Back to Journals » Degenerative Neurological and Neuromuscular Disease » Volume 16
(Z)-Endoxifen as a Potential Modulator of Utrophin Pathways in Duchenne Muscular Dystrophy: A Mechanistic and Transcriptomic Perspective
Authors Remmel HL ![]() , Hammer SS
, Hammer SS ![]() , Blackburn SM, Quay SC
, Blackburn SM, Quay SC ![]()
Received 9 November 2025
Accepted for publication 7 May 2026
Published 20 May 2026 Volume 2026:16 574524
DOI https://doi.org/10.2147/DNND.S574524
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Müller
H Lawrence Remmel,1,2 Sandra S Hammer,1 Scott M Blackburn,1 Steven C Quay1
1Atossa Therapeutics Inc., Seattle, WA, USA; 2Department of Pathology, University Medical Center Utrecht, Utrecht, the Netherlands
Correspondence: H Lawrence Remmel, Email [email protected]
Abstract: Duchenne muscular dystrophy (DMD) is a fatal X-linked neuromuscular disorder caused by mutations in the dystrophin gene, leading to sarcolemmal instability, chronic calcium overload, mitochondrial dysfunction, inflammation, and fibrosis. These effects manifest clinically as muscle atrophy, loss of mobility, and respiratory and cardiac insufficiencies. Although gene-targeted approaches such as exon skipping and microdystrophin delivery have advanced, their applicability is restricted by genotype and adverse effects. Thus, reasonably safe, effective therapies remain a critical unmet need. (Z)-endoxifen, the (Z)-isomer of endoxifen, is the principal active metabolite of tamoxifen, exhibiting higher potency in protein kinase C (PKC) inhibition than tamoxifen and regulating various protein activities differently than tamoxifen. This pharmacologic profile positions endoxifen to address multiple DMD pathological mechanisms simultaneously. Recent literature supports the prior hypothesis that endoxifen exerts pleiotropic benefits in DMD. Transcriptomic and preclinical evidence shows that endoxifen may have superior effects to tamoxifen in reversing DMD signatures given available dosing regimens. Additionally, pathway-level insights support endoxifen activation of myogenesis, oxidative phosphorylation, estrogen receptor beta (ERβ) activation, and inhibition of pro-inflammatory and epithelial-to-mesenchymal transition (EMT) pathways in DMD. These findings suggest (Z)-endoxifen may act on utrophin-linked regulatory axes, thereby potentially contributing to functional compensation for dystrophin deficiency. This provides justification for endoxifen’s development as a mutation-agnostic DMD therapeutic approach.
Keywords: gene expression regulation, selective estrogen receptor modulators, skeletal muscle regeneration, pharmacologic action mechanism, Becker muscular dystrophy, utrophin
Introduction and DMD Pathophysiology
Many neuromuscular disorders affect skeletal muscle. Muscular dystrophies (MDs) are an important therapeutic class of inherited myogenic disorders with varying types,1 characterized by progressive muscle wasting and weakness of variable distribution and severity, where muscle maintenance is compromised.2 Duchenne muscular dystrophy (DMD) and Becker muscular dystrophy (BMD) are the most common forms of MDs,3–5 with prevalence rates estimated at 4.8 and 1.6 per 100,000 people, respectively.3
DMD is caused by mutations in the X-linked DMD gene encoding dystrophin, a 427-kDa cytoskeletal protein that links the intracellular actin cytoskeleton to the extracellular matrix (ECM) via the dystrophin-associated glycoprotein complex (DGC).6–9 Loss of dystrophin disrupts this linkage, rendering the cell membranes (sarcolemma) surrounding skeletal muscle fibers and cardiomyocytes mechanically fragile.6,9 Contraction-induced microtears allow unregulated calcium influx, activating calpains and other proteases, triggering mitochondrial permeability transition, and leading to myofiber necrosis.10,11 Damaged fibers are replaced by fibrotic and adipose tissue, and over time the regenerative capacity of muscle declines, culminating in progressive weakness, loss of ambulation, cardiomyopathy, respiratory failure, and premature death.6,9,12 Current standard-of-care corticosteroids, such as prednisone and deflazacort, improve survival, prolong ambulation, and slow functional decline in patients with DMD; however, their long-term efficacy remains limited and treatment is associated with significant adverse effects, including weight gain, osteoporosis, growth delay, and behavioral changes.6,9,13 As a result, corticosteroid therapy primarily mitigates disease progression rather than addressing the disease’s underlying genetic defect. In recent years, mutation-specific therapies such as exon-skipping oligonucleotides have been developed, although these approaches are applicable to only a subset of patients, with approximately 27% of individuals with DMD amenable to currently FDA-approved exon-skipping therapies.14 Additional therapeutic strategies under investigation include other mutation-specific approaches, such as nonsense read-through therapies, as well as mutation-agnostic approaches aimed at improving muscle function or modifying disease progression, including gene therapy, gene editing, and utrophin modulation. Collectively, these limitations underscore the urgent need for safe, broadly applicable disease-modifying or curative therapies for DMD.
Like DMD, BMD, a less severe dystrophy, involves dystrophinopathies with an X-linked inheritance. Patients with BMD produce inadequate amounts of dystrophin, though they produce more dystrophin than patients with DMD. BMD and DMD belong to the dystrophinopathy spectrum as they share the same pathogenic gene and characteristics such as progressive muscle degeneration and dilated cardiomyopathy.15,16 Whereas BMD patients with in-frame mutations retain truncated dystrophin production, DMD patients lack functional protein production. A significant portion, approximately 20%, of DMD and BMD recessive carriers have clinical manifestations of DMD/BMD. If left ventricle involvement is included, the proportion of carriers with symptoms increases to approximately 40%.17 Because DMD and BMD share similar pathology, it is possible that therapies that benefit DMD patients would also benefit BMD patients,1 and may also benefit manifesting DMD and BMD recessive carriers.
Recent advances in mutation-specific therapies, such as antisense oligonucleotide (ASO)-mediated exon skipping and adeno-associated virus (AAV)-delivered, mutation-independent microdystrophin, have provided proof-of-concept that restoring dystrophin can improve outcomes.18–20 However, these approaches are constrained by antibody responses, other adverse effects, and uncertainties about long-term expression and durability.6,21 Although these approaches are in the clinic, their efficacy is limited and they generally only slow disease progression.22 Consequently, there is a pressing need for mutation-agnostic therapies that address shared downstream pathophysiological mechanisms.6
Tamoxifen, a selective estrogen receptor modulator (SERM), has demonstrated functional and histological benefits in mdx mouse models of DMD and in early clinical trials.23 Its mechanisms include membrane stabilization, anti-inflammatory effects, and attenuation of fibrosis. Importantly, chronic treatment with 4-OHT, a tamoxifen metabolite, in a bioengineered human iPSC-derived cardiomyocyte model slowed beating rate, increased contractile force, corrected calcium dysregulation, and improved survival of DMD cardiomyocytes, supporting tamoxifen’s potential as a cardioprotective therapy in DMD.24
(Z)-endoxifen, the primary active metabolite of tamoxifen, is generated in large part via CYP2D6 metabolism and has been shown to inhibit protein kinase C (PKC) isoforms, particularly PKC beta-1 (PKCβ1).25 PKCβ1 is implicated in pro-fibrotic transforming growth factor beta (TGF-β) signaling and immune activation in dystrophic muscle, making it an attractive target for modulation.26
Beyond estrogen receptor and PKC pathways, transcriptomic analyses indicate that endoxifen more effectively normalizes DMD-associated gene expression than tamoxifen.27 Bulk ribonucleic acid sequencing (RNA-seq) from dystrophic muscle treated with tamoxifen or endoxifen reveals significant activation of myogenic programs,28,29 mitochondrial oxidative phosphorylation,25,30 and estrogen-responsive signaling.25,31 These changes are consistent with a shift toward a regenerative, anti-fibrotic phenotype that could preserve muscle function.6
One pathway that integrates these effects is utrophin upregulation.6,32–34 Utrophin, which is widely expressed throughout all fetal stages, shares structural and functional homology with dystrophin and binds similar partners within the DGC.23,35 In healthy muscle, following birth, its expression is largely restricted to the neuromuscular and myotendinous junctions. In the absence of dystrophin, utrophin can be redistributed along the sarcolemma, partially compensating for the structural deficit.6,33,34,36 Importantly, genetic overexpression or pharmacologic upregulation of utrophin in mdx mice ameliorates DMD pathology without developmental toxicity.6,33,34
The promise of utrophin-based strategies in DMD is underpinned by evidence that utrophin can functionally compensate for dystrophin deficiency, that its upregulation is effective and non-toxic, and that earlier expression yields greater therapeutic benefit.33,34,37–39 Despite this promise, clinical translation of direct utrophin activators such as SMT C1100 (ezutromid) has been limited by suboptimal pharmacokinetics and insufficient target engagement.6
Endoxifen’s combined pharmacologic actions—PKCβ1 inhibition, normalization of calcium-handling proteins (eg, sarcoplasmic/endoplasmic reticulum calcium ATPase 1 and 2 [SERCA1/2], ryanodine receptors), and suppression of inflammation—are all mechanistically aligned with enhancing utrophin expression and stability. In mouse models, endoxifen creates a cellular context conducive to utrophin transcription and incorporation into the sarcolemmal complex by restoring myogenic transcription factors such as myoblast determination protein 1 (MyoD) and myocyte enhancer factor 2 (MEF2), reducing antagonistic inflammatory signaling, and improving mitochondrial support.27,31
Given the multifactorial pathogenesis of DMD, therapies that modulate multiple converging pathways may offer the best potential for durable functional improvement.6,19 Accordingly, we conducted a hypothesis-driven, integrative review of transcriptomic, mechanistic, and preclinical evidence to explore the theoretical rationale for (Z)-endoxifen as a mutation-agnostic, multi-target therapeutic candidate in DMD, with utrophin pathway activation proposed as a central mechanism and potential biomarker for clinical translation.
