")
Back to Journals » Journal of Blood Medicine » Volume 12
Why is Misdiagnosis of von Willebrand Disease Still Prevalent and How Can We Overcome It? A Focus on Clinical Considerations and Recommendations
Authors Colonne CK, Reardon B, Curnow J , Favaloro EJ
Received 1 June 2021
Accepted for publication 31 July 2021
Published 17 August 2021 Volume 2021:12 Pages 755—768
DOI https://doi.org/10.2147/JBM.S266791
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Chanukya K Colonne,1,* Benjamin Reardon,1,* Jennifer Curnow,2– 4 Emmanuel J Favaloro1,4,5
1Department of Haematology, Institute of Clinical Pathology and Medical Research (ICPMR), NSW Health Pathology, Westmead Hospital, Sydney, NSW, Australia; 2Department of Clinical Haematology, Westmead Hospital, Sydney, NSW, Australia; 3Faculty of Medicine and Health, University of Sydney, Sydney, NSW, Australia; 4Sydney Centres for Thrombosis and Haemostasis, Sydney, NSW, Australia; 5School of Biomedical Sciences, Charles Sturt University, Wagga Wagga, NSW, Australia
*These authors contributed equally to this work
Correspondence: Emmanuel J Favaloro
Department of Haematology, Institute of Clinical Pathology and Medical Research (ICPMR), Westmead, Sydney, NSW, 2145, Australia
Tel +612 8890 6618
Fax +612 9689 2331
Email [email protected]
Abstract: Despite von Willebrand disease (VWD) being the most common inherited bleeding disorder, its accurate diagnosis is frequently shrouded by diagnostic pitfalls. VWD is frequently under-diagnosed, over-diagnosed and misdiagnosed, leading to significant avoidable patient morbidity and health care system burden. At the heart of this dilemma lies the heterogeneity and complexity of von Willebrand factor (VWF) and associated defects, and the necessity of coalescing clinical and laboratory features to obtain an accurate diagnosis. Common pitfalls include poor clinical and scientific understanding and familiarity with VWD, incomplete clinical history and lack of routine use of standardised bleeding assessment tools (BAT), difficulty in accessing a comprehensive repertoire of laboratory tests, significant pre-analytical, analytical and post-analytical issues, and lack of expertise in laboratory testing and interpretation. Errors, resulting in under-diagnosis, over-diagnosis, and misdiagnosis of VWD, are presented and discussed. Strategies to minimise errors include better education of clinicians and laboratory staff on VWD, routine use of validated BAT, utilising a comprehensive gamut of laboratory investigations according to a standardised algorithm, and repeating testing to minimise pre-analytical errors. Recommendations on appropriate patient selection for VWD testing, how VWD should be investigated in the laboratory, and how to ensure test results are accurately interpreted in the correct clinical context are detailed.
Keywords: von Willebrand disease, VWD, diagnosis
Introduction
von Willebrand disease (VWD) is the most common inherited bleeding disorder.1,2 Despite this, VWD is one of the most commonly misdiagnosed or overlooked entities in everyday clinical practice. Of interest, VWD may be both over- and under-diagnosed, as well as misdiagnosed, either as another entity or as a different subtype of the disorder. This review will detail the causes underlying the diagnostic uncertainties overshadowing VWD and provide solutions on how to overcome these.
von Willebrand Disease
VWD was first described by Finnish physician Erik Adolf von Willebrand in 1926, following the presentation of a young girl with recurrent episodes of bleeding, which were clinically distinct from haemophilia.3 VWD is caused by quantitative or qualitative deficiencies in a plasma protein now called von Willebrand factor (VWF). VWF is a large, complex protein that has essential roles in primary and secondary hemostasis.4 High-molecular-weight VWF multimers mediate platelet adhesion at sites of vascular injury in primary hemostasis by binding to connective tissue and platelets.4 VWF also plays a key role in secondary hemostasis, acting as a chaperone to clotting factor VIII (FVIII) by binding to and stabilizing it in the circulation.4 The multifunctional nature of VWF explains the heterogeneity in clinical symptoms and bleeding risk seen in VWD, as well as the resulting diagnostic challenges.5
VWD is autosomally inherited and arises due to mutations in VWF, mapped at 12p13.3 and comprised of 52 exons.6 Approximate exon locations have been mapped to binding locations on the VWF protein with corresponding defects. The majority of mutations in VWD are missense, with some having variable penetrance, particularly in Type 1 VWD.4
VWD is classified into three major types (summarised in Table 1). Type 1 reflects a mild to moderate reduction in functionally normal VWF; Type 2 involves the expression of functionally abnormal VWF; and, Type 3 is the (near) complete absence of VWF.4 65–80% of identified VWD cases are Type 1, whereas Type 2 and 3 account for 20–35% and <1% of cases, respectively.7 A potential subtype of Type 1 called VWD Type 1C has been proposed. In contrast to classic VWD type 1, where there is a reduction in the production of VWF and/or a mild increased clearance, VWD Type 1C is characterised by a significant increase in clearance of VWF resulting in low VWF levels and an exaggerated but short-lived response to desmopressin.8 Type 2 VWD is separated into four ‘subtypes’ (2A, 2B, 2M, 2N), according to the functional defect. A separate defect affecting the platelet receptor for VWF, namely glycoprotein Ib (GPIb) is called platelet type (PT-) VWD.
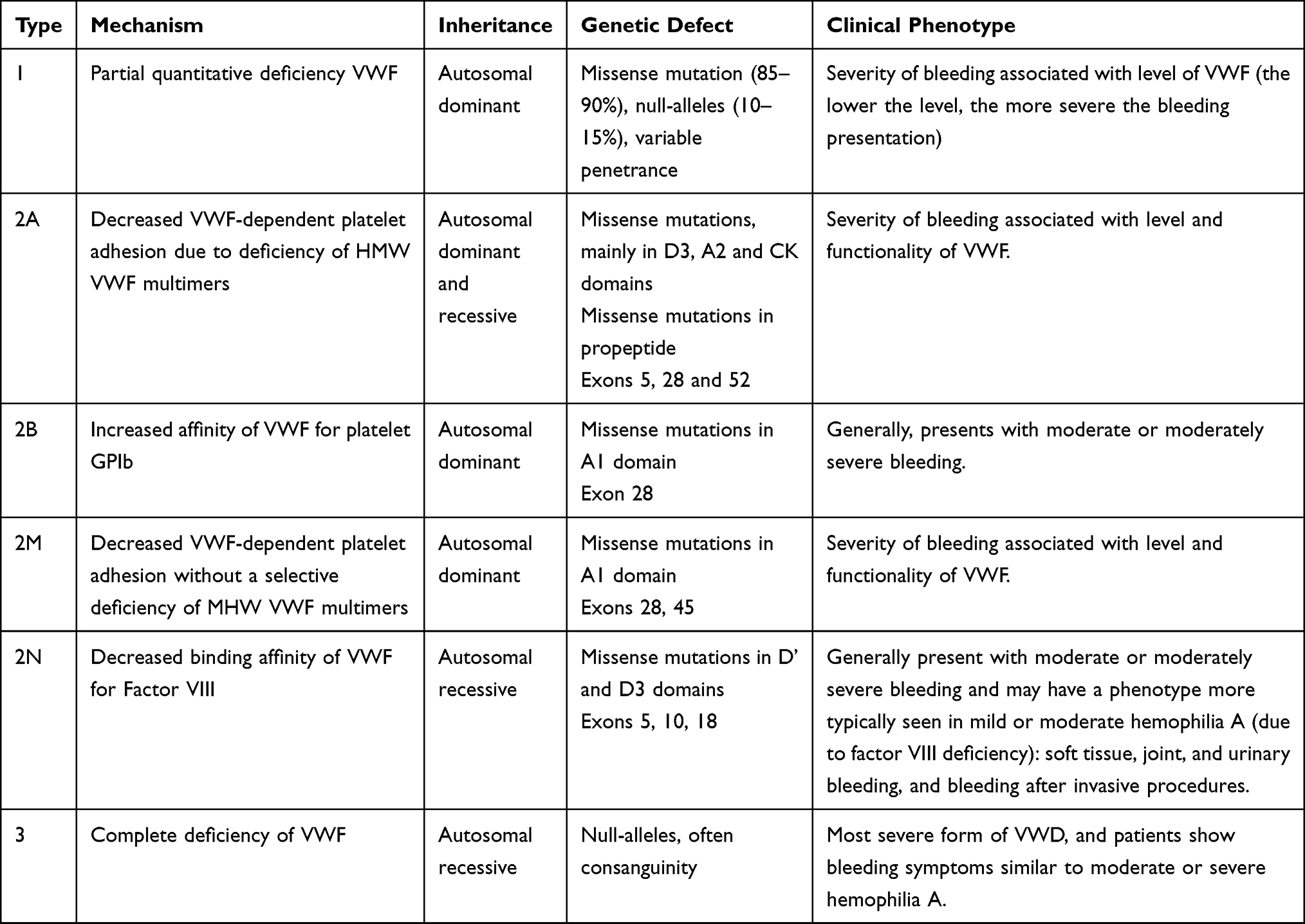 |
Table 1 Classification Scheme for von Willebrand Disease, Inheritance, Genetic Defect and Phenotypic Presentation |
VWD diagnosis requires the presence of both clinical features, such as a personal (typically lifelong) history of primarily mucocutaneous bleeding, and laboratory evidence of absence, deficiency, or defect in VWF. This is usually accompanied by a family history of the disease.
