Back to Archived Journals » Gastrointestinal Cancer: Targets and Therapy » Volume 4
WNT/β-catenin pathway activation in hepatocellular carcinoma: a clinical perspective
Authors Ma L, Wei W, Chua M ![]() , So S
, So S
Received 4 December 2013
Accepted for publication 24 January 2014
Published 10 April 2014 Volume 2014:4 Pages 49—63
DOI https://doi.org/10.2147/GICTT.S44256
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Li Ma, Wei Wei, Mei-Sze Chua, Samuel So
Asian Liver Center and Department of Surgery, Stanford University School of Medicine, Stanford University, Stanford, CA, USA
Abstract: Hepatocellular carcinoma (HCC) is a significant global health concern which requires multidisciplinary approaches in its management. However, apart from surgical resection of the tumor, molecularly targeted therapeutics remains limited to sorafenib. New targets and drugs are urgently needed to broaden the currently limited treatment options for HCC to allow more efficacious clinical interventions and ultimately to improve the overall survival of HCC patients. The WNT/β-catenin pathway controls multiple biological functions throughout embryonic development and adult homeostasis; its dysregulation underlies a wide range of pathologies including cancer. In particular, many lines of evidence suggest that hyperactivation of this pathway is associated with the initiation and development of HCC. The critical role of the WNT to WNT/β-catenin pathway in HCC lends itself to rationally designed approaches to intervene with various aberrant loci along its signaling cascade to achieve therapeutic effects in HCC. Here, we review the current state of knowledge on WNT/β-catenin pathway deregulation in HCC, and how this pathway may be exploited for therapeutic interventions.
Keywords: WNT/β-catenin signaling, hepatocarcinogenesis, β-catenin/TCF complexes, targeted therapy
Pathophysiology of liver cancer
Hepatocellular carcinoma (HCC), the most common adult liver malignancy, is the sixth most common cancer and the third most frequent cause of cancer-related death worldwide.1 Most HCC cases (80%) occur in Eastern and Southeastern Asia, and sub-Saharan Africa, where the dominant risk factor is chronic infection with hepatitis B virus (HBV), together with exposure to aflatoxin B1. The relative risk of developing HCC is 100 fold among chronic HBV carriers compared with noncarriers.2 Most cases of chronic HBV infection occur in early childhood.3 The main pathogenic mechanisms for HBV associated-HCC are immune-mediated chronic liver damage, HBV genome integration, and the HBV X protein (HBx)-associated oncogenic transformation.4
In recent years, the incidence of HCC has been increasing in developed countries, including North America, Europe, and Japan, where the dominant risk factor is chronic infection with hepatitis C virus (HCV), together with other risk factors, such as alcohol abuse and nonalcoholic fatty liver disease.2,3 The relative risk of developing HCC is 15–20 fold in HCV carriers compared with noncarriers; the risk is increased in HCV carriers with cirrhosis.5 The main pathogenic mechanisms for HCV-associated HCC are immune-mediated chronic liver damage, and non-structural proteins NS3- and NS5-associated oncogenic transformation.4
Long-term, chronic, viral hepatitis caused by either virus usually leads to the progression of cirrhosis, which is present in 80%–90% of HCC patients.3 Cirrhosis reduces hepatic functional reserve, which results in symptoms such as portal hypertension and ascites. Most patients die directly from cirrhosis in the advanced stage.6 Moreover, cirrhotic patients have a higher risk of developing HCC with an annual incidence of 2.0%–6.6%, compared with noncirrhotic patients with an annual incidence of 0.4%.7,8
Current treatment options and patient outcomes
Surgical resection, local ablation, and liver transplantation are curative options when the tumor is detected in the early stage (<20% of HCC patients). Patients with early stage disease who receive surgical resection and local ablation have 5-year survival rates of 60%–70%.2 However, even in developed countries, only 25% of these early stage patients are suitable for these interventions due to restrictions imposed by tumor size, liver function impairment, and portal hypertension.9 Moreover, approximately 70% of patients develop recurrent tumors after resection and ablation within 5 years, which usually arises within the first 2 years.10 As the most promising curative treatment, liver transplantation offers 4-year survival of 75% for selected patients, with recurrence rates below 15%.11,12 However, patients have to be carefully selected, and transplantation is limited due to shortage of organ donation.
Patients with intermediate-stage HCC are asymptomatic, with multinodular tumors but without vascular invasion or extrahepatic spread, and are candidates for transarterial chemoembolization (TACE).13 Conventional TACE provides locoregional treatment with intra-arterial infusion of conventional antineoplastic drugs such as doxorubicin or cisplatin, followed by embolization of the blood vessel with embolic agents, and increases median survival time by 20–25 months.14 However, conventional TACE comes with strong systemic toxicity, which has been reduced by improvements in embolization devices such as drug elution beads.15,16 Even so, systemic toxicity remains a severe problem for patients with limited hepatic functional reserve as conventional antineoplastic drugs are nonselective.
Most HCC patients remain asymptomatic until the disease is far advanced. Patients with advanced-stage HCC present with cancer-related symptoms such as pain or malaise, together with evidence of vascular invasion or extrahepatic spread. These patients are candidates for systemic treatment with small molecule antineoplastic drugs such as tyrosine kinase inhibitors (TKI).17 Sorafenib (Nexavar®) (Bayer; Leverkusen, North Rhine-Westphalia, Germany; and Onyx Pharmaceuticals; South San Francisco, CA, USA), the only TKI approved by the US Food and Drug Administration for HCC therapy,18 is the standard of care for patients with advanced HCC.19 Sorafenib targets tyrosine kinases and blocks different signaling pathways, including those that regulate cell proliferation (via RAS mitogen-activated protein kinase [MAPK]) and angiogenesis (via vascular endothelial growth factor receptor and platelet derived growth factor receptor).18 Phase III clinical trial indicated that sorafenib treatment increased survival time of patients with advanced HCC from 7.9 to 10.7 months only.20 Despite the recent development of more TKIs for advanced HCC treatment, none has been shown to be more effective than sorafenib in prolonging patients’ survival,21 underscoring the need for more efficacious, molecularly-targeted drugs.
The poor prognosis of patients with intermediate-stage and advanced stage HCC (>80% of HCC patients) prompts the identification of new therapeutic targets and the development of new therapeutic approaches for its treatment. Major signaling pathways that are critical to the complex and multistep process of hepatocarcinogenesis have recently been identified,22 and the targeting of these pathways offers multiple opportunities to develop novel therapies for HCC. This review discusses the potential of targeting one of these major pathways, the WNT/β-catenin pathway, which is commonly deregulated in HCC.
Introduction to the WNT/β-catenin pathway
WNT is a hybrid name (for Wingless-related integration site) used to denote genes belonging to the int1/Wingless family, which replaced the original confusing nomenclature used for the Int genes.23 2012 marks the 30th anniversary for the discovery of int1 (now known as WNT1). Int1 was first identified as an protooncogene transcriptionally activated by the mouse mammary tumor virus in mouse mammary tumor.24 So far, a family of 19 WNT-related genes has been characterized in vertebrates. WNTs are cysteine rich proteins around 40 kDa in size25 that participate in the complex WNT/β-catenin signaling cascade together with multiple other components. The WNT/β-catenin pathway plays crucial roles in many diverse cellular processes, including cell proliferation, cell fate decision, cell migration, and stem cell maintenance.26 As an important developmental signaling pathway, WNT/β-catenin pathway deregulation has been closely linked to cancer.23 For a history of this field and detailed biochemical mechanisms of this pathway, we refer the readers to recent reviews.27
The WNT/β-catenin pathway can be classified into canonical (β-catenin dependent) and noncanonical (β-catenin independent) pathways. WNT proteins trigger the signaling cascades in both these pathways by binding to seven-transmembrane frizzled receptors (FZD) and their coreceptors, the low density lipoprotein receptor-related proteins 5 and 6 (LRP5/6), or the orphan receptor tyrosine kinase Ror2, respectively.28–30 In the inactivated state, β-catenin is attached to cadherin in maintaining cell–cell adhesion.31 In the canonical pathway, cytoplasmic β-catenin is sequestered by the multiprotein “destruction complex” that comprises adenomatous polyposis coli (APC), AXIN, and two kinases, glycogen synthase kinase-3 (GSK3) and casein kinase α/β, which sequentially phosphorylate β-catenin. Phosphorylated β-catenin is then recognized by the E3 ubiquitin ligase complex-gamma-TrCP and degraded in the cytoplasm by proteasomes. The overall net effect is low β-catenin levels within the cells. In the activated states or upon WNT binding, the dishevelled protein inhibits GSK3 phosphorlyation activity, and the destruction complex becomes disintegrated. As a result, β-catenin accumulates in the cytoplasm and translocates to the nucleus, where it interacts with the transcription factors T-cell factor (TCF) 4/lymphoid enhancer-binding factor (LEF) to transcriptionally activate downstream target genes, eg, c-Myc,32 Cyclin D1,33 and AXIN2,34 to promote cell proliferation. However, Li et al35 recently proposed an alternative mechanism to the conventional view; in the activated state, the destruction complex does not disassociate, but rather, becomes saturated by accumulated phospho-β-catenin, which leads to accumulation of newly synthesized β-catenin in the cytoplasm.
As the name suggests, the noncanonical (or β-catenin independent) pathways do not rely on β-catenin transcriptional activity and include diverse pathways, the main ones of which are the WNT-calcium (Ca2+) and WNT polarity pathways.30 These pathways all contribute to planar cell polarity, early vertebrate gastrulation, or neuronal and epithelial cell migration.30,36 In the WNT-Ca2+ pathway, the prototype noncanonical WNT ligand, WNT5a, stimulates intracellular Ca2+ levels via binding to FZD and coreceptors, knypek (Kny) or Ror2,37,38 leading to activation of phospholipase C (PLC) and the release of intracellular Ca2+. Ca2+ then acts as the secondary messenger to relay signaling via downstream pathways mediated through calcium/calmodulin-dependent protein kinase II and protein kinase C.39,40 The WNT polarity pathway signals through the guanosine triphosphate hydrolase RhoA and Rac. Activation of Rho in turn activates Rho-associated kinase, whereas activation of Rac stimulates c-Jun N-terminal kinase, both of which exert effects on the actin cytoskeleton to control cellular polarity.40,41
Deregulation of the WNT/β-catenin pathway in cancer
An aberrant WNT/β-catenin pathway has been reported in various types of cancers42 and can broadly be classified as deregulations in the ligand-independent components (such as mutations involving APC, AXIN, β-catenin, and TCF4) or in the ligand-dependent components (involving WNT proteins and their FZD).
