")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 18
Zanubrutinib in Treating Waldenström Macroglobulinemia, the Last Shall Be the First
Authors Deshpande A , Munoz J
Received 9 March 2022
Accepted for publication 23 May 2022
Published 23 June 2022 Volume 2022:18 Pages 657—668
DOI https://doi.org/10.2147/TCRM.S338655
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Anagha Deshpande,1 Javier Munoz2
1Mayo Clinic Alix School of Medicine, Scottsdale, AZ, USA; 2Division of Hematology and Oncology, Mayo Clinic, Phoenix, AZ, USA
Correspondence: Anagha Deshpande, Mayo Clinic Alix School of Medicine, 13400 E Shea Blvd, Scottsdale, AZ, 85259, USA, Tel +1 480 251-0289, Email [email protected]
Abstract: In Waldenström macroglobulinemia (WM), a lymphoplasmacytic lymphoma characterized by monoclonal immunoglobulin M (IgM) gammopathy, aberrant Bruton tyrosine kinase (BTK) signaling has been identified as one mechanism of pathogenesis. For this reason, selective BTK inhibiting therapies have emerged as an attractive option for treatment within the therapeutic landscape also comprising chemotherapy, monoclonal antibodies, proteasome inhibitors, and B-cell lymphoma 2 (BCL2) inhibitors. The first BTK inhibiting therapy, ibrutinib, showed great efficacy in treating WM. However, response rates were dependent on whether patients had the CXCR4 mutation, a molecular aberration that may confer resistance to BTK inhibitors. Furthermore, ibrutinib’s toxicities, most notably hypertension and atrial arrhythmia, led to dose reductions or discontinuation. The toxicity profile of ibrutinib can be attributed to the inhibition of additional kinases that are structurally related to BTK. Therefore, the next-generation highly selective zanubrutinib was developed to address the concerns regarding toxicity and tolerance related to ibrutinib therapy. Based on the results of the randomized, open-label Phase 3 ASPEN (NCT03053440) trial, the Food and Drug Administration (FDA) approved zanubrutinib for treating WM. This trial directly compared zanubrutinib to ibrutinib in patients with treatment-naïve or relapsed/refractory WM, and the results showed stronger responses with zanubrutinib. More importantly, patients responded strongly to zanubrutinib therapy regardless of CXCR4 mutation status. Additionally, zanubrutinib was associated with fewer grade 3 or higher toxicities and was generally better tolerated. Another Phase 1/2 study has been conducted with just zanubrutinib in WM showcasing high efficacy with few toxicities as well. Even though zanubrutinib has been the third and last BTK inhibitor to currently penetrate the market for B-cell lymphoproliferative malignancies, we highlight the emergence of zanubrutinib as a key player in the forefront of the therapeutic landscape in WM.
Keywords: zanubrutinib, Waldenström macroglobulinemia, Bruton tyrosine kinase inhibitor, ibrutinib, rituximab, efficacy, safety, atrial fibrillation
Introduction
B-cell development, survival, and expansion is critically dependent on normally functioning and tightly regulated B-cell receptor (BCR) signaling pathways. Key mediators of the BCR pathway include the proximal kinases – Bruton tyrosine kinase (BTK), phosphatidyl inositol 3 kinase (PI3K), and spleen tyrosine kinase (SYK).1 A member of the Tec protein kinase (TEC) family, BTK is an important regulator of B-cell proliferation and survival.1 In the BCR pathway, BTK gets activated by upstream Src-family kinases; once activated, the enzyme goes on to stimulate downstream cell survival pathways including nuclear factor kappa-light-chain-enhancer of activated B-cells (NF-κB) and mitogen activated protein kinase (MAPK).2 Defective BTK was first identified in 1993 in X-linked hypogammaglobulinemia, an inherited immunodeficiency disease.3 More recently, aberrant BTK signaling has been implicated in the pathogenesis of a variety of B-cell malignancies including chronic lymphocytic leukemia (CLL),4–7 mantle cell lymphoma (MCL),5,8,9 diffuse-large B-cell lymphoma (DLBCL),10 marginal zone lymphoma (MZL),11 and Waldenström macroglobulinemia (WM).5,12 For these reasons, BTK inhibitors (BTKi) have emerged as an important and effective treatment option for difficult-to-manage B-cell malignancies. One BTKi available to treat B-cell malignancies is zanubrutinib. This next-generation highly specific BTKi has shown strong efficacy in treating a multitude of B-cell malignancies including CLL, MCL, MZL, and WM. Herein, we will discuss the particularly available data regarding the use of zanubrutinib in WM.
Waldenström Macroglobulinemia Overview
Waldenström macroglobulinemia (WM) is a rare lymphoplasmacytic lymphoma occurring in the bone marrow that is characterized by a monoclonal immunoglobulin M (IgM) gammopathy. This disease is presumed to be primarily sporadic with an unknown cause. However, the genetic landscape of WM has identified genetic mutations in the MYD88 and CXCR4 genes in 90% and 30% of cases, respectively.13,14 A genetic basis for disease pathogenesis has been further supported by analysis of 257 unrelated patients with WM revealing that 48% of patients with WM had a first-degree relative having either WM or another B-cell disorder.15 Additionally, a family history of WM or another plasma disorder led to a diagnosis of WM at a younger age with greater bone marrow involvement.15 Ultimately though, the greatest risk factor for developing WM is a pre-existing monoclonal gammopathy of undetermined significance (MGUS).16
Patients with WM develop symptoms due to disease infiltration of the lymph nodes, bone marrow, or spleen, effects of elevated IgM levels in the blood, or a combination. For disease infiltration, excessive bone marrow involvement and infiltration crowds out the marrow space and creates cytopenias, mainly anemia, thrombocytopenia, and neutropenia.16 Lytic bone disease is highly uncommon in WM, differentiating it from multiple myeloma. However, the disease does usually spread to lymph nodes, the liver, and spleen – creating lymphadenopathy, hepatomegaly, and splenomegaly.16 Some rare cases of pulmonary parenchymal infiltration have been documented, and these have presented as diffuse pulmonary infiltrates, nodules, or effusions.17 Infiltration of the stomach, bowel, skull base, and orbit has also been reported.18 An extremely rare neurologic complication of WM is Bing-Neel syndrome, the manifestation of direct central nervous system disease involvement that presents with a variety of symptoms including headache, ataxia, vertigo, nystagmus, diplopia, and more seriously, coma.19 BTK inhibitors like ibrutinib and zanubrutinib, have demonstrated CNS penetrance, and there are some case reports of BTKi use in Bing-Neel syndrome.20,21
The effects of elevated IgM levels in the blood are due to serum hyperviscosity, tissue deposition of IgM, or the autoantibody activity of IgM. Increased concentration of the monoclonal protein increases serum viscosity, and thus vascular resistance. This leads to the downstream effects of constitutional symptoms, bleeding, high-output cardiac failure, and ocular or neurological complications.16 The IgM can also deposit in a variety of tissues leading to complications such as proteinuria, diarrhea, and macroglobulinemia cutis.16 Furthermore, the immunoglobulin light chains of IgM can precipitate primary amyloidosis in a variety of organs such as the heart, peripheral nerves, kidney, liver, and lungs.16 Finally, autoimmune reactions can be triggered by high levels of IgM in the serum. At cold temperatures, autoimmune hemolytic anemias can occur with elevated cold agglutination titers.16 Symmetric polyneuropathies are also commonly reported.16 Clinical evidence of any of these adverse effects with significant symptoms serves as indication for initiating active treatment.