VWD has a variable clinical presentation due to the heterogeneity of the disease.9 VWD is mainly associated with mucocutaneous bleeding, although there are more severe bleeding phenotypes, which carry significant morbidity and mortality.10 Mucocutaneous bleeding may present as spontaneous or minimally provoked bruising, excessive bleeding from minor wounds, gum bleeding, epistaxis, menorrhagia and bleeding from the gastrointestinal tract.11 Site of bleeding may also change with patient age, with higher rates of gastrointestinal bleeding reported in older age groups.12 Other than spontaneous bleeding, patients with VWD also experience bleeding after invasive procedures, such as surgery or tooth extraction and during hemostatic challenges, such as childbirth or trauma.6 The severity of VWD ranges from very mild, with bleeding only after major procedures, up to spontaneous bleeding, including muscle and joint bleeding, in the most severe cases.
Clinical presentation and bleeding history can also assist with diagnosis (see Table 1). Type 1 VWD has a range of clinical presentations, from mild to severe, with bleeding phenotype broadly corresponding to the level of plasma VWF. Type 3 presents as the most severe phenotype of VWD, and is sometimes likened to moderate to severe hemophilia A. Type 2A and 2M presentations correspond to the quantity and functionality of VWF. Types 2B and 2N VWD generally present as a moderate to severe bleeding phenotype. Type 2N may also present with joint bleeding or urinary bleeding and presents as phenotypically similar to mild-to-moderate hemophilia A, but instead is due to the inability of VWF to appropriately bind to (and thus protect) Factor VIII.6
The assessment of bleeding history is the most important initial step in the analysis of a suspected bleeding tendency, as laboratory evaluation will only be initiated after the appropriate clinical suspicion.9 The severity of bleeding diathesis can be judged by the frequency of bleeding events as well as the age of onset.11 A patient’s bleeding phenotype can be identified and quantified with the use of various tools including the International Society on Thrombosis and Haemostasis (ISTH) bleeding assessment tool (BAT) and should be used to aid in clinical suspicion of a diagnosis of VWD (Table 2).13 A lack of understanding of the difference between “normal” and “abnormal” bleeding symptoms is a common reason for misdiagnosis of VWD.13
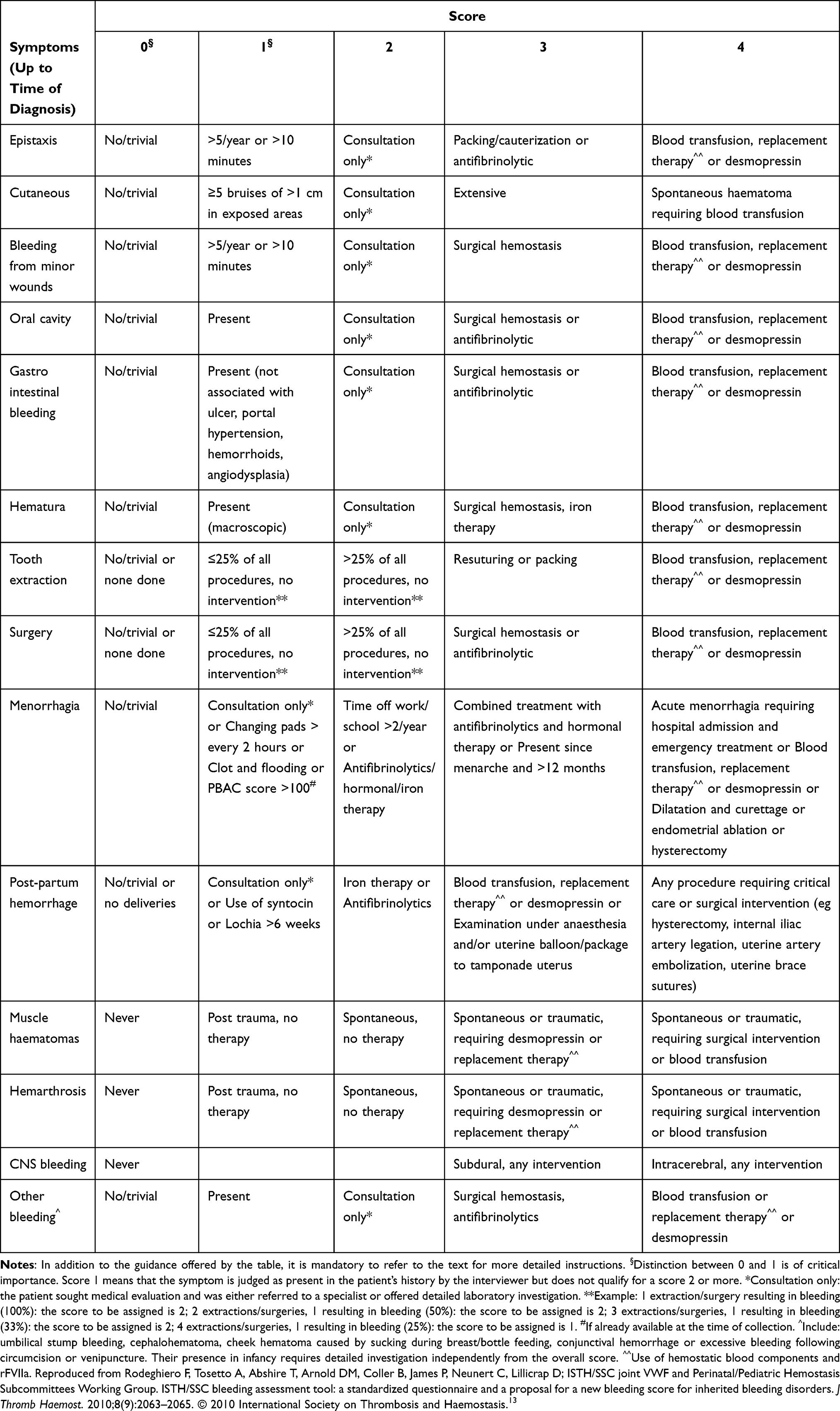 |
Table 2 ISTH Bleeding Assessment Tool (BAT) |
Bleeding frequency and severity are influenced by many factors, which must be carefully evaluated to avoid diagnostic oversight. Important considerations for diagnosis are the age and gender of the patient, their comorbidities, their family history of bleeding diathesis and diet (for relative vitamin C intake). It is essential to ascertain whether the patient is using any anticoagulant or antiplatelet medications, as well as otherwise undisclosed over-the-counter or herbal remedies, such as glucosamine, ginkgo, garlic, ginseng, fish oil, primrose oil, echinacea, dong quai or feverfew, all of which can dampen hemostasis and thus increase bleeding risk.14 Concurrent diagnoses such as joint hypermobility may also exaggerate joint-based symptoms.
