Back to Journals » Drug Design, Development and Therapy » Volume 20
(+)-JQ1 Upregulates SIRT3 to Suppress cGAS/STING Pathway-Mediated Neuronal Inflammation and Ferroptosis After Hypoxic-Ischemic Encephalopathy
Authors Yu Q, Ou J, Luo Y, Shen M, Lou J ![]() , Jiang S, Lin L, Zhang S, Zhu J, Lin Z
, Jiang S, Lin L, Zhang S, Zhu J, Lin Z ![]() , Fang M
, Fang M
Received 29 November 2025
Accepted for publication 16 February 2026
Published 25 February 2026 Volume 2026:20 578815
DOI https://doi.org/10.2147/DDDT.S578815
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Yan Zhu
Qianqian Yu,1,2,* Jiahao Ou,1,2,* Yuqi Luo,1,2,* Ming Shen,1,2 Jia Lou,3 Saizhi Jiang,4,5 Lizi Lin,5 Shutian Zhang,5 Jianghu Zhu,1,2 Zhenlang Lin,1,2 Mingchu Fang1,2
1Department of Neonatology, The Second Affiliated Hospital and Yuying Children’s Hospital of Wenzhou Medical University, Wenzhou, Zhejiang, People’s Republic of China; 2Key Laboratory of Perinatal Medicine of Wenzhou, Wenzhou, Zhejiang, People’s Republic of China; 3Department of Pediatrics, Dongyang People’s Hospital, Dongyang, Zhejiang, People’s Republic of China; 4Department of Pediatrics, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou, Zhejiang, People’s Republic of China; 5Wenzhou Medical University, Wenzhou, Zhejiang, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Zhenlang Lin; Mingchu Fang, Email [email protected]; [email protected]
Background: Neonatal hypoxic-ischemic encephalopathy (HIE) is a leading cause of neurological disability and mortality in newborns, with limited therapeutic options beyond hypothermia. Bromodomain and extra-terminal domain (BET) proteins function as epigenetic readers that regulate gene expression by recognizing acetylated lysine residues on histones. Among BET inhibitors, (+)-JQ1 (JQ1) has recently garnered attention due to its potent anti-inflammatory and antioxidant properties. This study aims to investigate the neuroprotective effects of JQ1 and elucidate the underlying mechanisms in the context of HIE brain injury.
Methods: We established an in vivo model via the modified Rice-Vannucci method, alongside an in vitro model using oxygen-glucose deprivation (OGD) in HT22 cells. Transcriptomic changes in cortical tissues during the acute phase after HIE were profiled through RNA sequencing. Western blot, immunofluorescence, immunohistochemistry, and transmission electron microscopy were employed to measure the levels of neuroinflammation and ferroptosis. Furthermore, SIRT3-knockdown HT22 cells under OGD conditions were used to validate the JQ1-mediated protective mechanisms.
Results: JQ1 treatment significantly reduced cerebral infarction, edema, and neuronal loss, while improving emotional behavior and cognitive functions after HIE. It inhibited the cGAS-STING pathway, and alleviated ferroptosis by restoring GPX4 and system Xc⁻ activity while reducing iron overload. These effects were reversed by the SIRT3 inhibitor 3-TYP or SIRT3 siRNA.
Conclusion: JQ1 exerts neuroprotective effects in neonatal hypoxic-ischemic encephalopathy by attenuating neuroinflammation and suppressing ferroptosis. We demonstrate that SIRT3 upregulation in the brain underlies the neuroprotective role of JQ1.
Keywords: hypoxic-ischemic encephalopathy, JQ1, SIRT3, ferroptosis, cGAS-STING pathway, oxidative stress
Introduction
Hypoxic-ischemic encephalopathy (HIE) is a brain injury from perinatal asphyxia, occurring in 1–8 per 1000 births in developed nations but with higher mortality in developing regions.1,2 The condition often leads to irreversible neurological deficits, including intellectual disability and epilepsy. Despite therapeutic hypothermia being the sole proven treatment,3,4 its narrow time window necessitates new strategies to improve long-term outcomes.
Epigenetic mechanisms, particularly chemical modifications that modulate gene expression without altering DNA sequence, plays a significant role in the pathogenesis of HIE. The Bromodomain and Extra-Terminal (BET) family (including BRD2, BRD3, BRD4, and BRDT) constitutes crucial epigenetic regulators. Among them, BRD4 is widely expressed in the brain and has been extensively studied. It acts as an epigenetic “reader” by recognizing acetylated histones and recruiting transcriptional complexes.5 (+)-JQ-1 (hereinafter referred to as JQ1) is a BRD4-specific inhibitor with excellent blood-brain barrier penetration and cell membrane permeability.6 While JQ1 is known for its anti-inflammatory and glial cell-modulating effects in other diseases,7–9 its direct neuroprotective potential and therapeutic value in HIE remain unexplored.
Hypoxic-ischemic (HI) injury induces severe mitochondrial dysfunction, disrupts energy metabolism, and triggers excessive production of reactive oxygen species (ROS), ultimately results in neuronal death. Mitochondria are both a major source of ROS and a sensitive target of oxidative damage.10 Excessive ROS accumulation further impairs mitochondrial integrity, creating a vicious cycle. This process exacerbates oxidative stress and promotes ferroptosis—an iron-dependent form of cell death driven by lipid peroxidation.11–13 Ferroptosis is closely associated with dysfunction of the cystine/glutamate antiporter system (system xc⁻), which leads to glutathione (GSH) depletion and glutathione peroxidase 4 (GPX4) inactivation, thereby impairing the clearance of lipid peroxides.
Furthermore, mitochondrial damage can cause leakage of mitochondrial DNA (mtDNA) into the cytoplasm, where it activates the cGAS-STING innate immune pathway. Subsequent TANK-binding kinase 1 (TBK1) phosphorylation and Interferon regulatory factor 3 (IRF3) translocation drive transcription of type I interferons (IFNs) and pro-inflammatory cytokines, exacerbating neuroinflammation.14 Following HIE, the cGAS–STING pathway has been demonstrated to be significantly upregulated.15–17 Notably, JQ1 demonstrates the capacity to suppress cGAS–STING pathway activation and attenuate neuroinflammatory responses.18 Additionally, it may mitigate oxidative stress and inhibit ferroptosis,19 thereby promoting the restoration of mitochondrial function. These mechanisms collectively highlight the potential of JQ1 as a promising neuroprotective strategy for HIE.
Sirtuins are a class of NAD⁺-dependent deacetylases that regulate protein activity by removing acetyl groups from lysine residues. Sirtuin 3 (SIRT3), the primary mitochondrial deacetylase, plays a central role in maintaining mitochondrial homeostasis.20,21 This includes regulating energy metabolism, biogenesis, dynamics, and oxidative stress responses. Research indicates that SIRT3 deficiency leads to elevated acetylation of mitochondrial proteins, reduced activity of various enzymes and protein complexes, and ultimately impaired oxidative phosphorylation. Intriguingly, recent studies suggest that BET inhibition may enhance the accessibility of acetylated histone sites to sirtuin-mediated deacetylation, thereby upregulating SIRT activity.22,23 Based on their potential interplay in gene regulation, we propose that JQ1 may mitigate neuronal injury after HIE by concurrently targeting BET proteins and SIRT3.
In this study, we evaluate the effects of JQ1 on both brain injury and neurological recovery following HIE. By applying JQ1 in a rat Rice-Vannucci model and a neuronal oxygen-glucose deprivation (OGD) model, we demonstrated that JQ1 exerts neuroprotective effects via SIRT3 upregulation, thereby inhibiting the activation of the cGAS-STING signaling pathway and modulating the release of inflammatory cytokines and ferroptosis. Our findings may identify a novel epigenetics-based therapeutic strategy for mitigating neonatal brain injury and improving long-term neurological outcomes.
Materials and Methods
Animals and Treatment
Sprague-Dawley (SD) rats (200–250 g) used in this study were purchased from Zhejiang Laboratory Animal Center. The animals were housed under specific pathogen-free (SPF) conditions with a 12-hour light/dark cycle, provided with free access to food and water. All experimental procedures and protocols (Ethics Approval No: WIUCAS24020501) were approved by the Laboratory Animal Ethics Committee of Wenzhou Institute, University of Chinese Academy of Sciences. A total of 287 neonatal rats were used in this study, with a survival rate of 94.1%.
Healthy 7-day-old male neonatal rats were selected for experiments, excluding singletons and low birth weight individuals. After marking, the pups were randomly assigned to experimental groups. The Rice-Vannucci model was employed as follows: Pups were anesthetized with isoflurane, and the left common carotid artery was exposed, ligated and transected within 5 minutes. After regaining consciousness, the pups were returned to their dams. Two hours later, they were placed in a hypoxia chamber (XBS-03+, AIPU, China) maintained at 37°C with 8% oxygen concentration and 5 L/min gas flow for 2.5 hours. Following hypoxia exposure, all pups were returned to their dams for subsequent experiments. The sham-operated group underwent left carotid artery exposure only, without ligation or hypoxia treatment.