Utrophin in DMD: The Rationale for Pharmacologic Upregulation
Utrophin is a structural homolog of dystrophin that shares considerable sequence and functional similarity.35,40–42 Unlike dystrophin, which is absent or truncated in DMD, utrophin is expressed ubiquitously during fetal development and later becomes restricted primarily to the neuromuscular junction and myotendinous junctions in mature muscle fibers.43,44 In DMD, a notable pathophysiological feature is the upregulation of utrophin in revertant fibers and regenerating myofibers, suggesting that utrophin can at least partially compensate for the absence of dystrophin.35,43 Observational studies in DMD have consistently shown that higher levels of utrophin are associated with milder clinical phenotypes and improved muscle integrity, even in the absence of functional dystrophin.6,44
The ability of utrophin to substitute for dystrophin has been further highlighted in natural history studies and animal models.35,45 For instance, utrn−/− knockout mice are viable and develop normally, demonstrating that utrophin is not essential for embryonic development.35 This finding demonstrated its favorable safety profile as a therapeutic target, since utrophin modulation would not be expected to result in deleterious developmental effects. Conversely, overexpression of utrophin in dystrophin-deficient mdx mice substantially reduces pathology, improves sarcolemmal stability, and enhances muscle function.39,46 Together, these findings establish that utrophin possesses a wide dosing range therapeutic potential, making it an attractive target for pharmacologic and genetic interventions.23,47
The p38 mitogen-activated protein kinase (MAPK) pathway plays a central role in regulating skeletal muscle differentiation and myogenic gene expression. During myogenesis, activation of p38 signaling promotes myoblast cell-cycle exit and enhances the activity of key muscle-specific transcription factors, including MyoD and MEF2, thereby driving the transcriptional programs required for myofiber formation and repair. In addition to its role in differentiation, p38 MAPK signaling has been implicated in the regulation of utrophin expression. Pharmacologic activation of this pathway with anisomycin, a potent p38 activator, has been shown to increase utrophin protein levels in C2C12 myoblasts and mdx primary myoblasts, as well as elevate utrophin expression in the diaphragm of mdx mice. These findings support a role for p38 signaling in the transcriptional control of utrophin in skeletal muscle.
Tamoxifen and its active metabolite, endoxifen, have also been reported to activate p38 MAPK signaling, raising the possibility that their ability to upregulate utrophin is mediated, at least in part, through this pathway. By engaging intracellular signaling cascades associated with myogenic differentiation, tamoxifen and endoxifen may enhance utrophin expression and thereby contribute to improved sarcolemmal stability in dystrophin-deficient muscle.
More recent work further refines the mechanistic basis of utrophin compensation in Duchenne muscular dystrophy (DMD), demonstrating that mutant DMD mRNA decay can drive UTRN upregulation through transcriptional adaptation in human cells. In this study, inhibition of nonsense-mediated decay reduced UTRN upregulation, whereas splice-switching antisense oligonucleotides (ASOs) that induced out-of-frame exon skipping increased UTRN expression. Conversely, restoration of the DMD reading frame using ASOs in DMDΔE52 myotubes attenuated utrophin upregulation. Together, these findings suggest that utrophin compensation is influenced not only by the absence of dystrophin, but also by mutation class and transcript fate—an important consideration when interpreting natural history variability and therapeutic responses in dystrophinopathies.48
Several strategies have been explored to harness utrophin as a therapeutic surrogate for dystrophin.40 One of the most extensively investigated pharmacologic approaches has been the development of small molecules that increase utrophin transcription.35 SMT C1100, an orally bioavailable utrophin modulator, demonstrated target engagement by elevating utrophin expression in preclinical models and advanced into clinical trials.49,50 However, despite promising mechanistic rationale, SMT C1100 failed to achieve consistent clinical efficacy in human trials and was ultimately discontinued due to poor bioavailability and limited therapeutic benefit.32,51 This underscores the challenges in achieving sufficient systemic exposure and sustained upregulation in muscle tissue.6
Beyond SMT C1100, other pharmacologic agents have been reported to modulate utrophin expression, including heregulin, biglycan, histone deacetylase inhibitors, and certain antibiotics.48,52 These approaches have shown variable success in preclinical studies but have yet to progress into late-stage clinical evaluation, in part due to concerns about specificity, systemic toxicity, or limited translational potential.6 Nonetheless, these agents collectively demonstrate the utility of utrophin transcriptional pathways and continue to serve as a foundation for future discovery efforts.
In parallel with pharmacologic modulation, gene therapy approaches have explored the delivery of engineered micro-utrophin constructs to restore dystrophin-like functionality.40,53 Micro-utrophin, by virtue of its reduced size relative to full-length dystrophin, is amenable to packaging into AAV vectors.54 Preclinical studies in mdx mice and canine DMD models have demonstrated that micro-utrophin can localize to the sarcolemma, interact with the dystrophin-associated glycoprotein complex, and ameliorate muscle pathology.33,34,38,55 Despite these advances, gene therapy with micro-utrophin is not without limitations: challenges in vector manufacturing, host immune responses, and the requirement for systemic delivery remain significant barriers to translation.54 Additionally, while micro-utrophin can partially substitute for dystrophin, its functional capacity may not be entirely equivalent, raising questions about long-term durability of clinical benefit.56
Collectively, the body of preclinical and early clinical evidence strongly supports utrophin as a viable therapeutic surrogate for dystrophin in DMD.35,40 Its ability to ameliorate pathology, non-essentiality in normal development, and favorable therapeutic potential make it a uniquely attractive target.6 Additionally, the presence of developmental myosin such as myosin heavy chain-embryonic (MyHC-emb) and neonatal myosin represents a useful marker of muscle regeneration, with utrophin upregulated in these fibers to help stabilize the sarcolemma. Developmental myosin is a meaningful indicator of muscle damage, which correlates with the clinical severity in DMD and BMD patients.57 In a therapeutic context, researchers are exploring utrophin upregulation strategies to mimic dystrophin function, and developmental myosin often serves as a readout for whether treatments improve regeneration.
While prior pharmacologic efforts such as SMT C1100 illustrate the difficulties in achieving clinical efficacy, the expanding toolbox of drug discovery, combined with advances in gene therapy, ensures that utrophin upregulation remains a critical and rational strategy for addressing the broad unmet needs in DMD.
Transcriptomic Insights into Endoxifen’s Action in DMD Skeletal Muscle
High-resolution transcriptomic profiling provides a systems-level perspective on how pharmacologic interventions influence disease-associated gene expression. In DMD, bulk RNA sequencing (RNA-seq) of dystrophic muscle consistently reveals activation of inflammatory and fibrotic pathways, downregulation of oxidative metabolism, and disruption of myogenic regulatory networks. Given endoxifen’s modulation of estrogen receptor (ER) signaling and inhibition of PKC and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathways, transcriptomic analyses represent a useful framework for evaluating potential shifts in inflammatory, fibrotic, and myogenic gene programs relative to healthy controls. Although a public endoxifen bulk RNA-seq dataset from dystrophic muscle is not yet available, transcriptomic datasets from related biological contexts indicate modulation of ER-responsive signaling, myogenic regulatory networks, oxidative phosphorylation pathways, and cytokine- and fibrosis-associated gene expression. Through ER-mediated signaling and anti-inflammatory pathway modulation, these transcriptomic patterns are consistent with modulation of pathways involving key myogenic transcription factors including MyoD, MYOG, and MEF2 within muscle gene expression networks. Key pathways normalized by endoxifen include myogenesis, oxidative phosphorylation, and estrogen-responsive signaling.29,30,58 Myogenesis-related transcripts, such as MYOD1, MYOG, and MEF2C, are upregulated by endoxifen toward levels seen in healthy controls.6 Oxidative phosphorylation genes encoding components of complexes I–V of the mitochondrial respiratory chain show coordinated restoration, indicative of improved metabolic capacity.6 Estrogen-responsive gene sets, which intersect with both metabolic and structural pathways, are also modulated, reflecting endoxifen’s potent estrogen receptor beta (ERβ) binding activity.6,59
Simultaneously, endoxifen suppresses several maladaptive transcriptional programs.27,28 Pathways associated with epithelial–mesenchymal transition (EMT), ECM remodeling, and fibrosis—driven by genes such as COL1A1, FN1, and MMP2—are significantly downregulated.25,60,61 Pro-inflammatory signaling molecules, including IL-6/JAK/STAT3 and NF-κB target genes, are attenuated, likely reflecting the compound’s combined PKCβ1 inhibition and ERβ activation.28 Genes governing apoptotic pathways and cell-cycle checkpoints, which can impede terminal myogenic differentiation, are also reduced in expression.25
When examined in the context of DMD’s regenerative deficits, these transcriptomic effects take on possible functional significance. Upregulation of myogenic regulators facilitates satellite cell activation and progression through the myogenic program, while suppression of inflammatory and fibrotic mediators creates a permissive microenvironment for effective regeneration.62 Normalization of mitochondrial gene expression supports the high energetic demands of repair and maintenance in dystrophic fibers.63
Comparison with tamoxifen-treated transcriptomes reveals that while both compounds share certain gene expression changes, endoxifen uniquely influences a broader array of calcium-handling, metabolic, and ECM-related genes.58 This includes stronger induction of ATP2A1/2 (SERCA1/2) and stabilization of ryanodine receptor transcripts, changes that directly relate to calcium homeostasis and downstream signaling relevant to muscle function.25 Enhanced suppression of fibrosis-related transcripts further distinguishes endoxifen, consistent with its more potent inhibition of PKCβ1—a kinase linked to TGF-β-driven fibrogenesis.25 Notably, SERMs do not bind directly to or regulate myosin itself, but influence muscle regeneration and fiber composition, leading to increased developmental myosin expression, preservation of adult myosin isoforms through reduced degeneration, and improved functional performance of myosin-driven contraction in impaired muscle.