Differential diagnoses for an individual who presents with symptoms consistent with VWD include mild haemophilia A or mild haemophilia B, vitamin C deficiency, platelet function disorder, or deficiency of other coagulation factors (II, VII, X or XI). In addition, should VWD be identified, the type of VWD should also be characterised. Optimal management of patients depends on an accurate diagnosis, and an error in diagnosis will potentially lead to inappropriate testing and compromise patient management, including over-diagnosis and unnecessary treatment.
Laboratory Testing
While thorough clinical evaluation is essential to guide appropriate investigations in the workup of VWD, a lack of expertise in laboratory testing and interpretation remains a common reason for diagnostic inaccuracies.
The first step to a correct laboratory diagnosis is ensuring the most appropriate tests are performed for the correct indication. Testing for VWD should be considered when there is a bleeding history, a positive family history, a mild unexplained thrombocytopenia, a mildly prolonged activated partial thromboplastin time (APTT), or an apparent hemophilia A in a female. Even males “diagnosed” with hemophilia A should be considered for VWD testing, as Type 3 or Type 2N VWD can sometimes be misdiagnosed as hemophilia A.
An approach to testing is outlined in Table 3.6,15 Laboratory assessment should commence with a review of the full blood count and blood film to investigate any platelet deficiencies or clumping. Blood group assessment is also important, given group O individuals are known to have a lower baseline VWF level.15 Routine coagulation tests, including prothrombin time (PT) and APTT are often useful as a baseline screen.
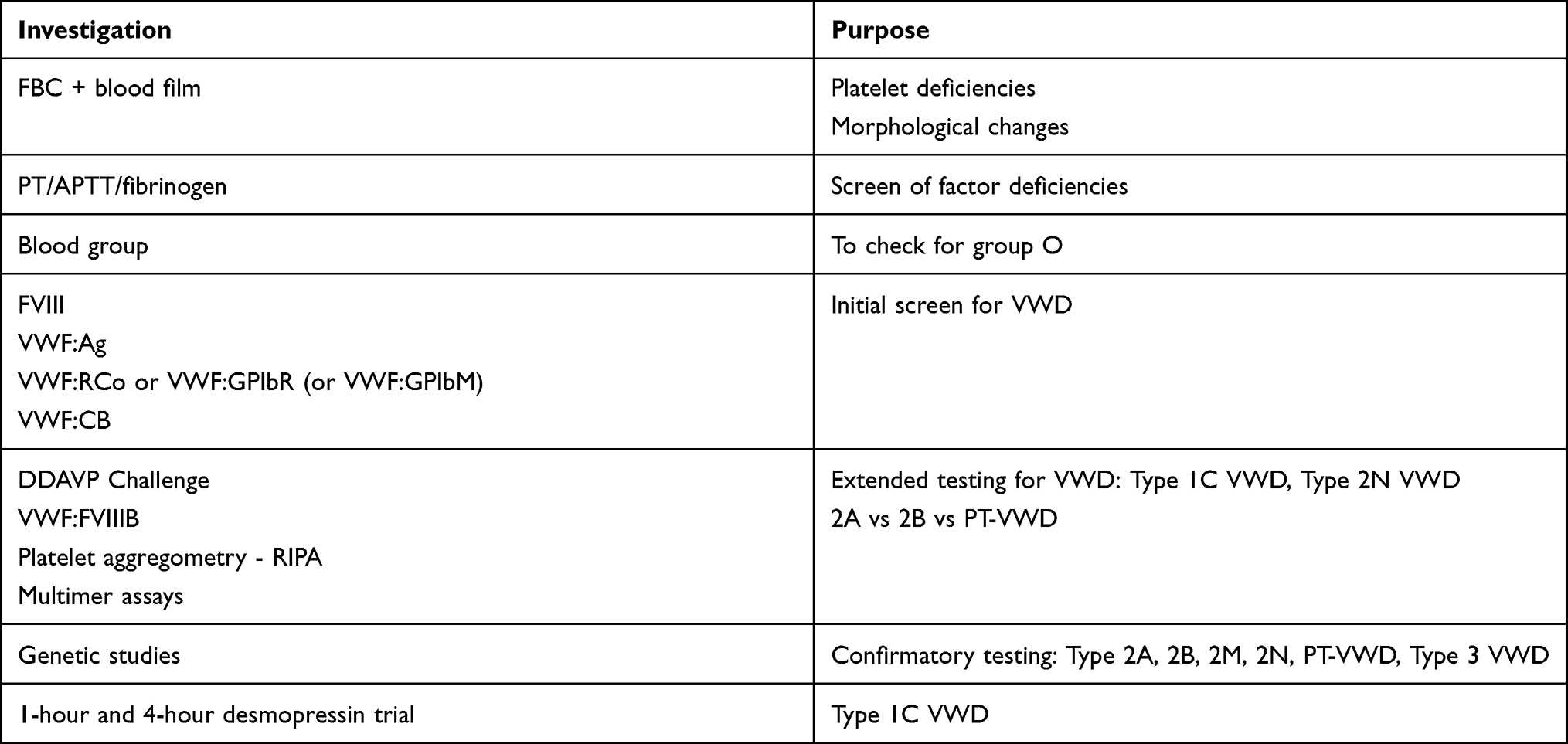 |
Table 3 Laboratory Work Up of a Patient with Suspected VWD |
Most importantly, a group of tests useful for evaluating VWD should also be performed. At a bare minimum, the VWD screening tests should include a factor VIII activity assay, a VWF protein level assessment (VWF:Ag; “antigen”), and sufficient tests to properly investigate the activity of VWF. Most laboratories utilise ELISA (enzyme-linked immunosorbent assay) or LIA (latex immunoassay) based methods to test for VWF:Ag, both offering low variability, high sensitivity, and full automation capabilities.6 ELISA assays may predominate in research laboratories due to the common use of ELISA methodology for other research analysis. LIA assays tend to dominate in diagnostic laboratories. The newest addition of CLIA (chemiluminescent immunoassays)-based testing methods is less readily available, although they offer the best low-level sensitivity and lowest assay variation in VWF:Ag testing.6 Importantly, no VWF:Ag assay identifies any functional parameter for VWF.
The activity assays for VWF should incorporate both an assessment of the ability of VWF to bind platelets and collagen, and where indicated also to FVIII. Thus, a ristocetin cofactor (VWF:RCo) or platelet GPIb recombinant surrogate to ristocetin cofactor assay (VWF:GPIbR) is required to assess for VWF platelet binding and a collagen binding assay (VWF:CB) is required to assess for VWF collagen binding; in our view, this pair of assays is required for all VWD investigations. An alternate gain of function “mutant” GPIb assay (VWF:GPIbM) is used in some laboratories instead of VWF:RCo or VWF:GPIbR.6 In general, VWF:RCo, VWF:GPIbR and VWF:GPIbM all represent platelet GPIb binding assays, and in diagnostic laboratories, they are usually performed as agglutination assays, including LIA-based.6 VWF:GPIbR is also available using CLIA. VWF:CB is usually performed by ELISA or increasingly by CLIA. Again, research labs may also perform some VWF activity assays, including VWF:GPIbM, by ELISA.