JQ1 (purity 99.80%; CAS No. 1268524-70-4, MedChemExpress, China) was administered intraperitoneally (i.p) at doses of 10, 25, or 50 mg/kg24,25 at 2 hours post-hypoxia, with repeat doses every 24 hours (Figure 1A). For SIRT3 inhibition, HIE group pups received intraperitoneal injection of 3-TYP (10 mg/kg; CAS 120241-79-4, MedChemExpress, China) 2 hours prior to hypoxia exposure.
 |
Figure 1 JQ1 therapy attenuates HI-Induced neuronal damage. (A) The chemical structure of JQ1. (B) Diagrammatic illustration of the HI modeling procedure. (C) Laser speckle contrast image measuring CBF 48h after HIE. (D) Calculation of the difference in rCBF between the bilateral ROI (%), n=9 per group. (E) Representative images of TTC-stained coronal brain sections of rats in the Sham group, HI group, HI+10mg/kg JQ1 group, HI+25mg/kg JQ1 group, and HI+50mg/kg JQ1 group 48h after HIE. (F) Quantification of the coronal brain infarct volume (%), n=5 per group. (G) Quantification of the brain dry-wet ratio (%), n=6 per group. (H) Representative gross morphology of the brains from each group 7 days after HI injury. (I) Quantification of the residual brain volume (%), n=5 per group. (J) Representative image of HE staining in the cortex, hippocampal CA1, CA3, and DG region 7 days after HI injury. The scale bar represents 100μm. (K–N) Quantitative analysis of cell numbers in the CA1, CA3, and DG region and cortex in each group, n=5 per group. Data are expressed as mean ± SD independent replicates: ###P < 0.001 versus Sham group; *P < 0.05, **P < 0.01, ***P < 0.001 versus HI+25mg/kg JQ1 group. |
Laser Speckle Blood Flow Imaging
Cerebral blood flow (CBF) was measured using the RFLSI ZW Laser Speckle Blood Flow Imaging System (RWD Life Technology, China). 48 hours after the surgery, rats were anesthetized with isoflurane. The skin along the midline of the head and neck was incised to fully expose the skull. Rats were positioned prone in a stereotaxic frame, and raw speckle images were captured. During acquisition, the exposed area was kept clean and the environment was maintained warm. Using RFLSI ANALYSIS software (RWD Life Technology, China), left cerebral hemisphere was selected as the region of interest (ROI) to analyze CBF changes. Following measurement, the scalp was sutured, and the pups were returned to their mothers.
Infarct Volume Measurement
Cerebral infarct volume was quantified using 2,3,5-triphenyltetrazolium chloride (TTC) staining. At 48 hours post-surgery, rats from each experimental group were anesthetized, and fresh brain tissues were harvested. Coronal sections (2-mm thickness) were immersed in 2% TTC solution (G3005, Solarbio, China) and incubated at 37°C for 30 minutes with gentle agitation every 10 minutes. Tissues were subsequently fixed in 4% paraformaldehyde for 24 hours. All procedures were performed under light-protected conditions. Fixed tissue slices were sequentially arranged on absorbent pads according to their anatomical orientation and photographed. Infarct areas were analyzed using Image J software (National Institutes of Health, USA). Infarct volume ratio (%) = [(contralateral normal hemispheric volume − ipsilateral normal hemispheric volume) / contralateral normal hemispheric volume] × 100%.
Brain Water Content Measurement
Following established protocols for cerebral edema quantification, rats from each experimental group were deeply anesthetized, and fresh left cerebral hemispheres were rapidly excised. Tissue specimens were immediately weighed to obtain wet weight (WW) using a calibrated analytical balance (±0.1 mg precision). Subsequently, samples were dehydrated in a forced-air drying oven at 65°C for 5 consecutive days until reaching constant mass, recorded as dry weight (DW). The brain water content was calculated using the formula: Brain water content (%) = [(WW - DW) / WW] × 100%.
Histopathological Staining
For histopathological evaluation, rats from each experimental group were transcardially perfused with 4% PFA under deep anesthesia at 48 hours and 7 days post-surgery. Brains were harvested and post-fixed in 4% PFA overnight at 4°C, followed by gradient dehydration, clearing, and paraffin embedding. Coronal sections (5-μm thickness) were cut using a microtome, mounted on adhesive slides, and dried at 60°C for subsequent staining procedures. Following deparaffinization and rehydration, sections were subjected to Hematoxylin and Eosin (HE) Staining (G1120, Solarbio, China) and Nissl Staining (G1120, Solarbio, China). Whole-slide images were acquired using a high-throughput slide scanner (KF-PRO-005-EX, KFBIO, China) equipped with bright-field optics. Regional morphological details were captured under an upright microscope (Eclipse Ni-U, Nikon, Japan). And ImageJ software was used for the analysis.
Immunohistochemistry (IHC) and Immunofluorescence (IF) Staining
Following dewaxing and rehydration, tissue sections underwent antigen retrieval by boiling in citrate buffer (pH 6.0; C1010, Solarbio, China) for 15 min, then cooled to room temperature. For IHC, endogenous peroxidase was blocked with 3% H2O2 (10 min, RT). All sections were blocked with 10% goat serum (SL038, Solarbio, China) at 37°C for 1 h, then incubated with primary antibodies (listed in Table S1) at 4°C overnight. The next day, sections were treated with either HRP-conjugated (for IHC) or fluorophore-conjugated (for IF) secondary antibodies at 37°C for 1 h. IHC-stained sections were developed with DAB and mounted with neutral resin, while IF-stained sections were covered using antifade mounting medium with DAPI (P0131-5mL, Beyotime, China). Images were acquired with an optical microscope and analyzed using ImageJ software.
Western Blot Analysis
Cells/tissues were lysed in RIPA buffer containing 1% PMSF (ST506, Beyotime, China) and 1% phosphatase inhibitors (P1260, Solarbio, China) on ice. Protein concentrations were determined using a BCA assay kit (ZJ102, Epizyme, China). Equal amounts of protein (20–40μg) were separated by SDS-PAGE and transferred onto 0.22-μm PVDF membranes (Millipore, USA). After blocking with 5% skim milk for 2 h at room temperature, the membranes were incubated with primary antibodies (listed in Table S1) overnight at 4°C. Following TBST washes, HRP-conjugated secondary antibodies were applied for 1 h. Blots were visualized using an ECL Plus chemiluminescence reagent kit (P10060, NCM Biotech, China). β-Actin was used as a loading control, while the phosphorylation levels of target proteins were calculated by normalizing the signal intensity of each phosphorylated form to that of its corresponding total (non-phosphorylated) protein. And band intensities were quantified with ImageJ software.
Behavioral Tests
All rats were treated to several behavioral tests 40 days after operation, including the Open-field test (OFT) and the Morris water maze (MWM).26
For OFT, the rats were placed in the testing cage for a 2-hour acclimation period prior to experimentation. Following this, each rat began testing at the midpoint of a 100cm×100cm×50cm black square arena, and all movement trajectories within 5 minutes were tracked. Quantitative analysis focused on three key metrics: central zone crossing frequency, the central time ratio (calculated as [central region duration/300s] × 100%), and the central distance ratio (derived from [central path distance/total path distance] × 100%). After each test, the arena was cleaned with 75% ethanol.
The MWM was consisted of a circular pool (1.6 m diameter, 22±1°C) containing a hidden platform in one quadrant, with visual cues positioned around the apparatus.27 During the 5-day training phase, each rat underwent four daily trials with randomized starting quadrants. Escape latency (time to reach the platform) was measured for each trial. Rats failing to locate the platform within 60s were gently guided to it and assigned 60s latency. On day 6, the platform was removed for probe testing. Rats were released from the opposite quadrant, and their swimming trajectories within 60s were tracked: (1) initial escape latency, (2) time/distance ratio in the target quadrant, and (3) platform crossing frequency. Between trials, animals were towel-dried and maintained in a temperature-controlled recovery area.
RNA Sequencing (RNA-Seq)
Total RNA was isolated from rat cerebral cortex tissues, quantified using NanoDrop-2000 (Thermo Fisher, USA). RNA purity was verified by A260/A280 (1.8–2.0) and A260/A230 (>2.0) ratio. The ligated products underwent PCR amplification and the final complementary DNA (cDNA) was screened for 300±50 bp fragments. Following library construction, paired-end sequencing of 150-bp fragments was performed on the Illumina Novaseq™ 6000 Sequencing Platform (LC-Bio Technology, China).
Quantitative Real-Time PCR (qRT-PCR)
The total RNA was extracted from rat tissues and HT22 cells at specific time points post-modeling using TRIzol Reagent (Invitrogen, USA). Then, equal amounts of RNA (1μg) were reverse-transcribed into cDNA using the PrimeScript RT Master Mix kit (Takara Bio Inc., Japan). Quantitative real-time PCR (qRT-PCR) was performed on the CFX96 Real‐Time PCR system (Bio-Rad, USA) with the TB Green Premix Ex Taq II kit (Takara Bio Inc., Japan) following the manufacturer’s instructions. Cycle threshold (Ct) values were recorded and normalized to β-actin levels. The relative mRNA expression levels of target genes were further calculated using the 2−ΔΔCt method. Primers were designed via the NCBI Primer- Blast tool, with specific forward and reverse primers listed in Table S2.
Assessment of Oxidative Stress and Ferrous Ion (Fe2⁺) Levels
Oxidative stress parameters and ferrous ion content were quantitatively analyzed using standardized commercial assay kits. Malondialdehyde (MDA) levels were determined using the Lipid Peroxidation MDA Assay Kit (S0131M, Beyotime, China), while glutathione peroxidase (GPx) activity was measured with the Cellular Glutathione Peroxidase Assay Kit with DTNB (S0057S, Beyotime, China). Ferrous ion (Fe2⁺) content was quantified using the Ferrous Ion Content Assay Kit (BC5415, Solarbio, China).
Briefly, at 48 hours post-HI, the injured cerebral hemisphere was homogenized and centrifuged. The supernatant was collected, and the assays were performed according to the manufacturers’ instructions.
Transmission Electron Microscopy
To directly examine the ultrastructural morphology of mitochondria, transmission electron microscopy (JEM-1400, JEOL, Japan) was employed. Briefly, brain cortex samples from the injured side of rats were dissected and immediately immersed in 2.5% glutaraldehyde for primary fixation at room temperature for 2 hours, followed by further fixation in the same solution at 4°C overnight. The following day, samples were rinsed with 1% osmium tetroxide for post-fixation, subjected to gradient dehydration using acetone, infiltrated with Epon 812 resin, embedded, and sectioned. Ultrathin sections were stained with uranyl acetate and lead citrate, and subsequently examined under transmission electron microscopy to assess mitochondrial morphology within the cortex.
