Back to Journals » Biologics: Targets and Therapy » Volume 19
Whole Transcriptome-Based ceRNA Regulatory Network Analysis of Radiation-Induced Esophageal Epithelial Cell Injury
Authors Lin H, Feng Y, Liu H, Zhang J, Zhang X, Ying X, Shi Y, Tan H, Tu W
Received 23 October 2024
Accepted for publication 11 April 2025
Published 22 April 2025 Volume 2025:19 Pages 231—249
DOI https://doi.org/10.2147/BTT.S496064
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Shein-Chung Chow
Hongyu Lin,1,2,* Yahui Feng,1,3,* Hangfeng Liu,1 Jinkang Zhang,1,2 Xiaolin Zhang,1 Xue Ying,4 Yuhong Shi,1 Hao Tan,1,2 Wenling Tu1– 3
1The Second Affiliated Hospital of Chengdu Medical College, Nuclear Industry 416 Hospital, Chengdu, 610051, People’s Republic of China; 2School of Bioscience and Technology, Chengdu Medical College, Chengdu, 610500, People’s Republic of China; 3NHC Key Laboratory of Nuclear Technology Medical Transformation (Mianyang Central Hospital), Mianyang, 621099, People’s Republic of China; 4Key Laboratory of Sichuan Province for Specific Structure of Small Molecule Drugs, School of Pharmacy, Chengdu Medical College, Chengdu, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Wenling Tu, The Second Affiliated Hospital of Chengdu Medical College, Nuclear Industry 416 Hospital, No. 4, Section 4, North Ring Road, Chenghua District, Chengdu, Sichuan Province, 610051, People’s Republic of China, Email [email protected] Hao Tan, The Second Affiliated Hospital of Chengdu Medical College, Nuclear Industry 416 Hospital, No. 4, Section 4, North Ring Road, Chenghua District, Chengdu, Sichuan Province, 610051, People’s Republic of China, Email [email protected]
Introduction: Esophageal epithelial cells are essential for esophageal homeostasis and defense against harmful stimuli, but the mechanisms of radiation-induced injury in these cells are poorly understood. The competitive endogenous RNA (ceRNA) network, involved in various physiological processes and diseases, may also play a role in radiation-induced injury, although its mechanism remains unclear. This study aimed to investigate the effects of ionizing radiation on human esophageal epithelial cells and explore the role of the ceRNA network in this injury.
Methods: Cellular phenotype experiments assessed the effects of ionizing radiation on human esophageal epithelial cells. Whole transcriptome sequencing (lncRNA, circRNA, miRNA, and mRNA) was performed on cells exposed to 0, 2, and 4 Gy radiation. Differentially expressed RNAs (dd-DERs) were identified through differential expression analysis and dose-dependent screening. A ceRNA network was constructed using co-expression analysis and binding site prediction. Real-time quantitative PCR validated the expression levels of selected dd-DERs, and gene set enrichment analysis explored affected pathways.
Results: We identified 41 lncRNAs, 18 miRNAs, and 192 mRNAs as dose-dependent differentially expressed RNAs. A ceRNA network comprising 10 lncRNAs, 5 miRNAs, and 55 mRNAs was established. Real-time PCR confirmed the expression levels of 8 dd-DERs within the network. Gene set enrichment analysis showed that radiation disrupted channel activity, cell replication, repair, and immune response. Functional enrichment analysis revealed modulation of metabolic pathways, particularly involving UGT1A family members.
Discussion: This study established a ceRNA network related to radiation-induced esophageal epithelial cell injury, advancing our understanding of its pathophysiology. The ceRNA network may mediate injury through metabolic pathway modulation. Future work should focus on elucidating specific ceRNA interactions and exploring therapeutic potential for mitigating radiation-induced esophageal injury.
Keywords: ionizing radiation, esophageal epithelial cells, radiation-induced injury, whole transcriptome, competitive endogenous RNA network
Introduction
Radiation-induced esophageal injury (RIEI) is a significant complication observed during radiation therapy for esophageal tumors or tumors located in close proximity to the esophagus, such as lung cancer.1 Clinically, RIEI is characterized by esophageal inflammation, mucosal congestion, edema, difficulties in swallowing, and has been associated with the development of esophageal cancer.2–4 Concurrent chemoradiation therapy (CCRT) enhances survival rates in patients with locally advanced non-small cell lung cancer, but it increases toxicity, particularly radiation esophagitis (RE). Among 1082 patients who underwent CCRT, the development of RE was prevalent, with grade 2 observed in 348 patients (32.2%), grade 3 in 185 (17.1%), and grade 4 in 10 (0.9%). There were no deaths related to RE.5 The damage from the ionizing radiation to biological targets may occur due to direct, indirect, and bystander effects. Direct effects occurs due to targeted assault on intracellular biomolecules such as DNA bases, as a result of damage, double-strand breaks, and alterations in protein structure.6 Indirect effects occur due to the production of reactive oxygen species (ROS) and free radicals, leading to oxidative stress and subsequent cellular damage at molecular-level.7 Bystander effects occur when neighboring normal cells respond to signals from damaged cells, inducing toxic effects on adjacent healthy tissues.8 Following radiation exposure, these three above-mentioned mechanisms collectively contribute to the onset of RIEI; however, the precise molecular mechanisms underlying them remain to be fully elucidated. During the acute inflammatory response of RIEI, there is an upregulation of free radicals (ROS), interferon-alpha (IFN-α), interleukin-6 (IL-6), tumor necrosis factor-alpha (TNF-α), interferon gamma-inducing factor (IFNGR), and interferon gamma (IFN-γ), contributing to pathogenic damage.9–13 Additionally, our previous research has revealed the roles of cyclooxygenase-2 (COX-2) – mediated arachidonic acid metabolism and IL-17 signaling-mediated inflammation in RIEI.14,15 Nevertheless, additional research is required to elucidate the exact molecular mechanisms responsible for RIEI.