Therapeutic Landscape
Multiple treatment options are available for WM including chemotherapy, monoclonal antibodies, proteasome inhibitors, BTK inhibitors (like ibrutinib, acalabrutinib, or zanubrutinib), and B-cell lymphoma 2 (BCL2) inhibitors. Treatment is usually initiated when patients present with progressive anemia, hyperviscosity complications, significant fatigue, or other significant constitutional symptoms. The development of novel therapeutic modalities such as BTKis and BCL2is has already altered the therapeutic armamentarium available for B-cell malignancies.22 The selection of an optimal therapeutic regimen is an individualized decision that is tailored by the clinician and the patient while considering a myriad of factors including patient characteristics (eg, age, performance status, other medical problems, other medications), disease characteristics (eg, mutational status, prognostic markers, bulky disease, geographic area of involvement, tumor lysis syndrome risk), and treatment characteristics (eg, cost, toxicity, convenience).22 We prefer enrolling any patient in a clinical trial when available. In the standard of care realm, we are respectful of the label as instructed by the Food and Drug Administration (FDA) approval and/or regimens mentioned in reputable national cancer guidelines after a prolonged discussion with the patient regarding pros and cons. Of note, BTKi appear in National Comprehensive Cancer Network (NCCN) guidelines as preferred options both for primary therapy and for previously treated WM as of the published guidelines accessed on May 03, 2022.23 We align ourselves with NCCN guidelines in the way that we shy away as much as possible from chemotherapeutic regimens in lieu of favoring novel non-chemotherapy agents; nevertheless, the ultimate therapeutic decision is individualized. In NCCN guidelines, both ibrutinib plus or minus rituximab and zanubrutinib appear as category 1 recommendations for primary therapy and for previously treated WM. Acalabrutinib does not currently appear as an option for primary therapy for WM in NCCN; nevertheless, acalabrutinib does appear as a part of other recommended regimens available for previously treated WM. Herein, we provide additional granularity regarding these multiple treatment options.
Plasmapheresis
A notable complication of the monoclonal gammopathy seen in WM is peripheral neuropathy that can be attributed to demyelination and widened myelin lamellae.24 Additionally, elevated IgM levels can create the hyperviscosity syndrome presenting with the classic triad of mucosal bleeding, visual changes, and neurologic complications. For these patients, plasmapheresis has been shown to lower serum IgM levels, improve the symptoms associated with hyperviscosity, and reduce the progression of neuropathy.25–27 However, plasmapheresis should only be used for symptom management until systemic therapy decreases tumor burden.28
Rituximab
Rituximab is a chimeric monoclonal antibody that targets the CD20 antigen expressed on all B-cells that leads to lymphocyte lysis after binding. In terms of rituximab monotherapy, one study was conducted on 27 patients with WM who were treated with rituximab intravenously for 4 weeks.29 Twelve patients (44%) achieved a partial response (PR) at a median time to response of 3.3 months.29 Treatment was generally well-tolerated, but 25% of patients experienced a mild infusion reaction mostly manifesting as fever and chills.29 Furthermore, at a median follow-up of 15.7 months, 9 of the 12 patients who responded to treatment remained disease progression free.29 Based on these findings, during the Second International Workshop on WM held in September 2002 in Athens, Greece, a consensus panel recommended using combinations of alkylating agents, nucleoside analogs, and rituximab for treating WM.30 Since then, many clinical studies have been conducted to evaluate the efficacy and safety of rituximab combination regimens in treating WM.31 A pooled meta-analysis found that rituximab combination therapies result in an objective response rate (ORR) of 84%, major response rate (MRR) 71%, and complete response (CR) 7% in patients with symptomatic WM.31 Rituximab plus conventional alkylating agents that containing chemotherapy resulted in an ORR of 86%, MRR 74%, and CRR 8%.31 On the other hand, rituximab plus a purine analog produced an ORR of 85%, MRR 74%, and CRR 9%.31 Finally, rituximab plus a proteasome inhibitor yielded an ORR of 86%, MRR 68%, and CRR 7%.31 In terms of safety, low grade adverse events related to the first infusion of rituximab were the most common; these included dyspnea, hypertension, angioedema, bronchospasm, cough, pyrexia, chills, rash, and vomiting.31 The most common grade 3 or higher adverse events were neutropenia, thrombocytopenia, and anemia; it should be noted that these adverse events generally did not lead to treatment discontinuation.31
One clinical challenge with rituximab therapy in WM is the paradoxical rise in IgM levels after initial infusion. One study evaluating rituximab in 54 patients with WM found that 54% had a rise in IgM levels between infusion and the first scheduled measurement time point.32 Two-months later, 59% of 22 evaluable patients continued to have elevated IgM levels.32 Finally, 4 months after therapy, 4 out of 15 patients still had elevated IgM levels.32 The IgM flare can either precipitate the hyperviscosity syndrome in patients or worsen existing hyperviscosity symptoms, thus creating the need for emergent plasmapheresis. Another challenge is that rituximab intolerance has been identified in a higher proportion of patients with WM when compared to other B-cell malignancies.33 An analysis on 85 patients intolerant to rituximab revealed that 34% developed intolerance during the first-line of treatment while 15% developed intolerance during the first year of exposure to rituximab.33 With both single-agent rituximab and rituximab-containing regimens, the monoclonal antibody had to be discontinued for worsening infusion-related reactions.33 The most common reactions, occurring in greater than 10% of patients, were anaphylaxis, chills and rigors, hives, hypotension, and shortness of breath.33 Thus, rituximab intolerance presents a unique challenge for treating WM. Nevertheless, we are usually able to circumvent these toxicities when using rituximab in B-cell malignancies.
Chemoimmunotherapy
Two commonly prescribed chemoimmunotherapy regimens for treating WM are bendamustine-rituximab (BR) and dexamethasone-rituximab-cyclophosphamide (RCD). For BR, the STiLNHL1–2003 phase 3 trial first highlighted this regimen’s efficacy in treating indolent non-Hodgkin lymphoma (including 41 patients with lymphoplasmacytic lymphoma) when compared to rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP).34 At a median follow-up of 45 months, the median progression-free survival (PFS) was longer in the BR group compared to the R-CHOP group.34 ORRs were 93% and 91% in the BR and R-CHOP groups, respectively.34 Furthermore, BR was better tolerated with lower rates of alopecia, hematological toxicity, infections, peripheral neuropathy, and stomatitis.34 More recently, at longer median 5.9 years follow-up, median PFS with BR was reported as 65.3 months.35 Furthermore, the study compared two groups who had been given BR induction therapy where one group received 2-year rituximab monotherapy afterwards, while the other was just observed.35 No significant difference in PFS or OS was found between the two groups – confirming the highly effective nature of induction with BR therapy.35
For the other regimen, RCD, a Phase II trial on symptomatic treatment-naïve WM patients found that 83% achieved a response (7% complete, 67% partial, and 9% minor).36 For all patients, the 2-year PFS rate was 67%; for patients who responded to RCD treatment, this rate was 80%.36 Thirty-two percent of patients experienced an IgM flare, however, levels dropped below baseline after RCD treatment was completed.36 For toxicities, only 7 of 72 patients experienced a grade 3 or 4 neutropenia with no patients having thrombocytopenia.36 Other adverse events reported were related to the infusion of rituximab and included chills, fevers, and headaches.36 For these reasons, chemoimmunotherapy remains a front-runner in the first-line treatments of WM.