Deregulations in ligand-independent components of the WNT/β-catenin pathway
Mutations in the ligand-independent WNT/β-catenin pathway enhance abnormal accumulation of free β-catenin or enhance the transcriptional activity of β-catenin/TCF4 in the nucleus.43 For example, inactive truncations of APC occur in more than 80% of colon cancer patients44,45 and are associated with increased levels of active nuclear β-catenin,46 which represents the initial stage of oncogenic transformation.47 Mutations and defective truncations of AXIN1 and AXIN2, which are negative regulators of β-catenin, have also been reported in many cancers, including HCC,48 colon cancer,49 gastric tumor,50 adrenocortical carcinoma,51 and prostate cancer.52 Most of the mutation sites of AXIN genes exist in the β-catenin-interaction domain, resulting in up-regulation of transcriptional activity of β-catenin.53
In addition, activating mutations and truncations of CTNNB1 have been found in various types of cancers, such as colon cancer,54,55 melanoma,56 HCC,57,58 gastrointestinal tumors,59 thyroid cancers,60 ovarian tumor,61 sporadic desmoid tumor,62 and Wilms tumors.63 Most of the reported mutations of CTNNB1 are found in the N-terminus, which contains the phosphorylation sites Ser37, Thr41, and Ser45. These mutations in the N-terminus produce deletion of the N-terminal fragment or alter the phosphorylation of β-catenin, thereby protecting β-catenin from degradation by the “destruction complex.”42,53 Recently, another central factor of the WNT/β-catenin pathway, TCF4, has been reported with numerous alternative splice variants and frequent frameshift mutations in the C-terminus due to microsatellite instability in colon cancer,64,65 renal cell carcinoma,66 brain tumors,67 and gastric and endometrial carcinoma.68 These different isoforms and mutations reduce the ability of TCF4 to interact with the negative functional regulator (c-terminal binding protein), thus enhancing the β-catenin/TCF4 transcriptional activity.65
Deregulations in ligand-dependent components of the WNT/β-catenin pathway
In the ligand-dependent WNT/β-catenin pathway, clinical studies have identified overexpression of most WNT proteins and FZD in various types of cancers, including breast cancer,69 brain and neurological cancer,70,71 head and neck cancer,72 acute lymphoblastic leukemia,73 renal cell carcinoma,74 prostate cancer,75 esophageal carcinoma,76 HCC,77 colon cancer,78 and synovial sarcomas.79 Moreover, epigenetic studies have revealed that frequent promoter hypermethylation and gene silencing of the secreted frizzled related proteins (sFRPs) occur in colon cancer.80 The downregulation of sFRPs has also been reported in melanoma, HCC, cervical carcinoma, breast carcinoma, ovary and kidney carcinomas.81–85 Such epigenetic downregulation of sFRP genes frees active WNT proteins and allows constitutive WNT signaling in cancer cells.86
Other regulators of the WNT/β-catenin signaling pathway
In addition to aberrant regulations directly involving components of the WNT/β-catenin signaling pathway, external regulators of this pathway also contribute toward its dysregulation that leads to cancer. For example, the transcription factor c-Myc is a classic downstream target gene of the WNT/β-catenin signaling pathway that was reported to transcriptionally repress the WNT inhibitors dickkopf (DKK) 1 and sFRP1, resulting in activated WNT/β-catenin signaling in breast cancer.87 c-Myc was also reported to activate B lymphoma Mo-MLV insertion region 1 homolog (BMI1), a polycomb group protein, that represses members of the DKK family. As a result of this repression, stimulated WNT/β-catenin pathway increases the c-Myc level, which further feeds into a feedback loop to activate the WNT/β-catenin signaling pathway.88 Since c-Myc is also frequently overexpressed in other types of cancers, including HCC, it is likely that similar mechanistic activation of the WNT/β-catenin signaling pathway occurs and contributes toward carcinogenesis of these cancers.
Specific to HCC, HBx is a transactivator of several cellular signaling pathways, including the WNT/β-catenin pathway. A functional screen for host factors involved in the transactivational properties of HBx identified APC as a binding partner of HBx; the competitive binding of HBx to APC displaces β-catenin from the destruction complex, resulting in β-catenin upregulation in the nucleus and the activation of WNT/β-catenin signaling.89 The HCV core protein was also found to significantly enhance TCF-dependent transcriptional activity induced by WNT3A in HCC cell lines.90 It additionally increased β-catenin levels in the Huh7 HCC cell line through inactivation of GSK 3β. As a result, downstream target genes such as c-Myc and cyclin D1 were upregulated, leading to increased cell proliferation via the canonical WNT/β-catenin pathway.
Another class of external regulators is the micro ribonucleic acids (RNAs) (miRNAs), which are small noncoding RNAs between 20 to 23 nucleotides in length that regulate diverse cellular processes. They function through forming a complementary complex with the 3′-untranslated region of the target messenger RNA (mRNA), resulting in mRNA degradation or inhibition of mRNA translation, ultimately causing posttranslational gene silencing.91 Many miRNAs have been reported to interact with members of the WNT/β-catenin signaling pathway,92 eg, miR-200a directly targets the β-catenin mRNA to inhibit its translation thereby blocking WNT/β-catenin signaling.93 The downregulation of miR-200a was correlated with upregulation of β-catenin in meningiomas, and in part contributes toward meningioma development.93 Other miRNAs such as miR-21, miR-34a, and let-7e appear to regulate WNT1 at the functional level by controlling the amount of protein expressed.94 Tumor suppressor oncomirs, mir-34a-3p and mir-34a-5p (which are direct downstream targets of tumor suppressor p53) were shown to directly target the untranslated regions of multiple genes in the WNT/β-catenin pathway, including WNT1, WNT3, LRP6, AXIN2, and β-catenin.95 In colon cancer, miR-34a targets multiple binding sites within the 5′ and 3′ untranslated region of AXIN2, causing AXIN2 suppression and increasing nuclear GSK-3β abundance.96 In HCC, miR-214 can either directly or indirectly target CTNNB1, thereby modulating the β-catenin transcriptional activity.97 Additionally, HCV was reported to induce and upregulate miRNA-155 via nuclear factor-kappa beta; miRNA-155 then markedly suppressed APC levels to activate WNT/β-catenin signaling, promoting cell proliferation and tumor growth of HCC cells.98
The WNT/β-catenin signaling pathway in HCC
Hyperactivation of the WNT/β-catenin signaling pathway is a major contributor to the pathogenesis of HCC.99 Reported aberrant expressions of various components of this pathway in HCC are summarized in Table 1.