In cases of suspected 2N VWD, an additional assay, the factor VIII binding assay (VWF:FVIIIB) is also recommended. These tests will allow identification of the two quantitative deficiencies (Type 1 and Type 3 VWD), as well as all the qualitative defects.
In brief, a low level of VWF:Ag with a similar level of activity (VWF:RCo, VWF:GPIbR, VWF:GPIbM, VWF:CB), suggests Type 1 VWD; a level of VWF:Ag and activity of <5% likely indicates Type 3 VWD; a disproportion of VWF activity and VWF:Ag, suggests a qualitative (Type 2) VWD. If Type 1 VWD is suspected, a VWF propeptide assay may be considered in those who do not have a sustained response to DDAVP to confirm Type 1C subtype, indicative of reduced survival of VWF in plasma.16 A provisional exclusion or diagnosis of VWD may require further testing for confirmation. This may include repeat testing of the same panel of tests or extending the test panel. For example, while a discordant low ratio of RCo/Ag and CB/Ag provides a likely indication of Type 2A, 2B, or PT-VWD, separating out these disorders, or diagnosis of difficult cases, requires an extended test repertoire, such as utilisation of the ristocetin induced platelet agglutination assay (RIPA), genetic testing or VWF multimer analysis. An appropriate testing algorithm is shown in Figure 1. Genetic testing is sometimes also useful in Type 3 VWD cases.
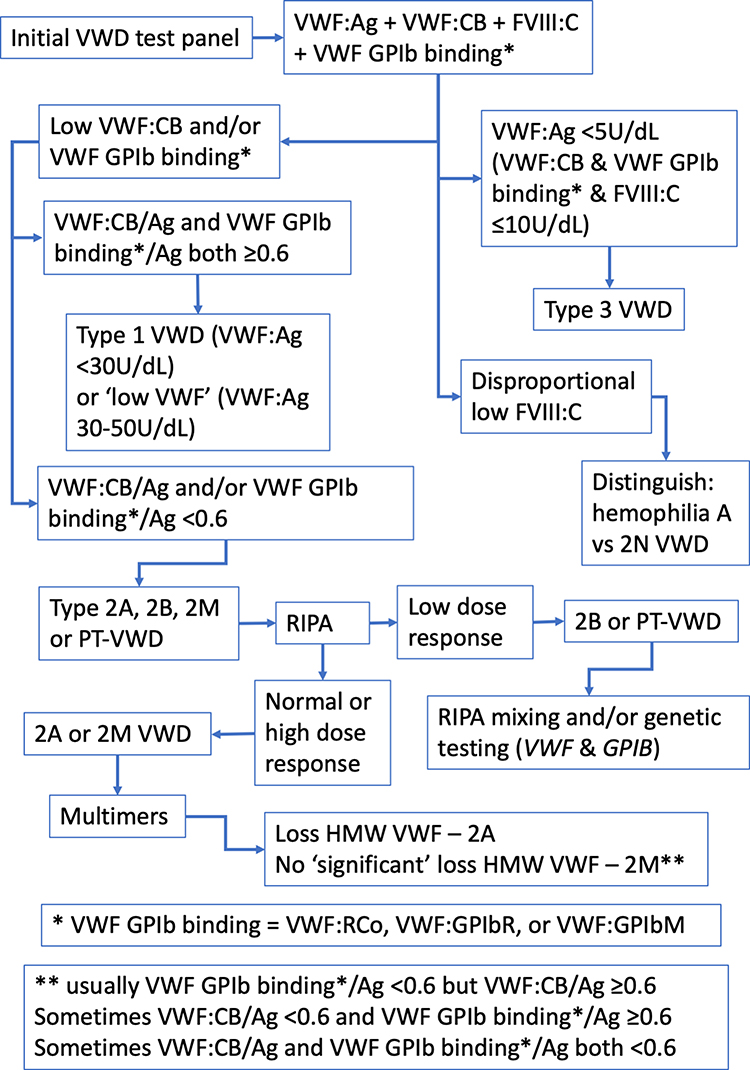 |
Figure 1 A simplified algorithm that describes the VWD diagnostic process using laboratory testing. This considers the differential utility of different VWF methods, as well as VWF multimers, and potentially genetic testing. Abbreviations: Ag, antigen; CB, collagen binding; FVIII, factor VIIII; GPIb, glycoprotein Ib (the platelet VWF receptor); GPIbM, GPIb mutation-based assay; GPIbR, recombinant GPIb-based assay; HMW, high molecular weight (VWF); RCo, ristocetin cofactor; RIPA, ristocetin induced platelet aggregation; VWF, von Willebrand factor; VWD, von Willebrand disease. |
Though not as well recognised, it is important to consider Type 1C VWD when subtyping VWD. Accurate identification of Type 1C VWD has significant therapeutic implications, as management of bleeding or prophylaxis against bleeding will likely require treating with VWF concentrates rather than desmopressin, given the short half-life of native VWF in these patients. Type 1C VWD can be identified by the abnormally high ratio of VWF pro-peptide to VWF:Ag associated with this condition due to the increased clearance of the mature VWF molecule compared with the pro-peptide.17,18 Pro-peptide measurement is not readily available in clinical practice, and in some cases, the ratio can be normal despite rapid clearance of VWF.19 Another method for detection of Type 1C VWF is via a desmopressin trial with a 1- and 4-hour post-infusion VWF level measurement, with Type 1C VWD patients expected to show a >30% decrease of VWF levels from peak levels at 4 hours post desmopressin infusion.16,19
The current definition of VWD is not restricted to those with VWF gene mutations.4 Underscoring this is the complexity, limited availability, high cost, and low clinical utility of genetic testing generally associated with VWD.5 A significant proportion of patients have no identifiable VWF mutations on readily available genetic methods.4 In particular, quantitative defects, such as seen in Type 1 VWD, often bear unsuccessful genetic analyses, despite undertaking evaluations of the entire VWF gene, and therefore also proving very costly.4,5 A positive result is more likely attainable for qualitative VWF defects or Type 2 VWD, as often a more directed analysis of the VWF gene can be performed, thereby making this more cost-effective.5 The diagnostic uncertainty that generally accompanies Type 2 VWD further increases the utility of genetic testing in these scenarios. In general, it is recommended to only perform genetic testing in select investigations, such as for confirmatory testing of Type 2A, 2B, 2M, 2N, PT-VWD given their diagnostic difficulty, and selected cases of Type 3 VWD, for example, to assist with pre-natal diagnoses (Table 3).5
Causes of Misdiagnosis
The complexity and breadth of tests required for the proper diagnosis of VWD leads itself to being an entity that is commonly under-diagnosed, misdiagnosed, or over-diagnosed.5,6,15,20–22 Both clinical and laboratory-related issues contribute to this. This is summarized in Table 4.