Taken together, transcriptomic profiling highlights endoxifen as a multi-pathway modulator with the potential to influence gene expression networks disrupted in DMD. Its broader transcriptional impact compared with tamoxifen, particularly across metabolic and extracellular matrix–associated pathways, is consistent with its enhanced biochemical potency and multi-target pharmacology. These transcriptomic patterns intersect with established mechanistic drivers of DMD pathology, including calcium dysregulation, chronic inflammation, fibrosis, and impaired regeneration. As such, they provide a useful framework for understanding the potential biological effects of endoxifen and may help inform the development of pharmacodynamic biomarkers for future clinical studies.
Effects of Endoxifen on Calcium Homeostasis and Protein Kinase C Signaling
As noted previously, disruption of intracellular calcium homeostasis is a central pathogenic feature of DMD, arising from sarcolemmal instability and abnormal ion channel regulation.11,28,64 Loss of dystrophin leads to microtears in the sarcolemma during contraction, allowing uncontrolled calcium influx that overwhelms buffering systems.10,41 Excess cytosolic calcium activates calpains, phospholipases, and mitochondrial permeability transition pores, promoting myofiber necrosis.65,66 Chronic calcium overload also perturbs excitation–contraction coupling and impairs regenerative signaling.64,67 Restoring calcium balance is therefore a critical therapeutic goal in DMD.64,68
While it is unknown whether endoxifen exerts direct effects on calcium-handling proteins, it targets signaling pathways that modulate calcium homeostasis indirectly.28 As noted earlier, one of endoxifen’s most potent biochemical actions is inhibition of PKCβ1, which is a calcium-dependent kinase that becomes chronically activated in DMD muscle due to persistent calcium elevation.25,28 This hyperactivation drives phosphorylation of cytoskeletal and signaling proteins, enhancing membrane instability and promoting pro-fibrotic TGF-β signaling.25 PKCβ1 also amplifies inflammatory cascades via NF-κB activation, further aggravating myofiber damage.69
By inhibiting PKCβ1 at nanomolar concentrations—significantly more potently than tamoxifen—endoxifen interrupts this pathogenic feedback loop.25 Downstream consequences include reduced transcription of fibrosis-associated genes (eg, COL1A1, CTGF) and attenuation of inflammatory mediators such as tumor necrosis factor alpha (TNF-α) and interleukin 6 (IL-6).25,30,70 Independent of endoxifen exposure, PKC inhibition may also indirectly support calcium homeostasis by stabilizing store-operated calcium entry mechanisms, which are dysregulated in dystrophic muscle.71
While endoxifen potently inhibits PKCβ1 and induces its degradation, its effect on PKC theta (PKCθ) is notably different and less potent. That said, reduced PKCθ activity not only limits fibrosis and inflammation in an mdx mouse model but also improves the regenerative microenvironment, facilitating effective satellite cell differentiation.72,73
The combined normalization of calcium-handling protein expression and suppression of PKC-driven pathological signaling has several functional implications.10 Improved sarco/endoplasmic reticulum calcium ATPase (SERCA) activity and ryanodine receptor stability enhance contractile efficiency and reduce energy cost per contraction.74 Lower cytosolic calcium minimizes activation of calcium-dependent proteases, slowing myofiber degeneration.10 Endoxifen’s modulation of calcium homeostasis and PKC signaling intersects mechanistically with utrophin biology.28 Stable calcium levels preserve cytoskeletal organization and sarcolemmal integrity, creating a favorable context for utrophin anchorage along the membrane.75 Furthermore, suppression of PKC-mediated inhibitory phosphorylation events on transcription factors may directly enhance utrophin gene expression.25 In this way, calcium and PKC modulation serve both protective and compensatory roles, sustaining fibers that express utrophin while promoting conditions for its upregulation.25
When compared to tamoxifen, endoxifen demonstrates broader and more potent effects on calcium and PKC pathways, likely due to its higher binding affinity for PKCβ1 and stronger ERα degradation.25 These differences may underlie the greater transcriptomic gene-reversal rate observed with endoxifen, particularly for genes involved in calcium cycling and ECM remodeling.
Given the centrality of calcium and PKC dysregulation in DMD pathophysiology, the dual modulation achieved by endoxifen represents a compelling therapeutic advantage. Future studies could integrate in vivo calcium imaging, PKC activity assays, and utrophin quantification to fully delineate the interplay between these pathways and functional outcomes in dystrophin-deficient muscle.
Single-Cell and Pseudotemporal Myogenesis Trajectory Analysis
Single-cell RNA sequencing (scRNA-seq) offers an unprecedented view of cellular heterogeneity and dynamic state transitions in dystrophic muscle.76 In DMD, scRNA-seq of muscle tissue reveals profound alterations in the composition and transcriptional programs of myogenic and non-myogenic cell populations.77 Satellite cells, the primary muscle stem cells, display impaired activation and differentiation, while fibro-adipogenic progenitors (FAPs) and immune cells adopt pro-fibrotic and pro-inflammatory phenotypes.78,79 These changes disrupt the coordinated myogenesis necessary for effective regeneration.
Application of pseudotemporal trajectory inference to dystrophic muscle datasets allows reconstruction of the myogenic continuum from quiescent satellite cells through activated progenitors, committed myoblasts, and differentiated myofibers.80 In untreated DMD muscle, trajectories are truncated, with a bottleneck at the transition from committed myoblast to mature myofiber.81,82 This reflects both intrinsic defects in myogenic programming and extrinsic inhibition from the inflammatory and fibrotic milieu.
Based on known pathway interactions, endoxifen has been shown to induce transcriptomic changes at the single-cell level in breast cancer cells, although comparable analyses have not yet been performed in dystrophic muscle. In DMD, single-cell transcriptomic studies demonstrate that satellite cells capable of effective regeneration display increased expression of activation markers such as MYF5 and MYOD1, reflecting exit from quiescence and entry into the myogenic program. Downstream in this trajectory, committed progenitors exhibit elevated expression of myogenin (MYOG) and structural genes including ACTA1 and TNNT1, which are associated with progression toward terminal differentiation and myofiber formation. In addition to promoting myogenic progression, endoxifen may reshape the non-myogenic cell landscape. FAPs in treated muscle downregulate pro-fibrotic genes including COL1A1, POSTN, and TNC, while upregulating ECM-remodeling inhibitors such as tissue inhibitor metalloproteinases 3 (TIMP3).83 This shift may reduce pathological ECM deposition, creating a more permissive environment for myofiber regeneration. Similarly, based on its known anti-inflammatory and estrogen receptor–mediated effects, endoxifen may influence macrophage polarization; however, direct evidence demonstrating a shift from pro-inflammatory (M1-like) to reparative (M2-like) phenotypes in dystrophic muscle is currently lacking.73,84 Such immune modulation can directly enhance satellite cell function and survival.
When compared to tamoxifen, endoxifen produces broader single-cell transcriptional changes, particularly in non-myogenic populations.25,30 This includes stronger suppression of pro-fibrotic FAP programs and greater induction of oxidative and contractile gene networks in myogenic cells.28,30,31 Such effects may derive from endoxifen’s more potent PKCβ1 inhibition, ERβ activation, and ERα degradation, leading to deeper modulation of both myogenic and stromal signaling pathways.
By simultaneously enhancing satellite cell activation, promoting differentiation, suppressing fibrosis, modulating immune responses, and supporting vascular remodeling, endoxifen addresses multiple cellular bottlenecks in DMD muscle regeneration. These multi-cellular effects are likely to act in concert with endoxifen’s utrophin-enhancing actions to stabilize muscle structure and function over time. Future studies should include scRNA-seq in endoxifen-treated mdx muscle to corroborate this mechanism.
Integration with Utrophin Regulation Pathways
As noted previously, utrophin is a structural and functional homolog of dystrophin, sharing many of its binding partners in the DGC.40–42 In healthy muscle following birth, utrophin expression is largely restricted to the neuromuscular and myotendinous junctions.85,86 In the absence of dystrophin, utrophin is redistributed along the sarcolemma, where it can partially compensate for the structural deficit.23 Preclinical studies have shown that even modest increases in utrophin can confer significant functional benefit in dystrophin-deficient models, without the developmental toxicity seen with dystrophin overexpression.35,40
Pharmacologic activation of utrophin has been pursued as a therapeutic strategy for DMD for decades. Compounds such as heregulin, biglycan, and the small molecule SMT C1100 have demonstrated utrophin upregulation in vitro and in animal models but have failed to achieve robust and durable increases in clinical trials.23,44 Limitations have included insufficient potency, poor pharmacokinetics, and lack of pleiotropic benefit beyond utrophin induction.35,44
(Z)-endoxifen offers a distinct mechanistic profile in this context. Rather than acting solely through a single utrophin-promoter activation pathway, endoxifen exerts broad transcriptomic and signaling changes that create a permissive environment for utrophin expression and stabilization. Available transcriptomic and pathway analyses suggest possible upregulation of UTRN itself alongside key transcriptional activators such as MEF2C, GA-binding protein alpha (GABPα), and NRF2.35,47 These factors integrate inputs from estrogen receptor modulation, PKC inhibition, and metabolic regulators like peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), all of which are influenced by endoxifen.
ERα degradation has been hypothesized to relieve transcriptional repression at the UTRN locus. In cardiac muscle cells, ERα has been implicated in modulating MEF2 activity, and its downregulation can enhance MEF2-dependent transcription of structural genes.87 Similarly, PKCβ1 inhibition might reduce inhibitory phosphorylation of transcription factors and co-activators involved in utrophin regulation. The net result would be a transcriptional landscape more favorable to sustained utrophin expression.