Competing endogenous RNA (ceRNA) is a relatively newly discovered RNA regulatory mechanism, where non-coding RNAs (such as lncRNA and circRNA) compete with mRNA for binding to miRNA, thereby modulating mRNA function at the post-transcriptional level.16,17 Studies conducted over recent years have implicated ceRNA networks in the pathogenesis of diverse diseases, such as Parkinson’s disease,18 Alzheimer’s disease,19 intervertebral disc degeneration,20 depression,21 and esophageal cancer.22,23 Several studies in the past few years, have unraveled abnormal RNA expression profiles in the RIEI animal model. Sun et al investigated the relationship between differentially expressed 36 lncRNAs and 853 mRNAs, using RNA-seq data obtained from both normal and radiation-treated rat esophageal tissues.24 Luo et al examined the association between miRNAs and circRNAs in a rat RIEI model.25 Likewise, in our previous study, we investigated an immune-related ceRNA network involving lncRNAs, miRNAs, and mRNAs in the esophageal tissues of radiation-exposed rats.26 Their findings unveiled potential regulatory associations and provided insights into the pathogenesis of RIEI. Nonetheless, these aforementioned studies focused primarily on investigating tissue-level mechanisms underlying disease progression in a rat model, irrespective of interspecies differences between rats and humans. Additionally, they did not explore potential relevant cellular-level mechanisms specific to esophageal tissues. Liu et al found that enhanced expression of miR-132-3p intensifies radiation-induced damage in esophageal epithelial cells.27 Sun et al examined the role of terminal uridylation enzyme 4 (TUT4) in modifying miRNA sequences and demonstrated the impact of TUT4 deficiency on downstream miRNA uridylation, thereby influencing damage to radiation-induced esophageal epithelial cells.28 Their cellular-level studies focused solely on the impacts of miRNAs and their modifications on injury caused by radiation-induced esophageal epithelial cells; however, they did not incorporate lncRNAs, circRNAs, miRNAs, and mRNAs to investigate potential underlying mechanisms.
In this study, we performed phenotypic experiments on human esophageal epithelial cells following different radiation exposure and subsequently extracted RNA for whole transcriptome sequencing (lncRNA, miRNA, mRNA, and circRNA). Through whole transcriptome analysis, we identified differentially expressed RNAs depending on radiation dosage. Furthermore, we constructed a ceRNA network involving lncRNAs, miRNAs, and mRNAs in radiation-induced injury of esophageal epithelial cells which may contribute to the development of RIEI in humans. These findings may provide and information for understanding the pathogenesis and novel insights for developing treatment strategies for RIEI.
Material and Methods
Cell Culture and Irradiation
Human esophageal epithelial cells (Het-1A) were purchased from ATCC (Catalog number: CRL-2692), the cells were cultured in Dulbecco’s modified Eagle medium supplemented with 10% fetal bovine serum (VivaCell, Shanghai, China) and 1% penicillin-streptomycin (VivaCell, Shanghai, China) under at 37 °C and 5% CO2. Based on the evaluation of the morphology and density of Het-1A cells at various doses and time points (Supplementary Figure 1), the Het-1A cells were ultimately irradiated with 0, 2, and 4 Gy using an X-ray linear accelerator (KUBTEC XCELL 320, Milford, CT) at a fixed dose rate of 1.7 Gy/min.29
Cell Apoptosis Assay
Het-1A cells were seeded in 6-well plates in triplicate, cultured overnight, and subsequently irradiated. After 6, 24 and 48 h of radiation, the cells were stained with fluorescein isothiocyanate-conjugated Annexin V and propidium iodide (AV/PI; Yeason, Shanghai, China). Flow cytometry analysis was performed using the FACS Celesta flow cytometer (Becton Dickinson, Franklin Lakes, NJ), and data were analyzed using FlowJoTM software (Version 10.7). In the AV/PI co-staining experiment, viable cells showed low fluorescence intensity. The apoptosis rate was estimated as the sum of the early apoptosis rate (lower-right quadrant) and late apoptosis rate (upper-right quadrant).
Lactate Dehydrogenase (LDH) Release Assay
The toxicity in Het-1A cells following exposure to various radiation doses was evaluated by quantifying the release of LDH into the supernatant using an LDH cytotoxicity assay kit (Beyotime, Nantong, China) following the manufacturer’s instructions. The results were reported as the mean ± standard error of the mean of absorbance measured at 490 nm using a microplate reader (Biotek, Winooski, VT).
Cell Viability Assay
Het-1A cells were seeded in a 96-well plate at a density of 5×103 cells/ well, followed by treatment with predetermined radiation doses. After 6, 24 and 48 h of radiation, cell viability was evaluated using the Cell Counting Kit-8 assay kit (APExBIO). The optical density was quantified at 450 nm using a microplate reader (Biotek, Winooski, VT).