Bortezomib
The proteosome is a 26S nuclear/cytosolic complex that regulates protein degradation, and thus, influences various cellular pathways including the cell cycle. Bortezomib is a small molecule that selectively and reversibly inhibits the proteosome to influence these cellular pathways. In 27 patients with WM who had previously failed at least one line of therapy, bortezomib was found to significantly reduce serum IgM levels from 4660 to 2092 mg/dL.37 An ORR of 85% was achieved with therapy at a median of 1.4 months.37 Within those who responded, 48.1% achieved a MR.37 The most common grade 3 or higher toxicities that occurred were sensory neuropathies, leukopenia, neutropenia, dizziness, and thrombocytopenia – occurring in 22.2%, 18.5%, 14.8%, 11.1%, and 7.4%, respectively.37 All events of neuropathy resolved with cessation of therapy in this particular study.37 Of note, response rates to bortezomib were found to not be negatively impacted by the CXCR4 mutation, a molecular aberration that has conferred resistance to other lines of treatment such as ibrutinib, which will be discussed later.38 Although bortezomib monotherapy has shown improved clinical outcomes in patients with WM, the associated toxicity of sensory neuropathy presents a clinical challenge. In fact, another study showed bortezomib’s efficacy at treating WM, but 74% developed new or worsening neuropathy, and this was the most common cause for dose reduction or therapy cessation.39
Venetoclax
BCL2, a regulatory molecule of the intrinsic apoptosis pathway, is overexpressed in WM, especially in patients with the MYD88L265P mutation, regardless of CXCR4 mutation status.40 Venetoclax is a BCL2 inhibitor that was approved for the treatment of CLL and acute myeloid leukemia.40 A phase 1 study was conducted to evaluate the efficacy and safety of venetoclax in 33 patients with relapsed/refractory (R/R) WM.40 Sixteen (50%) of these patients had previously been treated with BTKi therapy and were either intolerant to the therapy or had disease progression.40 The MYD88L265P mutation was found in all patients, but the CXCR4WHIM mutation was found in 17 patients.40 This study reported an ORR and MRR of 84% and 81%.40 No patient achieved a complete response.40 The median time to achieve either a minor or major response was 1.9 months.40 At a median follow-up of 33 months, 19 patients had progression of disease, and 12 had to start an additional treatment.40 The median progression-free survival (PFS) at 12, 24, and 30 months was 83%, 80%, and 95%.40 No significant difference in PFS was found based on prior BTKi therapy or CXCR4 mutational status.40 Twenty-two percent of patients had to reduce their dose of treatment because of neutropenia (n=2), pancytopenia (n=1), fatigue (n=1), diarrhea (n=1), multiple grade 2 events (n=1), and self-reduction (n=1).40 However, the dose reductions were not associated with any significant difference in response rates.40 Thirty-one (94%) patients experienced a grade 2 or higher adverse event.40 Only 7 patients experienced a grade 4 adverse event, and these were neutropenia (n=6) and febrile neutropenia (n=1).40
Ibrutinib
Ibrutinib is a BTKi that was approved for the treatment of WM by the FDA in January of 2015.41 Ibrutinib, in addition to its active metabolite PCI-45227, covalently and irreversibly binds to Cysteine 481 in the kinase domain of BTK to inhibit its activity.41 In WM, BTK forms a complex with the mutated MYD88 protein to create a constantly activated BTK molecule.41 Thus, ibrutinib has shown significant activity in managing WM. FDA approval followed the landmark phase II trial showing an ORR of 90.5%, MRR of 73%, 2-year PFS of 69.1%, and 2-year overall survival (OS) rates of 95.2% in 63 patients with symptomatic WM refractory to at least one prior treatment.42 The MYD88L265P mutation was present in 89% while the CXCR4WHIM mutation – a mutation that confers resistance to ibrutinib – was present in 34%.42 Responses to treatment were also assessed by genomic group: MYD88L265P CXCR4WT, MYD88L265P CXCR4WHIM, and MYD88WT CXCR4WT. ORR and MRR were 100% and 91.2%, 85.7% and 61.9%, and 71.4% and 28.6% in these groups, respectively.42 At long-term median 59-month follow-up, ORR and MRR were 90.5% and 79.4%, respectively.43 When looking at the best response to treatment, serum IgM levels declined from 3520 to 821 mg/dL, bone marrow disease involvement decreased from 60% to 20%, and hemoglobin levels rose from 10.3 to 14.2 g/dL.43 Patients with the MYD88L265P CXCR4WT genotype showed the best response to treatment with MRR of 97.2% and shortest time to major response duration of 1.8 months.43 For all patients, the 5-year survival rate was 87%.43 For toxicities, 15.9% experienced a grade 3 or higher neutropenia, 11.1% experienced a grade 3 or higher thrombocytopenia, and 3.2% experienced a pneumonia.43 Additionally, 8 patients (12.7%) experienced atrial arrhythmia, and of these, one had a grade 3 arrhythmia.43 A total of 5 patients had to discontinue ibrutinib due to hematoma, thrombocytopenia, influenza-related pneumonia, streptococcal endocarditis, and atrial fibrillation.43 Twelve patients had to reduce the dose of ibrutinib due to cytopenias, dermatitis or rash, stomatitis, leg edema, myalgias, and atrial fibrillation.43 Overall, this study showed the efficacy and durability of response to ibrutinib without any unexpected toxicities.
Since the efficacy of ibrutinib monotherapy showed dependence on MYD88 and CXCR4 mutation status, the iNNOVATE trial was conducted to assess the efficacy and safety of ibrutinib in combination with rituximab in treating WM in 150 patients who had either received no prior treatment (n=68) or who had disease recurrence after prior systemic therapy (n=82).44 Of those who had previously received systemic therapy, 70 patients had received a rituximab-containing regimen.44 In this trial, 150 patients were randomly assigned to receive ibrutinib and rituximab or a placebo and rituximab.44 At follow-up of 30 months, the PFS was higher in the ibrutinib-rituximab group at 82% compared to 28% in the groups who received placebo-rituximab.44 MRR was also higher in the ibrutinib-rituximab group at 73% versus 41% in the placebo-rituximab group.44 Furthermore, the higher response rates noted in the ibrutinib-rituximab group were independent of MYD88 and CXCR4 mutation status.44 In terms of safety, the most common any-grade adverse events reported in the ibrutinib-rituximab group were infusion-related reactions, nausea, diarrhea, and arthralgias, and these occurred more frequently in this group compared to the placebo-rituximab group.44 Grade 3 or higher adverse events that occurred more frequently in the ibrutinib-rituximab group included hypertension (13% vs 4%) and atrial fibrillation (12% vs 1%).44 On the other hand, grade 3 or higher adverse events that occurred more frequently in the placebo-rituximab group included anemia (17% vs 11%) and infusion-related reactions (16% vs 1%).44 The placebo-rituximab group also had higher incidences of any grade of IgM flare (47% vs 8%).44 Major hemorrhage rates were the same in both groups at 4%.44 At longer follow-up of 50 months, the median PFS was longer in the ibrutinib-rituximab group compared to the placebo-rituximab group, and the results showed a 75% reduction in risk of disease progression or death with ibrutinib-rituximab.45 Estimated 54-month PFS rates were 68% and 25% in the ibrutinib-rituximab and placebo-rituximab groups, respectively.45 PRR was also higher in the ibrutinib-rituximab group at 76% versus 31%.45 Seventy-seven percent of patients who received ibrutinib-rituximab had sustained hemoglobin improvement compared to 43% in the placebo-rituximab group.45 For safety, the prevalence of grade 3 or higher toxicities decreased over time.45 A total of 15% experienced a grade 3 or higher hypertension, and 16% experienced a grade 3 or higher atrial fibrillation.45 However, these events mostly occurred in the first 2–3 years of treatment, except for one event of grade 3 or higher hypertension occurring in the fifth year of treatment.45 No major hemorrhage events were reported in the last 2 years of treatment.45 Only one death was reported from a pneumonia, but this was deemed to be unrelated to the study drug.45 Out of 75, a total of 8 patients discontinued ibrutinib and 17 patients reduced the dose of ibrutinib due to adverse events.45 Most importantly, the final results from the iNNOVATE trial highlight that the combination of ibrutinib-rituximab greatly improves clinical outcomes regardless of MYD88 and CXCR4 genotype as well as prior treatment status.45
Acalabrutinib
A next-generation highly selective BTKi, acalabrutinib has less interaction with non-BTK signaling pathways, which contributes to a less severe and different side effect profile compared to ibrutinib.46 For these reasons, a single-arm, multicenter, international, Phase 2 study was conducted to evaluate the efficacy and safety of acalabrutinib monotherapy in WM.46 A total of 122 patients were studied with 14 being treatment naïve and 92 having R/R disease.46 At a median follow-up of 27.4 months, 93% of treatment naïve patients and 93% of R/R disease patients achieved an OR.46 Median duration of response, PFS, and OS were not reached.46 It should be noted that a limitation of this study is that responses were not analyzed based on MYD88 and CXCR4 mutational status via central laboratory assessment.46 Thus, the impact of CXCR4 mutational status regarding acalabrutinib use is currently preliminary.46 In terms of toxicities, grade 3 or 4 adverse events occurred in more than five percent of patients, and this included neutropenia (16%) and pneumonia (7%).46 Only one patient (1%) had grade 3 or 4 atrial fibrillation/flutter and three (3%) had grade 3 or 4 hemorrhage.46 One patient died from an intracranial hematoma that was deemed to be treatment related.46
Zanubrutinib in WM
Overview
Zanubrutinib (BGB-3111) is a next-generation BTKi that was developed to achieve BTK inhibiting efficacy while reducing the off-target adverse events documented with ibrutinib therapy.47,48 With high specificity for the BTK adenosine triphosphate binding site for cysteine 481, zanubrutinib covalently binds to this site, resulting in irreversible, sustainable kinase inhibition.47,48 Compared to ibrutinib, zanubrutinib binds to the EGFR, JAK3, human EGFR2, ITK, and TEC protein kinases with 3.5, 58, 28, 19, and 3.5 times less affinity, respectively.47 Additionally, the IC50 for zanubrutinib is 0.5 nM compared to 1.5 nM for ibrutinib.49 Currently, Zanubrutinib is FDA approved for the treatment of MCL, MZL, and WM.