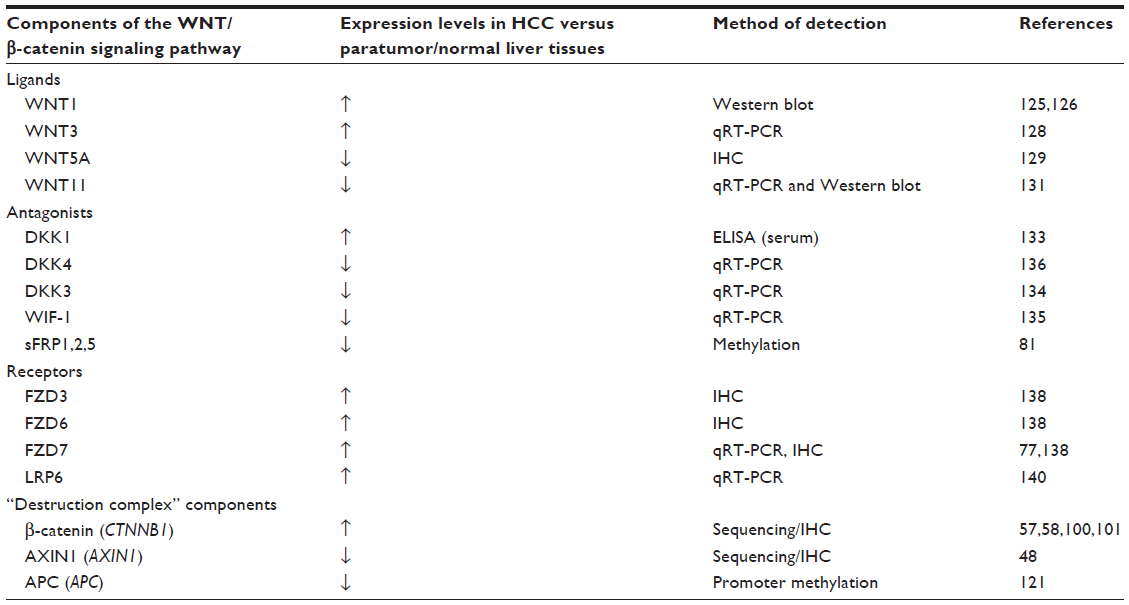 | Table 1 Clinical evidence of altered the WNT/β-catenin signaling pathway components in HCC |
Genetic and epigenetic regulation of CTNNB1 encoding β-catenin
On average, approximately one third of HCC are shown to harbor mutations in CTNNB1, which encodes the β-catenin protein.100–102 Stabilizing CTNNB1 mutations are primarily located in exon 3, on the Ser/Thr residues that render β-catenin phosphorylated by GSK3β.103,104 Somatic mutations in this exon result in accumulation of nonphosphorylated, active β-catenin in the nuclei, thereby activating the WNT/β-catenin signaling pathway. Indeed, aberrant nuclear β-catenin staining was associated with tumor proliferation and with poor prognosis of HCC patients.104 Specific mutations reported in CTNNB1 in HCC,100–102,105–110 and their functional consequences are listed in Table 2. However, inconsistency exists in the purported role of β-catenin in disease initiation and progression of HCC. In a cohort consisting mostly of HBV-related HCC, CTNNB1 mutation was found in 16% of HCC patients, but CTNNB1 mutation was neither observed in low grade dysplastic nodules nor high grade dysplastic nodules, suggesting that β-catenin is neither an early event gene nor the gatekeeper gene in HCC development.111 However, another study concluded that β-catenin was accumulated in the cytoplasm and the nuclei in precancerous lesions of the liver and might contribute, at least in part, to hepatic tumor genesis.112
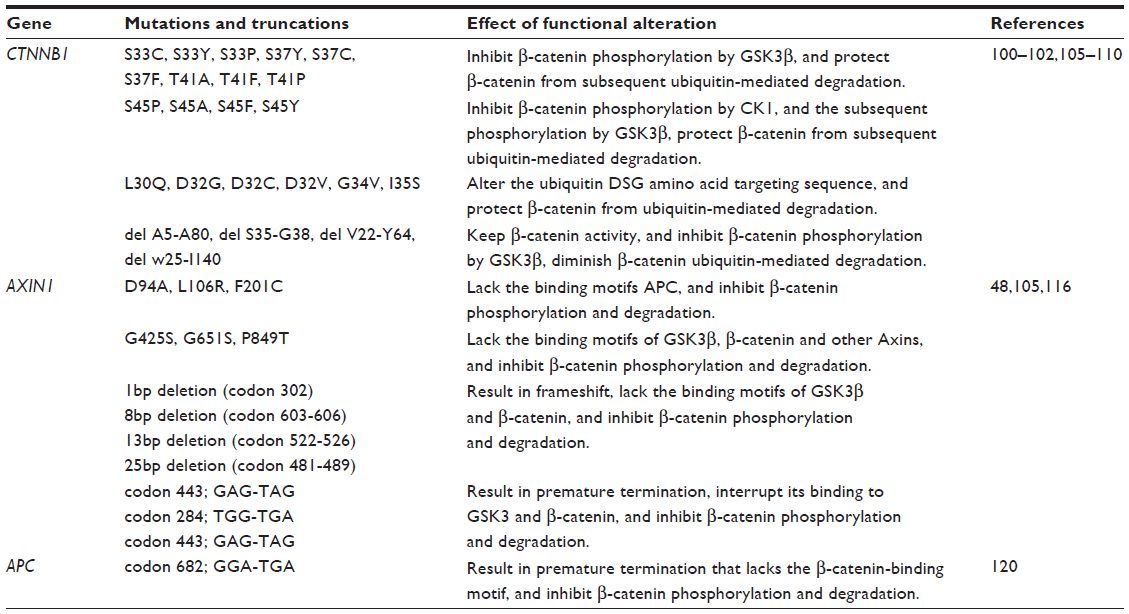 | Table 2 Mutations of key components involved in the WNT/β-catenin pathway in HCC |
Expression of β-catenin in HCC may be subject to multilayered regulation other than mutation-mediated stabilization, as evidenced by greater percentage of β-catenin nuclear staining in comparison to lower β-catenin mutation rates detected in HCC patients.102,103 Indeed, CTNNB1 is targeted by miR-214, either directly or indirectly through enhancer of zeste homolog 2 (EZH2), a member of the polycomb repression complex 2 complex,97 leading to aberrant WNT/β-catenin signaling in HCC. More recently, Jung et al reported that upon WNT signaling activation, proliferating cell nuclear antigen (PCNA)-associated factor dissociates from PCNA and binds directly to β-catenin and then recruits EZH2 to the β-catenin transcriptional complex to specifically enhance WNT target gene transactivation, independently of EZH2’s methyltransferase activity.113
Mutations in components of the “destruction complex”
AXIN determines the rate of β-catenin protein degradation and confers a tumor suppressor role.114 Functional studies demonstrated that conditional disruption of AXIN1 leads to development of liver tumors in mice,115 whereas restoration of AXIN1 by adenovirus-mediated gene transfer of wild-type AXIN1 suppressed HCC cell growth.48 Mutations in AXIN1 were detected in 6.2% of HCCs but not in low grade dysplastic nodules or high grade dysplastic nodules, suggesting that AXIN1 mutation might be a late event for malignant progression rather than an early genetic event involved in HCC initiation. The overall effects of AXIN1 mutations in HCC are impaired β-catenin phosphorylation and degradation (Table 2),48,105,116 resulting in β-catenin accumulation and activation of the WNT/β-catenin signaling pathway.
In contrast to CTNNB1 and AXIN, APC is rarely mutated in HCC populations,99 being observed only occasionally as secondary mutations in patients with familial adenomatous polyposis or Gardner syndrome.117–119 Specific somatic biallelic inactivation of the APC gene was first reported in a case of sporadic HCC, and it was associated with inability to phosphorylate and degrade β-catenin, and thus, malignant transformation via activation of the WNT/β-catenin pathway (Table 2).120 Instead, APC is frequently hypermethlyated in HCC.121 Based on two independent genome-wide methylation profiling studies performed on HCC patients,122,123 APC appears to be one of the top five most frequently hypermethylated genes in HCC and may account for the major mechanism in the loss of APC function in HCC, especially in HBV-related HCC. When bisulfite sequencing by polymerase chain reaction was performed to analyze the methylation status of both sense and antisense strands of the APC gene in HCC, it was found that hypermethylation of the C—phosphate—G sites on the sense strand only is specific for HCC.124 This result accounted for the observed variations in the specificity of the methylation of the APC gene for HCC. Importantly, 40% of HCC patients with nondiagnostic levels of alpha-fetoprotein (AFP) (<20 ng/mL) presented with methylation of the sense strand of APC, suggesting that sense strand methylation of APC may be useful in conjunction with AFP for improved HCC diagnosis.
Altered expression of WNTs and antagonists in HCC
Several WNT proteins have dysregulated expression in HCC. For example, WNT1 expression was elevated in both HBV- and HCV-related HCC.125,126 While it was observed to be upregulated by HCV core proteins,127 its regulation by HBV is less well established.125 In HBV-induced HCC, WNT3 and downstream β-catenin target genes were upregulated in tumor and peritumoral tissues compared to normal liver.128 Functional interaction between WNT3 and FZD7 was demonstrated to cause activation of the WNT/β-catenin signaling pathway in HCC cells,128 which may play a role during hepatocarcinogenesis. Compared to nontumor liver, WNT5a protein expression (as determined by immunohistochemistry) was increased in chronic hepatitis, cirrhosis, and dysplastic liver cells but reduced in HCC.129 WNT5a, which acts in the noncanonical pathway, functions as an antagonist of canonical WNT/β-catenin signaling in HCC cells, especially in poorly differentiated cell lines.130 Its reduced expression in HCC, therefore, allows uninhibited WNT/β-catenin signaling to promote cell proliferation. Additionally, the noncanonical WNT11 mRNA and protein levels are also downregulated in HCC.131 WNT11 has been implicated as a tumor suppressor during hepatocarcinogenesis because loss of expression promotes the malignant phenotype via both canonical and noncanonical WNT/β-catenin signaling pathways. Conversely, overexpression of WNT11 activates protein kinase C signaling, which then promotes phosphorylation and degradation of β-catenin and WNT/β-catenin signaling.
Overexpression of the WNT antagonist DKK1 in HCC patients was reported to correlate with β-catenin cytoplasmic/nuclear staining and to predict poor survival.132 The ability of DKK1 to inhibit the WNT/β-catenin pathway through a negative feedback loop seems to be abrogated in HCC, most likely because genetic alterations disrupt the central multiprotein complex that controls the stability of β-catenin. In a large cohort study, elevated DKK1 in serum was validated and suggested as complement to AFP in HCC diagnosis.133 Other WNT antagonists such as DKK3 and WNT inhibitory factor (WIF)-1 were found to be significantly reduced due to hypermethylation in HCC compared with adjacent noncancerous tissues and normal control tissues, and might contribute towards activated WNT/β-catenin signaling and HCC initiation.134 The mRNA expression of DKK3 in HCC was decreased as the pathological grade increased. Reduced expression of WIF-1 mRNA was detected in HCC, and this downregulation was generally dependent on the degree of methylation at its promoter region. In vitro assays indicated that WIF-1 can inhibit cell growth by blocking WNT signaling in HCC cells.135 Additionally, DKK4 acts as a negative regulator of the WNT/β-catenin pathway, and reduced expression of its mRNA was found in 47% (38/81) of HCC.136 Decreased expression of DKK4 was associated with β-catenin accumulation in HCC tissues. Ectopic expression of DKK4 in two HCC cell lines, PLC/PRF/5 (PLC) and MHCC97L (97L), attenuated β-catenin responsive luciferase activity and decreased both β-catenin and cyclin D1 protein levels. Additionally, overexpression of DKK4 negatively affected cell proliferation, colony formation, cell migration, and in vivo tumorigenicity.