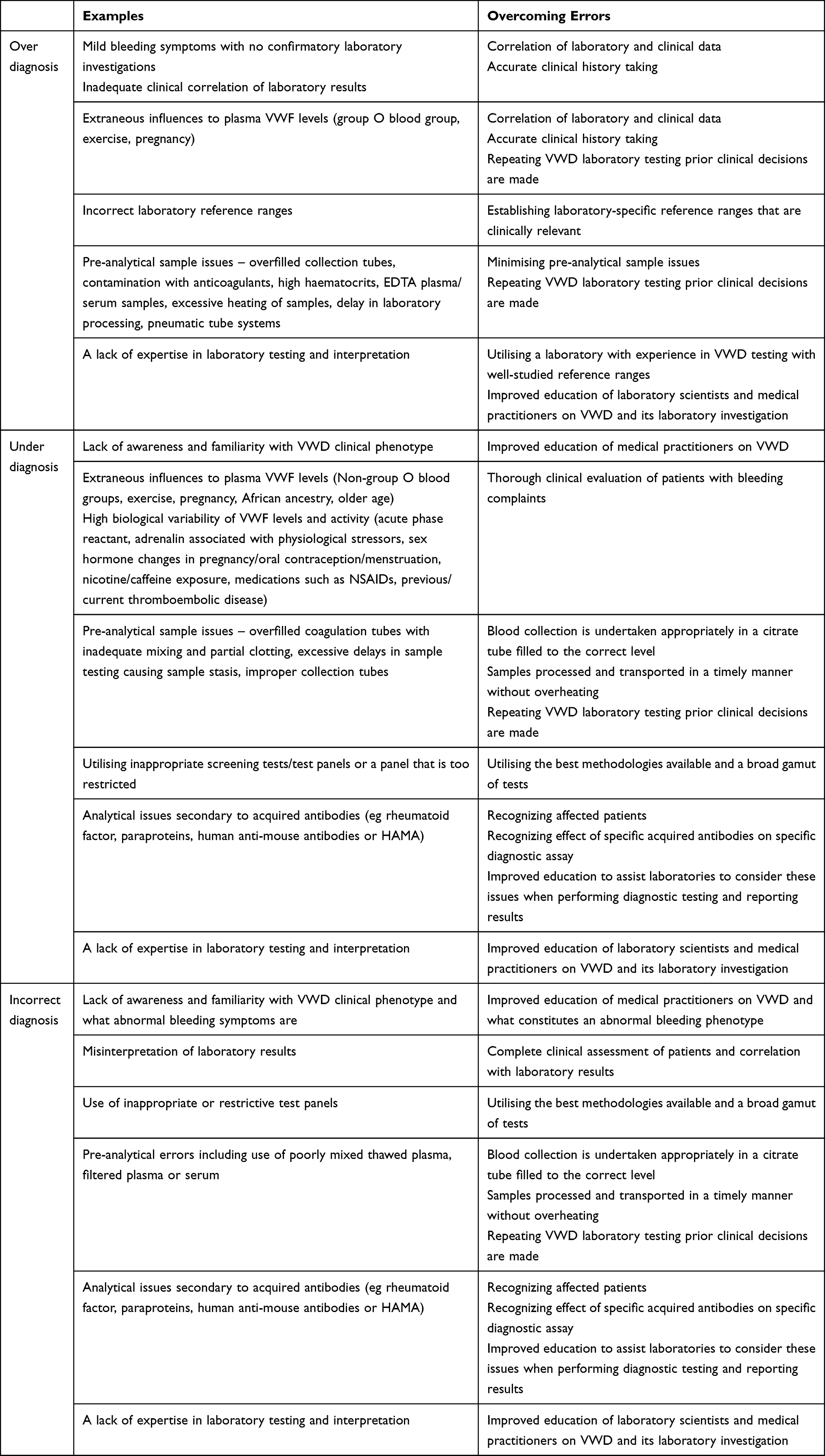 |
Table 4 Summary of Diagnostic Errors Encountered in von Willebrand Disease and How to Avoid Them |
Overdiagnosis of von Willebrand Disease
Overdiagnosis has been extensively described in the medical literature, and commonly identifies “problems” that were never destined to cause morbidity, or results in over-medicalisation of normal physiology through expanded definitions of disease.23 Mild bleeding symptoms may be a common feature of normal life and are thus often identified by the general public, particularly with regard to mucosal bleeding from dental procedures, epistaxis or menorrhagia.7 Such symptoms may lead to an overdiagnosis of VWD, particularly when VWD has been described in as much as 1% of the population, whereas only 0.05% actually present for investigation.6 In many of these patients, a true haemostatic challenge may never be obtained, and they may be labelled as von Willebrand-like bleeding without confirmatory laboratory investigations. Asking questions to distinguish normal from abnormal bleeding, together with the use of an appropriate bleeding assessment tool, is critical. This is particularly important in patients who have a family history of VWD, but have a normal bleeding phenotype, therefore not meeting diagnostic criteria for VWD.
Among those having laboratory investigations, there are several factors that may lead to an overdiagnosis of VWD, such as laboratory reference ranges, extraneous influences on plasma VWF levels (such as the ABO system) and current high availability of laboratory testing.6 Laboratories, which are not performing the full repertoire of tests, sometimes incorrectly interpret results, especially given recent changes in test methods used.6 Patients with an O-type blood group have up to 25% less plasma VWF than non-O type, and are therefore more likely to be falsely diagnosed as having VWD.15
Pre-analytical factors may also contribute to overdiagnosis, including inadequate patient history. Accurate history taking is considered one of the best screening tests for the risk of bleeding.13 Overfilled collection tubes, samples contaminated with anticoagulants, samples with high haematocrit, EDTA plasma or serum samples can all result in falsely low levels of VWF and/or its activity. Excessive heating of collected samples (eg, due to poor sample transport), delay in time taken to reach a laboratory, and use of pneumatic tube systems causing platelet activation and VWF adhesion may also affect VWF testing and are recommended to be avoided.24
Overdiagnosis of VWD can lead to increased patient morbidity and health care system burden. Overdiagnosis can result in unnecessary intervention for patients who require urgent or elective surgery by restricting the locations of appropriate interventions (such as dedicated bleeding centre facilities). Overdiagnosis can lead to unnecessary medical anxiety and extended familial testing, in addition to potentially exposing patients to an increased thrombotic risk with therapies.
Underdiagnosis of von Willebrand Disease
Underdiagnosis of VWD can also occur, and, for example, result from under-recognition of the VWD clinical phenotype and/or a lack of awareness of VWD, both resulting in appropriate VWD diagnostic investigations not being performed. A mild bleeding phenotype, such as with Type 1 VWD in a male, may not be clinically apparent, especially if there have been limited haemostatic challenges, such as surgery, dental work, or trauma. Females presenting with excessive uterine bleeding are often not tested for VWD, as menorrhagia may not be recognised as a “bleeding disorder” symptom.
High biological variability of VWF levels and activity also contributes to diagnostic uncertainty of VWD (especially in Type 1 and Type 2). VWF levels can increase transiently as an acute-phase reactant in inflammatory conditions, with increased adrenalin associated with exercise or other physiological stressors, recent caffeine exposure, and with changes in sex hormones as seen with pregnancy, the oral contraceptive pill, and menstruation.25,26 VWF levels hit a nadir on days 1 to 4 of the menstrual cycle, and testing at this phase of menses, when VWF levels are at their lowest and least likely to be elevated due to hormonal effects, is recommended to avoid underdiagnosis of VWD.27 Furthermore, there is an increase in VWF levels with increasing age, with African ancestry, and non-group O blood groups.22,28 These factors, if unappreciated, can result in underdiagnosis of VWD, especially if laboratory testing is not repeated or appropriately timed, and correlated with clinical bleeding history.
Pre-analytical issues related to test sample integrity are also an important factor leading to missed diagnoses of VWD. Using an overfilled coagulation tube may lead to inadequate mixing with partial clotting that can falsely elevate FVIII levels.22 Excessive delays in testing with sample stasis can result in potential shortening of routine APTT and factor activation that may result in false high levels.22 Ensuring blood collection is undertaken appropriately in a citrate tube filled to the correct level, with samples being processed and transported in a timely manner will assist with avoiding issues related to sample integrity.