Beyond transcription, endoxifen influences post-transcriptional and post-translational mechanisms relevant to utrophin biology.25,88 Normalization of calcium handling via SERCA upregulation and ryanodine receptor stabilization reduces calcium-dependent proteolysis of cytoskeletal proteins.89 This may prolong utrophin’s half-life at the sarcolemma. Endoxifen also attenuates fibrosis and chronic inflammation, limiting the ECM thickening and immune-mediated damage that can disrupt utrophin anchorage.25
In metabolic terms, activation of PGC-1α and mitochondrial oxidative phosphorylation pathways by endoxifen may indirectly enhance utrophin expression. PGC-1α not only drives mitochondrial biogenesis but has been linked to oxidative fiber type specification, which correlates with higher utrophin levels.90–92 Shifting the muscle transcriptome toward an oxidative phenotype could therefore reinforce both functional and structural stability.
From a spatial perspective, immunohistochemical analyses in preclinical models have shown that endoxifen treatment increases utrophin distribution along the extrasynaptic sarcolemma, not just at junctional sites.28 This widespread localization is critical in compensating for the loss of dystrophin across the fiber surface. In parallel, improvements in sarcolemmal integrity due to calcium normalization and reduced PKC activity may enhance the functional integration of utrophin-containing complexes.44,64
When compared with tamoxifen, endoxifen appears to achieve higher utrophin expression levels and more consistent extrasynaptic localization.28 These differences likely reflect its greater potency in both ERα degradation and PKCβ1 inhibition,25 theoretically leading to deeper modulation of the transcriptional and signaling networks that converge on utrophin regulation.
The integration of utrophin pathway activation with other protective mechanisms—calcium homeostasis, fibrosis suppression, metabolic optimization—positions endoxifen as more than just a utrophin activator.11 It functions as a multi-target modulator, leveraging utrophin enhancement as one element within a broader therapeutic program (Table 1 and Figure 1).28,59 This convergence may yield functional benefits greater than the sum of individual pathway effects, particularly in the heterogeneous and progressive context of DMD.
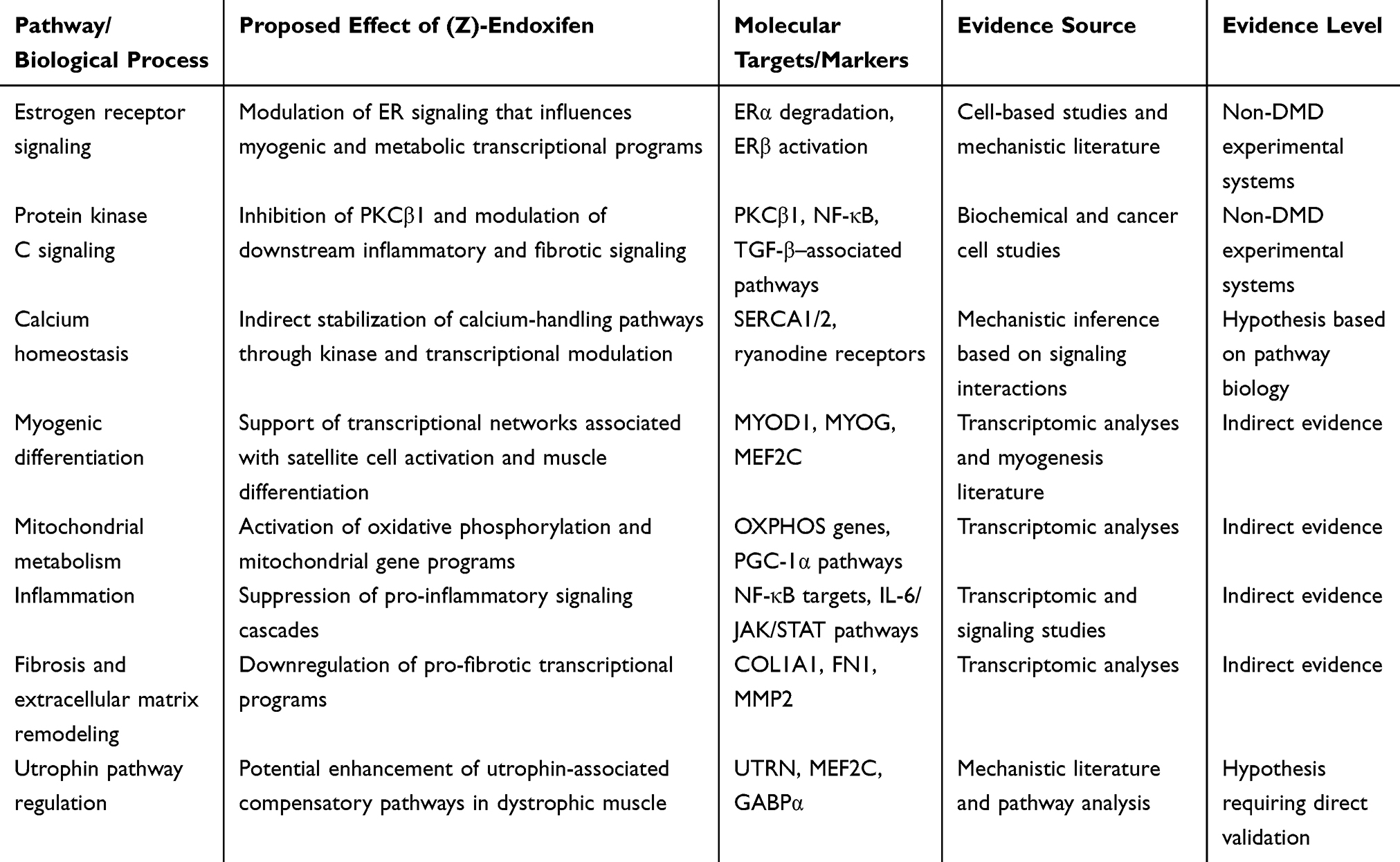 |
Table 1 Proposed Mechanisms of Action of (Z)-Endoxifen in Duchenne Muscular Dystrophy and Supporting Evidence |
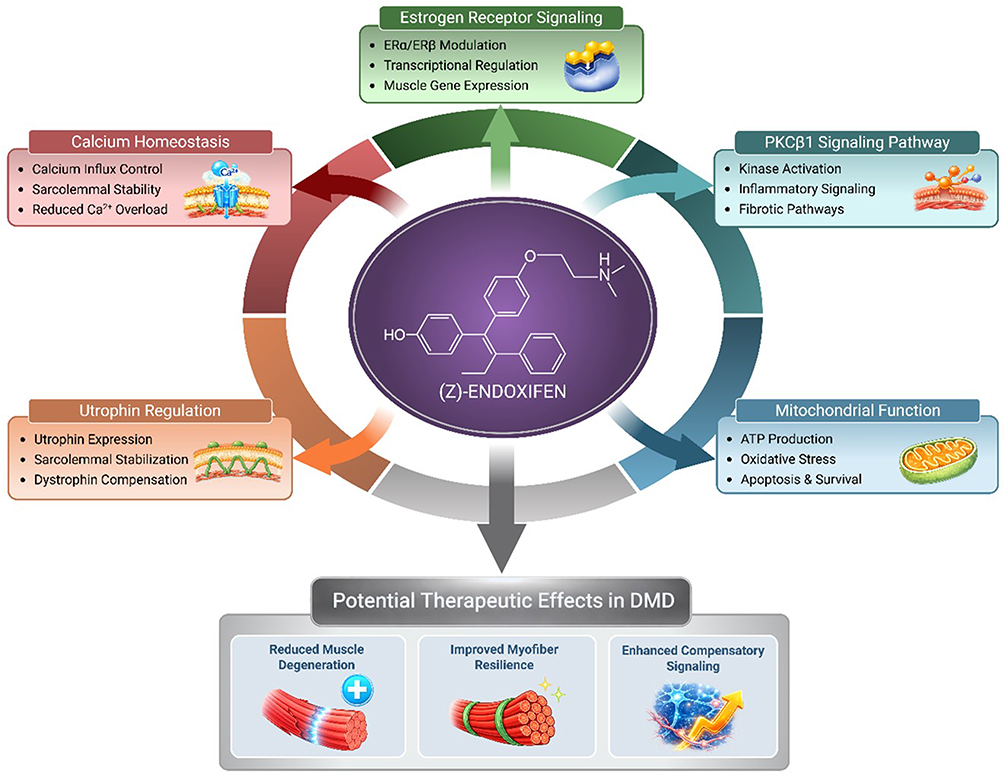 |
Figure 1 Proposed mechanisms of action of (Z)-endoxifen in Duchenne muscular dystrophy (DMD). Schematic overview of the potential molecular pathways through which (Z)-endoxifen may exert therapeutic effects in dystrophic muscle. (Z)-Endoxifen modulates estrogen receptor (ERα/ERβ) signaling, leading to transcriptional regulation of genes involved in muscle function and repair. The compound may also influence the PKCβ1 signaling pathway, contributing to kinase inhibition/modulation and downstream modulation of inflammatory and fibrotic signaling. In parallel, (Z)-endoxifen is proposed to support mitochondrial function, enhancing ATP production, reducing oxidative stress, and promoting cell survival. Additional effects include utrophin regulation, which may increase utrophin expression and improve sarcolemmal stability, providing functional compensation for dystrophin deficiency. (Z)-Endoxifen may also contribute to calcium homeostasis by improving calcium influx control, stabilizing the sarcolemma, and reducing pathological Ca2⁺ overload. Together, these mechanisms may lead to reduced muscle degeneration, improved myofiber resilience, and enhanced compensatory signaling, representing potential therapeutic benefits in DMD. Abbreviations: ATP, Adenosine Triphosphate; ERα, Estrogen Receptor Alpha; ERβ, Estrogen Receptor Beta; PGC-1α, Peroxisome Proliferator-Activated Receptor Gamma Coactivator 1-alpha; MYOD1, Myogenic Differentiation 1; MYOG, Myogenin; MEF2C, Myocyte Enhancer Factor 2C. |
Looking ahead, utrophin quantification could be a primary pharmacodynamic endpoint in early-phase trials of endoxifen. This includes assessing both transcript and protein levels, as well as spatial distribution along the sarcolemma. Correlating these measures with changes in calcium handling, PKC activity, and metabolic gene expression will help elucidate the mechanistic links between endoxifen’s multi-pathway actions and clinical benefit. Such integrative biomarker strategies can also guide dose selection and identify potential responders, maximizing the therapeutic potential of this approach.