Efficacy
FDA approval for zanubrutinib in WM was granted on August 31, 2021 following the results of the ASPEN (NCT03053440) trial.50 This trial was a randomized, open-label phase 3 study.51 All patients enrolled had either R/R disease after at least one prior line of therapy or treatment-naïve disease unsuitable for standard chemotherapy based on documented comorbidities or risk factors.51 In the first cohort, the study compared the efficacy and safety of zanubrutinib to ibrutinib in patients with MYD88L265P disease.51 In the second cohort, the study evaluated the efficacy and safety of just zanubrutinib in MYD88WT disease.51 For cohort 1, patients were randomized to receive either 160mg of zanubrutinib BID (n=102) or 420mg of ibrutinib QD (n=99).51 For cohort 2, patients only received 160mg of zanubrutinib BID.51 In cohort 1 within the group receiving zanubrutinib, 91 patients (89%) had the MYD88L265P/CXCR4WT genotype, and 11 (11%) had the MYD88L265P/CXCR4WHIM genotype.51 Within the group receiving ibrutinib, 90 patients (91%) had the MYD88L265P/CXCR4WT genotype, 8 (8%) had the MYD88L265P/CXCR4WHIM genotype, and 1 (1.0%) had the MYD88L265P/CXCR4UNK genotype.51 The two groups in cohort 1 were generally balanced well per baseline characteristics, but more patients assigned to receive zanubrutinib were above the age of 75 (33% vs 22%), and more were anemic (hemoglobin ≤110 g/L in 66% vs 54%).51
In terms of responses, at median follow-up of 19.5 months, no patient achieved a complete response (CR).51 A very good partial response (VPGR) was achieved in 28% of zanubrutinib patients and 19% of ibrutinib patients, but this difference was not statistically different (p=0.09).51 MRRs were 77% and 78% in the zanubrutinib and ibrutinib groups, respectively.51 One patient in each group with the CXCR4WHIM genotype achieved a VGPR.51 On the other hand, 31% of zanubrutinib-treated patients and 20% of ibrutinib-treated patients with the CXCR4WT genotype achieved a VGPR.51 Median time to a major response was 2.8 months for both groups with the CXCR4WT genotype.51 However, in patients with the CXCR4WHIM genotype, median time to major response was 6.6 months with ibrutinib and 3.1 months with zanubrutinib.51 At median follow-up of 18 months, 85% and 84% of zanubrutinib and ibrutinib patients were disease progression free, respectively.51 Median PFS and duration of response (DOR) were not reached.51 Estimated OS was 97% and 93% in the zanubrutinib and ibrutinib groups, respectively.51 The primary end point for the ASPEN trial was the proportion of patients achieving a CR or VGPR by independent review.51 Of note, this trial failed to show statistical superiority of zanubrutinib compared to ibrutinib based on the chosen primary end point.51 Nevertheless, the safety profile seemed to benefit zanubrutinib instead of ibrutinib, hence we will discuss it in detail in the toxicity section.51
For median IgM levels, these were reduced by 79% and 72% with zanubrutinib and ibrutinib, respectively.51 However, it should be noted that zanubrutinib showcased significantly greater efficacy at reducing IgM levels, as well as sustaining lowered IgM levels, based on analysis with the repeated-measures mixed-effect model and area under the IgM × time curve comparisons.51 For the zanubrutinib and ibrutinib groups, 69% and 73% patients had a reduction in bone marrow infiltration, with median maximal reductions of 10% and 15%, respectively.51
Overall, the ASPEN study showed a greater frequency of VGPRs in zanubrutinib-treated patients compared to ibrutinib-treated patients.51 It should be noted that in this study, post hoc analysis using next-generation sequencing for CXCR4 mutation detection instead of the original Sanger sequencing revealed 28% of patients with the CXCR4WHIM genotype compared to the originally reported 19%.51 However, VGPR rates within this analysis were comparable to the originally discussed results.51 Furthermore, this study showed that zanubrutinib therapy resulted in higher response rates among all patients (29% vs 21% in zanubrutinib-treated and ibrutinib-treated patients, respectively), among CXCR4WHIM genotype patients (18% vs 10%, respectively), and among CXCR4WT genotype patients (34% vs 24%, respectively) – showcasing the drug’s efficacy regardless of CXCR4 mutational status.51 At longer follow-up of a median of 24.2 months, the combined CR and VGPR was 30.4% and 18.2% in the zanubrutinib-treated group and ibrutinib-treated group, respectively.52
Another phase 1/2 study on zanubrutinib in 77 patients with treatment-naïve or R/R WM had patients take 160 mg twice daily (n = 50) or 320 mg once daily (n = 23).53 At a long-term median of 36-months follow-up, ORR and VGPR were reported to be 96% and 45%.53 Estimated 3-year PFS and OS were reported as 80.5% and 84.8%.53 In terms of genotype responses, 49.1% and 25% of patients with the MYD88L265 and MYD88WT genotypes achieved a VGPR/CR.53 Additionally, 59% and 27.3% of patients with the MYD88L265P/CXCR4WT and MYD88L265P/CXCR4WHIM genotypes achieved a VGPR/CR.53 However, MRRs were comparable (87.2% vs 90.9%) in the groups with and without the CXCR4 mutation.53 Ultimately, this study showed long-term durable responses with single-agent zanubrutinib in WM.53
Finally, a single-arm, multicenter, phase II study investigated the efficacy and safety of 160mg of zanubrutinib twice daily in 44 Chinese patients with R/R WM.54 The focus on an Asian population for this study was done due to the underrepresentation of this population in prior investigations.54 At a median follow-up of 33 months, MRR and VPGR rates were reported as 69.8% and 32.6%, respectively.54 Additionally, patients with the MYD88L265 and MYD88WT genotypes achieved MRRs of 73% and 50%, respectively.54 Overall, the study found higher response rates in the MYD88L265P/CXCR4WT group when compared to the other groups.54 For all patients, the 24-month PFS and OS were reported as 60.5% and 87.8%, respectively.54 Overall, this study displayed zanubrutinib’s efficacy in treating WM in Asian populations.