Cell Culture and Treatment
HT22 cells were purchased from the Cell Bank of Chinese Academy of Sciences (Shanghai, China). They were cultured in high-glucose DMEM (Gibco, USA) containing 10% FBS (Gibco, USA) and 1% penicillin/streptomycin (NCM Biotech, China) at 37°C in a humidified incubator with 5% CO2.
To simulate the in vitro pathology of HIE, we established an oxygen-glucose deprivation (OGD) model. After 12 hours of adherent culture, the HT22 cells were washed with phosphate-buffered saline and incubated in glucose-free DMEM (Gibco, USA). Subsequently, they were exposed to a hypoxic environment (1% O2, 5% CO2, and 94% N2) in a tri-gas chamber (Thermo Fisher, USA) for specified durations. Following OGD, the cells were returned to the standard culture conditions for 2 hours before sample collection.
Throughout the OGD experiment, the JQ1 treatment group received 500nM JQ1 dissolved in glucose-free medium.6,28,29 Control cells were maintained under normoxic conditions until all experimental procedures were completed. To inhibit STING, the cells were pre-stimulated with 1 µM C-176 (HY-112906, MedChemExpress, China) before cultured in hypoxic condition.30
Cell Viability Assay
Cell viability was assessed using the Cell Counting Kit-8 (CCK8; NCM Biotech, China). HT22 cells were seeded in 96-well plates at a density of 1 × 104 cells per well. To evaluate the potential cytotoxicity of JQ1, various concentrations of the compound were administered to the cells prior to OGD treatment. Following hypoxia induction, 10% (by volume) CCK8 solution was added according to the manufacturer’s instructions. The cells were incubated at 37 °C for 2 hours in the dark. The optical density at 450 nm was measured using a microplate reader (Varioskan LUX, Thermo Fisher, USA).
Short-Interfering RNA (siRNA) Transfection
The knockdown efficiency of mouse-specific SIRT3 siRNAs (GenePharma, China) was validated by Western blot and qRT-PCR. The most effective sequence was selected from three designed candidates (listed in Table S3) for subsequent experiments. When HT22 cells reached 60% confluence during stable adherent growth, they were transfected with si-SIRT3 or control siRNA using Lipofectamine 3000 transfection kit (Invitrogen, USA) and Opti-MEM (Gibco, USA), according to the manufacturer’s protocols. After 8 hours, the medium was replaced with normal culture medium, followed by an additional 24 hours of incubation before further modeling and drug treatment.
Measurement of ROS
The intracellular ROS levels were detected using a Reactive Oxygen Species Assay Kit (S0033S, Beyotime, China). According to the manufacturer’s instructions, the HT22 cells were incubated with DCFH-DA (10μM) and Hoechst at 37°C in the dark for 20 minutes. Then, the cells were washed three times with serum-free DMEM. Images were captured using a fluorescence inverted microscope (Keyence, China), and the fluorescence intensity was quantified using Image J software. For flow cytometry analysis, cells were first incubated with 10μM DCFH-DA for staining, followed by washing. After digestion and collection, the labeled cells were detected using a flow cytometer (CytoFLEX LX, Beckman, USA), and data were analyzed with FlowJo software.
Mitochondrial Fluorescence Staining
According to the manufacturer’s instructions, Mito-Tracker Green (C1048, Beyotime, China) and Hoechst 33342 (C1028, Beyotime, China) were diluted in fresh culture medium to prepare working solutions. After thorough mixing, the solution was added to the cells. The cells were then incubated at 37 °C for 20 minutes under light-protected conditions. Following washing, images were acquired using a confocal laser scanning microscope (STELLARIS 5, Leica, Germany).
Statistical Analysis
All statistical analyses were conducted using GraphPad Prism 8.0 (GraphPad software, USA). Data were obtained from at least three independent experiments and were presented as mean ± SD. Normality was assessed using the Shapiro–Wilk test, and homogeneity of variance was evaluated with Levene’s test. For comparisons between two groups, Student’s t-test was applied, whereas one-way ANOVA was used for multi-group comparisons. If the data deviated from normal distribution, nonparametric tests were employed. A p value < 0.05 was considered statistically significant.
Results
JQ1 Attenuates Acute Injury After HIE
To validate successful model establishment (Figure 1B), CBF was dynamically monitored using laser speckle contrast imaging. At 48 hours post operation, the left hemisphere (ipsilateral to carotid ligation) showed significantly reduced perfusion compared to the contralateral side (Figure 1C and D). Quantitative analysis of cerebral infarction volume with TTC staining revealed that all three doses of JQ1 (10,25, and 50mg/kg) provided neuroprotective effects (Figure 1E and F). Among them, 25 mg/kg most significantly reduced infarction volume and was therefore selected for further study. JQ1 treatment also markedly alleviated cerebral edema, as shown by the wet-dry weight ratio of the injured hemisphere (Figure 1G). Together, these findings suggest that JQ1 provides acute-phase protection after HIE, an effect could be partially reversed by the SIRT3 inhibitor 3-TYP.
JQ1 Treatment Mitigated Brain Structural Damage Following HI Injury
Seven days post HI, macroscopic examination and quantitative results of residual brain volume revealed cerebral atrophy in the left hemisphere of HI-induced rats, which was alleviated by JQ1 administration (Figure 1H and I). HE staining revealed two key pathological changes. It indicated inflammatory cell infiltration and disrupted cellular arrangement in the cerebral cortex and hippocampal CA1, CA3, and DG regions (Figure 1J–N). Nissl staining showed a marked reduction and near disappearance of Nissl bodies (Figure S1). JQ1 treatment reduced neuronal loss and promoted morphological recovery after injury, whereas 3-TYP exacerbated somatic swelling.
IHC and Western blot analysis demonstrated decreased expression of Microtubule-associated protein-2 (MAP-2) and Myelin basic protein (MBP) following HI. Significant MAP2 loss occurred in the cortex and hippocampal CA3 region, while MBP reduction was observed in the corpus callosum and striatum (Figure 2A–F). JQ1 partially restored cortical MAP2 and preserved MBP in both areas, suggesting a role in stabilizing microtubule function and promoting long-term myelination. Additionally, HI injury led to reduced Post-Synaptic Density Protein 95 (PSD95) expression and increased glial fibrillary acidic protein (GFAP), indicating impaired synaptic plasticity and astrocyte activation, which impeded recovery (Figure 2G–K). JQ1 counteracted these pathological changes, whereas 3-TYP exerted opposite effects.
 |
Figure 2 JQ1 promotes neuronal structural integrity after HIE. (A) The IHC staining of MAP-2 in brain cortex and hippocampal CA3 region 7 days after HI injury. The scale bar represents 100μm. (B) The IHC staining of MBP in brain callosum and striatum region 7 days after HI injury. The scale bar represents 100μm. (C and D) Quantitative analysis of MAP-2-positive area in the corresponding brain regions, n=5 per group. (E and F) Quantitative analysis of MBP-positive area in the corresponding brain regions, n=5 per group. (G) Western blot analysis of the protein levels of MAP-2, MBP, PSD95, and GFAP 7 days after HI injury. (H–K) Quantification of the relative protein levels of MAP-2, MBP, PSD95, and GFAP (normalized to β-Actin), n=5 per group. Data are expressed as mean ± SD independent replicates: ##P < 0.01, ###P < 0.001 versus Sham group; *P < 0.05, **P < 0.01, ***P < 0.001 versus HI+JQ1 group. |
JQ1 Ameliorates Anxiety-Like Behavior and Cognitive Deficits in HIE Rats
To assess the long-term effects of JQ1 pretreatment on emotion and cognitive functions in rats post-operatively, we performed open-field test and Morris water maze tests.31,32
The OFT was employed to evaluate the impact of JQ1 on anxiety-like behaviors induced by HI injury. As shown in the Figure 3A–D, the HI group rats displayed more corner-seeking behavior, marked by reduced crossings into the central zone, decreased central movement distance, and significantly less time spent in the center. In contrast, treatment with JQ1 effectively ameliorated these deficits, promoting exploratory behavior and reducing depression-like tendencies. However, administration of 3-TYP resulted in further deterioration of open-field performance.
 |
Figure 3 JQ1 ameliorates anxiety-like behavior and cognitive deficits in HIE rats. (A) Representative image of route traces in OFT. (B) Quantitative analysis of mean crossing frequency in OFT, n=6 per group. (C) Quantitative analysis of central area duration (%) in OFT, n=6 per group. (D) Quantitative analysis of central distance ratio (%) in OFT, n=6 per group. (E) Representative swim route traces of rats from each group during the MWM acquisition phase. (F) Quantitative analysis of mean escape latency(s) during the MWM acquisition phase, n=8 per group. (G) Quantitative analysis of mean escape latency(s) during the MWM probe trial, n=8 per group. (H) Representative swim route traces of rats from each group during the MWM probe trial. (I) Quantitative analysis of platform crossing frequency, n=8 per group. (J) Quantitative analysis of platform quadrant duration (%), n=8 per group. (K) Quantitative analysis of platform quadrant distance ratio (%), n=8 per group. Data are expressed as mean ± SD independent replicates: ##P < 0.01, ###P < 0.001 versus Sham group; *P < 0.05, **P < 0.01, ***P < 0.001 versus HI+JQ1 group; &&P < 0.01, &&&P < 0.001 versus HI+JQ1+3-TYP group. |
During the MWM acquisition phase, HI rats showed prolonged escape latency to locate the submerged platform, reflecting impaired spatial learning (Figure 3E and F). JQ1 treatment notably improved this deficit, whereas co-administration of 3-TYP attenuated this improvement. In the subsequent probe trial, there was no significant difference in average swimming speed between groups (Figure S2A). Sham group rats exhibited efficient spatial memory retention through accurate quadrant navigation and frequent platform location crossings (Figure 3G–I). Conversely, HI rats showed marked spatial memory impairment, characterized by decreased swimming distance and duration in target quadrant (Figure 3J and K). While JQ1 therapy provided partial cognitive restoration, this beneficial outcome was abolished by 3-TYP co- treatment.