Perspective: Current Evidence, Mechanistic Rationale, and Remaining Knowledge Gaps for Endoxifen in Duchenne Muscular Dystrophy
(Z)-endoxifen has emerged as a promising mutation-agnostic therapeutic candidate for Duchenne muscular dystrophy (DMD), based on a convergence of mechanistic insights, pharmacologic properties, and emerging transcriptomic evidence. Unlike gene-targeted strategies that address specific mutational contexts, endoxifen targets downstream pathophysiologic pathways shared across all DMD genotypes. Through a combination of estrogen receptor-β (ERβ) activation, protein kinase C beta-1 (PKCβ1) inhibition, modulation of TGF-β signaling, restoration of calcium homeostasis, satellite cell regulation, and transcriptional reprogramming favoring utrophin pathway activation, endoxifen has the potential to influence multiple interconnected drivers of dystrophic pathology. Anticipated therapeutic effects include reductions in muscle damage, fibrosis, and inflammation, along with improvements in muscle regeneration and structural stability.
The strongest current support for endoxifen in DMD derives from mechanistic plausibility, pharmacologic comparisons with tamoxifen, and emerging transcriptomic analyses rather than from a mature body of direct efficacy studies in dystrophin-deficient systems. Endoxifen possesses a compelling biochemical profile, including potent modulation of estrogen receptors and inhibition of PKCβ1, with downstream effects relevant to fibrosis, inflammatory signaling, mitochondrial function, calcium handling, and myogenic differentiation. These properties align with several therapeutic objectives in DMD, particularly the promotion of a regenerative and less fibrotic muscle environment that could facilitate utrophin expression and sarcolemmal stabilization. However, it remains important to distinguish between biologic rationale and demonstrated therapeutic activity in disease-relevant in vivo models.
Several lines of evidence support continued investigation of endoxifen in dystrophinopathies. First, tamoxifen itself has demonstrated beneficial effects in DMD-relevant preclinical models and early translational studies, including improvements in skeletal and cardiac phenotypes, thereby providing precedent for selective estrogen receptor modulation as a therapeutic strategy in dystrophin deficiency. Second, endoxifen has shown greater pharmacologic potency than tamoxifen in several systems, particularly with respect to PKCβ1 inhibition and estrogen receptor-α degradation, suggesting that it may achieve deeper pathway modulation at clinically achievable exposures. Third, DMD-focused transcriptomic analyses have generated a coherent hypothesis that endoxifen may normalize disease-associated transcriptional programs involving myogenesis, oxidative phosphorylation, inflammatory signaling, extracellular matrix remodeling, and utrophin-associated compensatory pathways. Together, these observations justify endoxifen as a plausible multi-pathway therapeutic candidate, although they do not yet establish the magnitude, durability, or tissue specificity of benefit required for clinical translation.
A particularly compelling aspect of endoxifen’s proposed activity is its potential to influence multiple molecular and cellular systems involved in dystrophic pathologies. Transcriptomic analyses suggest activation of key myogenic regulators, including MYOD1, MYOG, and MEF2C, along with normalization of mitochondrial oxidative phosphorylation pathways and suppression of pro-fibrotic and pro-inflammatory signaling networks. These coordinated changes could theoretically facilitate satellite cell activation, enhance myofiber differentiation, improve metabolic resilience, and reduce extracellular matrix deposition. Restoration of calcium homeostasis represents another important component of the proposed mechanism. By increasing expression of SERCA1 and SERCA2 and stabilizing ryanodine receptor function, endoxifen may reduce pathological cytosolic calcium accumulation, thereby limiting activation of calcium-dependent proteases and preventing downstream mitochondrial dysfunction. Concurrent inhibition of PKCβ1 may further interrupt a calcium-driven feedback loop that exacerbates membrane instability, fibrosis, and inflammation.
Integration with utrophin pathway activation may represent the key unifying mechanism underlying these effects. Utrophin has long been recognized as a functional analog of dystrophin capable of stabilizing the dystrophin-glycoprotein complex (DGC) and protecting muscle fibers from contraction-induced injury. The potential ability of endoxifen to enhance utrophin expression both directly through transcriptional modulation and indirectly through normalization of the surrounding biochemical environment distinguishes it from previous single-pathway utrophin activators. By simultaneously improving calcium regulation, mitochondrial function, inflammatory signaling, and fibrotic remodeling, endoxifen could create a permissive cellular environment in which utrophin-mediated structural compensation becomes more effective.
Beyond effects on myofibers themselves, endoxifen may also influence the broader multicellular environment that governs muscle regeneration. In dystrophic muscle, fibro-adipogenic progenitors often drive fibrotic remodeling, macrophage populations adopt chronic inflammatory phenotypes, and endothelial dysfunction impairs vascular support for regeneration. Emerging evidence suggests that endoxifen may shift fibro-adipogenic progenitors toward less fibrotic phenotypes, promote reparative macrophage polarization, and enhance angiogenic signaling in endothelial cells. Such coordinated multi-cellular modulation is particularly relevant in DMD, where extrinsic inhibitory signals frequently limit intrinsic myogenic potential. By addressing both intrinsic regenerative capacity and the extrinsic tissue environment, endoxifen may offer broader correction of the dystrophic milieu.
From a translational standpoint, several pharmacologic characteristics further strengthen the rationale for investigation. Compared with tamoxifen, endoxifen demonstrates greater potency in ERβ activation, ERα degradation, and PKCβ1 inhibition, allowing deeper pathway engagement at lower doses. Its oral bioavailability and pharmacokinetic properties have already been characterized in oncology settings, providing a valuable clinical pharmacology foundation for repurposing in neuromuscular disease. Nevertheless, the exposure-response relationships relevant to skeletal and cardiac muscle remain unknown. Key unanswered questions include the extent of drug penetration into dystrophic muscle, the concentrations required for sustained target engagement in vivo, and the relationship between systemic exposure and downstream biomarkers such as utrophin expression or PKC pathway activity.
Despite the strong mechanistic rationale, several important knowledge gaps remain. The most significant limitation is the relative scarcity of direct endoxifen efficacy data in canonical dystrophin-deficient models. Dedicated studies demonstrating reproducible functional improvement in mdx, D2-mdx, and other dystrophic models exist but are still limited, particularly for clinically relevant endpoints such as grip strength, force generation, resistance to eccentric contraction, respiratory performance, and cardiac function.93 Similarly, although utrophin pathway activation is a central element of the proposed therapeutic model, there is still insufficient direct evidence demonstrating that endoxifen consistently increases utrophin transcript abundance, protein levels, and extrasynaptic sarcolemmal localization across multiple muscle groups and developmental stages. Whether any observed utrophin effects are primary, indirect, or contingent upon broader remodeling of inflammation and calcium homeostasis remains to be clarified.
Additional uncertainty relates to pharmacokinetics and pharmacodynamics in dystrophic muscle. While endoxifen’s pharmacology has been extensively studied in oncology, the dose-response relationships required for sustained modulation of muscle-specific pathways in DMD are unknown. Pediatric physiology, disease-related alterations in muscle perfusion, concomitant corticosteroid and other DMD therapies and supportive pharmacological management therapies such as ACE inhibitors may all influence drug disposition and pharmacologic activity. These factors are particularly important given that DMD therapies typically require long-term administration beginning early in life.
Mechanistic understanding at the cellular level also remains incomplete. Much of the proposed model for endoxifen in DMD assumes coordinated action across myofibers, satellite cells, fibro-adipogenic progenitors, immune cells, and vascular compartments, yet this integrated framework has not been directly validated using single-cell or spatial approaches in treated dystrophic muscle. Important mechanistic questions therefore remain unresolved, including whether endoxifen primarily enhances intrinsic myogenic progression, suppresses extrinsic fibrotic and inflammatory barriers to regeneration, or exerts stage-specific effects depending on disease progression. Likewise, the interactions between endoxifen and established DMD modifiers such as mitochondrial dysfunction, calcineurin/NFAT signaling, TGF-β activity, and regenerative exhaustion have not yet been systematically mapped.
Translational development will also require robust biomarker strategies capable of linking molecular effects to functional outcomes. Potential biomarkers include utrophin protein quantification, developmental myosin expression, serum muscle injury markers, transcriptomic reversal signatures, calcium-handling protein levels, and imaging-based assessments of muscle composition using magnetic resonance imaging. Multi-modal biomarker integration will likely be necessary to capture the full therapeutic impact of a multi-pathway modulator such as endoxifen. These measurements will be critical for demonstrating target engagement, informing dose selection, and guiding early clinical trial design.
Long-term safety considerations must also be addressed. Although tamoxifen and endoxifen have established safety profiles in other clinical settings, DMD introduces distinct considerations, including pediatric exposure, pubertal development, bone health, endocrine effects, cardiomyopathy, and chronic co-administration with corticosteroids or emerging gene-based therapies. These issues do not preclude development but underscore the importance of indication-specific safety evaluation rather than simple extrapolation from oncology experience.