The sFRPs also act as WNT antagonists. In HCC, methylation of sFRP genes is an early event in liver carcinogenesis.81 Ectopic expression of sFRPs downregulated TCF/LEF transcriptional activity in liver cancer cells, whereas overexpression of a mutant β-catenin and depletion of sFRP1 using small interfering RNA (siRNA) synergistically upregulated TCF/LEF transcriptional activity.81 Additionally, epigenetic silencing of sFRP1 induced by the HCV core protein may lead to the activation of the WNT/β-catenin pathway and could contribute to metastasis in HCC progression.137
Aberrant expression of WNT receptors
WNT receptors FZD3 and FZD6 are upregulated in HCC.138 FZD7 was found to be overexpressed in nearly 90% of tumors during the early development of HCC, especially in those related to chronic HBV infection.77 FZD7 overexpression was observed to occur early in the disease process and contributed to stabilized wild-type β-catenin levels and enhanced tumor cell migration. Additional studies confirm that activated WNT3/FZD7 canonical pathway has a role in the early stages of hepatocarcinogenesis by promoting the acquisition of a malignant phenotype with features of epithelial-mesenchymal transition, although these cells lack tumor initiation ability in vivo.139
The LRP6 coreceptor is frequently overexpressed (in about 45% of HCC patients), and its elevated expression contributes to hyperactivation of the WNT/β-catenin signaling pathway in human HCCs, with consequent elevated levels of β-catenin protein, enhanced cell proliferation, migration, and invasion in vitro as well as enhanced tumorigenicity in vivo.140
Targeting the WNT/β-catenin pathway in HCC
Abundant evidence in clinical settings have implicated the critical role of aberrant WNT/β-catenin signaling in HCC initiation and development, underscoring the potential of targeting this pathway for therapeutic intervention of this typically hard-to-treat cancer. The WNT/β-catenin pathway is often considered difficult to drug due to the lack of obvious enzyme targets.141 Nevertheless, there has been a repertoire of small molecules and natural compounds reported to inhibit the WNT/β-catenin signaling pathway at multiple points along the pathway.142 For example, a panel of antibodies and biologics developed by OncoMed Pharmaceuticals (Redwood City, CA, USA) have already entered early phase clinical trials;143 of those, antibodies targeting FZD7/8 were found to inhibit tumor growth in multiple cancer types using human patient derived xenograft models to reduce tumor-initiating cell frequency and to exhibit synergistic activity with standard-of-care chemotherapeutic agents.144 Additionally, β-catenin (both wild-type and activated mutants) has been suggested as a prime target for therapeutic intervention.145 Current attempts in targeting WNT/β-catenin signaling in HCC using biologics and small molecules remain restricted to preclinical data, as summarized in Figure 1 and Table 3.
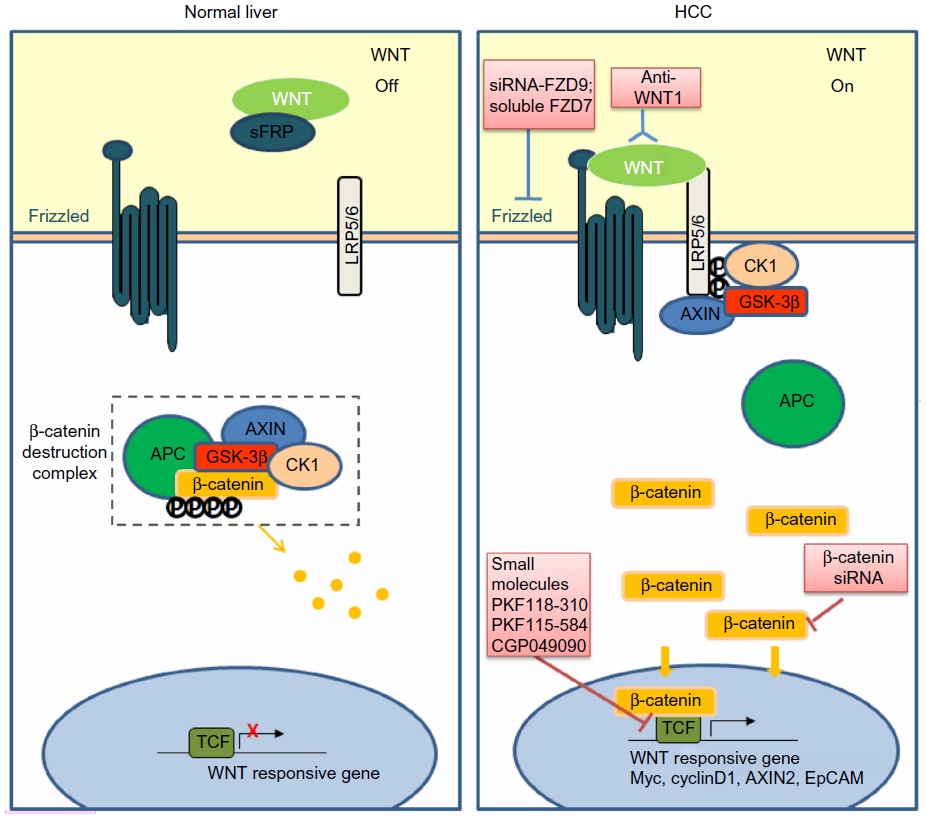 | Figure 1 Targeting the WNT/β-catenin pathway in HCC. |
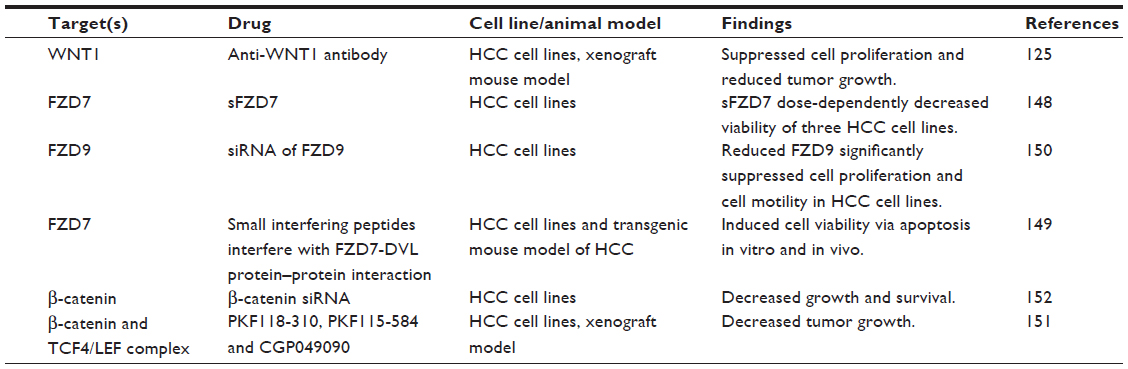 | Table 3 Experimental approaches in targeting the WNT/β-catenin signaling pathway in HCC |
Targeting WNT ligands
Since WNT ligands are direct activators of the WNT/β-catenin pathway, preventing ligand–receptor interactions or neutralizing WNT activators represent straightforward therapeutic approaches to antagonize WNT/β-catenin signaling. WNT1 was found to be overexpressed in a subgroup of HCC patients. The inhibition of WNT1-mediated signal activation using a specific anti-WNT1 antibody effectively repressed HCC cell proliferation in vitro and in vivo, with accompanying decreases in β-catenin/TCF4 transcriptional activities and downregulation of target genes c-Myc, Cyclin D1, and survivin.126 WNT1 expression was also reported to be downregulated by miR-122, a liver-enriched miRNA frequently lost in a subset of primary HCC patients.146 The ectopic overexpression of miR-122 resulted in antitumor effects of reduced cell proliferation and enhanced cell apoptosis in HCC cell lines,147 suggesting that miR-122 may be potentially useful in blocking the functions of WNT1-mediated signaling.
Targeting WNT receptors
Targeting of FZD has also met with encouraging preclinical success. For example, a soluble ectodomain of the FZD7 receptor (sFZD7) was reported to inhibit WNT/β-catenin signaling, likely via competitive inhibition of WNT ligand binding to FZD7.148 Furthermore, sFZD7 sensitized HCC cells to doxorubicin, suggesting possible combination therapy with conventional chemotherapeutic agents in patients who are less responsive to these conventional agents.148 Nambotin et al additionally designed small interfering peptides that competitively antagonized the cytoplasmic domain of FZD7, which resulted in apoptosis of cells and slowed tumor progression in a HCC mouse model.149 Knockdown of FZD9 using siRNA was also shown to suppress proliferation and motility of hepatoma cells.150
Targeting transcription complexes
β-catenin, the multifunctional effector protein, complexes with the transcription cofactors TCF4/LEF in the final relay of signals in the WNT/β-catenin pathway. Targeting this complex provides the most specific means of halting the activation of downstream target genes. Wei et al showed that three small molecule antagonists of the TCF4/β-catenin complex (namely PKF118-310, PKF115-584, and CGP049090) significantly inhibited proliferation of HCC cells in vitro and delayed tumor growth in vivo.151 These effects were accompanied by decreased expressions of downstream target genes such as Cyclin D1 and survivin. Zeng et al showed that siRNA-mediated β-catenin knockdown in human HCC cells decreased growth and survival of these cells, regardless of mutations in the CTNNB1 gene.152 Likewise, Wang et al observed significantly suppressed cell proliferation and cell cycle arrest in HepG2 cells upon siRNA-mediated β-catenin knockdown, together with changes in apoptosis- and angiogenesis-related genes.153 These studies provide convincing proof-of-principle findings that inhibiting β-catenin and its interaction with TCF4/LEF are effective ways of thwarting the downstream effects of WNT/β-catenin signaling, regardless of CTNNB1 mutation status and, therefore, represent a feasible approach for treating the heterogeneous subtypes of HCC.
Targeting enzymes in the WNT/β-catenin pathway
Recently, targeting of tankyrase enzymes has emerged as a promising therapeutic approach in cancers that rely on the WNT/β-catenin pathway for their growth.154 Tankyrases are members of the poly(adenosine diphosphate-ribose) polymerase (PARP) family that have multiple functions in various cellular processes, including regulation of telomere length by interacting with the telomeric repeat binding factor 1, regulation of mitosis, and regulation of the posttranslational modification (PARsylation) that leads to AXIN1 degradation and eventual activation of the WNT/β-catenin pathway.154 In a large scale chemical screen for tankyrase inhibitors, Huang et al identified XAV-939 as a potent inhibitor of tankyrase 1 (TANK1/PARP5A) and tankyrase 2 (TANK2/PARP5B),155 which stabilized AXIN levels in colon cancer cell lines, thereby promoting degradation of β-catenin and inhibiting WNT/β-catenin signaling. A number of derivatives based on XAV-939 have since been synthesized by different groups and have demonstrated antitumor efficacy in breast,156 colon,157,158 and lung159 cancers.