Differential Gene Expression in the Rat Cortex Following HIE
To investigate the mechanism underlying the neuroprotective effects of JQ1, RNA-seq was performed on cortex samples from the Sham, HI, and HI+JQ1 groups. The analysis identified a subset of differentially expressed genes (DEGs) that were changed in response to HI injury and exhibited reversed expression patterns following JQ1 treatment (Figure 4A–D). Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses of these genes indicated associations with inflammatory response, oxidative stress, and activation of the cGAS-STING signaling pathway (Figure 4E and F). Western blot analysis further confirmed these findings. It showed increased Cyclic Gmp–Amp Synthase (CGAS) and Stimulator Of Interferon Genes (STING) protein level at 48 and 72 hours after HI injury (Figure 4G–I), consistent with the transcriptomic data and previously published studies.
 |
Figure 4 Enrichment analysis of DEGs associated with HI and JQ1 treatment. (A) Volcano plot of DEGs between the HI and Sham groups, n=3 per group. (B) Volcano plot of DEGs between the HI+JQ1 and HI groups, n=3 per group. (C and D) Venn diagram depicting intersecting genes of HI+JQ1, HI, and Sham groups. (E) The bubble chart of KEGG enrichment based on DEGs. (F) The bubble chart of GO enrichment based on DEGs. Red boxes highlight key biological pathways enriched in the analysis, including reactive oxygen species metabolic process, cGAS/STING signaling pathway and immune receptor activity. (G) Western blot analysis of the protein levels of cGAS and STING at 24, 48, and 72 hours after HI injury. (H and I) Quantification of the relative protein levels of cGAS and STING (normalized to β-Actin), n=5 per group. (J–L) The mRNA expression of the inflammatory factors IL1β, IL6, and TNFα (normalized to β-Actin) in the brain tissues 48h after HIE, n=5 per group. Data are expressed as mean ± SD independent replicates: #P < 0.05, ##P < 0.01, ###P < 0.001 versus Sham group; *P < 0.05, **P < 0.01 versus HI+JQ1 group. |
JQ1 Confers Protection Against HI-Induced Inflammation and Oxidative Stress
qRT-PCR analysis of key inflammatory markers revealed a robust acute inflammatory response after HI injury, characterized by significantly elevated mRNA expression of IL-6, IL-1β, and TNF-α (Figure 4J–L). In line with our transcriptome sequencing results, JQ1 treatment effectively downregulated HI-induced expression of these inflammatory biomarkers in rat brain tissue. However, this anti-inflammatory effect was partially abrogated by co-administration of the SIRT3 inhibitor 3-TYP.
Compared to the Sham group, HI injury significantly increased the levels of malondialdehyde (MDA), a marker of oxidative stress, while inhibiting the activity of GPx, a key antioxidant enzyme. However, treatment with JQ1 substantially mitigated oxidative damage by reducing MDA formation and restoring GPx activity (Figure 5A and B). Together, these findings demonstrate that JQ1 confers dual protective effects against both neuroinflammation and oxidative stress in HI-injured brain tissue, thereby promoting cellular homeostasis. Again, the beneficial effects of JQ1 were partially reversed by 3-TYP, further supporting the involvement of SIRT3 in mediating its neuroprotective functions.
 |
Figure 5 JQ1 suppresses oxidative stress and ferroptosis processes. (A) Quantitative analysis of MDA levels in the brain tissues 48h after HIE, n=5 per group. (B) Quantification of the relative GPx activity in the brain tissues 48h after HIE, n=6 per group. (C and D) Quantification of the relative protein levels of SLC7A11 and SLC3A2 (normalized to β-Actin), n=5 per group. (E) Representative Western blot images of TFR1, 4-HNE, SLC7A11, SLC3A2, and GPX4 expression 48h after HIE. (F) Quantification of the relative protein levels of GPX4 (normalized to β-Actin), n=5 per group. (G and H) The mRNA expression of the ferroptosis factors SLC7A11 and GPX4 (normalized to β-Actin) in the brain tissues 48h after HIE, n=5 per group. (I) Quantification of the relative protein levels of 4-HNE (normalized to β-Actin), n=5 per group. (J) Quantitative analysis of Fe2⁺ in the brain tissues 48h after HIE, n=5 per group. (K) Quantification of the relative protein levels of TFR1 (normalized to β-Actin), n=5 per group. (L) Representative images of transmission electron microscope discover mitochondria damage (red arrows). The scale bar represents 1μm in the up image and 200nm in the magnification image. Data are expressed as mean ± SD independent replicates: ###P < 0.001 versus Sham group; *P < 0.05, **P < 0.01, ***P < 0.001 versus HI+JQ1 group. |
JQ1 Alleviates Ferroptosis After HIE
The system Xc⁻ complex, composed of SLC7A11 and SLC3A2, facilitates the uptake of extracellular cystine necessary for glutathione synthesis.33 GSH is then utilized by glutathione peroxidase 4 (GPX4) to detoxify lipid peroxides (eg, PL-OOH) into harmless lipid alcohols (PL-OH), thereby directly suppressing ferroptosis. Following HI injury, both protein and mRNA expression levels of SLC7A11, SLC3A2, and GPX4 were significantly downregulated (Figure 5C–H). We also observed elevated levels of 4-hydroxynonenal (4-HNE), a toxic end product of lipid peroxidation, indicating pronounced ferroptotic activity (Figure 5I). Compared to the Sham group, the HI group exhibited elevated iron levels and dysregulated iron metabolism in brain tissue, as detected by ferrous ion assay (Figure 5J). Furthermore, increased transferrin receptor 1 (TFR1) expression enhanced cellular iron import, aggravating iron overload and oxidative damage (Figure 5K). JQ1 treatment effectively restored the expression of these key anti-ferroptotic components, enhanced system Xc⁻ activity, attenuated GPX4 dysfunction, and reduced iron accumulation. Conversely, co-treatment with the SIRT3 inhibitor 3-TYP further aggravated transcriptional downregulation of system Xc⁻ subunits and worsened ferroptotic damage.
We also employed transmission electron microscopy (TEM) to examine neuronal mitochondrial morphology. As shown in Figure 5L, the HI group displayed shrunken mitochondria, increased membrane density, and loss of cristae. In contrast, JQ1 treatment notably ameliorated HI-induced mitochondrial fragmentation and maintained relatively intact ultrastructure in neurons, exhibiting only mild swelling and slightly blurred yet discernible cristae. These results demonstrated that JQ1 protects neurons from ferroptosis and mitochondrial structural damage after HIE.
JQ1 Negatively Regulates the cGAS-STING Signaling Pathway: SIRT3 Was Identified as the Key Mediator
Western blot and qRT-PCR analyses demonstrated that, compared to the HI group, JQ1 treatment significantly up-regulated expression levels of SIRT3 and Mn-SOD, while suppressing the expression of cGAS and STING (Figure 6A–E, Figure S2B–D). Subsequently, HI injury markedly induced phosphorylation of the downstream signaling molecule IRF3 (Figure 6F and G). JQ1 administration effectively suppressed this activation. To further investigate the neuroprotective effects of JQ1, we conducted immunofluorescence co-staining for Neuronal Nuclei (NeuN) along with cGAS and STING. Prominent neuronal damage was observed in the CA3 region and cortex following HI, manifested as somatic shrinkage and synaptic loss (Figure 6H–K). JQ1 treatment effectively alleviated these morphological deficits and concurrently reduced the activation of both STING and CGAS (Figure S2E–H). These protective effects were reversed upon co-treatment with the SIRT3 inhibitor 3-TYP.
 |
Figure 6 JQ1 exerts a protective effect through modulation of the cGAS-STING signaling pathway. (A) Representative Western blot images of SIRT3, Mn-SOD, cGAS, STING, p-IRF3, and IRF3 expression 48h after HIE. (B–E) Quantification of the relative protein levels of SIRT3, Mn-SOD, cGAS, and STING (normalized to β-Actin), n=5 per group. (F) Quantification of the relative protein levels of p-IRF3 (normalized to IRF3), n=5 per group. (G) Quantification of the relative protein levels of IRF3 (normalized to β-Actin), n=5 per group. (H and I) Representative images of immunofluorescence staining for STING (green), NeuN (red), and DAPI (blue) in the cortex and hippocampal CA3 of each group 48h after HIE. The scale bar represents 100μm. (J and K) Representative images of immunofluorescence staining for cGAS (green), NeuN (red), and DAPI (blue) in the cortex and hippocampal CA3 of each group 48h after HIE. The scale bar represents 100μm. Data are expressed as mean ± SD independent replicates: ns: not significant; ###P < 0.001 versus Sham group; *P < 0.05, **P < 0.01, ***P < 0.001 versus HI+JQ1 group. |
Ionized calcium-binding adapter molecule 1 (Iba1) is a marker of activated microglia, while GFAP indicates reactive astrogliosis.34,35 To evaluate SIRT3 expression across neuronal and glial cells, we performed multiplex immunofluorescence staining co-labeling SIRT3 with NeuN, Iba1, and GFAP (Figure 7A–F and Figure S2I). The results demonstrated elevated expression of Iba1 and GFAP in the cerebral cortex after HI injury, suggesting neuroinflammatory responses and repair processes. Multiplex immunofluorescence further revealed an overall reduction in SIRT3 expression following HIE. The spatial correlation between SIRT3 and Iba1⁺ microglia is increased, while its correlation with neurons (NeuN⁺ cells) is significantly decreased. In contrast, no significant colocalization of SIRT3 was detected with astrocytes (GFAP⁺ cells). Nevertheless, the precise relationship between SIRT3 and glial cells still require further validation. Treatment with JQ1 effectively attenuated the loss of SIRT3 fluorescence signal and improved neuronal integrity, as reflected by more preserved NeuN morphology in the hippocampal CA3 region after HI injury (Figure 7G and H). Consistent with our earlier Western blot findings, these results indicated that JQ1 restored SIRT3 expression, an effect that was blocked by co-administration of 3-TYP.