Cellular Reactive Oxygen Species Assay
Het-1A cells were seeded in triplicate within 6-well plates and cultured overnight. Subsequently, the cells were irradiated with radiation doses of 0, 2, or 4 Gy. Following 6, 24 and 48 h of irradiation, the levels of ROS in Het-1A cells were evaluated using the ROS-sensitive dye, 2’,7’-dichlorofluorescein diacetate (DCF-DA). The cells were incubated with 10 mM DCF-DA at 37°C for 20 min and washed with phosphate-buffered saline. Subsequently, ROS levels were measured using the FACS Celesta flow cytometer (Beyotime, Nantong, China), and data were analyzed using FlowJoTM software (Version 10.7).
Colony Formation Assay
For the colony formation assay, 600 cells were plated into 6-well plates and incubated for 11 days. Colonies were then fixed with methanol and stained with crystal violet solution when they reached an adequate size for visualization. Stained colonies were photographed and counted by imageJ software. Each group was tested independently in triplicate.
Cellular DNA Damage Detection
Het-1A cells were seeded into confocal dishes and allowed to adhere overnight. Subsequently, the cells were exposed to ionizing radiation at doses of 0, 2, and 4 Gy. To evaluate DNA damage, γ-H2AX expression levels were quantified using a γ-H2AX Immunofluorescence Kit (Beyotime, Nantong, China) at three time points (6, 24, and 48 hours) post-irradiation, following the manufacturer’s protocol. Fluorescence imaging and data acquisition were performed using a Zeiss LSM710 confocal microscope (ZEISS, Germany).
Whole Transcriptome Sequencing (miRNA, lncRNA, mRNA, and circRNA)
Total RNA was extracted using TRIzol reagent (Thermo Fisher Scientific, USA) according to the manufacturer’s instructions. The quantity and purity of total RNA were estimated using a Bioanalyzer 2100 and RNA 6000 Nano LabChip Kit (Agilent, CA, USA). To remove rRNA, approximately 5 μg of total RNA was treated with the Ribo-Zero Gold rRNA Removal Kit (Illumina, San Diego, CA, USA). The remaining RNA was fragmented of shorter lengths using the NEBNext® Magnesium RNA Fragmentation Module (New England BioLabs). The steps for library preparation and sequencing of the four types of RNA are as follows:
Sequencing of mRNA and lncRNA
RNA fragments were reverse transcribed into cDNA employing SuperScript™ II Reverse Transcriptase (Invitrogen, Carlsbad, CA, USA). Subsequently, the second strand of DNA with U-label was synthesized. Next, the U-labeled second strand DNA underwent removal treatment, followed by PCR amplification of the resulting products. Finally, paired-end sequencing (2×150 bp) was performed on the Illumina Novaseq™ 6000 platform. The generated reads were filtered and trimmed using Cutadapt software (version: cutadapt-1.9). The filtered reads were assessed for quality using the FastQC tool (version 0.11.9).
Reads from all samples were aligned to the reference genome using HISAT2 software (version: hisat2-2.2.1). Subsequently, the transcript was reconstructed using a combination of HISAT2 and StringTie software (version: stringtie-2.1.6). The resulting transcripts were categorized using gffcompare software (version: gffcompare-0.9.8). Transcripts overlapping with known mRNAs, lncRNAs, and those shorter than 200 bp were filtered out. Novel transcripts with coding potential were predicted using CPC0.9-r2 and CNCI2.0 tools. Transcripts with Coding Potential Calculator (CPC) scores < 0.5 and CNCI scores < 0 were retained and considered as novel lncRNAs. The levels of all transcripts were estimated using StringTie and ballgown software, and the expressions were analyzed using Fragments Per Kilobase of transcript per Million mapped reads (FP) values.
Sequencing of circRNA
First, the Cutadapt tool was used to remove reads containing adaptor contamination, low-quality bases, and undetermined bases. Circular RNA was then assembled using a combination of CIRCExplorer2 (version 2.2.6) and CIRI (version 2.0.2) with the aligned reads. Back-spliced reads among the unmapped reads were identified using Tophat-fusion, CIRCExplorer2, or CIRI. We then normalized the back-spliced reads supporting circular RNAs based on their read length and the number of mapped reads (splice-aligned reads per billion mapped, SRPBM) to facilitate quantitative comparisons between different RNA-seq experiments. The SRPBM value for each circular RNA in a sample was calculated by dividing the number of back-spliced reads by the total number of mapped reads (in billions) and the read length. SRPBM = number of circular reads / number of mapped reads (in billions) / read length.
Sequencing of miRNAs
The miRNA library was prepared after isolating total RNA from the samples and purifying using the provided protocol. The amplified products were then purified and enriched using polyacrylamide gel electrophoresis for miRNA library preparation. Single-end sequencing (SE50) was performed on the Illumina HiSeq 2500 platform through standard procedures. The raw reads were initially processed using an in-house developed tool, the ACGT101-miR (v4.2) program, to remove low-quality reads and eliminate any biases introduced during library preparation or sequencing. Subsequently, unique sequences with lengths in the range of 18 to 26 nucleotides were aligned to specific species precursors in miRBase 22.1 using BLAST search; this allowed for the identification of known miRNAs and novel 3p- and 5p-derived miRNAs. Unique sequences aligning to mature miRNA precursors of specific species were classified as known miRNAs.
Principal Component Analysis (PCA) and t-Distributed Stochastic Neighbor Embedding (t-SNE)
The “stats” and “Rtsne” R packages were employed to conduct Principal Component Analysis (PCA) and t-Distributed Stochastic Neighbor Embedding (t-SNE), respectively. These analyses were performed to assess the expression differences of long non-coding RNAs (lncRNAs), circular RNAs (circRNAs), microRNAs (miRNAs), and messenger RNAs (mRNAs) across distinct irradiation groups (0 Gy, 2 Gy, and 4 Gy).