Through another chemical screen, Chen et al identified two new groups of chemical inhibitors of enzymes involved in the WNT/β-catenin pathway, known as inhibitors of WNT production (IWP) and inhibitors of WNT response (IWR).160 IWP compounds inhibit the activity of porcupine, a membrane-bound O-acyltransferase that is essential to the production of WNT proteins, by adding a palmitoyl group to enable their signaling abilities. IWP compounds therefore block all WNT-dependent biochemical changes downstream of WNT ligand/receptor interaction. However, IWR compounds appear to only promote β-catenin destruction likely by promoting stability of AXIN-scaffolded destruction complexes. The efficacy of tankyrase inhibitors, IWPs, and IWRs remain to be tested in HCC.
Other effects of targeting the WNT/β-catenin pathway in HCC
The WNT/β-catenin pathway plays integral roles in the development of the embryo as well as in the maintenance, self-renewal, and differentiation of adult mammalian tissue stem cells.40 Recent evidence suggests that aberrant regulation of this pathway in adult stem cells leads to neoplastic proliferation and tumor formation.161,162 Indeed, the WNT/β-catenin pathway has been implicated in the maintenance of “stem-like” properties of liver cancer stem cells (LCSCs).163 In particular, the LCSC marker epithelial cell adhesion molecule (EpCAM)164 has been reported to be a target gene of WNT/β-catenin signaling.165 Specifically, nuclear accumulation of β-catenin induced, whereas degradation of β-catenin or inhibition of TCF/β-catenin complex formation reduced, EpCAM gene expression in cultured normal human hepatocytes and HCC cell lines. Moreover, two TCF binding elements were identified in the EpCAM promoter. EpCAM+ HCC cells were more sensitive to TCF/β-catenin inhibitors than EpCAM− HCC in vitro, suggesting that EpCAM expression may allow effective stratification of patients with predicted pharmacologic responses to WNT/β-catenin signaling antagonists. In a separate study, the activation of WNT/β-catenin signaling enriched the EpCAM+ cell population in HCC cell lines, whereas RNA interference-based blockage of EpCAM attenuated the self-renewing, differentiation, and invasive abilities of these cells.166 Additionally, LCSCs that are enriched for the cell surface marker CD133167 were found to express 21 proteins that are involved in the WNT/β-catenin pathway.168 WNT/β-catenin activity measured by TOPflash luciferase reporter assay (Millipore Corporation, Billerica, MA, USA) was concomitantly higher in the CD133+ cells compared to that in the CD133− cells isolated from the Huh7 HCC cell line.168 Thus, targeting the WNT/β-catenin pathway offers an indirect but effective way of eradicating the functions of at least two subpopulations of LCSCs, which may potentially inhibit hepatocarcinogenesis, recurrence, and also sensitize HCC cells to conventional radio- and chemotherapies.
Coincidentally, the WNT/β-catenin signaling pathway is associated with chemoresistance in HCC, and may in part mediate the chemoresistance of LCSCs. For example, positive EpCAM expression was observed only in nonresponders to interferon-alpha/5-fluorouracil therapy, suggesting that EpCAM is a potentially useful marker of resistance to such therapy.169 In vitro activation of WNT/β-catenin signaling by GSK-3 inhibitor (6-bromoindirubin-3’-oxime) was found to induce chemoresistance to interferon-alpha/5-fluorouracil. Additionally, cisplatin resistance in HCC was reported to be associated with an increase in miR-130a levels, which was shown to directly inhibit expression of tumor suppressor gene RUNX3, resulting in activation of WNT/β-catenin signaling and increased drug resistance.170 Overexpression of miR-130a contributed to cisplatin resistance in Huh7 cells, whereas knockdown of miR-130a overcame cisplatin resistance in cisplatin-resistant Huh7 cells. These data suggest that miR-130a/RUNX3/WNT/β-catenin signaling represents a novel pathway regulating chemoresistance, and offers a novel approach to sensitizing HCC cells to chemotherapy.
Perspectives and conclusion
Given the established role of WNT/β-catenin signaling in the initiation and development of HCC and its documented dysregulation in subsets of HCC patients, the targeting of multiple components of this pathway offers new molecularly targeted approaches for potentially more efficacious and personalized treatment of HCC patients. In support of this, recent experimental and preclinical studies with investigational agents targeting multiple points along this pathway have shown encouraging antitumor effects in HCC cells. However, rigorous and systematic evaluation of their safety, toxicity, and preclinical efficacies are warranted. Like other molecularly targeted therapies, toxicities may arise from off-target effects. For example, because β-catenin has important functions in cell adhesion, complete abrogation of β-catenin function may cause detrimental effects on cellular homeostasis. Also, normal organ/tissue turnover is highly dependent on the WNT/β-catenin signaling pathway; the inhibition of this pathway may cause potential toxicity in organs such as the gut and hair follicles. As demonstrated by a study using the tankyrase inhibitor XAV-939 in colon cancer cells, high doses resulted in severe intestinal damage.158 The heterogeneous pathophysiology of HCC also suggests that combined treatment of WNT/β-catenin pathway inhibitors and other conventional or molecularly targeted agents such as sorafenib may provide a more complete antitumor response. Of note, sorafenib was shown to modulate the WNT/β-catenin pathway and β-catenin level in HCC cell lines with dysregulated WNT/β-catenin signaling (characterized by upregulation of liver-specific WNT targets and nuclear β-catenin).171 It is probable that combined treatment of sorafenib with other WNT/β-catenin inhibitors may enhance the current clinical efficacy of sorafenib.
Taken together, continued efforts in understanding the detailed molecular events underlying WNT/β-catenin-mediated hepatocarcinogenesis and in the design and development of inhibitors of this pathway will likely impact on the clinical management and outcome of HCC patients globally.
Disclosure
The authors report no conflicts of interest in this work.
References
Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127(12):2893–2917. | |
Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet. 2003;362(9399):1907–1917. | |
El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365(12):1118–1127. | |
Arzumanyan A, Reis HM, Feitelson MA. Pathogenic mechanisms in HBV- and HCV-associated hepatocellular carcinoma. Nat Rev Cancer. 2013;13(2):123–135. | |
Donato F, Tagger A, Gelatti U, et al. Alcohol and hepatocellular carcinoma: the effect of lifetime intake and hepatitis virus infections in men and women. Am J Epidemiol. 2002;155(4):323–331. | |
Ginès P, Cárdenas A, Arroyo V, Rodés J. Management of cirrhosis and ascites. N Engl J Med. 2004;350(16):1646–1654. | |
Beasley RP, Hwang LY, Lin CC, Chien CS. Hepatocellular carcinoma and hepatitis B virus. A prospective study of 22,707 men in Taiwan. Lancet. 1981;2(8256):1129–1133. | |
Fattovich G, Giustina G, Schalm SW, et al. Occurrence of hepatocellular carcinoma and decompensation in western European patients with cirrhosis type B. The EUROHEP Study Group on Hepatitis B Virus and Cirrhosis. Hepatology. 1995;21(1):77–82. | |
Avila MA, Berasain C, Sangro B, Prieto J. New therapies for hepatocellular carcinoma. Oncogene. 2006;25(27):3866–3884. | |
Llovet JM, Schwartz M, Mazzaferro V. Resection and liver transplantation for hepatocellular carcinoma. Semin Liver Dis. 2005;25(2):181–200. | |
Mazzaferro V, Regalia E, Doci R, et al. Liver transplantation for the treatment of small hepatocellular carcinomas in patients with cirrhosis. N Engl J Med. 1996;334(11):693–699. | |
Jonas S, Bechstein WO, Steinmüller T, et al. Vascular invasion and histopathologic grading determine outcome after liver transplantation for hepatocellular carcinoma in cirrhosis. Hepatology. 2001;33(5):1080–1086. | |
Lencioni R. Loco-regional treatment of hepatocellular carcinoma. Hepatology. 2010;52(2):762–773. | |
Llovet JM, Bruix J. Systematic review of randomized trials for unresectable hepatocellular carcinoma: Chemoembolization improves survival. Hepatology. 2003;37(2):429–442. | |
Varela M, Real MI, Burrel M, et al. Chemoembolization of hepatocellular carcinoma with drug eluting beads: efficacy and doxorubicin pharmacokinetics. J Hepatol. 2007;46(3):474–481. | |
Salem R, Lewandowski RJ, Mulcahy MF, et al. Radioembolization for hepatocellular carcinoma using Yttrium-90 microspheres: a comprehensive report of long-term outcomes. Gastroenterology. 2010;138(1):52–64. | |
Villanueva A, Llovet JM. Targeted therapies for hepatocellular carcinoma. Gastroenterology. 2011;140(5):1410–1426. | |