Within the broader therapeutic landscape, endoxifen could potentially complement existing and emerging treatments for DMD and Becker muscular dystrophy (BMD), as well as for manifesting female carriers. Combination with exon-skipping therapies or microdystrophin gene therapy could provide additive or synergistic benefits by addressing both the primary genetic defect and downstream pathological processes. For patients who are not eligible for gene-targeted therapies, a mutation-agnostic pharmacologic approach such as endoxifen may represent an important strategy for slowing disease progression and preserving muscle function.
Taken together, the present body of evidence positions endoxifen as a compelling mechanistic candidate rather than a clinically de-risked therapy for DMD. While mechanistic, pharmacologic, and transcriptomic data provide a strong scientific rationale, definitive validation of therapeutic efficacy remains necessary. In particular, rigorous evaluation in established dystrophin-deficient models such as mdx and D2-mdx mice will be required to determine whether endoxifen meaningfully improves muscle function, reduces pathology, and promotes utrophin-associated compensatory mechanisms in vivo. Such studies will be critical to confirm whether the hypothesized multi-pathway effects of endoxifen translate into measurable therapeutic benefit and to inform biomarker development, dose optimization, and future clinical trial design.
If these knowledge gaps can be addressed, endoxifen may ultimately progress from a biologically attractive pathway modulator to a credible therapeutic strategy capable of benefiting patients across the full genetic spectrum of dystrophinopathies.
Conclusion
In conclusion, the mechanistic and multiomic evidence presented here supports a compelling preclinical rationale for advancing (Z)-endoxifen into clinical evaluation for DMD and other dystrophies. By targeting converging pathological processes including calcium dysregulation, PKC hyperactivation, inflammation, fibrosis, and impaired regeneration, while promoting utrophin expression, endoxifen represents a comprehensive and mutation-agnostic therapeutic approach. However, direct validation of these effects in dystrophin-deficient models remains necessary to confirm the proposed mechanisms and therapeutic potential in the context of DMD. The next steps will therefore require rigorous preclinical evaluation, thoughtful clinical trial design, robust biomarker integration, and exploration of combination strategies to fully assess and realize the potential of endoxifen to improve outcomes for patients with DMD and other dystrophies.
Generative AI
ChatGPT was used to assist with literature source identification and statement verification.
Abbreviations
AAV, adeno-associated virus; ARG1, arginase 1; ASO, antisense oligonucleotide; BMD, Becker muscular dystrophy; DGC, dystrophin-associated glycoprotein complex; DMD, Duchenne muscular dystrophy; ECM, extracellular matrix; EMT, epithelial-to-mesenchymal transition; ER, estrogen receptor; ERβ, estrogen receptor beta; FAP, fibro-adipogenic progenitors; GABPα, GA-binding protein alpha; IL-10, interleukin-10; iPSC-CM, induced pluripotent stem cell-derived cardiomyocyte; MD, muscular dystrophy; MEF2, myocyte enhancer factor 2; MRI, magnetic resonance imaging; MyHC-emb, myosin heavy chain-embryonic; MyoD, myoblast determination protein 1; MYOG, myogenin; NF‑κB, nuclear factor kappa-light-chain-enhancer of activated B cells; PGC-1α, peroxisome proliferator-activated receptor gamma coactivator 1-alpha; PKC, protein kinase C; PKCβ1, protein kinase C beta-1; PKCθ, protein kinase C theta; RNA-seq, ribonucleic acid sequencing; scRNA-seq, single-cell ribonucleic acid sequencing; SERCA, sarco/endoplasmic reticulum calcium ATPase; SERM, selective estrogen receptor modulator; TIMP3, tissue inhibitor metalloproteinases 3; TGF-β, transforming growth factor beta; TNF-α, tumor necrosis factor alpha.
Data Sharing Statement
Data sharing is not applicable to this article as no data sets were generated or analyzed.
Acknowledgments
The authors wish to acknowledge with gratitude input on the manuscript provided by Dame Professor Kay E. Davies, CBE, FRS, Acad. Med. Sci., Emeritus Fellow Hertford College, Oxford.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
Mr. H. Lawrence Remmel holds shares in Atossa Therapeutics, Inc.; he is the lead independent director of Atossa Therapeutics, Inc., and conducts research partially sponsored by Atossa Therapeutics, Inc. Dr. Sandra S. Hammer and Scott Blackburn receive a salary, bonus, and stock from Atossa Therapeutics Inc. Dr. Kay Davies is a paid consultant with Atossa Therapeutics for her expertise in DMD pathogenesis and utrophin biology. Dr. Steven C. Quay reports personal fees from Atossa Therapeutics Inc, outside the submitted work. In addition, Dr Steven Quay has multiple patents licensed to Atossa Therapeutics, Inc. and he is the Founder and CEO of Atossa Therapeutics, Inc. He receives a salary, bonus, and stock options from Atossa. He is also the inventor of multiple patents and patent applications related to endoxifen and its clinical use. The authors report no other conflicts of interest in this work.
A preprint draft version of the manuscript has been published on Zenodo: https://doi.org/10.5281/zenodo.18551587.
References
1. Luna-Angulo A, Landa-Solís C, Escobar-Cedillo RE, et al. Pharmacological treatments and therapeutic targets in muscle dystrophies generated by alterations in dystrophin-associated proteins. Medicina. 2024;60(7):1060. doi:10.3390/medicina60071060
2. Bargiela A, Hernández-Torres F. Editorial: muscular dystrophies: current therapeutic advances to improve and restore muscle homeostasis. Front Cell Dev Biol. 2022;3(10):1009439. doi:10.3389/fcell.2022.1009439
3. Salari N, Fatahi B, Valipour E, et al. Global prevalence of Duchenne and Becker muscular dystrophy: a systematic review and meta-analysis. J Orthop Surg Res. 2022;17(1):96. doi:10.1186/s13018-022-02996-8
4. Crisafulli S, Sultana J, Fontana A, Salvo F, Messina S, Trifirò G. Global epidemiology of Duchenne muscular dystrophy: an updated systematic review and meta-analysis. Orphanet J Rare Dis. 2020;15(1):141. doi:10.1186/s13023-020-01430-8
5. Giegerich ESM. Duchenne muscular dystrophy prevalence in the U.S.: a novel incidence-based modeling approach using system dynamics. Value Health. 2019;22:S244.
6. Guiraud S, Edwards B, Babbs A, et al. The potential of utrophin and dystrophin combination therapies for Duchenne muscular dystrophy. Hum Mol Genet. 2019;28(13):2189–16. Erratum in: Hum Mol Genet. 2023 Oct 4;32(20):3026. PMID: 30990876; PMCID: PMC6586144. (Guiraud 2019a). doi:10.1093/hmg/ddz049
7. Blake DJ, Weir A, Newey SE, Davies KE. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol Rev. 2002;82(2):291–329. doi:10.1152/physrev.00028.2001
8. Porter GA, Dmytrenko GM, Winkelmann JC, Bloch RJ. Dystrophin colocalizes with beta-spectrin in distinct subsarcolemmal domains in mammalian skeletal muscle. J Cell Biol. 1992;117(5):997–1005. doi:10.1083/jcb.117.5.997
9. Hoffman EP, Brown RH Jr, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51(6):919–928. doi:10.1016/0092-8674(87)90579-4
10. Mareedu S, Million ED, Duan D, Babu GJ. Abnormal Calcium Handling in Duchenne Muscular Dystrophy: mechanisms and Potential Therapies. Front Physiol. 2021;9(12):647010. doi:10.3389/fphys.2021.647010
11. Goonasekera SA, Lam CK, Millay DP, et al. Mitigation of muscular dystrophy in mice by SERCA overexpression in skeletal muscle. J Clin Invest. 2011;121(3):1044–1052. doi:10.1172/JCI43844
12. Duan D, Goemans N, Takeda S, Mercuri E, Aartsma-Rus A. Duchenne muscular dystrophy. Nat Rev Dis Primers. 2021;7(1):13. doi:10.1038/s41572-021-00248-3
13. Matthews E, Brassington R, Kuntzer T, Jichi F, Manzur AY. Corticosteroids for the treatment of Duchenne muscular dystrophy. Cochrane Database Syst Rev. 2016;2016(5):CD003725. doi:10.1002/14651858.CD003725.pub4
14. Leckie J, Zia A, Yokota T. An updated analysis of exon-skipping applicability for duchenne muscular dystrophy using the UMD-DMD database. Genes. 2024;15(11):1489. PMID: 39596689; PMCID: PMC11593839. doi:10.3390/genes15111489
15. Greiner E, Breaux A, Kasten J, et al. Cardiac atrial pathology in Duchenne muscular dystrophy. Muscle Nerve. 2024;69(5):572–579. doi:10.1002/mus.28072
16. Johansson C, Schrama EJ, Kotol D, et al. Contrasting Becker and Duchenne muscular dystrophy serum biomarker candidates by using data independent acquisition LC-MS/MS. Skelet Muscle. 2025;15(1):15. doi:10.1186/s13395-025-00385-3
17. Hoogerwaard EM, Bakker E, Ippel PF, et al. Signs and symptoms of Duchenne muscular dystrophy and Becker muscular dystrophy among carriers in the Netherlands: a cohort study. Lancet. 1999;353(9170):2116–2119. doi:10.1016/s0140-6736(98)10028-4
18. Chwalenia K, Feng VY, Hemmer N, et al. AAV microdystrophin gene replacement therapy for Duchenne muscular dystrophy: progress and prospects. Gene Ther. 2025. doi:10.1038/s41434-025-00561-6
19. D’Ambrosio ES, Mendell JR. Evolving therapeutic options for the treatment of Duchenne muscular dystrophy. Neurotherapeutics. 2023;20(6):1669–1681. doi:10.1007/s13311-023-01423-y
20. van Deutekom JC, Bremmer-Bout M, Janson AA, et al. Antisense-induced exon skipping restores dystrophin expression in DMD patient derived muscle cells. Hum Mol Genet. 2001;10(15):1547–1554. doi:10.1093/hmg/10.15.1547
21. Byrne BJ, Flanigan KM, Matesanz SE, et al. Current clinical applications of AAV-mediated gene therapy. Mol Ther. 2025;33(6):2479–2516. doi:10.1016/j.ymthe.2025.04.045
22. Roberts TC, Wood MJA, Davies KE. Therapeutic approaches for Duchenne muscular dystrophy. Nat Rev Drug Discov. 2023;22(11):917–934. doi:10.1038/s41573-023-00775-6
23. Guiraud S, Roblin D, Kay DE. The potential of utrophin modulators for the treatment of Duchenne muscular dystrophy. Expert Opin Orphan Drugs. 2018;6(3):179–192. doi:10.1080/21678707.2018.1438261
24. Birnbaum F, Eguchi A, Pardon G, Chang ACY, Blau HM. Tamoxifen treatment ameliorates contractile dysfunction of Duchenne muscular dystrophy stem cell-derived cardiomyocytes on bioengineered substrates. NPJ Regen Med. 2022;7(1):19. PMID: 35304486; PMCID: PMC8933505. doi:10.1038/s41536-022-00214-x
25. Jayaraman S, Wu X, Kalari KR, et al. Endoxifen downregulates AKT phosphorylation through protein kinase C beta 1 inhibition in ERα+ breast cancer. NPJ Breast Cancer. 2023;9(1):10125.