Gene Set Enrichment Analysis (GSEA)
Enrichment analysis involved gene conversion using the “org.Hs.eg.db” and “clusterProfiler” R packages, and the GSEA method was applied. According to NES values, pathways were ranked in descending order, and the top five activated and inhibited pathways were selected for presentation.
Identification of Dose-Dependent Differentially Expressed RNAs
Differential expression was analyzed using the “DESeq2” R package to examine the variations in RNA expression between different groups for each RNA type: 2 Gy vs 0 Gy, 4 Gy vs 0 Gy, and 4 Gy vs 2 Gy. The threshold for differential conditions was set at |log2Fold Change| ≥ 1 and P-value < 0.05. The differentially expressed RNAs (DERs) included differentially expressed circRNAs (DECs), lncRNAs (DELs), miRNAs (DEMs), and mRNAs (DEGs). Following the differential analysis, we identified differentially expressed RNAs (referred to as dd-DERs) exhibiting dose-dependent upregulation or downregulation. The filtering criteria for differentially expressed RNAs (dd-DERs) were based on our previous study,26 upregulated dd-DERs were defined as 4 Gy ≥ 2 Gy > 0 Gy, and downregulated dd-DERs were defined as 4 Gy ≤ 2 Gy < 0 Gy.
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway Analysis
GO and KEGG pathway analyses were conducted on the differentially expressed genes employing the R package “clusterProfiler”. The GO analysis focused solely on the Biological Process (BP) category. A P-value < 0.05 indicated statistical significance.
Construction of Competing Endogenous RNA (ceRNA) Network
In section 2.9 of the research methodology, we could identify a limited number of differentially expressed circRNAs. Subsequent screening revealed no circRNAs with dose-dependent regulation. Hence, for the construction of, we focused exclusively on utilizing dose-dependent differentially expressed lncRNAs (dd-DELs), dd-DEMs, and dose-dependent differentially expressed genes (dd-DEGs) to construct the ceRNA network. The criteria for correlation significance were defined as a P-value < 0.05 and a correlation coefficient < −0.7. For this assessment, we conducted a co-expression analysis. The sequences of the co-expressed RNAs were compiled, and target gene was predicted using the OmicStudio Target Gene Prediction Cloud Analysis platform (https://www.omicstudio.cn). Predictions were generated by integrating outcomes from TargetScan (version 5.0) and miRanda (version 3.3a) softwares, employing the threshold for target selection as TargetScan_score ≥ 50 and miranda_Energy < −20, and the overlapping results from both prediction tools were considered. This approach enabled the identification of dd-DELs and dd-DEGs, potentially interacting with dd-DEMs. Subsequently, a ceRNA network was constructed by integrating co-expression analysis and predicted binding sites using shared dd-DELs, dd-DEMs, and dd-DEGs.26 The ceRNA network was visualized using Cytoscape software (version 3.10.1).
Reverse Transcription Quantitative Polymerase Chain Reaction (RT-qPCR)
Total RNAs were extracted from rat esophagus using TRIzol reagent (Thermo Fisher Scientific, USA), and three biological replicates were used for each sample group. Total RNA was reverse transcribed into cDNA using the miRNAs 1st Strand cDNA Synthesis Kit (Vazyme, China) and FastKing RT Kit (TIANGEN, China). Subsequently, RT-qPCR was performed on a BioRad real-time fluorescent quantitative PCR instrument employing the miRNA Universal SYBR qPCR Master Mix (Vazyme, China) and SuperReal PreMix Plus (TIANGEN, China). The relative expression levels of RNAs were assessed using the 2−ΔΔCt method. All primer sequences for the experiment are provided in Supplementary material 1.
Statistical Analysis
Statistical analysis and data visualization were performed using R version 4.3.1 and GraphPad version 9.5.1. The threshold for statistical significance was set at P-value < 0.05. For comparisons between two groups, independent samples t-tests were employed, while for comparisons involving multiple groups, either one-way ANOVA or Kruskal–Wallis tests were utilized.