Wilhelm SM, Adnane L, Newell P, Villanueva A, Llovet JM, Lynch M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol Cancer Ther. 2008;7(10):3129–3140. | |
Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet. 2012;379(9822):1245–1255. | |
Llovet JM, Ricci S, Mazzaferro V, et al; SHARP Investigators Study Group. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378–390. | |
Villanueva A, Hernandez-Gea V, Llovet JM. Medical therapies for hepatocellular carcinoma: a critical view of the evidence. Nat Rev Gastroenterol Hepatol. 2013;10(1):34–42. | |
Moeini A, Cornellà H, Villanueva A. Emerging signaling pathways in hepatocellular carcinoma. Liver Cancer. 2012;1(2):83–93. | |
Clevers H, Nusse R. WNT/β-catenin signaling and disease. Cell. 2012;149(6):1192–1205. | |
Nusse R, Varmus HE. Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome. Cell. 1982;31(1):99–109. | |
Papkoff J, Brown AM, Varmus HE. The int-1 proto-oncogene products are glycoproteins that appear to enter the secretory pathway. Mol Cell Biol. 1987;7(11):3978–3984. | |
Anastas JN, Moon RT. WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer. 2013;13(1):11–26. | |
Nusse R, Varmus H. Three decades of WNTs: a personal perspective on how a scientific field developed. EMBO J. 2012;31(12):2670–2684. | |
Bhanot P, Brink M, Samos CH, et al. A new member of the frizzled family from Drosophila functions as a Wingless receptor. Nature. 1996;382(6588):225–230. | |
Wehrli M, Dougan ST, Caldwell K, et al. Arrow encodes an LDL-receptor-related protein essential for wingless signalling. Nature. 2000;407(6803):527–530. | |
Grumolato L, Liu G, Mong P, et al. Canonical and noncanonical WNTs use a common mechanism to activate completely unrelated coreceptors. Genes Dev. 2010;24(22):2517–2530. | |
Takeichi M. Cadherins: a molecular family important in selective cell-cell adhesion. Annu Rev Biochem. 1990;59:237–252. | |
He TC, Sparks AB, Rago C, et al. Identification of c-MYC as a target of the APC pathway. Science. 1998;281(5382):1509–1512. | |
Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398(6726):422–426. | |
Yan D, Wiesmann M, Rohan M, et al. Elevated expression of axin2 and hnkd mRNA provides evidence that WNT/beta -catenin signaling is activated in human colon tumors. Proc Natl Acad Sci U S A. 2001;98(26):14973–14978. | |
Li VS, Ng SS, Boersema PJ, et al. WNT signaling through inhibition of β-catenin degradation in an intact Axin1 complex. Cell. 2012;149(6):1245–1256. | |
Moon RT, Campbell RM, Christian JL, McGrew LL, Shih J, Fraser S. XWNT-5A: a maternal WNT that affects morphogenetic movements after overexpression in embryos of Xenopus laevis. Development. 1993;119(1):97–111. | |
Slusarski DC, Yang-Snyder J, Busa WB, Moon RT. Modulation of embryonic intracellular Ca2+ signaling by WNT-5A. Dev Biol. 1997;182(1):114–120. | |
Oishi I, Suzuki H, Onishi N, et al. The receptor tyrosine kinase Ror2 is involved in non-canonical WNT5a/JNK signalling pathway. Genes Cells. 2003;8(7):645–654. | |
Kühl M, Sheldahl LC, Park M, Miller JR, Moon RT. The WNT/Ca2+ pathway: a new vertebrate WNT signaling pathway takes shape. Trends Genet. 2000;16(7):279–283. | |
Van Camp JK, Beckers S, Zegers D, Van Hul W. WNT Signaling and the Control of Human Stem Cell Fate. Stem Cell Rev. Epub December 10, 2013. | |
Lawrence PA, Struhl G, Casal J. Planar cell polarity: one or two pathways? Nat Rev Genet. 2007;8(7):555–563. | |
Klaus A, Birchmeier W. WNT signalling and its impact on development and cancer. Nat Rev Cancer. 2008;8(5):387–398. | |
Polakis P. WNT signaling in cancer. Cold Spring Harb Perspect Biol. 2012;4(5). | |
Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87(2):159–170. | |
Rowan AJ, Lamlum H, Ilyas M, et al. APC mutations in sporadic colorectal tumors: A mutational “hotspot” and interdependence of the “two hits”. Proc Natl Acad Sci U S A. 2000;97(7):3352–3357. | |
Ahmed Y, Hayashi S, Levine A, Wieschaus E. Regulation of armadillo by a Drosophila APC inhibits neuronal apoptosis during retinal development. Cell. 1998;93(7):1171–1182. | |
Jones S, Chen WD, Parmigiani G, et al. Comparative lesion sequencing provides insights into tumor evolution. Proc Natl Acad Sci U S A. 2008;105(11):4283–4288. | |
Satoh S, Daigo Y, Furukawa Y, et al. AXIN1 mutations in hepatocellular carcinomas, and growth suppression in cancer cells by virus-mediated transfer of AXIN1. Nat Genet. 2000;24(3):245–250. | |
Liu W, Dong X, Mai M, et al. Mutations in AXIN2 cause colorectal cancer with defective mismatch repair by activating beta-catenin/TCF signalling. Nat Genet. 2000;26(2):146–147. | |
Hidaka Y, Mitomi H, Saito T, et al. Alteration in the WNT/β-catenin signaling pathway in gastric neoplasias of fundic gland (chief cell predominant) type. Hum Pathol. 2013;44(11):2438–2448. | |
Chapman A, Durand J, Ouadi L, Bourdeau I. Identification of genetic alterations of AXIN2 gene in adrenocortical tumors. J Clin Endocrinol Metab. 2011;96(9):E1477–E1481. | |
Yardy GW, Bicknell DC, Wilding JL, et al. Mutations in the AXIN1 gene in advanced prostate cancer. Eur Urol. 2009;56(3):486–494. | |
Polakis P. WNT signaling and cancer. Genes Dev. 2000;14(15):1837–1851. | |
Korinek V, Barker N, Morin PJ, et al. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC-/- colon carcinoma. Science. 1997;275(5307):1784–1787. | |
Morin PJ, Sparks AB, Korinek V, et al. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 1997;275(5307):1787–1790. | |
Rubinfeld B, Robbins P, El-Gamil M, Albert I, Porfiri E, Polakis P. Stabilization of beta-catenin by genetic defects in melanoma cell lines. Science. 1997;275(5307):1790–1792. | |
de La Coste A, Romagnolo B, Billuart P, et al. Somatic mutations of the beta-catenin gene are frequent in mouse and human hepatocellular carcinomas. Proc Natl Acad Sci U S A. 1998;95(15):8847–8851. | |
Miyoshi Y, Iwao K, Nagasawa Y, et al. Activation of the beta-catenin gene in primary hepatocellular carcinomas by somatic alterations involving exon 3. Cancer Res. 1998;58(12):2524–2527. | |
Fujimori M, Ikeda S, Shimizu Y, Okajima M, Asahara T. Accumulation of beta-catenin protein and mutations in exon 3 of beta-catenin gene in gastrointestinal carcinoid tumor. Cancer Res. 2001;61(18):6656–6659. | |
Garcia-Rostan G, Tallini G, Herrero A, D’Aquila TG, Carcangiu ML, Rimm DL. Frequent mutation and nuclear localization of beta-catenin in anaplastic thyroid carcinoma. Cancer Res. 1999;59(8):1811–1815. | |
Palacios J, Gamallo C. Mutations in the beta-catenin gene (CTNNB1) in endometrioid ovarian carcinomas. Cancer Res. 1998;58(7):1344–1347. | |
Shitoh K, Konishi F, Iijima T, et al. A novel case of a sporadic desmoid tumour with mutation of the beta catenin gene. J Clin Pathol. 1999;52(9):695–696. | |
Koesters R, Ridder R, Kopp-Schneider A, et al. Mutational activation of the beta-catenin proto-oncogene is a common event in the development of Wilms’ tumors. Cancer Res. 1999;59(16):3880–3882. | |
Duval A, Gayet J, Zhou XP, Iacopetta B, Thomas G, Hamelin R. Frequent frameshift mutations of the TCF-4 gene in colorectal cancers with microsatellite instability. Cancer Res. 1999;59(17):4213–4215. | |
Duval A, Rolland S, Tubacher E, Bui H, Thomas G, Hamelin R. The human T-cell transcription factor-4 gene: structure, extensive characterization of alternative splicings, and mutational analysis in colorectal cancer cell lines. Cancer Res. 2000;60(14):3872–3879. | |
Shiina H, Igawa M, Breault J, et al. The human T-cell factor-4 gene splicing isoforms, WNT signal pathway, and apoptosis in renal cell carcinoma. Clin Cancer Res. 2003;9(6):2121–2132. | |
Howng SL, Huang FH, Hwang SL, et al. Differential expression and splicing isoform analysis of human Tcf-4 transcription factor in brain tumors. Int J Oncol. 2004;25(6):1685–1692. | |
Duval A, Iacopetta B, Ranzani GN, Lothe RA, Thomas G, Hamelin R. Variable mutation frequencies in coding repeats of TCF-4 and other target genes in colon, gastric and endometrial carcinoma showing microsatellite instability. Oncogene. 1999;18(48):6806–6809. | |
Milovanovic T, Planutis K, Nguyen A, et al. Expression of WNT genes and frizzled 1 and 2 receptors in normal breast epithelium and infiltrating breast carcinoma. Int J Oncol. 2004;25(5):1337–1342. | |
Flahaut M, Meier R, Coulon A, et al. The WNT receptor FZD1 mediates chemoresistance in neuroblastoma through activation of the WNT/beta-catenin pathway. Oncogene. 2009;28(23):2245–2256. | |
Zhang Z, Schittenhelm J, Guo K, et al. Upregulation of frizzled 9 in astrocytomas. Neuropathol Appl Neurobiol. 2006;32(6):615–624. | |
Rhee CS, Sen M, Lu D, et al. WNT and frizzled receptors as potential targets for immunotherapy in head and neck squamous cell carcinomas. Oncogene. 2002;21(43):6598–6605. | |
Khan NI, Bradstock KF, Bendall LJ. Activation of WNT/beta-catenin pathway mediates growth and survival in B-cell progenitor acute lymphoblastic leukaemia. Br J Haematol. 2007;138(3):338–348. | |