Laboratory-related analytical issues can also result in an underdiagnosis of VWD. Using inappropriate screening tests or an inappropriate test panel or a test panel that is too restricted is a commonly recognised issue.6,22 For example, performing only a VWF:Ag test as the initial screening test, has the potential of missing Type 2 VWD, as many Type 2 VWD have VWF:Ag within the normal range.22 Performing only the VWF:Ag and the highly variable classical VWF:RCo as screening tests with exclusion of VWF:CB, as is commonly practiced worldwide, can also result in missed VWD diagnosis, especially Type 2 VWD.29 A study of North American specialised coagulation laboratories found that only one third of laboratories correctly identified Type 2 VWD when using either VWF:Ag and VWF:RCo tests.30 The presence of various acquired antibodies, such as rheumatoid factor, paraproteins, and human anti-mouse antibodies or HAMA, can result in a false normal laboratory test result with some assay methods, especially in immune assays such as latex-based immunoturbidimetric assays (LIA) which are commonly utilised in the investigation of VWD.22 Another issue is not utilising the best methodologies available. Utilising VWF:Ag assays that lack a high degree of sensitivity to VWF can result in the underdiagnosis of Type 3 VWD, since falsely high VWF levels may be reported. The newer chemiluminescence latex immunoassay technology (CLIA) for ristocetin cofactor activity measurement using non-platelet recombinant GPIb receptor (VWF:GPIbR) has been shown to have superior sensitivity and reduced variability to other methods, and is recommended in preference to traditional VWF:RCo testing methods in current bleeding disorder guidelines.31–33 As another example, testing only for FVIII, and not testing for VWF parameters, may miss patients with Type 3 and Type 2N VWD, and falsely identify haemophilia A.
Underdiagnosis of VWD can lead to increased patient morbidity and mortality. Underdiagnosis may result in an increased uncorrected bleeding risk in patients when subjected to haemostatic challenges, such as surgery. Under-recognised VWD can cause ongoing preventable morbidity in patients, such as those with excessive uterine bleeding. Underdiagnosis of VWD can also result in missed identification of similarly affected family members given the variable expressivity and penetrance of genes encoding VWF, as well as lost opportunities for pre-pregnancy counselling of patients.7 Under-recognition of VWD also precipitates misdiagnosis or incorrect diagnoses of other bleeding disorders, which may result in patients receiving incorrect treatment.
Incorrect Diagnosis of von Willebrand Disease
Pre-analytical and analytical issues that potentiate underdiagnosis of VWD can also result in misdiagnosis of bleeding disorders. Lack of familiarity with the different VWD types, misinterpretation of laboratory results, or use of inappropriate or restrictive test panels, can lead to qualitative types of VWD (Type 2 VWD) being misdiagnosed as Type 1 VWD.6 A common issue is pre-analytical errors, such as poorly mixed thawed plasma, filtered plasma or serum, leading to identification of a false VWD phenotype. This is especially a problem that results in misdiagnosis of Type 1 VWD as Type 2 VWD.6 Analytical issues stemming from acquired antibodies, which can result in a false normal VWF:Ag on LIA test, in combination with a low activity assay, can also lead to a false Type 2 VWD diagnosis in a Type 1 VWD.
VWD may also be misdiagnosed as an entirely different bleeding disorder. Whilst haemophilia is well ingrained in the clinician’s mind, VWD, despite its higher prevalence, remains an esoteric entity to many, resulting in missed ordering of specific VWD tests. This can result in certain types of VWD being incorrectly diagnosed as haemophilia A. If VWD testing is completely disregarded, a patient with severe Type 3 VWD may be inadvertently diagnosed with haemophilia A, due to associated low FVIII activity found in both conditions. This potential pitfall should especially be considered when there is no family history of severe haemophilia A. Even if a select panel of screening VWD tests are performed, not performing an FVIII binding assay when a low FVIII activity is detected can result in the misdiagnosis of Type 2N VWD as haemophilia A. The correct diagnosis of Type 2N VWD can only be achieved by performing a VWF:FVIII binding assay or by genetic testing. Misdiagnosis of VWD as haemophilia A has significant negative clinical implications, as treating severe VWD patients with recombinant FVIII products devoid of any VWF will not alleviate their bleeding diathesis. Such a misdiagnosis can also result in significant issues when it comes to genetic and pre-pregnancy counselling, given the autosomal inheritance of VWD and X-linked inheritance of haemophilia A.
VWD may also be misdiagnosed as immune thrombocytopenic purpura (ITP) or an inherited platelet disorder, particularly in cases of Type 2B VWD associated with thrombocytopenia. It is important to consider VWD in any patient that presents with a thrombocytopenia, or with abnormal platelet function, especially if there is a family history or prolonged personal history of a bleeding phenotype. Accurate diagnosis is important to avoid unnecessary and potentially harmful treatments, such as immunosuppression, splenectomy and platelet transfusions. Alternatively, Type 2B VWD and PT-VWD may be misdiagnosed for the other. Here, RIPA testing with mixing studies will point to the correct type; and, genetic testing of VWF and GPIb may be required (Table 3).
Acquired von Willebrand Syndrome
Acquired von Willebrand syndrome (AVWS) is a rare, potentially underdiagnosed bleeding disorder that may also be misdiagnosed as congenital VWD (Table 5).34,35 Diagnosis of AVWS is challenged by the same problems already mentioned for congenital VWD. In addition, diagnosis of AVWS is further hampered by the lack of a single diagnostic test that can rule in or rule out this disorder.34 The main distinguishing features from inherited VWD are lack of family history, late onset bleeding phenotype, and presence of a suspicious underlying condition.34 In difficult cases, testing family members, genetic analysis, and specialised assays such VWF pro-peptide levels and VWF mixing studies may be helpful. Differentiating the two conditions, one acquired and the other congenital, is important, as the disease course, management principles, and prognosis may differ.34
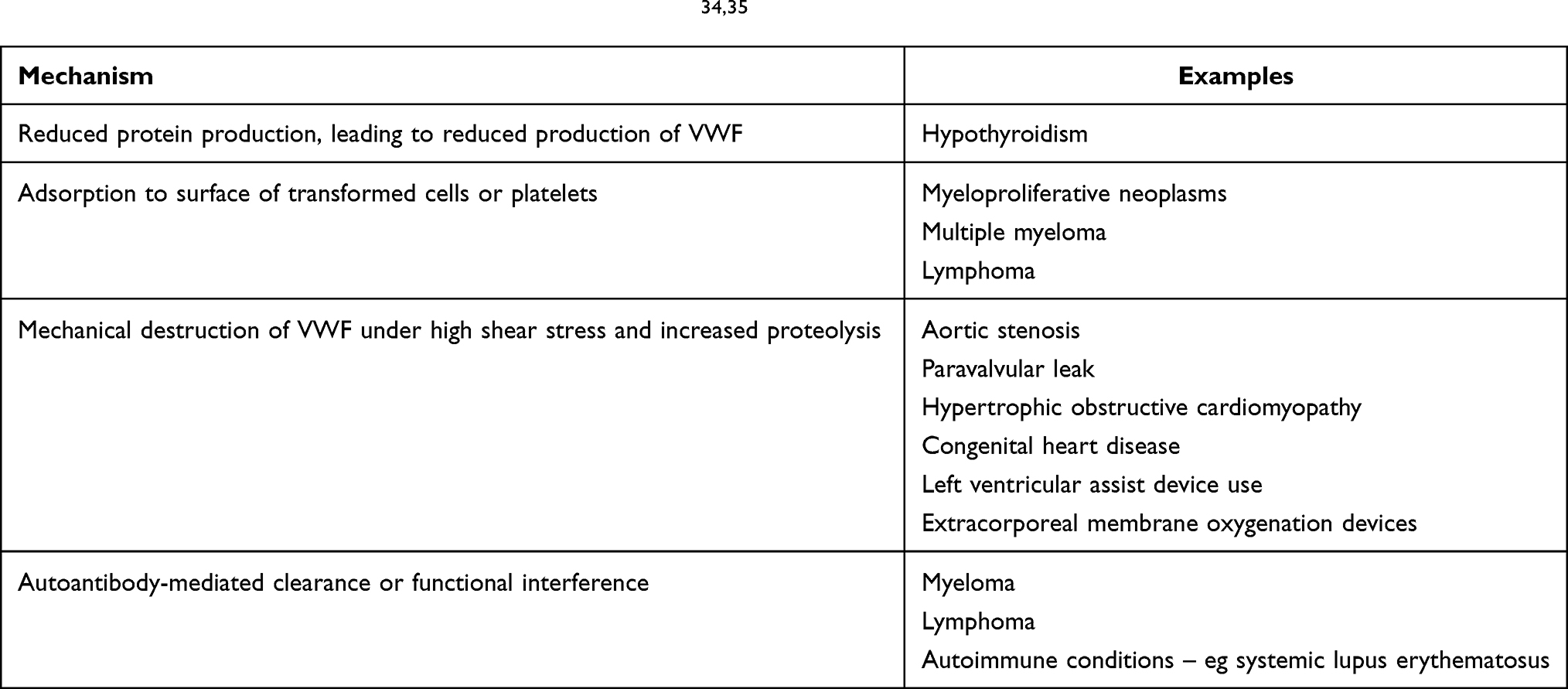 |
Table 5 Pathogenesis of Acquired von Willebrand Disease34,35 |
It is noted that several primary conditions can lead to AVWS, and there are several potential mechanisms involved in AVWS (Table 5). First, AVWS can be due to decreased protein production, such as in hypothyroidism, leading to a Type 1 deficiency of VWF. Second, VWF can be absorbed onto aberrant cells, such as in blood cancers, leading to clearance of plasma VWF. This may lead to either a Type 1 or 2 AVWS, depending on whether or not high molecular weight (HMW) is preferentially absorbed. Third, structural blood vessel deformities or the presence of artificial surfaces can lead to loss of VWF, typically HMW forms, again leading to a Type 2 AVWS. Absorption of VWF can also occur during extracorporeal membrane oxygenation. Fourth, antibodies against VWF may be formed in autoimmune disease and certain blood disorders, and thus also leading to clearance of VWF, or interference to its function.