Results
Cell Phenotype Analysis of Radiation-Induced Injury in Het-1A Cells
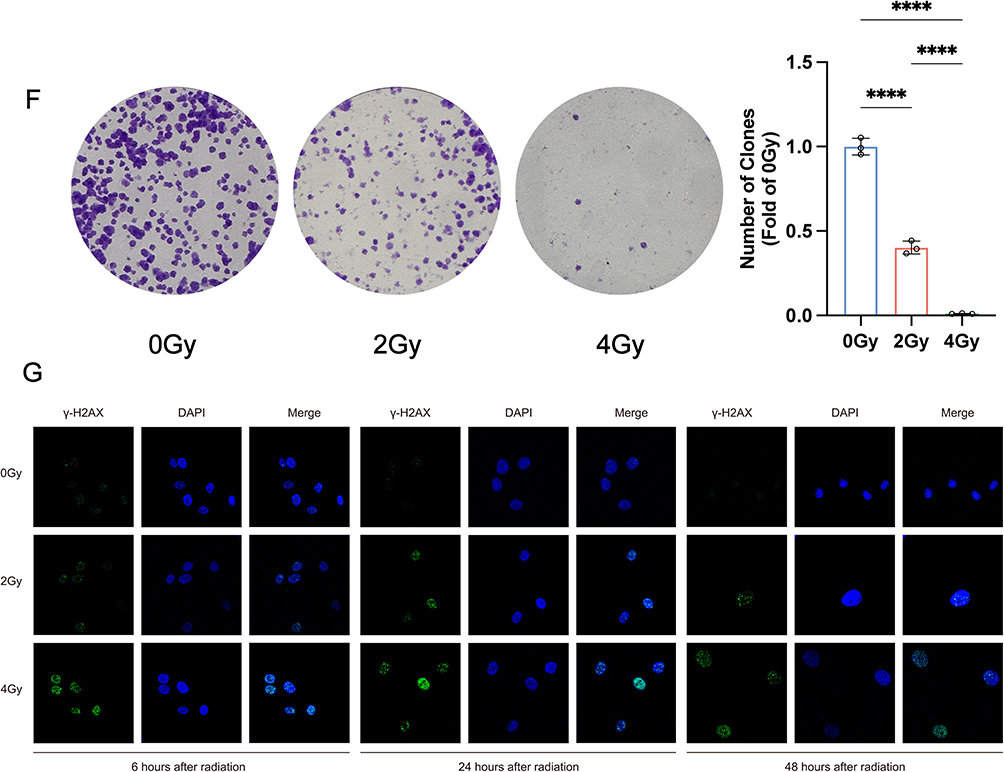
To systematically assess radiation-induced damage in Het-1A cells, we conducted a comprehensive series of phenotypic analyses following exposure to 0, 2, and 4 Gy radiation doses. Our observations revealed significant radiation-induced morphological alterations accompanied by a dose-dependent reduction in cell density (Figure 1A). Quantitative analyses demonstrated consistent dose-response relationships: apoptosis rates, intracellular LDH release, and ROS levels showed progressive increases, while cell viability exhibited a corresponding decline (Figure 1B–E). The clonogenic survival assay further confirmed the radiation sensitivity of Het-1A cells, with colony-forming capacity decreasing proportionally with escalating radiation doses (Figure 1F). Parallel evaluation of DNA damage through γ-H2AX foci quantification at multiple time points revealed a clear dose-dependent accumulation of DNA damage markers (Figure 1G). Collectively, these findings demonstrate that radiation doses of 2 and 4 Gy induce substantial cellular damage in Het-1A cells, with the severity of damage exhibiting a positive correlation with radiation dose. Figure 1 Continued. Figure 1 Phenotypic outcomes of radiation damage in Het-1A cells (A) Microscopic observation of cell morphology. (B) The apoptosis rate was detected at 6, 24, and 48 h after irradiation. (C) The release rate of lactate dehydrogenase (LDH) was detected at 6, 24, and 48 h after irradiation. (D) Cell viability was measured at 6, 24, and 48 h after irradiation. (E) Intracellular ROS levels were measured at 6, 24, and 48 h after irradiation. (F) Assay of colony formation after irradiation. (G) Images were taken by confocal microscopy with γ-H2AX. * P-value < 0.05; ** P-value < 0.01; *** P-value < 0.001; **** P-value < 0.0001. Data are presented as mean ± standard error (n = 3).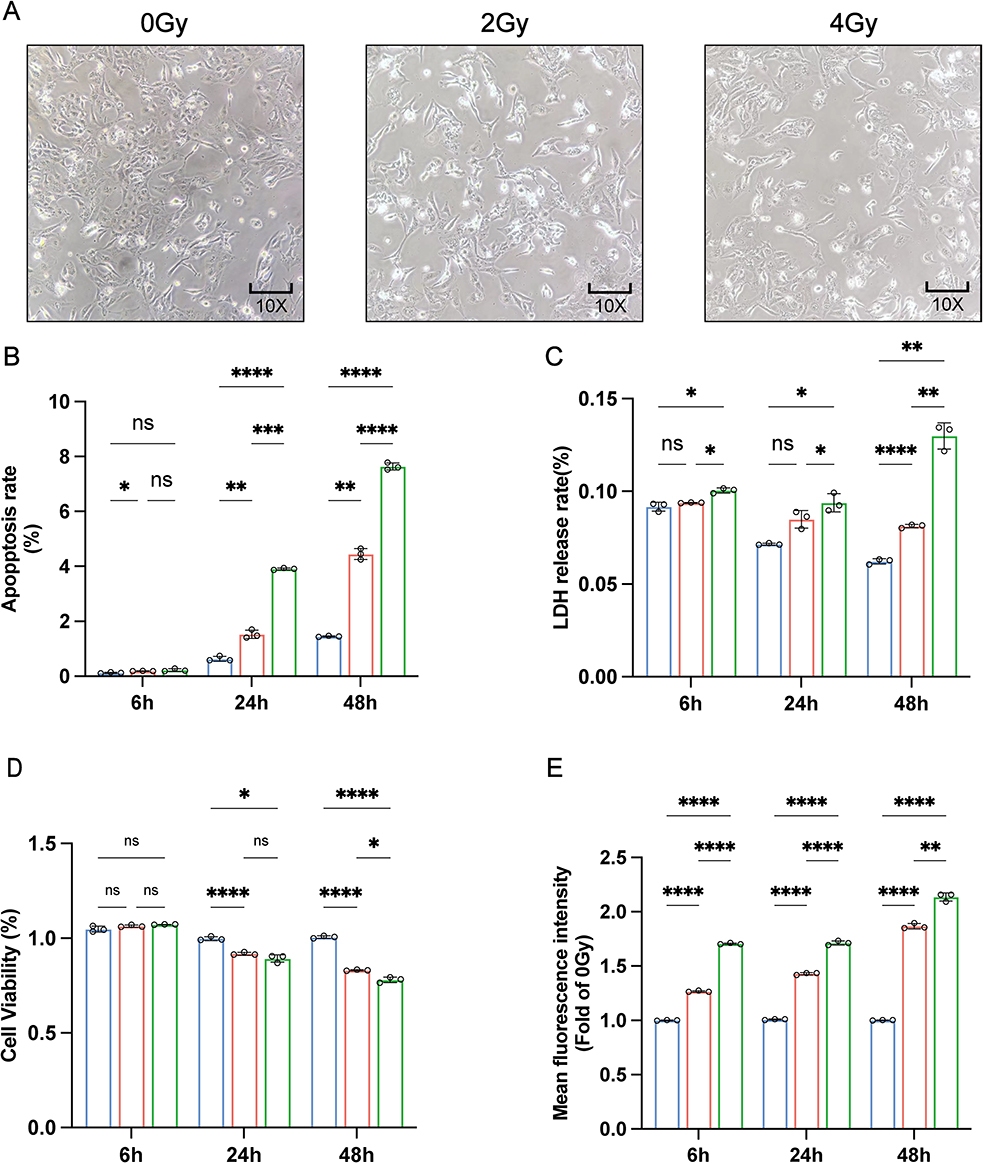
Whole-Transcriptome Expression Profiling of Radiation-Induced Esophageal Epithelial Cell Injury