 |
Figure 7 SIRT3 is identified as the key mechanism underlying JQ1’s therapeutic effects. (A and B) Representative images of immunofluorescence staining and corresponding statistical analysis for SIRT3 (green), Iba1 (red), and DAPI (blue) in the cortex of each group 48h after HIE. The scale bar represents 100μm in the left and 50μm in the magnification image. (C) Quantification of the Pearson’s correlation coefficient for SIRT3-Iba1 colocalization. (D and E) Representative images of immunofluorescence staining and corresponding statistical analysis for SIRT3 (green), NeuN (red), and DAPI (blue) in the cortex of each group 48h after HIE. The scale bar represents 100μm in the left and 50μm in the magnification image. (F) Quantification of the Pearson’s correlation coefficient for SIRT3- NeuN colocalization. (G and H) Representative images of immunofluorescence staining and corresponding statistical analysis for SIRT3 (green), NeuN (red), and DAPI (blue) in the hippocampal CA3 of each group 48h after HIE, n=5 per group. The scale bar represents 100μm. Data are expressed as mean ± SD independent replicates: #P < 0.05, ###P < 0.001 versus Sham group; ***P < 0.001 versus HI+JQ1 group. |
In summary, we propose that JQ1 upregulates SIRT3 expression, and negatively regulates the CGAS-STING signaling pathway, thereby attenuating neuroinflammation following HI brain injury.
JQ1 Reduces OGD-Induced Cell Injury in HT22 Cells
To simulate the pathophysiology of HIE in vitro, an OGD model was established in HT22 cells. CCK-8 assays indicated a significant time-dependent decrease in cell viability after OGD, demonstrating its detrimental effects on neuronal survival (Figure S3A). Following 9 hours of OGD, cell viability decreased by nearly half, accompanied by characteristic morphological alterations such as cellular shrinkage and membrane rounding; this time point was therefore selected for subsequent experiments (Figure S3B). Western blot analysis showed that OGD downregulated SIRT3 protein expression and upregulated cGAS and STING, suggesting activation of DNA damage response pathways (Figure 8A–C).
 |
Figure 8 JQ1 alleviates OGD-induced mitochondrial impairment in HT22 cells. (A and B) Representative Western blot images and quantification of SIRT3, cGAS, and STING expression (normalized to β-Actin) in HT22 cells, n=6 per group. (C) The mRNA expression of SIRT3 (normalized to β-Actin) in HT22 cells, n=5 per group. (D) Relative fluorescence intensity of DCFH-DA staining in HT22 cells of each group, n=5 per group. (E) The ROS generation of HT22 cells was detected by DCFH-DA (green) staining. Scale bar = 100μm. (F) Representative data showing ROS production by flow cytometry. (G and H) Representative Western blot images and quantification of SIRT3 expression (normalized to β-Actin) in HT22 cells, n=5 per group. (I) The mRNA expression of SIRT3 (normalized to β-Actin) in HT22 cells, n=5 per group. (J) Representative images of Mito-Tracker Green probe mitochondrial imaging, the scale bar represents 5μm. Data are expressed as mean ± SD independent replicates: ##P < 0.01, ###P < 0.001 versus Control group; ***P < 0.001 versus OGD+JQ1 group; @@P < 0.01, @@@P < 0.001 versus si-Nc group; &P < 0.05, &&P < 0.01, &&&P < 0.001 versus si-Sirt3-2 group. |
In control groups without OGD, JQ1 at concentrations of 500, 1000, and 1500nM showed no significant cytotoxicity toward HT22 cells, whereas doses above 2000nM progressively impaired cell survival (Figure S3C). Based on these results and previous studies, 500 nM JQ1 was chosen for all subsequent interventions and was applied concomitantly with OGD exposure. ROS represent a class of highly potent oxygen-containing oxidants. Both excessive and insufficient ROS levels are deleterious and contribute to pathology resulting from the dramatic change of redox environment. Under OGD conditions, we observed a substantial increase in ROS production, as quantified by the DCFH-DA fluorescence assay. This elevated oxidative burden reflects a disruption of cellular redox homeostasis and is indicative of pro-inflammatory activation. JQ1 treatment attenuated ROS generation and reduced oxidative stress (Figure 8D–F).
Furthermore, Western blot analysis showed that OGD downregulated key ferroptosis-related proteins, while JQ1 administration restored system Xc⁻ function and enhanced GPX4 activity. Collectively, these findings demonstrate that JQ1 effectively alleviates OGD-induced oxidative stress in HT22 cells, likely through mechanisms involving the regulation of ferroptosis.
SIRT3 Mediates the Protective Effects of JQ1 Against CGAS/STING Activation and Ferroptosis Following OGD
To further investigate whether SIRT3 mediates the protective role of JQ1, we transfected cells with siRNA targeting SIRT3 and evaluated its impact on downstream processes. The transfection efficiency was confirmed by Western blot and qRT-PCR, with siRNA2 showing the most significant SIRT3 knockdown; it was therefore selected for subsequent experiments (Figure 8G–I). Morphological analysis of mitochondria stained with Mito-Tracker revealed that under normal conditions, mitochondria displayed an elongated and interconnected architecture. In contrast, OGD exposure resulted in severe mitochondrial damage, characterized by pronounced fragmentation and shrinkage. JQ1 administration markedly ameliorated these alterations, preserving mitochondrial integrity. However, the beneficial effects of JQ1 were abolished in SIRT3-knockdown groups, indicating that SIRT3 is critically involved in the protective mechanism (Figure 8J).
Western blot results showed that JQ1 treatment upregulated SIRT3 expression while concurrently suppressing activation of the CGAS-STING pathway. Conversely, knockdown of SIRT3 elevated the protein levels of both CGAS and STING (Figure 9A and Figure S3D–F). These findings were supported by immunofluorescence staining: signal intensity of both molecules was enhanced in the cytoplasm following SIRT3 silencing (Figure 9B, C, and Figure S3G–H). Furthermore, Western blot analyses demonstrated that SIRT3 knockdown partially reversed the protective effects of JQ1 against OGD-induced ferroptosis, as evidenced by reduced expression of GPX4, SLC7A11, and SLC3A2 (Figure 9D–G).
 |
Figure 9 SIRT3 Mediates the Protective Effects of JQ1 Against CGAS/STING Activation and Ferroptosis Following OGD. (A) Representative Western blot images of SIRT3, cGAS, and STING expression in HT22 cells. (B) Representative images of immunofluorescence staining for STING (green) and DAPI (blue) in HT22 cells of each group. Scale bar = 25μm. (C) Representative images of immunofluorescence staining for cGAS (green) and DAPI (blue) in HT22 cells of each group. Scale bar = 25μm. (D–G) Representative Western blot images and quantification of SLC3A2, SLC7A11, and GPX4 expression (normalized to β-Actin) in HT22 cells. (H–K) Representative Western blot images and quantification of STING, SLC7A11, and GPX4 expression (normalized to β-Actin) in HT22 cells. Data are expressed as mean ± SD independent replicates: ns: not significant; ###P < 0.001 versus Control group; *P < 0.05, **P < 0.01, ***P < 0.001 versus OGD+JQ1+si-NC group; @P < 0.05, @@P < 0.01, @@@P < 0.001 versus OGD +C-176 group; &P < 0.05 versus OGD+JQ1 group. |
To further confirm the involvement of the cGAS‑STING pathway in mediating the anti‑ferroptotic effects of JQ1, C‐176 (STING inhibitor) were used to treat HT22 cells. The STING level in the OGD+JQ1+C‑176 group did not differ significantly from that in the OGD+C‑176 group, but it was lower than in the OGD+JQ1 group. This indicates that JQ1 and C‑176 synergistically suppress STING expression. Both JQ1 and C‑176 rescued the OGD‑induced down‑regulation of GPX4 and SLC7A11. JQ1 showed a stronger effect than C‑176 in up‑regulating GPX4, and the combination of the two agents yielded a greater rescue than C‑176 alone (Figure 9H–K).
Collectively, these results indicate that JQ1 modulates neuronal inflammation and ferroptosis via the cGAS‑STING pathway, and that SIRT3 serves as a critical negative regulator of this axis (Figure 10).