26. Biernacka A, Dobaczewski M, Frangogiannis NG. TGF-β signaling in fibrosis. Growth Factors. 2011;29(5):196–202. doi:10.3109/08977194.2011.595714
27. Wu X, Hawse JR, Subramaniam M, Goetz MP, Ingle JN, Spelsberg TC. The tamoxifen metabolite, endoxifen, is a potent antiestrogen that targets estrogen receptor alpha for degradation in breast cancer cells. Cancer Res. 2009;69(5):1722–1727. doi:10.1158/0008-5472.CAN-08-3933
28. Remmel HL, Hammer SS, Neff LA, et al. A hypothesized therapeutic role of (Z)-endoxifen in Duchenne Muscular Dystrophy (DMD). Degener Neurol Neuromuscul Dis. 2025;15:1–15. PMID: 40124418; PMCID: PMC11923445. doi:10.2147/DNND.S496904
29. Botti V, Menzel O, Staedler D. A state-of-the-art review of tamoxifen as a potential therapeutic for duchenne muscular dystrophy. Front Pharmacol. 2022;13:1030785. PMID: 36467064; PMCID: PMC9709317. doi:10.3389/fphar.2022.1030785
30. Hawse JR, Subramaniam M, Cicek M, et al. Endoxifen’s molecular mechanisms of action are concentration dependent and different than that of other anti-estrogens. PLoS One. 2013;8(1):e54613. PMID: 23382923; PMCID: PMC3557294. doi:10.1371/journal.pone.0054613
31. Dorchies OM, Reutenauer-Patte J, Dahmane E, et al. The anticancer drug tamoxifen counteracts the pathology in a mouse model of duchenne muscular dystrophy. Am J Pathol. 2013;182(2):485–504. PMID: 23332367. doi:10.1016/j.ajpath.2012.10.018
32. Babbs A, Chatzopoulou M, Edwards B, et al. From diagnosis to therapy in Duchenne muscular dystrophy. Biochem Soc Trans. 2020;48(3):813–821. doi:10.1042/BST20190282
33. Tinsley J, Deconinck N, Fisher R, et al. Expression of full-length utrophin prevents muscular dystrophy in mdx mice. Nat Med. 1998;4(12):1441–1444. doi:10.1038/4033
34. Tinsley JM, Potter AC, Phelps SR, Fisher R, Trickett JI, Davies KE. Amelioration of the dystrophic phenotype of mdx mice using a truncated utrophin transgene. Nature. 1996;384(6607):349–353. PMID: 8934518. doi:10.1038/384349a0
35. Wu R, Song Y, Wu S, Chen Y. Promising therapeutic approaches of utrophin replacing dystrophin in the treatment of Duchenne muscular dystrophy. Fundam Res. 2022;2(6):885–893. doi:10.1016/j.fmre.2022.07.004
36. Love DR, Hill DF, Dickson G, et al. An autosomal transcript in skeletal muscle with homology to dystrophin. Nature. 1989;339(6219):55–58. doi:10.1038/339055a0
37. Li B, Xiong W, Chang ACH, Chang ACY. Exploring new therapeutics for Duchenne muscular dystrophy and related cardiomyopathy. Rare Dis Orphan Drugs J. 2025;4:9. doi:10.20517/rdodj.2024.44
38. Rafael JA, Tinsley JM, Potter AC, Deconinck AE, Davies KE. Skeletal muscle-specific expression of a utrophin transgene rescues utrophin-dystrophin deficient mice. Nat Genet. 1998;19(1):79–82. doi:10.1038/ng0598-79
39. Deconinck N, Tinsley J, De Backer F, et al. Expression of truncated utrophin leads to major functional improvements in dystrophin-deficient muscles of mice. Nat Med. 1997;3(11):1216–1221. PMID: 9359695. doi:10.1038/nm1197-1216
40. Szwec S, Kapłucha Z, Chamberlain JS, Konieczny P. Dystrophin- and utrophin-based therapeutic approaches for treatment of duchenne muscular dystrophy: a comparative review. BioDrugs. 2024;38(1):95–119. Erratum in: BioDrugs. 2025 Jan;39(1):167. PMID: 37917377; PMCID: PMC10789850. doi:10.1007/s40259-023-00632-3
41. Wilson DGS, Tinker A, Iskratsch T. The role of the dystrophin glycoprotein complex in muscle cell mechanotransduction. Commun Biol. 2022;5(1):1022. doi:10.1038/s42003-022-03980-y
42. Blake DJ, Tinsley JM, Davies KE. Utrophin: a structural and functional comparison to dystrophin. Brain Pathol. 1996;6(1):37–47. doi:10.1111/j.1750-3639.1996.tb00781.x
43. Angulski ABB, Hosny N, Cohen H, et al. Duchenne muscular dystrophy: disease mechanism and therapeutic strategies. Front Physiol. 2023;14:1183101. doi:10.3389/fphys.2023.1183101
44. Andrysiak K, Ferdek PE, Sanetra AM, et al. Upregulation of utrophin improves the phenotype of Duchenne muscular dystrophy hiPSC-derived CMs. Mol Ther Nucleic Acids. 2024;35(3):102247. doi:10.1016/j.omtn.2024.102247
45. Perkins KJ, Davies KE. Alternative utrophin mRNAs contribute to phenotypic differences between dystrophin-deficient mice and Duchenne muscular dystrophy. FEBS Lett. 2018;592(11):1856–1869. doi:10.1002/1873-3468.13099
46. Banks GB, Combs AC, Odom GL, Bloch RJ, Chamberlain JS. Muscle structure influences utrophin expression in mdx mice. PLoS Genet. 2014;10(6):e1004431. PMID: 24922526; PMCID: PMC4055409. doi:10.1371/journal.pgen.1004431
47. Wu R, Li P, Xiao P, et al. Activation of endogenous full-length utrophin by MyoAAV-UA as a therapeutic approach for Duchenne muscular dystrophy. Nat Commun. 2025;16(1):2398. doi:10.1038/s41467-025-57831-5
48. Falcucci L, Dooley CM, Adamoski D, et al. Transcriptional adaptation upregulates utrophin in Duchenne muscular dystrophy. Nature. 2025;639(8054):493–502. PMID: 39939773; PMCID: PMC11903304. doi:10.1038/s41586-024-08539-x
49. Soblechero-Martín P, López-Martínez A, de la Puente-Ovejero L, Vallejo-Illarramendi A, Arechavala-Gomeza V. Utrophin modulator drugs as potential therapies for Duchenne and Becker muscular dystrophies. Neuropathol Appl Neurobiol. 2021;47(6):711–723. doi:10.1111/nan.12735
50. Tinsley JM, Fairclough RJ, Storer R, et al. Daily treatment with SMTC1100, a novel small molecule utrophin upregulator, dramatically reduces the dystrophic symptoms in the mdx mouse. PLoS One. 2011;6(5):e19189. PMID: 21573153; PMCID: PMC3089598. doi:10.1371/journal.pone.0019189
51. Summit Therapeutics. Summit’s Letter to the Community; 2018. Available from: https://www.parentprojectmd.org/summit-therapeutics-announces-phaseout-dmd-did-not-meet-primary-endpoint/.