Toxicity
In the ASPEN study, in the group who received zanubrutinib, at a median follow-up of 19.4 months, the most common adverse events of any grade were neutropenia (29%), upper respiratory infection (24%), and diarrhea (21%).51 On the other hand, in the ibrutinib-treated group, the most common adverse events of any grade were diarrhea (32%), upper respiratory infection (29%), contusion (24%), and muscle spasms (24%).51 In the zanubrutinib and ibrutinib treated groups, grade 3 or higher adverse rates occurred in 58% and 63% of patients, respectively.51 Grade 3 or higher hypertension and pneumonia occurred more frequently in the ibrutinib-treated patients while a grade 3 or higher neutropenia occurred in the zanubrutinib-treated patients.51 Additionally, 40% and 41% of zanubrutinib and ibrutinib-treated patients experienced at least one serious adverse event, respectively.51 The most common serious adverse events in the zanubrutinib versus ibrutinib groups were pneumonia (seen in 1 vs 9 patients), neutropenia (3 vs 0), influenza (3 vs 1), and sepsis (2 vs 3).51 Atrial fibrillation/flutter occurred within 6 months of treatment in 1 zanubrutinib-treated patient and 7 ibrutinib-treated patients; beyond 12 months of treatment, this occurred in only an additional 4 ibrutinib-treated patients.51 Hypertension also occurred beyond 12 months of treatment in 1 zanubrutinib-treated patient and 6 ibrutinib-treated patients.51 However, zanubrutinib-treated patients had a much greater incidence of any grade neutropenia (25% vs 12%) and grade 3 or higher neutropenia (20% vs 8%) when compared to ibrutinib-treated patients.51 Two ibrutinib-treated patients died from complications related to sepsis, and one zanubrutinib-treated patient died from complications of cardiac arrest.51
At longer median 24.2 months of follow-up, the common adverse events of any grade to occur in the zanubrutinib-treated versus ibrutinib-treated groups were atrial fibrillation/flutter (3.0% vs 18.4%), bleeding (50.5% vs 60.2%), major hemorrhage (5.9% vs 10.2%), diarrhea (21.8% vs 32.7%), hypertension (12.9% vs 20.4%) and neutropenia (31.7% vs 15.3%).52 Zanubrutinib-treated patients had higher rates of grade 3 or higher neutropenia compared to ibrutinib-treated patients (22.8% vs 8.2%).52 However, grade 3 or higher infection rates were comparable between the zanubrutinib-treated and ibrutinib-treated patients (18.8% vs 23.5%).52 No additional patients had any adverse event that led to death.52
In the phase 1/2 study, at long-term median follow-up of 36 months, all patients had experienced at least one grade 1 adverse event, and 58.4% reported at least one grade 3 or higher adverse event.53 The most common adverse events of any grade were upper respiratory tract infection (51.9%), contusion (32.5%), and cough (22.1%).53 For the grade 3 or higher adverse events, the most commonly reported ones were neutropenia (15.6%), anemia (9.1%), basal cell carcinoma (5.2%), cellulitis (5.2%), hypertension (3.9%), pneumonia (3.9%), diarrhea (2.6%), headache (2.6%), fall (2.6%), and actinic keratosis (2.6%).53 A total of nine patients died during the study; two died from progressive disease, two died from unknown causes, and five died from adverse events.53 These adverse events leading to death included abdominal sepsis, bacterial arthritis, Scedosporium infection, gastric adenocarcinoma, and worsening bronchiectasis.53 However, it should be noted, that four of the five adverse events leading to death were deemed to be unrelated to the study drug.53
In the Asian population focused study, at median follow-up of 33 months, the most frequently reported treatment-emergent adverse events were neutropenia (59.1%), lymphopenia (31.8%), upper respiratory tract infection (29.5%), pneumonia (29.5%), and thrombocytopenia (29.5%).54 In terms of grade 3 or higher adverse events, the most reported ones were neutropenia (31.8%), thrombocytopenia (20.5%), pneumonia (20.5%), lymphopenia (11.4%), and hypertension (11.4%).54 It should be noted that none of the patients in this study experienced atrial fibrillation or tumor lysis syndrome.54 Serious adverse events occurred in 56.8% of patients and these were pneumonia (22.7%), upper respiratory tract infection (6.8%), skin infection (4.5%), and pleural effusion (4.5%).54 Seven patients (15.9%) died during the study.54 Three died from disease progression, three from adverse events, and one from severe pneumonia and respiratory failure.54 The adverse events leading to death were acute hepatitis B multi-organ dysfunction syndrome, and respiratory failure.54
Tolerability
In the ASPEN study, at median follow-up of 19.4 months, the median treatment durations for zanubrutinib and ibrutinib were 18.7 months and 18.6 months, respectively.51 For both groups, the median dose intensity was 98%. Due to adverse events, 23% of ibrutinib-treated patients needed to have their dose reduced, while 14% of zanubrutinib-treated patients required a dose reduction. Nine percent of ibrutinib-treated patients discontinued treatment altogether for adverse events of myocardial infarction, sepsis, death, drug-induced liver injury, hepatitis, interstitial lung disease, and pneumonia. On the other hand, 4% of zanubrutinib-treated patients discontinued treatment for adverse events of subdural hemorrhage, cardiac arrest, neutropenia, and IgA multiple myeloma. At a longer follow-up of 24.2 months, no additional zanubrutinib-treated patients discontinued treatment due to adverse events.52 However, 5 additional ibrutinib-treated patients discontinued treatment.52 Thus, these findings illustrated greater tolerability of zanubrutinib compared to ibrutinib.
In the long-term phase 1/2 study evaluating zanubrutinib alone, at a median 36-months of follow-up, only 72.7% of patients remained on treatment.53 Thirteen percent discontinued zanubrutinib because of adverse events, of which only one was attributed to the treatment.53 On the other hand, 10.4% had to discontinue treatment because of disease progression, and 3.9% discontinued for other reasons.53 At long-term follow-up, no patients required dose reduction of zanubrutinib.53
In the Asian population focused study, at a median follow-up of 33 months, seventeen patients (38.6%) had to have treatment interruption rather than reduction due to adverse events including pneumonia (13.6%), thrombocytopenia (6.8%), neutropenia (4.5%), and thrombocytopenia (4.5). After supportive treatment, most of these patients were able to resume treatment. However, six patients (16.3%) had to discontinue treatment altogether because of adverse events.54 These included pneumonia, laryngeal cancer, rapid WM disease progression with suspected transformation to aggressive lymphoma, intracranial mass, and acute hepatitis B with multi-organ dysfunctional syndrome.54 Only one patient in this study had to reduce their dose of the drug because of arthralgias, and these resolved with no recurrence.54
Future Trials
A search was conducted on May 03, 2022, on the current clinical trials on zanubrutinib in WM on clinicaltrials.gov.55 Studies not yet recruiting, recruiting, enrolling by invitation, active (not recruiting), suspended, and completed are summarized in Table 1. Studies with a status of terminated, unknown, and withdrawn were excluded.
