian 16, Revision C.01. Wallingford CT: Gaussian, Inc.; 2016.
21. Sanner MF. Python: a programming language for software integration and development. J Mol Graph Model. 1999;17(1):57–61. doi:10.1016/S1093-3263(99)99999-0
22. Rose PW, Prlić A, Bi C, et al. The RCSB protein data bank: views of structural biology for basic and applied research and education. Nucleic Acids Res. 2015;43(D1):D345–D356. doi:10.1093/nar/gku1214
23. Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera—A visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. doi:10.1002/jcc.20084
24. Morris GM, Goodsell DS, Halliday RS, et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comp Chem. 1998;19(14):1639–1662. doi:10.1002/(SICI)1096-987X(19981115)19:14<1639::AID-JCC10>3.0.CO;2-B
25. BIOVIA, Dassault Systèmes, Discovery Studio 2019. San Diego: Dassault Systèmes; 2019. Available from: https://www.3ds.com/products-services/biovia/.
26. Chung MC, Bosquesi PL, dos Santos JL. A prodrug approach to improve the physico-chemical properties and decrease the genotoxicity of nitro compounds. Curr Pharm Des. 2011;17(32):3515–3526. doi:10.2174/138161211798194512
27. Cambridge MedChem Consulting. Available from: https://www.cambridgemedchemconsulting.com/resources/bioisoteres/.
28. Szatylowicz H, Stasyuk OA, Fonseca Guerra C, Krygowski TM. Effect of intra- and intermolecular interactions on the properties of para-substituted nitrobenzene derivatives. Crystals. 2016;6(3):29–45. doi:10.3390/cryst6030029
29. Katsuno K, Burrows JN, Duncan K, et al. Hit and lead criteria in drug discovery for infectious diseases of the developing world. Nat Rev Drug Discov. 2015;14(11):751–758. doi:10.1038/nrd4683
30. Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 1997;23(1–3):3–25. doi:10.1016/S0169-409X(96)00423-1
31. Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45(12):2615–2623. doi:10.1021/jm020017n
32. Egan WJ, Merz KM, Baldwin JJ. Prediction of drug absorption using multivariate statistics. J Med Chem. 2000;43(21):3867–3877. doi:10.1021/jm000292e
33. Dong Y, Qui X, Shaw N, et al. Molecular basis for the inhibition of β-hydroxyacyl-ACP dehydratase HadAB complex from Mycobacterium tuberculosis by flavonoid inhibitors. Prot Cell. 2015;6(7):504–517. doi:10.1007/s13238-015-0181-1
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.