52. Hadwen J, Farooq F, Witherspoon L, Schock S, Mongeon K, MacKenzie A. Anisomycin activates utrophin upregulation through a p38 signaling pathway. Clin Transl Sci. 2018;11(5):506–512. doi:10.1111/cts.12562
53. Krishna L, Prashant A, Kumar YH, et al. Molecular and biochemical therapeutic strategies for duchenne muscular dystrophy. Neurol Int. 2024;16(4):731–760. doi:10.3390/neurolint16040055
54. Duan D. Systemic AAV micro-dystrophin gene therapy for duchenne muscular dystrophy. Mol Ther. 2018;26(10):2337–2356. doi:10.1016/j.ymthe.2018.07.011
55. Song Y, Morales L, Malik AS, et al. Non-immunogenic utrophin gene therapy for the treatment of muscular dystrophy animal models. Nat Med. 2019;25(10):1505–1511. doi:10.1038/s41591-019-0594-0
56. Duan D. Micro-utrophin therapy for duchenne muscular dystrophy. Mol Ther. 2019;27(11):1872–1874. doi:10.1016/j.ymthe.2019.10.011
57. Guiraud S, Edwards B, Squire SE, et al. Embryonic myosin is a regeneration marker to monitor utrophin-based therapies for DMD. Hum Mol Genet. 2019;28(2):307–319. Guiraud 2019b. doi:10.1093/hmg/ddy353
58. Remmel HL, Hammer SS, Singh H, et al. Comparative analysis of endoxifen, tamoxifen and fulvestrant: a bioinformatics approach to uncover mechanisms of action in breast cancer. bioRxiv. 2024. doi:10.1101/2024.10.02.616224
59. Jayaraman S, Reid JM, Hawse JR, Goetz MP. Endoxifen, an estrogen receptor targeted therapy: from bench to bedside. Endocrinology. 2021;162(12):bqab191. Erratum in: Endocrinology. 2021 Dec 1;162(12):bqab201. PMID: 34480554; PMCID: PMC8787422. doi:10.1210/endocr/bqab191
60. Fu C, Duan S, Zhou X, Meng Y, Chen X. Overexpression of COL11A1 confers tamoxifen resistance in breast cancer. NPJ Breast Cancer. 2024;10(1):38. doi:10.1038/s41523-024-00645-3
61. Lee O, Wang M, Hosseini O, et al. Z-Endoxifen prevents aggressive mammary cancers in mice by inhibiting cell proliferation and creating a tumor suppressive microenvironment. Biomed Pharmacother. 2023;162:114607. doi:10.1016/j.biopha.2023.114607
62. Kodippili K, Rudnicki MA. Satellite cell contribution to disease pathology in Duchenne muscular dystrophy. Front Physiol. 2023;14:1180980. doi:10.3389/fphys.2023.1180980
63. Dong H, Tsai SY. Mitochondrial properties in skeletal muscle fiber. Cells. 2023;12(17):2183. doi:10.3390/cells12172183
64. Zabłocka B, Górecki DC, Zabłocki K. Disrupted calcium homeostasis in duchenne muscular dystrophy: a common mechanism behind diverse consequences. Int J Mol Sci. 2021;22(20):11040. doi:10.3390/ijms222011040
65. Carraro M, Bernardi P. The mitochondrial permeability transition pore in Ca2+ homeostasis. Cell Calcium. 2023;111:102719. doi:10.1016/j.ceca.2023.102719
66. Robichaux DJ, Harata M, Murphy E, Karch J. Mitochondrial permeability transition pore-dependent necrosis. J Mol Cell Cardiol. 2023;174:47–55. doi:10.1016/j.yjmcc.2022.11.003
67. Dowling P, Gargan S, Swandulla D, Ohlendieck K. Proteomic profiling of impaired excitation-contraction coupling and abnormal calcium handling in muscular dystrophy. Proteomics. 2022;22(23–24):e2200003. doi:10.1002/pmic.202200003
68. Law ML, Cohen H, Martin AA, Angulski ABB, Metzger JM. Dysregulation of calcium handling in duchenne muscular dystrophy-associated dilated cardiomyopathy: mechanisms and experimental therapeutic strategies. J Clin Med. 2020;9(2):520. doi:10.3390/jcm9020520
69. Akshaya R, Mohan S, Vellapandian C. A voyage on the role of nuclear factor Kappa B (NF-kB) signaling pathway in duchenne muscular dystrophy: an inherited muscle disorder. Cureus. 2024;16(8):e67901. doi:10.7759/cureus.67901
70. Lim Y, Li L, Desta Z, Rae JM, Flockhart DA, Skaar TC. Gene expression profiles of 4-hydroxy-N-desmethyl-tamoxifen (endoxifen)- and 4-hydroxy-tamoxifen (4OHTAM)-treated human breast cancer cells determined by CDNA microarray analysis. Clin Pharmacol Ther. 2004;75(2):49.
71. Sabourin J, Harisseh R, Harnois T, et al. Dystrophin/α1-syntrophin scaffold regulated PLC/PKC-dependent store-operated calcium entry in myotubes. Cell Calcium. 2012;52(6):445–456. doi:10.1016/j.ceca.2012.08.003
72. Marrocco V, Fiore P, Benedetti A, et al. Pharmacological inhibition of PKCθ counteracts muscle disease in a mouse model of duchenne muscular dystrophy. EBioMedicine. 2017;16:150–161. doi:10.1016/j.ebiom.2017.01.001
73. Madaro L, Pelle A, Nicoletti C, et al. PKC theta ablation improves healing in a mouse model of muscular dystrophy. PLoS One. 2012;7(2):e31515. doi:10.1371/journal.pone.0031515
74. Xu H, Van Remmen H. The SarcoEndoplasmic Reticulum Calcium ATPase (SERCA) pump: a potential target for intervention in aging and skeletal muscle pathologies. Skelet Muscle. 2021;11(1):25. doi:10.1186/s13395-021-00280-7
75. Gao QQ, McNally EM. The dystrophin complex: structure, function, and implications for therapy. Compr Physiol. 2015;5(3):1223–1239. doi:10.1002/cphy.c140048
76. Saleh KK, Xi H, Switzler C, et al. Single cell sequencing maps skeletal muscle cellular diversity as disease severity increases in dystrophic mouse models. iScience. 2022;25(11):105415. doi:10.1016/j.isci.2022.105415
77. Granet JA, Robertson R, Cusmano AA, et al. Muscle stem cells in Duchenne muscular dystrophy exhibit molecular impairments and altered cell fate trajectories impacting regenerative capacity. Cell Death Dis. 2025;16(1):437. doi:10.1038/s41419-025-07755-1
78. Koike H, Manabe I, Oishi Y. Mechanisms of cooperative cell-cell interactions in skeletal muscle regeneration. Inflamm Regen. 2022;42(1):48. doi:10.1186/s41232-022-00234-6
79. Collins BC, Kardon G. It takes all kinds: heterogeneity among satellite cells and fibro-adipogenic progenitors during skeletal muscle regeneration. Development. 2021;148(21):dev199861. doi:10.1242/dev.199861
80. Byun WS, Lee J, Baek JH. Beyond the bulk: overview and novel insights into the dynamics of muscle satellite cells during muscle regeneration. Inflamm Regen. 2024;44(1):39. doi:10.1186/s41232-024-00354-1
81. Cardone N, Taglietti V, Baratto S, et al. Myopathologic trajectory in Duchenne muscular dystrophy (DMD) reveals lack of regeneration due to senescence in satellite cells. Acta Neuropathol Commun. 2023;11(1):167. doi:10.1186/s40478-023-01657-z
82. Ausems CRM, van Engelen BGM, van Bokhoven H, Wansink DG. Systemic cell therapy for muscular dystrophies: the ultimate transplantable muscle progenitor cell and current challenges for clinical efficacy. Stem Cell Rev Rep. 2021;17(3):878–899. doi:10.1007/s12015-020-10100-y
83. Loomis T, Smith LR. Thrown for a loop: fibro-adipogenic progenitors in skeletal muscle fibrosis. Am J Physiol Cell Physiol. 2023;325(4):C895–C906. doi:10.1152/ajpcell.00245.2023
84. Deng B, Wehling-Henricks M, Villalta SA, Wang Y, Tidball JG. IL-10 triggers changes in macrophage phenotype that promote muscle growth and regeneration. J Immunol. 2012;189(7):3669–3680. doi:10.4049/jimmunol.1103180
85. Belanto JJ, Mader TL, Eckhoff MD, et al. Microtubule binding distinguishes dystrophin from utrophin. Proc Natl Acad Sci U S A. 2014;111(15):5723–5728. doi:10.1073/pnas.1323842111
86. Love DR, Morris GE, Ellis JM, et al. Tissue distribution of the dystrophin-related gene product and expression in the mdx and dy mouse. Proc Natl Acad Sci U S A. 1991;88(8):3243–3247. PMID: 2014247; PMCID: PMC51422. doi:10.1073/pnas.88.8.3243
87. Medrano JL, Naya FJ. The transcription factor MEF2A fine-tunes gene expression in the atrial and ventricular chambers of the adult heart. J Biol Chem. 2017;292(51):20975–20988. doi:10.1074/jbc.M117.806422
88. Péladeau C, Adam N, Bronicki LM, et al. Identification of therapeutics that target eEF1A2 and upregulate utrophin A translation in dystrophic muscles. Nat Commun. 2020;11(1):1990. doi:10.1038/s41467-020-15971-w
89. Nemirovskaya TL, Sharlo KA. Roles of ATP and SERCA in the regulation of calcium turnover in unloaded skeletal muscles: current view and future directions. Int J Mol Sci. 2022;23(13):6937. doi:10.3390/ijms23136937
90. Abu Shelbayeh O, Arroum T, Morris S, Busch KB. PGC-1α is a master regulator of mitochondrial lifecycle and ROS stress response. Antioxidants. 2023;12(5):1075. doi:10.3390/antiox12051075
91. Chan MC, Rowe GC, Raghuram S, Patten IS, Farrell C, Arany Z. Post-natal induction of PGC-1α protects against severe muscle dystrophy independently of utrophin. Skelet Muscle. 2014;4(1):2. doi:10.1186/2044-5040-4-2
92. Handschin C, Kobayashi YM, Chin S, Seale P, Campbell KP, Spiegelman BM. PGC-1alpha regulates the neuromuscular junction program and ameliorates Duchenne muscular dystrophy. Genes Dev. 2007;21(7):770–783. doi:10.1101/gad.1525107
93. Hammer S, Remmel HL, Neff L, Dorchies O, Quay SC. (Z)-endoxifen restores muscle performance and lowers damage biomarkers in mdx5Cv dystrophic mice. In:
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.