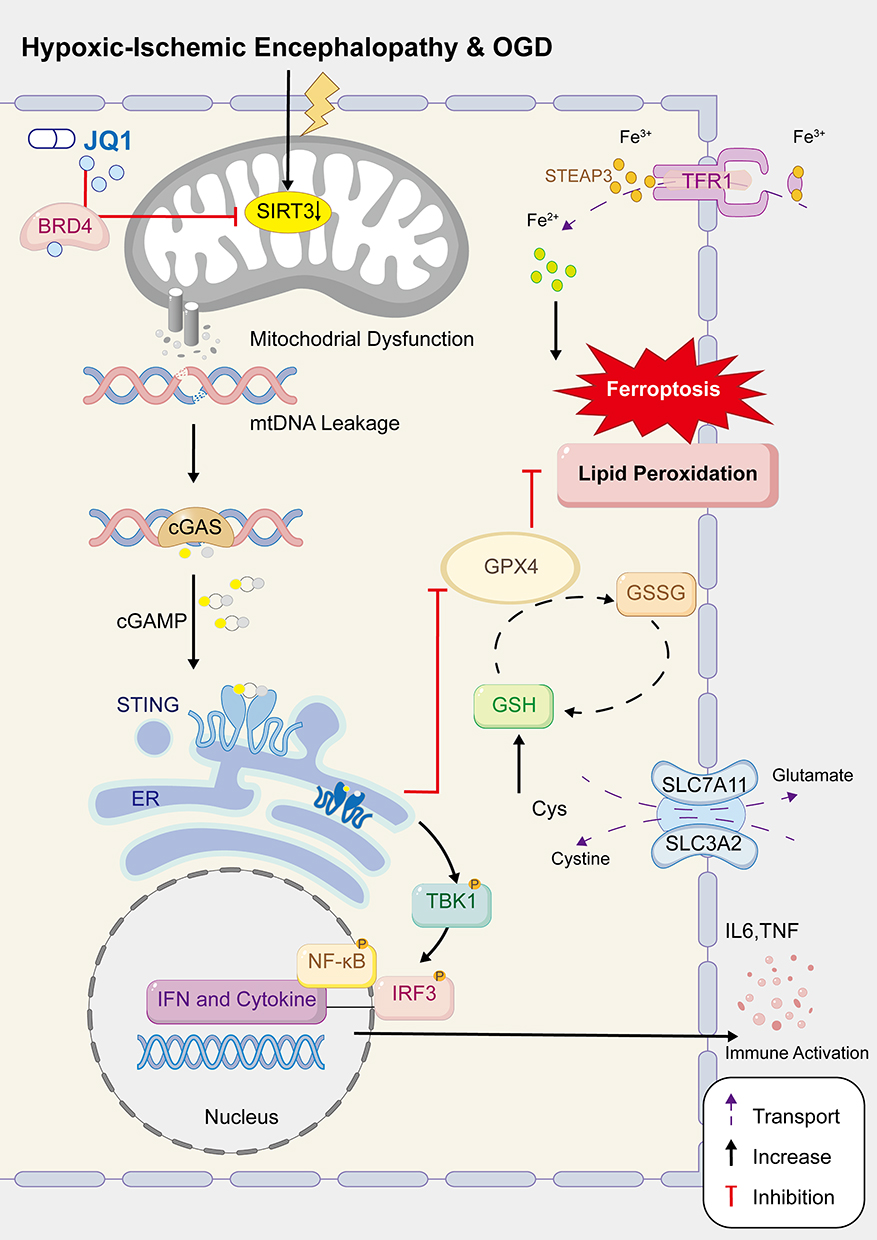 |
Figure 10 Schematic model for the mechanism of JQ1 in HIE. |
Discussion
In this study, we systematically evaluated the neuroprotective effects of the BET inhibitor JQ1 against HIE at both cellular and whole-animal levels. We demonstrated that JQ1 exerts neuroprotection primarily through upregulating SIRT3 expression—an effect supported epigenetically by a significant increase in SIRT3 mRNA. Conversely, these protective effects were abolished upon SIRT3 inhibition or siRNA-mediated knockdown, confirming the essential role of SIRT3 in JQ1-mediated neuroprotection.
Experimental results demonstrated that 48 hours after HIE induction, the injured brain regions exhibited significant cerebral infarction and typical ischemic histopathological alterations. JQ1 treatment markedly reduced cerebral infarct volume and alleviated pathological damage in both the cortex and hippocampus. By day 7 post-injury, the HI group showed pronounced cerebral atrophy, inflammatory cell infiltration, and loss of Nissl bodies, which were substantially reversed by JQ1 administration. Furthermore, JQ1 treatment significantly attenuated the loss of NeuN, MBP, PSD95, and MAP2, while also downregulating the expression of GFAP. These findings collectively confirm the acute neuroprotective role of JQ1 in HIE.
Several studies have reported that BET inhibitors can improve cognitive function. For instance, JQ1 has been shown to enhance brain plasticity,36 ameliorate cognitive deficits, and rescue hippocampal-dependent learning and memory impairments.37 However, other evidence suggests that JQ1 may adversely affect prefrontal cortex development and disrupt neuronal transcription and memory formation.38 In our study, JQ1-treated HIE rats exhibited reduced anxiety-like behaviors and increased exploratory activity in the open field test during adolescence. Moreover, the Morris water maze test revealed significant improvements in spatial learning and memory compared to the HI group, indicating that JQ1 contributes to the preservation of neurocognitive function after HIE, particularly in the acquisition and retrieval of spatial memory.
The pathological progression of HIE often involves neuroinflammation, oxidative stress, apoptosis, autophagy, and ferroptosis,39,40 accompanied by significant activation of microglia and astrocytes, as well as neuronal loss. In line with these established mechanisms, our study specifically focused on neuronal alterations under disease conditions. We confirmed that post-HIE, there was a marked activation of glial cells (evidenced by increased IBA1 and GFAP expression) alongside neuronal injury and a concomitant decrease in SIRT3 expression within neurons. Furthermore, we preliminarily observed an apparent increase in SIRT3 expression in IBA1-positive cells, although this particular finding requires further validation. High-throughput transcriptomic sequencing of cortical tissue revealed numerous differentially expressed genes after HIE, a subset of which were normalized by JQ1 treatment. Enrichment analysis indicated that these genes are primarily involved in biological processes such as inflammatory response, oxidative stress, glutathione metabolism, and the cGAS–STING signaling pathway, suggesting that JQ1 may mitigate neuronal damage through modulation of these pathways.
The cGAS–STING pathway has recently garnered attention as a crucial innate immune signaling cascade, activated in a cGAS-dependent manner by micronuclei, mitochondrial DNA, aberrant cell cycle progression, and cytoplasmic chromatin fragments.41–43 In addition to cGAMP synthesized by cGAS, various bacterial or viral cyclic dinucleotides can also directly activate STING. Although numerous inhibitors targeting this pathway have been reported for the treatment of autoimmune diseases44 and infectious diseases,45 their roles in central nervous system injury remain poorly understood. Consistent with previous reports, we observed activation of the cGAS–STING pathway following HIE. Importantly, JQ1 treatment significantly suppressed the expression of cGAS and STING at both transcriptional and protein levels and effectively reduced the release of key inflammatory cytokines, including IL-6 and TNF-α. These results suggest that JQ1 may exert anti-inflammatory effects in HIE by interfering with cGAS–STING pathway activation, thereby attenuating secondary neural damage.
Iron-mediated oxidative stress represents a core mechanism underlying ferroptosis. Specifically, the inhibition of system Xc⁻, depletion of GSH, and reduced activity of GPX4 collectively contribute to enhanced lipid peroxidation and the accumulation of phospholipid hydroperoxides.46 Fe2⁺ catalyze lipid peroxidation via the Fenton reaction, resulting in massive generation of ROS, disruption of intracellular redox homeostasis, and ultimately the induction of ferroptosis. Consistent with previous studies,47,48 we observed pronounced neuronal ferroptosis under hypoxic conditions, characterized by decreased protein expression of several glutathione pathway markers (eg, SLC7A11, SLC3A2, and GPX4), reduced GPx activity, and Fe2⁺ accumulation. Therefore, targeting ferroptosis-related alterations post-HIE may offer neuroprotective potential. Interestingly, the modulatory effect of JQ1 on ferroptosis appears cell-type specific: it inhibits erastin-induced ferroptosis in cardiomyocytes, thereby improving cardiac remodeling and dysfunction.19 Conversely, in tumor cells and senescent human dermal fibroblasts, JQ1 downregulate the expression of ferroptosis-resistance genes, promoting ferroptosis and suppressing tumor progression.49,50 In our in vivo and in vitro models, JQ1 significantly enhanced antioxidant pathway activity, suggesting its potential to mitigate ferroptosis-associated neural injury.
Maintaining normal mitochondrial function is crucial for neuronal survival. As the central hub of cellular energy metabolism, mitochondria play a key role in oxidative stress and cell death pathways. Following cerebral blood flow interruption due to HI injury, rapid depletion of ATP triggers a cascade of events including dysfunction of membrane ion pumps, sodium influx, potassium efflux, membrane depolarization, and voltage-dependent calcium influx leading to mitochondrial Ca2⁺ overload. These changes significantly increase the generation of free radicals and activate cell death signaling pathways and inflammatory mediators. Consistent evidence indicates that impairment of mitochondrial structure and function following traumatic brain injury contributes to cognitive deficits and neuronal death.51 In this study, we confirmed that neuronal mitochondrial damage occurs under hypoxic conditions: electron microscopy revealed atrophic changes in mitochondria within cortical neurons following HI injury, and in vitro OGD models demonstrated ultrastructural alterations such as mitochondrial swelling and rounding. Therefore, targeting mitochondria has emerged as a promising neuroprotective strategy for HI-induced brain injury. It is noteworthy that mitochondrial damage after HIE can also lead to mtDNA leakage and redox imbalance, thereby further activating downstream inflammatory responses and cell death signaling.
Emerging evidence reveals a complex bidirectional regulatory relationship between the cGAS-STING pathway and ferroptosis. On one hand, activation of STING promotes lipid peroxidation,52,53 inhibits system Xc⁻ function, and induces autophagic degradation of GPX4,54 thereby facilitating the progression of ferroptosis. For instance, STING enhances mitochondrial MFN1/2 via protein interactions to promote ferroptosis in pancreatic cancer.55 Its suppression underlies the anti-ferroptotic effect of moderate-intensity exercise in neuroprotection.56 Similarly, ICA69 knockout ameliorates LPS-induced cardiac injury by inhibiting STING-dependent inflammation and ferroptosis.57 Concurrently, sustained or excessive neuroinflammation driven by STING signaling disrupts microenvironmental homeostasis and exacerbates iron metabolic dysregulation. On the other hand, ferroptosis bidirectionally modulates the cGAS–STING pathway through mechanisms involving redox imbalance, iron dyshomeostasis, and metabolic reprogramming.58,59 Notably, in the context of pancreatic cancer development, oxidized nucleobases released upon GPX4 depletion activate the STING-dependent DNA-sensing pathway, thus promoting macrophage infiltration and activation.60 Within the neuroprotective mechanisms mediated by JQ1, our study primarily focuses on the role of the cGAS‑STING pathway in regulating ferroptosis. Whether ferroptosis conversely modulates the cGAS pathway remains to be further investigated.