In addition to potentially being underdiagnosed or misdiagnosed as congenital VWD, AVWS can also be over-diagnosed in certain conditions. For example, during extracorporeal membrane oxygenation, additional confounders may be present leading to an increased bleeding phenotype, such as anticoagulation, to provide blood compatibility with foreign surfaces.
Recommendations
In order to avoid the underdiagnosis, overdiagnosis and misdiagnosis of VWD, it is important to consider three central questions.
Who Should Have VWD Testing?
It is important to have VWD readily present in the repertoire of differential diagnoses one considers in order to avoid underdiagnosing and misdiagnosing this common inherited disorder. It is, however, equally important not to cast a mindless wide net of VWD testing without first considering the pre-test probability of the diagnosis. Testing in situations of low pre-test probability, especially if paired with an inadequate test panel and pre-analytical sample issues, sets the stage for inaccurate diagnoses including overdiagnosis and misdiagnosis that can lead to significant patient morbidity. VWD testing should be considered in patients with a significant rather than trivial bleeding history, a family history of significant bleeding or VWD, or in patients with laboratory abnormalities, such as a mild unexplained thrombocytopenia, a mild prolonged APTT or an apparent factor VIII deficiency.
How Should VWD Testing Be Performed?
The importance of a comprehensive test panel that utilises the best methodologies cannot be overstated. Having a basic understanding of the type of tests and methodologies utilised by local laboratories will help understand the potential pitfalls and rigor of the diagnostic process. A suitable diagnostic algorithm is provided (Figure 1).
How to Ensure VWD Test Results are Correctly Interpreted?
It is important to always repeat VWD studies on a fresh sample for confirmation of initial test results. VWD testing should always be supplemented by a full blood count, coagulation studies, and blood group analysis (Table 3). The results should be interpreted in context with any anticoagulant use, hormone therapy, pregnancy and potential physiological stressors, or testing timed to minimise the impact of these factors. Utilising the results of a desmopressin trial can also be helpful in providing supporting evidence of the diagnostic possibilities.6 Correlation between clinical phenotype and bleeding history is essential for an accurate diagnosis. Finally, family studies can be a valuable adjunct, especially when trying to disentangle differential diagnoses such as acquired bleeding disorders and X-linked hemophilias.
Conclusions
VWD is a complex, heterogenous condition, which lends itself to significant diagnostic dilemmas and inaccuracies, and in turn leads to significant avoidable patient harm including inappropriate testing, overdiagnosis, underdiagnosis and inadequate treatment. Utilising a structured approach, the careful consideration of differential diagnoses, the integration of accurate clinical and laboratory features, as well as ensuring tests are repeated to minimise pre-analytical errors, are all important aspects in ensuring the accuracy of the diagnosis of VWD.
Acknowledgments
The opinions in this review are those of the authors, and not necessarily those of NSW Health Pathology.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bowman M, Hopman WM, Rapson D, Lillicrap D, James P. The prevalence of symptomatic von Willebrand disease in primary care practice. J Thromb Haemost. 2010;8(1):213–216. Epub 2009 Oct 23. PMID: 19874468. doi:10.1111/j.1538-7836.2009.03661
2. Lavin M, O’Donnell JS. How I treat low von Willebrand factor levels. Blood. 2019;133(8):795–804. Epub 2018 Dec 21. PMID: 30578256. doi:10.1182/blood-2018-10-844936
3. von Willebrand EA. Hereditar pseudohemofili. Fin Laekaresaellsk Hand. 1926;68:87–112.