Janssens N, Andries L, Janicot M, Perera T, Bakker A. Alteration of frizzled expression in renal cell carcinoma. Tumour Biol. 2004;25(4):161–171. | |
Thiele S, Rauner M, Goettsch C, et al. Expression profile of WNT molecules in prostate cancer and its regulation by aminobisphosphonates. J Cell Biochem. 2011;112(6):1593–1600. | |
Tanaka S, Akiyoshi T, Mori M, Wands JR, Sugimachi K. A novel frizzled gene identified in human esophageal carcinoma mediates APC/beta-catenin signals. Proc Natl Acad Sci U S A. 1998;95(17):10164–10169. | |
Merle P, de la Monte S, Kim M, et al. Functional consequences of frizzled-7 receptor overexpression in human hepatocellular carcinoma. Gastroenterology. 2004;127(4):1110–1122. | |
Terasaki H, Saitoh T, Shiokawa K, Katoh M. Frizzled-10, up-regulated in primary colorectal cancer, is a positive regulator of the WNT – beta-catenin – TCF signaling pathway. Int J Mol Med. 2002;9(2):107–112. | |
Nagayama S, Fukukawa C, Katagiri T, et al. Therapeutic potential of antibodies against FZD 10, a cell-surface protein, for synovial sarcomas. Oncogene. 2005;24(41):6201–6212. | |
Suzuki H, Gabrielson E, Chen W, et al. A genomic screen for genes upregulated by demethylation and histone deacetylase inhibition in human colorectal cancer. Nat Genet. 2002;31(2):141–149. | |
Takagi H, Sasaki S, Suzuki H, et al. Frequent epigenetic inactivation of SFRP genes in hepatocellular carcinoma. J Gastroenterol. 2008;43(5):378–389. | |
Zhou Z, Wang J, Han X, Zhou J, Linder S. Up-regulation of human secreted frizzled homolog in apoptosis and its down-regulation in breast tumors. Int J Cancer. 1998;78(1):95–99. | |
Ko J, Ryu KS, Lee YH, et al. Human secreted frizzled-related protein is down-regulated and induces apoptosis in human cervical cancer. Exp Cell Res. 2002;280(2):280–287. | |
Ugolini F, Charafe-Jauffret E, Bardou VJ, et al. WNT pathway and mammary carcinogenesis: loss of expression of candidate tumor suppressor gene SFRP1 in most invasive carcinomas except of the medullary type. Oncogene. 2001;20(41):5810–5817. | |
Ekström EJ, Sherwood V, Andersson T. Methylation and loss of secreted Frizzled-related protein 3 enhances melanoma cell migration and invasion. PLoS One. 2011;6(4):e18674. | |
Kawano Y, Kypta R. Secreted antagonists of the WNT signalling pathway. J Cell Sci. 2003;116(Pt 13):2627–2634. | |
Cowling VH, D’Cruz CM, Chodosh LA, Cole MD. c-Myc transforms human mammary epithelial cells through repression of the WNT inhibitors DKK1 and SFRP1. Mol Cell Biol. 2007;27(14):5135–5146. | |
Cho JH, Dimri M, Dimri GP. A positive feedback loop regulates the expression of polycomb group protein BMI1 via WNT signaling pathway. J Biol Chem. 2013;288(5):3406–3418. | |
Hsieh A, Kim HS, Lim SO, Yu DY, Jung G. Hepatitis B viral X protein interacts with tumor suppressor adenomatous polyposis coli to activate WNT/β-catenin signaling. Cancer Lett. 2011;300(2):162–172. | |
Liu J, Ding X, Tang J, et al. Enhancement of canonical WNT/β-catenin signaling activity by HCV core protein promotes cell growth of hepatocellular carcinoma cells. PLoS One. 2011;6(11):e27496. | |
Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–297. | |
Huang K, Zhang JX, Han L, et al. MicroRNA roles in beta-catenin pathway. Mol Cancer. 2010;9:252. | |
Saydam O, Shen Y, Würdinger T, et al. Downregulated microRNA-200a in meningiomas promotes tumor growth by reducing E-cadherin and activating the WNT/beta-catenin signaling pathway. Mol Cell Biol. 2009;29(21):5923–5940. | |
Hashimi ST, Fulcher JA, Chang MH, Gov L, Wang S, Lee B. MicroRNA profiling identifies miR-34a and miR-21 and their target genes JAG1 and WNT1 in the coordinate regulation of dendritic cell differentiation. Blood. 2009;114(2):404–414. | |
Cha YH, Kim NH, Park C, Lee I, Kim HS, Yook JI. MiRNA-34 intrinsically links p53 tumor suppressor and WNT signaling. Cell Cycle. 2012;11(7):1273–1281. | |
Kim NH, Cha YH, Kang SE, et al. p53 regulates nuclear GSK-3 levels through miR-34-mediated Axin2 suppression in colorectal cancer cells. Cell Cycle. 2013;12(10):1578–1587. | |
Xia H, Ooi LL, Hui KM. MiR-214 targets β-catenin pathway to suppress invasion, stem-like traits and recurrence of human hepatocellular carcinoma. PLoS One. 2012;7(9):e44206. | |
Zhang Y, Wei W, Cheng N, et al. Hepatitis C virus-induced up-regulation of microRNA-155 promotes hepatocarcinogenesis by activating WNT signaling. Hepatology. 2012;56(5):1631–1640. | |
Takigawa Y, Brown AM. WNT signaling in liver cancer. Curr Drug Targets. 2008;9(11):1013–1024. | |
Huang H, Fujii H, Sankila A, et al. Beta-catenin mutations are frequent in human hepatocellular carcinomas associated with hepatitis C virus infection. Am J Pathol. 1999;155(6):1795–1801. | |
Hsu HC, Jeng YM, Mao TL, Chu JS, Lai PL, Peng SY. Beta-catenin mutations are associated with a subset of low-stage hepatocellular carcinoma negative for hepatitis B virus and with favorable prognosis. Am J Pathol. 2000;157(3):763–770. | |
Wong CM, Fan ST, Ng IO. Beta-catenin mutation and overexpression in hepatocellular carcinoma: clinicopathologic and prognostic significance. Cancer. 2001;92(1):136–145. | |
Nhieu JT, Renard CA, Wei Y, Cherqui D, Zafrani ES, Buendia MA. Nuclear accumulation of mutated beta-catenin in hepatocellular carcinoma is associated with increased cell proliferation. Am J Pathol. 1999;155(3):703–710. | |
Inagawa S, Itabashi M, Adachi S, et al. Expression and prognostic roles of beta-catenin in hepatocellular carcinoma: correlation with tumor progression and postoperative survival. Clin Cancer Res. 2002;8(2):450–456. | |
Zucman-Rossi J, Benhamouche S, Godard C, et al. Differential effects of inactivated Axin1 and activated beta-catenin mutations in human hepatocellular carcinomas. Oncogene. 2007;26(5):774–780. | |
Cieply B, Zeng G, Proverbs-Singh T, Geller DA, Monga SP. Unique phenotype of hepatocellular cancers with exon-3 mutations in beta-catenin gene. Hepatology. 2009;49(3):821–831. | |
Austinat M, Dunsch R, Wittekind C, Tannapfel A, Gebhardt R, Gaunitz F. Correlation between beta-catenin mutations and expression of WNT-signaling target genes in hepatocellular carcinoma. Mol Cancer. 2008;7:21. | |
Tornesello ML, Buonaguro L, Tatangelo F, Botti G, Izzo F, Buonaguro FM. Mutations in TP53, CTNNB1 and PIK3CA genes in hepatocellular carcinoma associated with hepatitis B and hepatitis C virus infections. Genomics. 2013;102(2):74–83. | |
Amit S, Hatzubai A, Birman Y, et al. Axin-mediated CKI phosphorylation of beta-catenin at Ser 45: a molecular switch for the WNT pathway. Genes Dev. 2002;16(9):1066–1076. | |
Yost C, Torres M, Miller JR, Huang E, Kimelman D, Moon RT. The axis-inducing activity, stability, and subcellular distribution of beta-catenin is regulated in Xenopus embryos by glycogen synthase kinase 3. Genes Dev. 1996;10(12):1443–1454. | |
Park JY, Park WS, Nam SW, et al. Mutations of beta-catenin and AXIN I genes are a late event in human hepatocellular carcinogenesis. Liver Int. 2005;25(1):70–76. | |
Murata M, Miyoshi Y, Ohsawa M, et al. Accumulation of beta-catenin in the cytoplasm and the nuclei during the early hepatic tumorigenesis. Hepatol Res. 2001;21(2):126–135. | |
Jung HY, Jun S, Lee M, et al. PAF and EZH2 induce WNT/β-catenin signaling hyperactivation. Mol Cell. 2013;52(2):193–205. | |
Lee E, Salic A, Krüger R, Heinrich R, Kirschner MW. The roles of APC and Axin derived from experimental and theoretical analysis of the WNT pathway. PLoS Biol. 2003;1(1):E10. | |
Feng GJ, Cotta W, Wei XQ, et al. Conditional disruption of Axin1 leads to development of liver tumors in mice. Gastroenterology. 2012;143(6):1650–1659. | |
Taniguchi K, Roberts LR, Aderca IN, et al. Mutational spectrum of beta-catenin, AXIN1, and AXIN2 in hepatocellular carcinomas and hepatoblastomas. Oncogene. 2002;21(31):4863–4871. | |
Bala S, Sulekova Z, Ballhausen WG. Constitutive APC exon 14 skipping in early-onset familial adenomatous polyposis reveals a dramatic quantitative distortion of APC gene-specific isoforms. Hum Mutat. 1997;10(3):201–206. | |
Gruner BA, DeNapoli TS, Andrews W, Tomlinson G, Bowman L, Weitman SD. Hepatocellular carcinoma in children associated with Gardner syndrome or familial adenomatous polyposis. J Pediatr Hematol Oncol. 1998;20(3):274–278. | |
Su LK, Abdalla EK, Law CH, Kohlmann W, Rashid A, Vauthey JN. Biallelic inactivation of the APC gene is associated with hepatocellular carcinoma in familial adenomatous polyposis coli. Cancer. 2001;92(2):332–339. | |
Katoh H, Shibata T, Kokubu A, et al. Genetic inactivation of the APC gene contributes to the malignant progression of sporadic hepatocellular carcinoma: a case report. Genes Chromosomes Cancer. 2006;45(11):1050–1057. | |
Csepregi A, Röcken C, Hoffmann J, et al. APC promoter methylation and protein expression in hepatocellular carcinoma. J Cancer Res Clin Oncol. 2008;134(5):579–589. | |