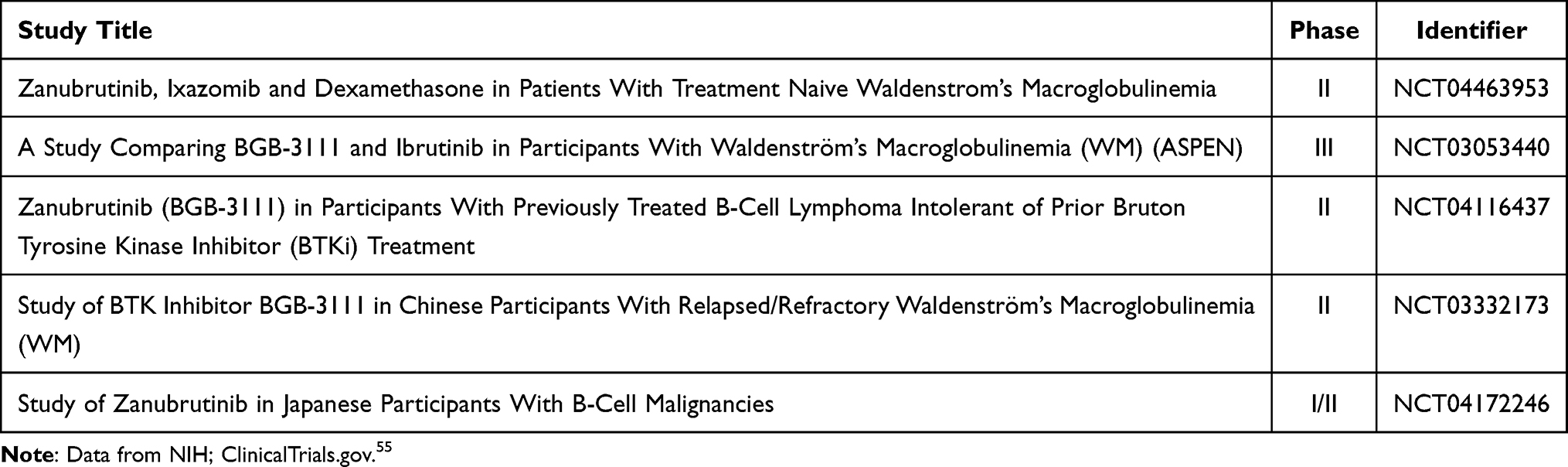 |
Table 1 A Comprehensive List of the Current (as of May 03, 2022) Clinical Trials Being Conducted on Zanubrutinib and WM |
Beyond the therapies described above, chimeric antigen receptor (CAR) T-cell therapies have shown great efficacy in treating a variety of B-cell malignancies. This therapy involves using a patient’s own T-cells and genetically engineering the cells to express a T-cell receptor that targets a specified tumor antigen.56 The current FDA approved CAR T-cell therapies include axicabtagene ciloleucel, tisagenlecleucel, lisocabtagene maraleucel, brexucabtagene autoleucel, idecabtagene vicleucel, and ciltacabtagene autoleucel. Clinical trials evaluating the efficacy of these therapies have shown strong ORR and CR in R/R B-cell malignancies. Although all the therapies discussed above have shown efficacy in treating WM, the studies eventually have reported disease relapse or progression. Additionally, the studies have discussed dose reduction or treatment cessation due to intolerance. Therefore, to address these issues, studies are looking to evaluate the efficacy and safety of CAR T-cell therapy in WM. So far, one study has reported that three patients with R/R WM treated with anti-CD19 CAR T-cell therapy had a clinical response to treatment.57 However, all three of them had disease recurrence within 3 to 26 months after the initial treatment.57 Of note, CAR T-cell therapy was well tolerated in all patients and no grade 3 or higher events of cytokine release syndrome or neurotoxicity occurred.57 We greatly look forward to further future studies evaluating the efficacy and safety of CAR T-cell therapy in WM.
Conclusions
Within the therapeutic landscape of WM, BTK inhibitors have emerged as greatly effective therapies. The first BTKi used in WM was ibrutinib; however, this treatment as a single agent displayed reduced efficacy in patients with the CXCR4 mutation. Additionally, ibrutinib was associated with many adverse events that led to drug dose reduction or discontinuation. Subsequently, the newer, highly specific BTKi zanubrutinib emerged. This drug showed efficacy in treating WM, regardless of CXCR4 mutation status, with a favorable toxicity profile. Table 2 summarizes the efficacy and safety data from the main clinical trials evaluating zanubrutinib in WM. Furthermore, randomized trials comparing zanubrutinib directly to ibrutinib in B-cell lymphoproliferative disorders such as CLL or WM showed that zanubrutinib conferred fewer toxicities and greater tolerability. Thus, the last of this drug class to become FDA approved in B-cell lymphoproliferative disorders, zanubrutinib, rises as a major player for the treatment of WM. Certainly, additional non-covalent BTKi are under development which, if proven to be safer and perhaps more efficacious than current options available, will reinforce the paradigm that the last shall be first as the therapeutic field continues to evolve.
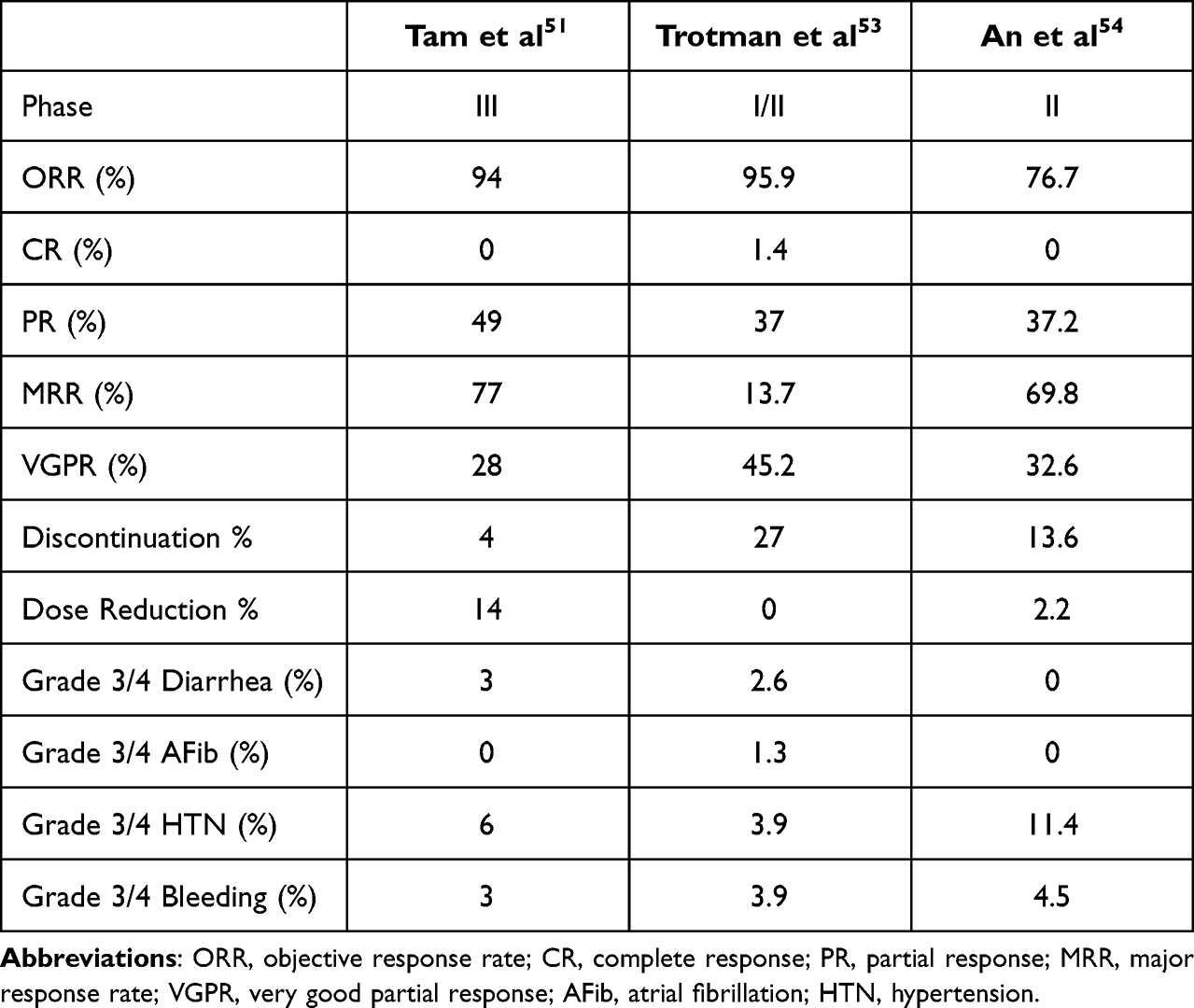 |
Table 2 A Summative Table on the Data Published in the Clinical Trials Evaluating Zanubrutinib in WM |
Disclosure
Javier Munoz reports personal fees from consulting for Pharmacyclics/AbbVie, Bayer, Gilead/Kite Pharma, Pfizer, Janssen, Juno/Celgene, BMS, Kyowa, Alexion, Fosunkite, Innovent, Seattle Genetics, Debiopharm, Karyopharm, Genmab, ADC Therapeutics, Epizyme, Beigene, Servier, Novartis, and Morphosys/Incyte, grants from research funding from Bayer, Gilead/Kite Pharma, Celgene, Merck, Portola, Incyte, Genentech, Pharmacyclics, Seattle Genetics, Janssen, Millennium, personal fees from Honoraria from Targeted Oncology, OncView, Curio, Kyowa, Physicians’ Education Resource, and Seattle Genetics, personal fees from speaker’s bureaus for Gilead/Kite Pharma, Kyowa, Bayer, Pharmacyclics/Janssen, Seattle Genetics, Acrotech/Aurobindo, Beigene, Verastem, AstraZeneca, Celgene/BMS, and Genentech/Roche, during the conduct of the study. The authors report no other potential conflicts of interest in relation to this work.