Whole transcriptome sequencing was performed on cell samples at 48 h after 0, 2, and 4 Gy irradiation to evaluate the differential expression of circRNA, lncRNA, miRNA, and mRNA. PCA and t-SNE evaluated the overall expression patterns of the four RNA types in each group and showed well-defined clusters of lncRNA (Figure 2A and Supplementary Figure 2A), miRNA (Figure 2B and Supplementary Figure 2B), mRNA (Figure 2C and Supplementary Figure 2C), and circRNA (Figure 2D and Supplementary Figure 2D) across the four groups. It was shown that the overall expression of these RNA types differed significantly between the groups.
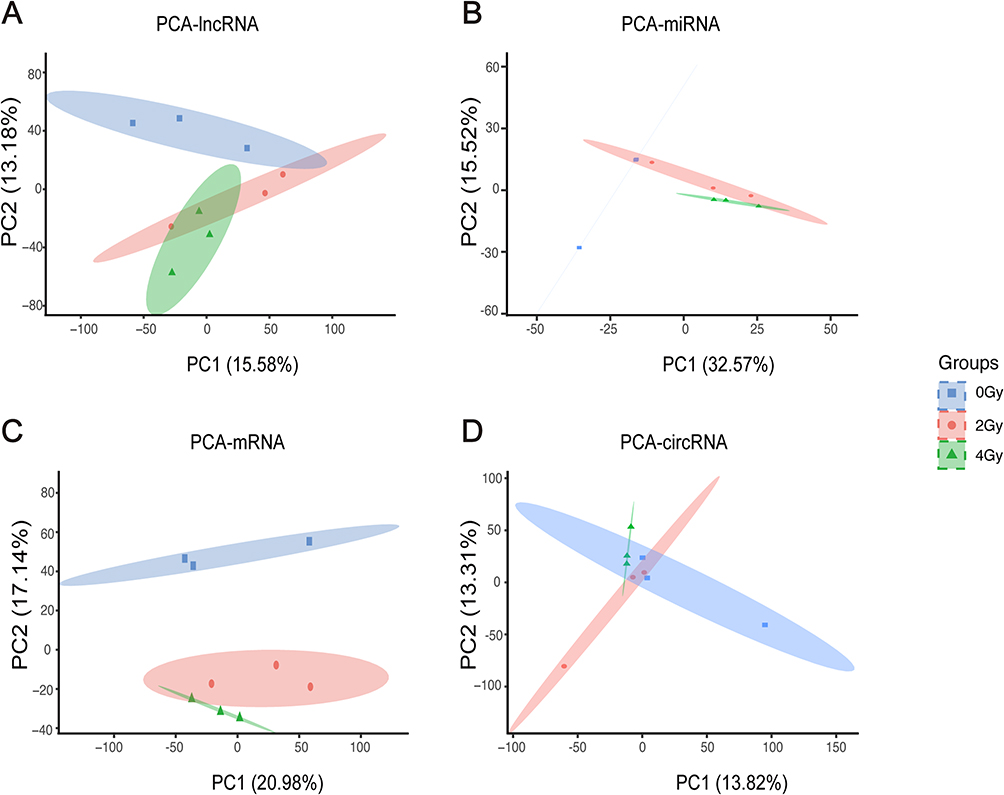 |
Figure 2 Principl Component Analysis plots illustrate the expression levels of lncRNA (A), miRNA (B), mRNA (C) and circRNA (D) in Het-1A cells exposed to 0, 2 and 4 Gy. |
Gene Set Enrichment Analysis of mRNA Expression Profiling in Radiation-Induced Esophageal Epithelial Cell Injury
GSEA was employed to investigate functional changes at the cellular-level by analyzing the expression profiles of all mRNAs. GSEA analyses were carried out on the three pairwise comparisons, including 2 Gy vs 0 Gy, 4 Gy vs 0 Gy, and 4 Gy vs 2 Gy. The GSEA analysis utilizing the GO gene set revealed significant enrichment in the activation of channel activity and mitotic inhibition following ionizing radiation exposure. Specifically, molecular functions (MFs) such as voltage-gated potassium channel activity, potassium channel activity, and monoatomic ion channel activity were upregulated (NES > 0), whereas pathways related to BP and cellular components (CCs), including sister chromatid cohesion, kinetochore, and condensed chromosome, centromeric region were downregulated (NES < 0) across all three comparative analyses (Figure 3A). The enrichment analysis using KEGG revealed radiation activated immune-related pathways involving viral protein interaction with cytokine and cytokine receptor, neuroactive ligand-receptor interaction, cytokine-cytokine receptor interaction, complement and coagulation cascades, and calcium signaling pathway (Figure 3B). Conversely, radiation-inhibited repair-related pathways, such as nucleotide excision repair, base excision repair, and nucleocytoplasmic transport were also noted (Figure 3B). The enrichment analysis of both gene sets mentioned above revealed significant disruptions in channel activity, cell replication, cellular repair, and immune response in Het-1A cells upon radiation exposure.