4. Sadler JE, Budde U, Eikenboom JC, et al.; Working Party on von Willebrand Disease Classification. Update on the pathophysiology and classification of von Willebrand disease: a report of the subcommittee on von Willebrand factor. J Thromb Haemost. 2006;4(10):2103–2114. Epub 2006 Aug 2. PMID: 16889557. doi:10.1111/j.1538-7836.2006.02146.x
5. Favaloro EJ. Diagnosing von Willebrand disease: a short history of laboratory milestones and innovations, plus current status, challenges, and solutions. Semin Thromb Hemost. 2014;40(5):551–570. Epub 2014 Jun 30. PMID: 24978322. doi:10.1055/s-0034-1383546
6. Favaloro EJ, Pasalic L, Curnow J. Laboratory tests used to help diagnose von Willebrand disease: an update. Pathology. 2016;48(4):303–318. Epub 2016 May 4. PMID: 27131932. doi:10.1016/j.pathol.2016.03.001
7. Lillicrap D. von Willebrand disease: advances in pathogenetic understanding, diagnosis, and therapy. Blood. 2013;122(23):3735–3740. Epub 2013 Sep 24. PMID: 24065240; PMCID: PMC3952678. doi:10.1182/blood-2013-06-498303
8. Ng C, Motto DG, Di Paola J. Diagnostic approach to von Willebrand disease. Blood. 2015;125(13):2029–2037. Epub 2015 Feb 23. PMID: 25712990; PMCID: PMC4375103. doi:10.1182/blood-2014-08-528398
9. De Jong A, Eikenboom J. Developments in the diagnostic procedures for von Willebrand disease. J Thromb Haemost. 2016;14(3):449–460. Epub 2016 Feb 12. PMID: 26714181. doi:10.1111/jth.13243
10. Bolton-Maggs PH, Lillicrap D, Goudemand J, Berntorp E. von Willebrand disease update: diagnostic and treatment dilemmas. Haemophilia. 2008;14(Suppl s3):56–61. PMID: 18510523. doi:10.1111/j.1365-2516.2008.01713.x
11. Siboni S, Biguzzi E, Caiani V, Mistretta C, Bucciarelli P, Peyvandi F. Baseline factor VIII plasma levels and age at first bleeding in patients with severe forms of von Willebrand disease. Haemophilia. 2016;22(4):564–569. doi:10.1111/hae.12900
12. Sanders YV, Giezenaar MA, Laros-van Gorkom BA, et al.; WiN study group. von Willebrand disease and aging: an evolving phenotype. J Thromb Haemost. 2014;12(7):1066–1075. PMID: 24750783. doi:10.1111/jth.12586
13. Rodeghiero F, Tosetto A, Abshire T, et al.; ISTH/SSC joint VWF and Perinatal/Pediatric Hemostasis Subcommittees Working Group. ISTH/SSC bleeding assessment tool: a standardized questionnaire and a proposal for a new bleeding score for inherited bleeding disorders. J Thromb Haemost. 2010;8(9):2063–2065. PMID: 20626619. doi:10.1111/j.1538-7836.2010.03975.x
14. Abebe W. Review of herbal medications with the potential to cause bleeding: dental implications, and risk prediction and prevention avenues. EPMA J. 2019;10(1):51–64. PMID: 30984314; PMCID: PMC6459456. doi:10.1007/s13167-018-0158-2
15. Castaman G, Montgomery RR, Meschengieser SS, Haberichter SL, Woods AI, Lazzari MA. von Willebrand’s disease diagnosis and laboratory issues. Haemophilia. 2010;16 Suppl 5(5):67–73. PMID: 20590859; PMCID: PMC4313748. doi:10.1111/j.1365-2516.2010.02296.x
16. James PD, Connell NT, Ameer B, et al. ASH ISTH NHF WFH 2021 guidelines on the diagnosis of von Willebrand disease. Blood Adv. 2021;5(1):280–300. PMID: 33570651; PMCID: PMC7805340. doi:10.1182/bloodadvances.2020003265
17. Sharma R, Haberichter SL. New advances in the diagnosis of von Willebrand disease. Hematology Am Soc Hematol Educ Program. 2019;2019(1):596–600. PMID: 31808831; PMCID: PMC6913428. doi:10.1182/hematology.2019000064
18. Sztukowska M, Gallinaro L, Cattini MG, et al. Von Willebrand factor propeptide makes it easy to identify the shorter Von Willebrand factor survival in patients with type 1 and type Vicenza von Willebrand disease. Br J Haematol. 2008;143(1):107–114. Epub 2008 Aug 7. PMID: 18691167. doi:10.1111/j.1365-2141.2008.07311.x
19. Haberichter SL, Castaman G, Budde U, et al. Identification of type 1 von Willebrand disease patients with reduced von Willebrand factor survival by assay of the VWF propeptide in the European study: molecular and clinical markers for the diagnosis and management of type 1 VWD (MCMDM-1VWD). Blood. 2008;111(10):4979–4985. Epub 2008 Mar 14. PMID: 18344424; PMCID: PMC2384129. doi:10.1182/blood-2007-09-110940
20. Koutts J. A short history of diagnostic tests for von Willebrand disease: in memory of Barry Firkin (1930 to 2001) and Ted Zimmerman (1937 to 1988). Semin Thromb Hemost. 2006;32(5):445–455. PMID: 16862517. doi:10.1055/s-2006-947858
21. Sadler JE. Von Willebrand disease type 1: a diagnosis in search of a disease. Blood. 2003;101(6):2089–2093. Epub 2002 Oct 31. PMID: 12411289. doi:10.1182/blood-2002-09-2892
22. Favaloro EJ, Lippi G. Preanalytical issues that may cause misdiagnosis in haemophilia and von Willebrand disease. Haemophilia. 2018;24(2):198–210. Epub 2017 Dec 22. PMID: 29271545. doi:10.1111/hae.13396
23. Brodersen J, Schwartz LM, Heneghan C, O’Sullivan JW, Aronson JK, Woloshin S. Overdiagnosis: what it is and what it isn’t. BMJ Evid Based Med. 2018;23(1):1–3. PMID: 29367314. doi:10.1136/ebmed-2017-110886
24. Magnette A, Chatelain M, Chatelain B, Ten Cate H, Mullier F. Pre-analytical issues in the haemostasis laboratory: guidance for the clinical laboratories. Thromb J. 2016;14(1):49. PMID: 27999475; PMCID: PMC5154122. doi:10.1186/s12959-016-0123-z
25. Lippi G, Maffulli N. Biological influence of physical exercise on hemostasis. Semin Thromb Hemost. 2009;35(3):269–276. Epub 2009 May 18. PMID: 19452402. doi:10.1055/s-0029-1222605
26. Brenner B. Haemostatic changes in pregnancy. Thromb Res. 2004;114(5–6):409–414. PMID: 15507271. doi:10.1016/j.thromres.2004.08.004
27. Kouides PA, Conard J, Peyvandi F, Lukes A, Kadir R. Hemostasis and menstruation: appropriate investigation for underlying disorders of hemostasis in women with excessive menstrual bleeding. Fertil Steril. 2005;84(5):1345–1351. PMID: 16275228. doi:10.1016/j.fertnstert.2005.05.035
28. Favaloro EJ, Soltani S, McDonald J, Grezchnik E, Easton L, Favaloro JW. Reassessment of ABO blood group, sex, and age on laboratory parameters used to diagnose von Willebrand disorder: potential influence on the diagnosis vs the potential association with risk of thrombosis. Am J Clin Pathol. 2005;124(6):910–917. Erratum in: Am J Clin Pathol. 2006 May;125(5):796.PMID: 16416741. doi:10.1309/W76QF806CE80CL2T
29. Favaloro EJ, Bonar RA, Meiring M, et al. Evaluating errors in the laboratory identification of von Willebrand disease in the real world. Thromb Res. 2014;134(2):393–403. Epub 2014 May 21. PMID: 24913998. doi:10.1016/j.thromres.2014.05.020
30. Chandler WL, Peerschke EI, Castellone DD, Meijer P; NASCOLA Proficiency Testing Committee. Von Willebrand factor assay proficiency testing. The North American specialized coagulation laboratory association experience. Am J Clin Pathol. 2011;135(6):862–869. PMID: 21571959. doi:10.1309/AJCPH5JK4ONENPAE
31. Verfaillie CJ, De Witte E, Devreese KM. Validation of a new panel of automated chemiluminescence assays for von Willebrand factor antigen and activity in the screening for von Willebrand disease. Int J Lab Hematol. 2013;35(5):555–565. Epub 2013 Apr 3. PMID: 23551532. doi:10.1111/ijlh.12087
32. Cabrera N, Moret A, Caunedo P, et al. Comparison of a new chemiluminescent immunoassay for von Willebrand factor activity with the ristocetin cofactor-induced platelet agglutination method. Haemophilia. 2013;19(6):920–925. Epub 2013 Jun 4. PMID: 23730809. doi:10.1111/hae.12203
33. de Maistre E, Volot F, Mourey G, et al. Performance of two new automated assays for measuring von Willebrand activity: hemosIL AcuStar and innovance. Thromb Haemost. 2014;112(4):825–830. Epub 2014 Aug 7. PMID: 25103956. doi:10.1160/TH14-02-0108
34. Tiede A, Rand JH, Budde U, Ganser A, Federici AB. How I treat the acquired von Willebrand syndrome. Blood. 2011;117(25):6777–6785. Epub 2011 May 3. PMID: 21540459. doi:10.1182/blood-2010-11-297580
35. Shetty S, Kasatkar P, Ghosh K. Pathophysiology of acquired von Willebrand disease: a concise review. Eur J Haematol. 2011;87(2):99–106. PMID: 21535159. doi:10.1111/j.1600-0609.2011.01636.x
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.