References
1. Fowler N, Davis E. Targeting B-cell receptor signaling: changing the paradigm. Hematology Am Soc Hematol Educ Program. 2013;2013(1):553–560. doi:10.1182/asheducation-2013.1.553
2. Herman SE, Gordon AL, Hertlein E, et al. Bruton tyrosine kinase represents a promising therapeutic target for treatment of chronic lymphocytic leukemia and is effectively targeted by PCI-32765. Blood. 2011;117(23):6287–6296. doi:10.1182/blood-2011-01-328484
3. Vetrie D, Vořechovský I, Sideras P, et al. The gene involved in X-linked agammaglobulinaemia is a member of the src family of protein-tyrosine kinases. Nature. 1993;361(6409):226–233. doi:10.1038/361226a0
4. Palma M, Mulder TA, Osterborg A. BTK inhibitors in chronic lymphocytic leukemia: biological activity and immune effects. Front Immunol. 2021;12:686768. doi:10.3389/fimmu.2021.686768
5. Tam CS, Dimopoulos M, Garcia-Sanz R, et al. Pooled safety analysis of zanubrutinib monotherapy in patients with B-cell malignancies. Blood Adv. 2022;6(4):1296–1308. doi:10.1182/bloodadvances.2021005621
6. Cull G, Burger JA, Opat S, et al. Zanubrutinib for treatment-naive and relapsed/refractory chronic lymphocytic leukaemia: long-term follow-up of the phase I/II AU-003 study. Br J Haematol. 2022;196(5):1209–1218. doi:10.1111/bjh.17994
7. Tam CS, Trotman J, Opat S, et al. Phase 1 study of the selective BTK inhibitor zanubrutinib in B-cell malignancies and safety and efficacy evaluation in CLL. Blood. 2019;134(11):851–859. doi:10.1182/blood.2019001160
8. Wang ML, Rule S, Martin P, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2013;369(6):507–516. doi:10.1056/NEJMoa1306220
9. Tam CS, Opat S, Simpson D, et al. Zanubrutinib for the treatment of relapsed or refractory mantle cell lymphoma. Blood Adv. 2021;5(12):2577–2585. doi:10.1182/bloodadvances.2020004074
10. Davis RE, Ngo VN, Lenz G, et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature. 2010;463(7277):88–92. doi:10.1038/nature08638
11. Noy A, de Vos S, Thieblemont C, et al. Targeting Bruton tyrosine kinase with ibrutinib in relapsed/refractory marginal zone lymphoma. Blood. 2017;129(16):2224–2232. doi:10.1182/blood-2016-10-747345
12. Hunter ZR, Yang G, Xu L, Liu X, Castillo JJ, Treon SP. Genomics, signaling, and treatment of waldenstrom macroglobulinemia. J Clin Oncol. 2017;35(9):994–1001. doi:10.1200/JCO.2016.71.0814
13. Castillo JJ, Moreno DF, Arbelaez MI, Hunter ZR, Treon SP. CXCR4 mutations affect presentation and outcomes in patients with Waldenstrom macroglobulinemia: a systematic review. Expert Rev Hematol. 2019;12(10):873–881. doi:10.1080/17474086.2019.1649132
14. Poulain S, Roumier C, Decambron A, et al. MYD88 L265P mutation in Waldenstrom macroglobulinemia. Blood. 2013;121(22):4504–4511. doi:10.1182/blood-2012-06-436329
15. Treon SP, Hunter ZR, Aggarwal A, et al. Characterization of familial Waldenström’s macroglobulinemia. Ann Oncol. 2006;17(3):488–494. doi:10.1093/annonc/mdj111
16. Vijay A, Gertz MA. Waldenström macroglobulinemia. Blood. 2007;109(12):5096–5103. doi:10.1182/blood-2006-11-055012
17. Fadil A, Taylor DE. The lung and Waldenstrom’s macroglobulinemia. South Med J. 1998;91(7):681–685. doi:10.1097/00007611-199807000-00017
18. Rosenthal JA, Curran WJ
19. Varettoni M, Defrancesco I, Diamanti L, Marchioni E, Farina LM, Pichiecchio A. Bing-Neel syndrome: illustrative cases and comprehensive review of the literature. Mediterr J Hematol Infect Dis. 2017;9(1):e2017061. doi:10.4084/mjhid.2017.061
20. Castillo JJ, Itchaki G, Paludo J, et al. Ibrutinib for the treatment of Bing-Neel syndrome: a multicenter study. Blood. 2019;133(4):299–305. doi:10.1182/blood-2018-10-879593
21. Hashmi H, Dhanoa JS, Emmons R. Rare case of Bing-Neel syndrome treated successfully with ibrutinib. BMJ Case Rep. 2019;12(6):e230067. doi:10.1136/bcr-2019-230067
22. Rosenthal A, Munoz J. Zanubrutinib for the treatment of B-cell malignancies. touchRev Oncol Haematol. 2022;18:1.
23. NCCN; National Comprehensive Cancer Network. NCCN Guidelines. Available from: https://www.nccn.org/guidelines/guidelines-detail?category=1&id=1475.
24. Vital C, Vital A, Deminiere C, Julien J, Lagueny A, Steck AJ. Myelin modifications in 8 cases of peripheral neuropathy with Waldenstrom’s macroglobulinemia and anti-MAG activity. Ultrastruct Pathol. 1997;21(6):509–516. doi:10.3109/01913129709016367
25. Chaudhry HM, Mauermann ML, Rajkumar SV. Monoclonal gammopathy-associated peripheral neuropathy: diagnosis and management. Mayo Clin Proc. 2017;92(5):838–850. doi:10.1016/j.mayocp.2017.02.003
26. Berkman EM, Orlin JB. Use of plasmapheresis and partial plasma exchange in the management of patients with cryoglobulinemia. Transfusion. 1980;20(2):171–178. doi:10.1046/j.1537-2995.1980.20280169957.x
27. Menke MN, Feke GT, McMeel JW, Treon SP. Effect of plasmapheresis on hyperviscosity-related retinopathy and retinal hemodynamics in patients with Waldenstrom’s macroglobulinemia. Invest Ophthalmol Vis Sci. 2008;49(3):1157–1160. doi:10.1167/iovs.07-1254
28. Stone MJ, Bogen SA. Role of plasmapheresis in Waldenstrom’s macroglobulinemia. Clin Lymphoma Myeloma Leuk. 2013;13(2):238–240. doi:10.1016/j.clml.2013.02.013
29. Dimopoulos MA, Zervas C, Zomas A, et al. Treatment of Waldenstrom’s macroglobulinemia with rituximab. J Clin Oncol. 2002;20(9):2327–2333. doi:10.1200/JCO.2002.09.039
30. Gertz MA, Anagnostopoulos A, Anderson K, et al. Treatment recommendations in Waldenstrom’s macroglobulinemia: consensus panel recommendations from the second International Workshop on Waldenstrom’s Macroglobulinemia. Semin Oncol. 2003;30(2):121–126. doi:10.1053/sonc.2003.50039
31. Zheng YH, Xu L, Cao C, et al. Rituximab-based combination therapy in patients with Waldenstrom macroglobulinemia: a systematic review and meta-analysis. Onco Targets Ther. 2019;12:2751–2766. doi:10.2147/OTT.S191179
32. Ghobrial IM, Fonseca R, Greipp PR, et al. Initial immunoglobulin M ‘flare’ after rituximab therapy in patients diagnosed with Waldenstrom macroglobulinemia: an Eastern Cooperative Oncology Group Study. Cancer. 2004;101(11):2593–2598. doi:10.1002/cncr.20658
33. Castillo JJ, Kanan S, Meid K, Manning R, Hunter ZR, Treon SP. Rituximab intolerance in patients with Waldenstrom macroglobulinaemia. Br J Haematol. 2016;174(4):645–648. doi:10.1111/bjh.13794
34. Rummel MJ, Niederle N, Maschmeyer G, et al. Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: an open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet. 2013;381(9873):1203–1210. doi:10.1016/S0140-6736(12)61763-2
35. Rummel MJ, Lerchenmüller C, Hensel M, et al. Two years rituximab maintenance vs. observation after first Line treatment with bendamustine plus rituximab (B-R) in patients with Waldenström’s Macroglobulinemia (MW): results of a prospective, randomized, multicenter phase 3 study (the StiL NHL7-2008 MAINTAIN trial). Blood. 2019;134:343.