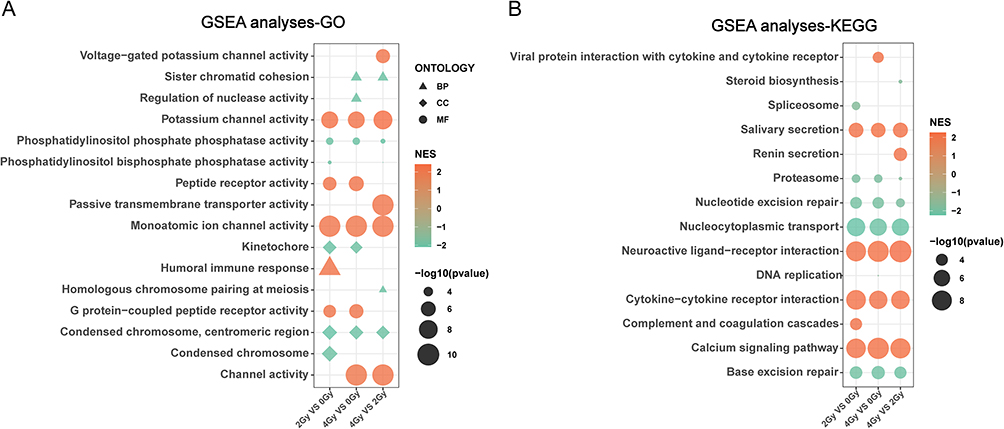 |
Figure 3 The Gene Set Enrichment Analysis of mRNA expression profiles based on the Gene Ontology (A) and Kyoto Encyclopedia of Genes and Genomes (B) gene sets. |
Identification of Differentially Expressed RNAs
DERs, including lncRNA, miRNA, mRNA, and circRNA, were analyzed in the three pairwise comparisons of radiation exposures: 2 Gy vs 0 Gy, 4 Gy vs 0 Gy, and 4 Gy vs 2 Gy. Pairwise comparisons of the three groups of lncRNAs revealed, respectively, 79 downregulated and 61 upregulated (Figure 4A and D), 91 downregulated and 86 upregulated (Figure 4B and D), and 55 downregulated and 73 upregulated DELs (Figure 4C and D). Considering miRNAs, we identified 19 downregulated and 20 upregulated DEMs under 2 Gy vs 0 Gy radiation doses (Figure 4E and H), 18 downregulated and 30 upregulated DEMs under 4 Gy vs 0 Gy (Figure 4F and H), and 9 downregulated and 6 upregulated DEMs in 4 Gy vs 2 Gy radiation doses (Figure 4G and H). Among mRNAs, we identified 182 downregulated and 161 upregulated DEGs in 2 Gy vs 0 Gy radiation doses (Figure 4I and L), 309 downregulated and 172 upregulated DEGs in 4 Gy vs 0 Gy (Figure 4J and L), and 62 downregulated and 69 upregulated DEGs under 4 Gy vs 2 Gy radiation doses (Figure 4K and L). Among circRNAs, we identified 25 downregulated and 35 upregulated DECs in 2 Gy vs 0 Gy radiation doses (Figure 4M and P), 45 downregulated and 21 upregulated DECs in 4 Gy vs 0 Gy (Figure 4N and P), and 22 downregulated and 23 upregulated DECs in 4 Gy vs 2 Gy radiation doses (Figure 4O and P). The human esophageal epithelial cells in the 4 Gy group exhibited a significantly higher number of DERs compared to those in the 2 Gy group, indicating a more pronounced disturbance at the transcriptome level. These cellular findings were in concordance with the previously documented pathological changes in the esophageal structure. The differentially expressed RNAs are shown in Supplementary Material 2.