There are still some shortcomings in our study. Future work could incorporate more detailed analyses of motor coordination, muscle strength, or gait to provide a more complete neurobehavioral profile. Additionally, while preliminary RNA-seq and electron microscopy data have been validated, expanding the sample size and performing deeper statistical analyses would make the evidence more solid. Systematic evaluation of sex differences in female models and translational studies is needed to assess the generalizability and clinical relevance of the therapeutic strategy for neonatal cerebral ischemia.
Conclusions
Our findings demonstrate that (+)-JQ1 exerts neuroprotective effects in neonatal hypoxic-ischemic encephalopathy by attenuating neuroinflammation and suppressing ferroptosis. These beneficial outcomes are likely mediated through the upregulation of SIRT3, which promotes neuronal survival.
Abbreviations
HIE, Hypoxic-Ischemic Encephalopathyl; HI, Hypoxic-Ischemic; BRD4, Bromodomain-Containing Protein 4; BET, Bromodomain And Extra-Terminal; JQ1, (+)-JQ-1; SIRT3, Sirtuin 3; mtDNA, Mitochondrial DNA; cGAS, Cyclic Gmp–Amp Synthase; cGAMP, Cyclic Gmp–Amp; STING, Stimulator Of Interferon Genes; TBK1, Tank-Binding Kinase 1; IRF3, Interferon Regulatory Factor 3; IFNs, Type I Interferons; qRT-PCR, Quantitative Real-Time PCR; Ct, Cycle Threshold; MDA, Malondialdehyde; GSH, Glutathione; GPx, Glutathione Peroxidase; GPX4, Glutathione Peroxidase 4; 4HNE, 4-Hydroxynonenal; OGD, Oxygen-Glucose Deprivation; CCK8, Cell Counting Kit-8; CBF, Cortical Surface Cerebral Blood Flow; ROI, Region Of Interest; HE, Hematoxylin And Eosin; TTC, 2,3,5-Triphenyltetrazolium Chloride; IHC, Immunohistochemistry; IF, Immunofluorescence; Iba1, Ionized calcium-binding adapter molecule 1; NeuN, Neuronal Nuclei; MAP-2, Microtubule-Associated Protein-2; MBP, Myelin Basic Protein; GFAP, Glial Fibrillary Acidic Protein; PSD-95, Post-Synaptic Density Protein 95; OFT, Open-Field Test; MWM, Morris Water Maze; RNA-seq, RNA sequencing; DEGs, Differentially Expressed Genes; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; TEM, Transmission Electron Microscopy; SPR, Surface Plasmon Resonance; ROS, Reactive Oxygen Species; siRNA, Short-Interfering RNA.
Data Sharing Statement
The data that support the findings of this study are available from the corresponding author Mingchu Fang upon reasonable request.
Ethics Approval and Informed Consent
All animal experiments were approved by the Laboratory Animal Ethics Committee of Wenzhou Institute, University of Chinese Academy of Sciences (Ethics Approval Numbers: WIUCAS24020501) and conducted in accordance with the Guidelines for the Care and Use of Laboratory Animals of the National Institutes of Health (NIH Publications).
Consent for Publication
All the co-authors and participants have given their consent for publication in Drug Design, Development and Therapy.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This project was supported by the National Natural Science Foundation of China (Grant Numbers: 82402013, 82271747, and 82571979) and Wu Jieping Medical Foundation (320.6750.2025-23-16).
Disclosure
The authors declare no competing financial interests or personal relationships that could have appeared to influence the work reported in this work.
References
1. Douglas-Escobar M, Weiss MD. Hypoxic-ischemic encephalopathy: a review for the clinician. JAMA Pediatr. 2015;169(4):397–25. doi:10.1001/jamapediatrics.2014.3269
2. Perin J, Mulick A, Yeung D, et al. Global, regional, and national causes of under-5 mortality in 2000-192000–19: an updated systematic analysis with implications for the Sustainable Development Goals. Lancet Child Adolesc Health. 2022;6(2):106–115. doi:10.1016/s2352-4642(21)00311-4
3. Arnautovic T, Sinha S, Laptook A. Neonatal hypoxic-ischemic encephalopathy and hypothermia treatment. Obstet Gynecol. 2024;143(1):67–81. doi:10.1097/aog.0000000000005392
4. Gluckman P D, Wyatt JS, Azzopardi D, et al. Selective head cooling with mild systemic hypothermia after neonatal encephalopathy: multicentre randomised trial. Lancet. 2005;365(9460):663–670. doi:10.1016/s0140-6736(05)17946-x
5. Hsu SC, Blobel GA. The role of bromodomain and extraterminal motif (bet) proteins in chromatin structure. Cold Spring Harb Symp Quant Biol. 2017;82:37–43. doi:10.1101/sqb.2017.82.033829
6. Filippakopoulos P, Qi J, Picaud S, et al. Selective inhibition of bet bromodomains. Nature. 2010;468(7327):1067–1073. doi:10.1038/nature09504
7. Matuszewska M, Wilkaniec A, Cieślik M, et al. The inhibition of bromodomain and extraterminal domain (bet) proteins protects against microglia-mediated neuronal loss in vitro. Biomolecules. 2025;15(4):528. doi:10.3390/biom15040528
8. Bai Q, Shao E, Ma D, et al. A human tau expressing zebrafish model of progressive supranuclear palsy identifies brd4 as a regulator of microglial synaptic elimination. Nat Commun. 2024;15(1):8195. doi:10.1038/s41467-024-52173-0
9. Palomés-Borrajo G, Badia J, Navarro X, et al. Nerve excitability and neuropathic pain is reduced by bet protein inhibition after spared nerve injury. J Pain. 2021;22(12):1617–1630. doi:10.1016/j.jpain.2021.05.005
10. Sinha K, Das J, Pal PB, et al. Oxidative stress: the mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch Toxicol. 2013;87(7):1157–1180. doi:10.1007/s00204-013-1034-4
11. Liu X, Luo Q, Zhao Y, et al. The ferroptosis-mitochondrial axis in depression: unraveling the feedforward loop of oxidative stress, metabolic homeostasis dysregulation, and neuroinflammation. Antioxidants. 2025;14(5):613. doi:10.3390/antiox14050613
12. Huo L, Fu J, Wang S, et al. Emerging ferroptosis inhibitors as a novel therapeutic strategy for the treatment of neonatal hypoxic-ischemic encephalopathy. Eur J Med Chem. 2024;271:116453. doi:10.1016/j.ejmech.2024.116453
13. Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–1072. doi:10.1016/j.cell.2012.03.042
14. Yang NS, Zhong WJ, Sha HX, et al. Mtdna-cgas-sting axis-dependent nlrp3 inflammasome activation contributes to postoperative cognitive dysfunction induced by sevoflurane in mice. Int J Biol Sci. 2024;20(5):1927–1946. doi:10.7150/ijbs.91543
15. Shen H, Lu H, Mao L, et al. Inhibition of cgas attenuates neonatal hypoxic-ischemic encephalopathy via regulating microglia polarization and pyroptosis. Transl Pediatr. 2024;13(8):1378–1394. doi:10.21037/tp-24-148
16. Song L, Shen H, Hong F, et al. Rorα-activated mitophagy attenuating hypoxic-ischemic encephalopathy via suppression of microglial cgas-sting axis. Front Immunol. 2025;16:1592737. doi:10.3389/fimmu.2025.1592737
17. Gamdzyk M, Doycheva DM, Araujo C, et al. Cgas/sting pathway activation contributes to delayed neurodegeneration in neonatal hypoxia-ischemia rat model: possible involvement of line-1. Mol Neurobiol. 2020;57(6):2600–2619. doi:10.1007/s12035-020-01904-7
18. Zou M, Ke Q, Nie Q, et al. Inhibition of cgas-sting by jq1 alleviates oxidative stress-induced retina inflammation and degeneration. Cell Death Differ. 2022;29(9):1816–1833. doi:10.1038/s41418-022-00967-4
19. Yang M, Wang T, Shao J, et al. (+)-jq-1 alleviates cardiac injury in myocardial infarction by inhibiting ferroptosis through the nampt/sirt1 pathway. Cell Death Dis. 2025;16(1):548. doi:10.1038/s41419-025-07880-x
20. Chelladurai P, Boucherat O, Stenmark K, et al. Targeting histone acetylation in pulmonary hypertension and right ventricular hypertrophy. Br J Pharmacol. 2021;178(1):54–71. doi:10.1111/bph.14932
21. Zhang J, Xiang H, Liu J, et al. Mitochondrial sirtuin 3: new emerging biological function and therapeutic target. Theranostics. 2020;10(18):8315–8342. doi:10.7150/thno.45922
22. Järvenpää J, Rahnasto-Rilla M, Lahtela-Kakkonen M, et al. Profiling the regulatory interplay of bet bromodomains and sirtuins in cancer cell lines. Biomed Pharmacother. 2022;147:112652. doi:10.1016/j.biopha.2022.112652
23. Ho L, Hossen N, Nguyen T, et al. Epigenetic mechanisms as emerging therapeutic targets and microfluidic chips application in pulmonary arterial hypertension. Biomedicines. 2022;10(1):170. doi:10.3390/biomedicines10010170
24. Bilecki W, Wawrzczak-Bargieła A, Majcher-Maślanka I, et al. Inhibition of bet proteins during adolescence affects prefrontal cortical development: relevance to schizophrenia. Int J Mol Sci. 2021;22(16):8710. doi:10.3390/ijms22168710