Hernandez-Vargas H, Lambert MP, Le Calvez-Kelm F, et al. Hepatocellular carcinoma displays distinct DNA methylation signatures with potential as clinical predictors. PLoS One. 2010; 5(3):e9749. | |
Song MA, Tiirikainen M, Kwee S, Okimoto G, Yu H, Wong LL. Elucidating the landscape of aberrant DNA methylation in hepatocellular carcinoma. PLoS One. 2013;8(2):e55761. | |
Jain S, Chang TT, Hamilton JP, et al. Methylation of the CpG sites only on the sense strand of the APC gene is specific for hepatocellular carcinoma. PLoS One. 2011;6(11):e26799. | |
Lee HH, Uen YH, Tian YF, et al. WNT-1 protein as a prognostic biomarker for hepatitis B-related and hepatitis C-related hepatocellular carcinoma after surgery. Cancer Epidemiol Biomarkers Prev. 2009;18(5):1562–1569. | |
Wei W, Chua MS, Grepper S, So SK. Blockade of WNT-1 signaling leads to anti-tumor effects in hepatocellular carcinoma cells. Mol Cancer. 2009;8:76. | |
Fukutomi T, Zhou Y, Kawai S, Eguchi H, Wands JR, Li J. Hepatitis C virus core protein stimulates hepatocyte growth: correlation with upregulation of WNT-1 expression. Hepatology. 2005;41(5):1096–1105. | |
Kim M, Lee HC, Tsedensodnom O, et al. Functional interaction between WNT3 and frizzled-7 leads to activation of the WNT/beta-catenin signaling pathway in hepatocellular carcinoma cells. J Hepatol. 2008;48(5):780–791. | |
Geng M, Cao YC, Chen YJ, Jiang H, Bi LQ, Liu XH. Loss of WNT5a and Ror2 protein in hepatocellular carcinoma associated with poor prognosis. World J Gastroenterol. 2012;18(12):1328–1338. | |
Yuzugullu H, Benhaj K, Ozturk N, et al. Canonical WNT signaling is antagonized by noncanonical WNT5a in hepatocellular carcinoma cells. Mol Cancer. 2009;8:90. | |
Toyama T, Lee HC, Koga H, Wands JR, Kim M. Noncanonical WNT11 inhibits hepatocellular carcinoma cell proliferation and migration. Mol Cancer Res. 2010;8(2):254–265. | |
Yu B, Yang X, Xu Y, et al. Elevated expression of DKK1 is associated with cytoplasmic/nuclear beta-catenin accumulation and poor prognosis in hepatocellular carcinomas. J Hepatol. 2009;50(5):948–957. | |
Shen Q, Fan J, Yang XR, et al. Serum DKK1 as a protein biomarker for the diagnosis of hepatocellular carcinoma: a large-scale, multicentre study. Lancet Oncol. 2012;13(8):817–826. | |
Ding Z, Qian YB, Zhu LX, Xiong QR. Promoter methylation and mRNA expression of DKK-3 and WIF-1 in hepatocellular carcinoma. World J Gastroenterol. 2009;15(21):2595–2601. | |
Deng Y, Yu B, Cheng Q, et al. Epigenetic silencing of WIF-1 in hepatocellular carcinomas. J Cancer Res Clin Oncol. 2010;136(8):1161–1167. | |
Fatima S, Lee NP, Tsang FH, et al. Dickkopf 4 (DKK4) acts on WNT/β-catenin pathway by influencing β-catenin in hepatocellular carcinoma. Oncogene. 2012;31(38):4233–4244. | |
Quan H, Zhou F, Nie D, et al. Hepatitis C virus core protein epigenetically silences SFRP1 and enhances HCC aggressiveness by inducing epithelial-mesenchymal transition. Oncogene. 2013, Epub ahead of print. | |
Bengochea A, de Souza MM, Lefrançois L, et al. Common dysregulation of WNT/Frizzled receptor elements in human hepatocellular carcinoma. Br J Cancer. 2008;99(1):143–150. | |
Nambotin SB, Tomimaru Y, Merle P, Wands JR, Kim M. Functional consequences of WNT3/Frizzled7-mediated signaling in non-transformed hepatic cells. Oncogenesis. 2012;1:e31. | |
Tung EK, Wong BY, Yau TO, Ng IO. Upregulation of the WNT co-receptor LRP6 promotes hepatocarcinogenesis and enhances cell invasion. PLoS One. 2012;7(5):e36565. | |
Garber K. Drugging the WNT pathway: problems and progress. J Natl Cancer Inst. 2009;101(8):548–550. | |
Voronkov A, Krauss S. WNT/beta-catenin signaling and small molecule inhibitors. Curr Pharm Des. 2013;19(4):634–664. | |
Development pipeline [webpage on the Internet]. Redwood City, CA:OncoMed Pharmaceuticals. Availalbe from: http://www.oncomed.com/Pipeline.html. Accessed November 21, 2013. | |
Gurney A, Axelrod F, Bond CJ, et al. WNT pathway inhibition via the targeting of Frizzled receptors results in decreased growth and tumorigenicity of human tumors. Proc Natl Acad Sci USA. 2012;109(29):11717–11722. | |
Polakis P. Drugging WNT signalling in cancer. EMBO J. 2012;31(12):2737–2746. | |
Coulouarn C, Factor VM, Andersen JB, Durkin ME, Thorgeirsson SS. Loss of miR-122 expression in liver cancer correlates with suppression of the hepatic phenotype and gain of metastatic properties. Oncogene. 2009;28(40):3526–3536. | |
Xu J, Zhu X, Wu L, et al. MicroRNA-122 suppresses cell proliferation and induces cell apoptosis in hepatocellular carcinoma by directly targeting WNT/β-catenin pathway. Liver Int. 2012;32(5):752–760. | |
Wei W, Chua MS, Grepper S, So SK. Soluble Frizzled-7 receptor inhibits WNT signaling and sensitizes hepatocellular carcinoma cells towards doxorubicin. Mol Cancer. 2011;10:16. | |
Nambotin SB, Lefrancois L, Sainsily X, et al. Pharmacological inhibition of Frizzled-7 displays anti-tumor properties in hepatocellular carcinoma. J Hepatol. 2011;54(2):288–299. | |
Fujimoto T, Tomizawa M, Yokosuka O. SiRNA of frizzled-9 suppresses proliferation and motility of hepatoma cells. Int J Oncol. 2009;35(4):861–866. | |
Wei W, Chua MS, Grepper S, So S. Small molecule antagonists of Tcf4/beta-catenin complex inhibit the growth of HCC cells in vitro and in vivo. Int J Cancer. 2010;126(10):2426–2436. | |
Zeng G, Apte U, Cieply B, Singh S, Monga SP. siRNA-mediated beta-catenin knockdown in human hepatoma cells results in decreased growth and survival. Neoplasia. 2007;9(11):951–959. | |
Wang XH, Sun X, Meng XW, et al. beta-catenin siRNA regulation of apoptosis- and angiogenesis-related gene expression in hepatocellular carcinoma cells: potential uses for gene therapy. Oncol Rep. 2010;24(4):1093–1099. | |
Riffell JL, Lord CJ, Ashworth A. Tankyrase-targeted therapeutics: expanding opportunities in the PARP family. Nat Rev Drug Discov. 2012;11(12):923–936. | |
Huang SM, Mishina YM, Liu S, et al. Tankyrase inhibition stabilizes axin and antagonizes WNT signalling. Nature. 2009;461(7264):614–620. | |
Bao R, Christova T, Song S, Angers S, Yan X, Attisano L. Inhibition of tankyrases induces axin stabilization and blocks Wnt signalling in breast cancer cells. PLoS One. 2012;7(11):e48670. | |
Waaler J, Machon O, Tumova L, et al. A novel tankyrase inhibitor decreases canonical WNT signaling in colon carcinoma cells and reduces tumor growth in conditional APC mutant mice. Cancer Res. 2012;72(11):2822–2832. | |
Lau T, Chan E, Callow M, et al. A novel tankyrase small-molecule inhibitor suppresses APC mutation-driven colorectal tumor growth. Cancer Res. 2013;73(10):3132–3144. | |
Busch AM, Johnson KC, Stan RV, et al. Evidence for tankyrases as antineoplastic targets in lung cancer. BMC Cancer. 2013;13:211. | |
Chen B, Dodge ME, Tang W, et al. Small molecule-mediated disruption of WNT-dependent signaling in tissue regeneration and cancer. Nat Chem Biol. 2009;5(2):100–107. | |
Ben-Porath I, Thomson MW, Carey VJ, et al. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet. 2008;40(5):499–507. | |
LaBarge MA. The difficulty of targeting cancer stem cell niches. Clin Cancer Res. 2010;16(12):3121–3129. | |
Yang W, Yan HX, Chen L, et al. WNT/beta-catenin signaling contributes to activation of normal and tumorigenic liver progenitor cells. Cancer Res. 2008;68(11):4287–4295. | |
Terris B, Cavard C, Perret C. EpCAM, a new marker for cancer stem cells in hepatocellular carcinoma. J Hepatol. 2010;52(2):280–281. | |
Yamashita T, Budhu A, Forgues M, Wang XW. Activation of hepatic stem cell marker EpCAM by WNT-beta-catenin signaling in hepatocellular carcinoma. Cancer Res. 2007;67(22):10831–10839. | |
Yamashita T, Ji J, Budhu A, et al. EpCAM-positive hepatocellular carcinoma cells are tumor-initiating cells with stem/progenitor cell features. Gastroenterology. 2009;136(3):1012–1024. | |
Suetsugu A, Nagaki M, Aoki H, Motohashi T, Kunisada T, Moriwaki H. Characterization of CD133+ hepatocellular carcinoma cells as cancer stem/progenitor cells. Biochem Biophys Res Commun. 2006;351(4):820–824. | |
Tsai ST, Tsou CC, Mao WY, et al. Label-free quantitative proteomics of CD133-positive liver cancer stem cells. Proteome Sci. 2012;10(1):69. | |
Noda T, Nagano H, Takemasa I, et al. Activation of WNT/beta-catenin signalling pathway induces chemoresistance to interferon-alpha/5-fluorouracil combination therapy for hepatocellular carcinoma. Br J Cancer. 2009;100(10):1647–1658. | |
Xu N, Shen C, Luo Y, et al. Upregulated miR-130a increases drug resistance by regulating RUNX3 and WNT signaling in cisplatin-treated HCC cell. Biochem Biophys Res Commun. 2012;425(2):468–472. | |
Lachenmayer A, Alsinet C, Savic R, et al. WNT-pathway activation in two molecular classes of hepatocellular carcinoma and experimental modulation by sorafenib. Clin Cancer Res. 2012;18(18):4997–5007. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.