36. Dimopoulos MA, Anagnostopoulos A, Kyrtsonis M-C, et al. Primary treatment of Waldenström macroglobulinemia with dexamethasone, rituximab, and cyclophosphamide. J Clin Oncol. 2007;25(22):3344–3349. doi:10.1200/JCO.2007.10.9926
37. Treon SP, Hunter ZR, Matous J, et al. Multicenter clinical trial of bortezomib in relapsed/refractory Waldenstrom’s macroglobulinemia: results of WMCTG Trial 03-248. Clin Cancer Res. 2007;13(11):3320–3325. doi:10.1158/1078-0432.CCR-06-2511
38. Sklavenitis-Pistofidis R, Capelletti M, Liu CJ, et al. Bortezomib overcomes the negative impact of CXCR4 mutations on survival of Waldenstrom macroglobulinemia patients. Blood. 2018;132(24):2608–2612. doi:10.1182/blood-2018-07-863241
39. Chen CI, Kouroukis CT, White D, et al. Bortezomib is active in patients with untreated or relapsed Waldenstrom’s macroglobulinemia: a phase II study of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007;25(12):1570–1575.
40. Castillo JJ, Allan JN, Siddiqi T, et al. Venetoclax in previously treated Waldenström Macroglobulinemia. J Clin Oncol. 2022;40(1):63–71. doi:10.1200/JCO.21.01194
41. Papanota AM, Ntanasis-Stathopoulos I, Kastritis E, Dimopoulos MA, Gavriatopoulou M. Evaluating ibrutinib in the treatment of symptomatic Waldenstrom’s macroglobulinemia. J Blood Med. 2019;10:291–300. doi:10.2147/JBM.S183997
42. Treon SP, Tripsas CK, Meid K, et al. Ibrutinib in previously treated Waldenström’s Macroglobulinemia. N Engl J Med. 2015;372(15):1430–1440. doi:10.1056/NEJMoa1501548
43. Treon SP, Meid K, Gustine J, et al. Long-term follow-up of ibrutinib monotherapy in symptomatic, previously treated patients with Waldenström Macroglobulinemia. J Clin Oncol. 2021;39(6):565–575. doi:10.1200/JCO.20.00555
44. Dimopoulos MA, Tedeschi A, Trotman J, et al. Phase 3 trial of ibrutinib plus rituximab in Waldenström’s Macroglobulinemia. N Engl J Med. 2018;378(25):2399–2410. doi:10.1056/NEJMoa1802917
45. Buske C, Tedeschi A, Trotman J, et al. Ibrutinib plus rituximab versus placebo plus rituximab for Waldenström’s Macroglobulinemia: final analysis from the randomized Phase III iNNOVATE study. J Clin Oncol. 2022;40(1):52–62. doi:10.1200/JCO.21.00838
46. Owen RG, McCarthy H, Rule S, et al. Acalabrutinib monotherapy in patients with Waldenström macroglobulinemia: a single-arm, multicentre, phase 2 study. Lancet Haematol. 2020;7(2):e112–e121. doi:10.1016/S2352-3026(19)30210-8
47. Guo Y, Liu Y, Hu N, et al. Discovery of Zanubrutinib (BGB-3111), a novel, potent, and selective covalent inhibitor of Bruton’s tyrosine kinase. J Med Chem. 2019;62(17):7923–7940. doi:10.1021/acs.jmedchem.9b00687
48. Ou YC, Liu L, Tariq B, et al. Population pharmacokinetic analysis of the BTK inhibitor zanubrutinib in healthy volunteers and patients with B-cell malignancies. Clin Transl Sci. 2021;14(2):764–772. doi:10.1111/cts.12948
49. Tam CS, Ou YC, Trotman J, Opat S. Clinical pharmacology and PK/PD translation of the second-generation Bruton’s tyrosine kinase inhibitor, zanubrutinib. Expert Rev Clin Pharmacol. 2021;14(11):1329–1344. doi:10.1080/17512433.2021.1978288
50. FDA approves zanubrutinib for Waldenström’s macroglobulinemia. U.S. Food & Drug Administration; 2021. Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-zanubrutinib-waldenstroms-macroglobulinemia.
51. Tam CS, Opat S, D’Sa S, et al. A randomized phase 3 trial of zanubrutinib vs ibrutinib in symptomatic Waldenström macroglobulinemia: the ASPEN study. Blood. 2020;136(18):2038–2050. doi:10.1182/blood.2020006844
52. BeiGene presents updated head to head results from phase 3 trial of zanubrutinib vs. ibrutinib in patients with Waldenström’s Macroglobulinemia at the 2020 American Society of Clinical Oncology (ASCO) virtual scientific program. BeiGene, LTD; 2020. Available from: https://ir.beigene.com/news-details/?id=f4dce275-d7c9-4f50-8f0d-2d2ce07db346.
53. Trotman J, Opat S, Gottlieb D, et al. Zanubrutinib for the treatment of patients with Waldenstrom macroglobulinemia: 3 years of follow-up. Blood. 2020;136(18):2027–2037. doi:10.1182/blood.2020006449
54. An G, Zhou D, Cheng S, et al. A Phase II trial of the Bruton tyrosine-kinase inhibitor zanubrutinib (BGB-3111) in patients with relapsed/refractory Waldenström Macroglobulinemia. Clin Cancer Res. 2021;27(20):5492–5501. doi:10.1158/1078-0432.CCR-21-0539
55. NIH. ClinicalTrials.gov; 2022. Available from: https://clinicaltrials.gov/ct2/results?cond=Waldenstrom+Macroglobulinemia&term=BGB-3111&cntry=&state=&city=&dist=&Search=Search&recrs=a&recrs=b&recrs=d&recrs=e&recrs=f&recrs=g.
56. Feins S, Kong W, Williams EF, Milone MC, Fraietta JA. An introduction to chimeric antigen receptor (CAR) T-cell immunotherapy for human cancer. Am J Hematol. 2019;94(S1):S3–S9. doi:10.1002/ajh.25418
57. Palomba ML, Qualls D, Monette S, et al. CD19-directed chimeric antigen receptor T cell therapy in Waldenström macroglobulinemia: a preclinical model and initial clinical experience. J Immunother Cancer. 2022;10(2):e004128. doi:10.1136/jitc-2021-004128
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.