25. Sartor GC, Powell SK, Brothers SP, et al. Epigenetic readers of lysine acetylation regulate cocaine-induced plasticity. J Neurosci. 2015;35(45):15062–15072. doi:10.1523/jneurosci.0826-15.2015
26. Lin WC, Wilbrecht L. Making sense of strengths and weaknesses observed in adolescent laboratory rodents. Curr Opin Psychol. 2022;45:101297. doi:10.1016/j.copsyc.2021.12.009
27. Morris R. Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods. 1984;11(1):47–60. doi:10.1016/0165-0270(84)90007-4
28. Ott CJ, Kopp N, Bird L, et al. Bet bromodomain inhibition targets both c-myc and il7r in high-risk acute lymphoblastic leukemia. Blood. 2012;120(14):2843–2852. doi:10.1182/blood-2012-02-413021
29. Zhao L, Li J, Fu Y, et al. Photoreceptor protection via blockade of bet epigenetic readers in a murine model of inherited retinal degeneration. J Neuroinflammation. 2017;14(1):14. doi:10.1186/s12974-016-0775-4
30. Kong L, Li W, Chang E, et al. Mtdna-sting axis mediates microglial polarization via irf3/nf-κb signaling after ischemic stroke. Front Immunol. 2022;13:860977. doi:10.3389/fimmu.2022.860977
31. Prut L, Belzung C. The open field as a paradigm to measure the effects of drugs on anxiety-like behaviors: a review. Eur J Pharmacol. 2003;463(1–3):3–33. doi:10.1016/s0014-2999(03)01272-x
32. Vorhees CV, Williams MT. Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat Protoc. 2006;1(2):848–858. doi:10.1038/nprot.2006.116
33. Koppula P, Zhang Y, Zhuang L, et al. Amino acid transporter slc7a11/xct at the crossroads of regulating redox homeostasis and nutrient dependency of cancer. Cancer Commun. 2018;38(1):12. doi:10.1186/s40880-018-0288-x
34. Liu F, Mccullough LD. Inflammatory responses in hypoxic ischemic encephalopathy. Acta Pharmacol Sin. 2013;34(9):1121–1130. doi:10.1038/aps.2013.89
35. Seifert G, Schilling K, Steinhäuser C. Astrocyte dysfunction in neurological disorders: a molecular perspective. Nat Rev Neurosci. 2006;7(3):194–206. doi:10.1038/nrn1870
36. Benito E, Ramachandran B, Schroeder H, et al. The bet/brd inhibitor jq1 improves brain plasticity in wt and app mice. Transl Psychiatry. 2017;7(9):e1239. doi:10.1038/tp.2017.202
37. Quezada E, Cappelli C, Diaz I, et al. Bet bromodomain inhibitors pfi-1 and jq1 are identified in an epigenetic compound screen to enhance c9orf72 gene expression and shown to ameliorate c9orf72-associated pathological and behavioral abnormalities in a c9als/ftd model. Clin Clin Epigenet. 2021;13(1):56. doi:10.1186/s13148-021-01039-z
38. Korb E, Herre M, Zucker-Scharff I, et al. Bet protein brd4 activates transcription in neurons and bet inhibitor jq1 blocks memory in mice. Nat Neurosci. 2015;18(10):1464–1473. doi:10.1038/nn.4095
39. Wu L, Chang E, Zhao H, et al. Regulated cell death in hypoxic-ischaemic encephalopathy: recent development and mechanistic overview. Cell Death Discov. 2024;10(1):277. doi:10.1038/s41420-024-02014-2
40. Beacom MJ, Gunn AJ, Bennet L. Preterm brain injury: mechanisms and challenges. Annu Rev Physiol. 2025;87(1):79–106. doi:10.1146/annurev-physiol-022724-104754
41. Hopfner KP, Hornung V. Molecular mechanisms and cellular functions of cgas-sting signalling. Nat Rev Mol Cell Biol. 2020;21(9):501–521. doi:10.1038/s41580-020-0244-x
42. Zhang Z, Zhang C. Regulation of cgas-sting signalling and its diversity of cellular outcomes. Nat Rev Immunol. 2025;25(6):425–444. doi:10.1038/s41577-024-01112-7
43. West AP, Khoury-Hanold W, Staron M, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature. 2015;520(7548):553–557. doi:10.1038/nature14156
44. Dai J, Huang YJ, He X, et al. Acetylation blocks cgas activity and inhibits self-DNA-induced autoimmunity. Cell. 2019;176(6):1447–60.e14. doi:10.1016/j.cell.2019.01.016
45. Vinuesa CG, Grenov A, Kassiotis G. Innate virus-sensing pathways in b cell systemic autoimmunity. Science. 2023;380(6644):478–484. doi:10.1126/science.adg6427
46. Costa I, Barbosa D, Benfeito S, et al. Molecular mechanisms of ferroptosis and their involvement in brain diseases. Pharmacol Ther. 2023;244:108373. doi:10.1016/j.pharmthera.2023.108373
47. Yuan Y, Zhai Y, Chen J, et al. Kaempferol ameliorates oxygen-glucose deprivation/reoxygenation-induced neuronal ferroptosis by activating nrf2/slc7a11/gpx4 axis. Biomolecules. 2021;11(7):923. doi:10.3390/biom11070923
48. Jin Y, Wang X, Xu X, et al. Dimethyl malonate preserves brain and neurobehavioral phenotype following neonatal hypoxia-ischemia by inhibiting fth1-mediated ferritinophagy. Redox Biol. 2025;86:103792. doi:10.1016/j.redox.2025.103792
49. Go S, Kang M, Kwon SP, et al. The senolytic drug jq1 removes senescent cells via ferroptosis. Tissue Eng Regen Med. 2021;18(5):841–850. doi:10.1007/s13770-021-00346-z
50. Fan C, Guo X, Zhang J, et al. Brd4 inhibitors broadly promote erastin-induced ferroptosis in different cell lines by targeting ros and fsp1. Discov Oncol. 2024;15(1):98. doi:10.1007/s12672-024-00928-y
51. Wu W, Wei Z, Wu Z, et al. Exercise training alleviates neuronal apoptosis and re-establishes mitochondrial quality control after cerebral ischemia by increasing sirt3 expression. Cell Biol Toxicol. 2024;41(1):10. doi:10.1007/s10565-024-09957-3
52. Chen S, Jiang X, Li X, et al. Stimulator of interferon genes regulates ferroptosis and airway inflammation via acyl-coa synthetase long-chain family member 4 in asthmatic mice. Inflammation. 2025. doi:10.1007/s10753-025-02338-w
53. Jiang GY, Yang HR, Li C, et al. Ginsenoside rd alleviates early brain injury by inhibiting ferroptosis through cgas/sting/dhodh pathway after subarachnoid hemorrhage. Free Radic Biol Med. 2025;228:299–318. doi:10.1016/j.freeradbiomed.2024.12.058
54. Wang X, Chen T, Chen S, et al. Sting aggravates ferroptosis-dependent myocardial ischemia-reperfusion injury by targeting gpx4 for autophagic degradation. Signal Transduct Target Ther. 2025;10(1):136. doi:10.1038/s41392-025-02216-9
55. Li C, Liu J, Hou W, et al. Sting1 promotes ferroptosis through mfn1/2-dependent mitochondrial fusion. Front Cell Dev Biol. 2021;9:698679. doi:10.3389/fcell.2021.698679
56. Chen J, Zhu T, Yu D, et al. Moderate intensity of treadmill exercise rescues tbi-induced ferroptosis, neurodegeneration, and cognitive impairments via suppressing sting pathway. Mol Neurobiol. 2023;60(9):4872–4896. doi:10.1007/s12035-023-03379-8
57. Kong C, Ni X, Wang Y, et al. Ica69 aggravates ferroptosis causing septic cardiac dysfunction via sting trafficking. Cell Death Discov. 2022;8(1):187. doi:10.1038/s41420-022-00957-y
58. Yao Y, He X, Zhu Y, et al. Intersection of immune signaling and cell death: the bidirectional regulatory mechanism of sting pathway and ferroptosis. Autoimmun Rev. 2025;24(10):103877. doi:10.1016/j.autrev.2025.103877
59. Jia M, Qin D, Zhao C, et al. Redox homeostasis maintained by gpx4 facilitates sting activation. Nat Immunol. 2020;21(7):727–735. doi:10.1038/s41590-020-0699-0
60. Dai E, Han L, Liu J, et al. Ferroptotic damage promotes pancreatic tumorigenesis through a tmem173/sting-dependent DNA sensor pathway. Nat Commun. 2020;11(1):6339. doi:10.1038/s41467-020-20154-8
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
The Emerging Role of Ferroptosis in Various Chronic Liver Diseases: Opportunity or Challenge
Zhu L, Luo S, Zhu Y, Tang S, Li C, Jin X, Wu F, Jiang H, Wu L, Xu Y
Journal of Inflammation Research 2023, 16:381-389
Published Date: 31 January 2023
Variation of Ferroptosis-Related Markers in HaCaT Cell Photoaging Models Induced by UVB
Zhang PC, Hong Y, Zong SQ, Chen L, Zhang C, Tian DZ, Ke D, Tian LM
Clinical, Cosmetic and Investigational Dermatology 2023, 16:3147-3155
Published Date: 1 November 2023
Ciprofol Ameliorates Myocardial Ischemia/Reperfusion Injury by Inhibiting Ferroptosis Through Upregulating HIF-1α

Ding J, Wang BY, Yang YF, Kuai LY, Wan JJ, Zhang M, Xia HY, Wang Y, Zheng Z, Meng XW, Peng K, Ji FH
Drug Design, Development and Therapy 2024, 18:6115-6132
Published Date: 18 December 2024
Enteric-Coated Aspirin Induces Small Intestinal Injury via the Nrf2/Gpx4 Pathway: A Promising Model for Chronic Enteropathy

Zhang M, Xia S, Feng L, Han X, Zhang Y, Huang Y, Liu Y, Zhao K, Guan J, Tian D, Liao J, Yu Y
Drug Design, Development and Therapy 2025, 19:891-910
Published Date: 11 February 2025
Vaccarin Ameliorates Renal Fibrosis by Inhibiting Ferroptosis via Nrf2/SLC7A11/GPX4 Signaling Pathway
Cui M, Xu Q, Duan L, Lu J, Hu J
Drug Design, Development and Therapy 2025, 19:1609-1626
Published Date: 6 March 2025