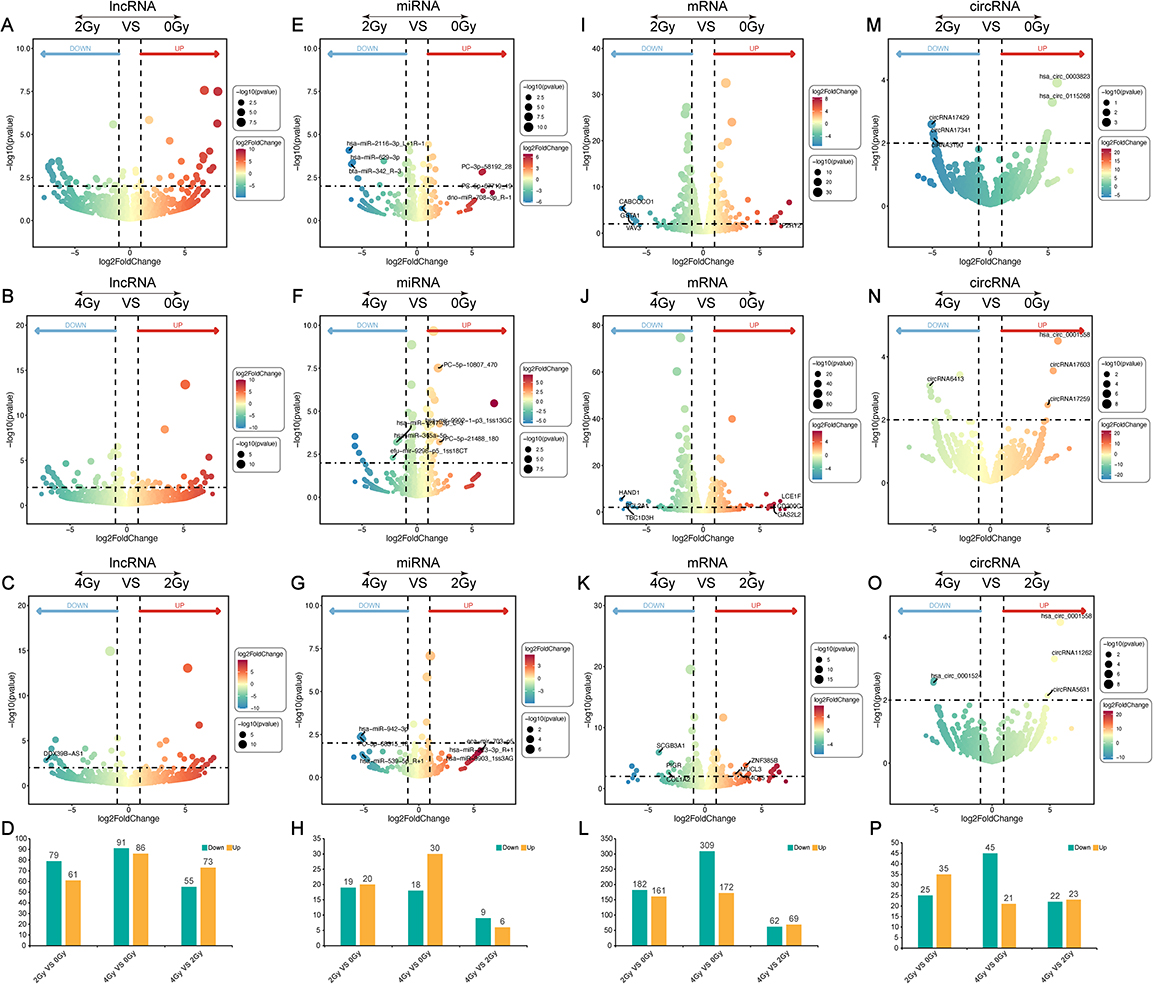 |
Figure 4 Differentially expressed RNAs (DERs) were identified among the three pairwise comparisons. (A–C). Volcano plots illustrating the differential expression of lncRNAs (DELs): (A) (2 Gy vs 0 Gy), (B) (4 Gy vs 0 Gy), and (C) (4 Gy vs 2 Gy). The bar graph shows the number of DELs in each comparison (D). Volcano plots (E–G) depict the differential expression of microRNAs (DEMs): (E) (2 Gy vs 0 Gy), (F) (4 Gy vs 0 Gy), and (G) (4 Gy vs 2 Gy), while the corresponding bar graph represents the number of DEMs in each comparison (H). Volcano plots (I–K) display the differential expression of genes (DEGs) of (I) (2 Gy vs 0 Gy), (J) (4 Gy vs 0 Gy), and (K) (4 Gy vs 2 Gy). The bar graph exhibits the number of DEGs in each group (L). Volcano plots (M–O) display the differential expression of circRNAs (DECs): (M) (2 Gy vs 0 Gy), (N) (4 Gy vs 0 Gy), and (O) (4 Gy vs 2 Gy). The bar graph presents the number of DECs in each group (P). |
Identification of Dose-Dependent Differentially Expressed RNAs
Utilizing the analysis performed in Section 3.4 as a foundation, We defined the criteria for radiation dose dependence as follows: either 4 Gy ≥ 2 Gy > 0 Gy or 4 Gy ≤ 2 Gy < 0 Gy. Subsequently, using these criteria, we screened and identified dd-DERs that were dose-dependent and exhibited either upregulation or downregulation. We noted 17 upregulated and 24 downregulated dd-DELs (Figure 5A), 11 upregulated and 7 downregulated dd-DEMs (Figure 5B), and 84 upregulated and 108 downregulated dd-DEGs (Figure 5C). A limited number of DECs were identified in the aforementioned differential RNA expression analysis; therefore, we did not screen out any dd-DECs. Consequently, dd-DECs were excluded from the subsequent construction of the ceRNA network. In summary, we identified a total of 41 dd-DELs, 18 dd-DEMs, and 192 dd-DEGs that potentially contribute to radiation-induced injury of esophageal epithelial cells. The Dose-Dependent Differentially Expressed RNAs are shown in Supplementary Material 3.
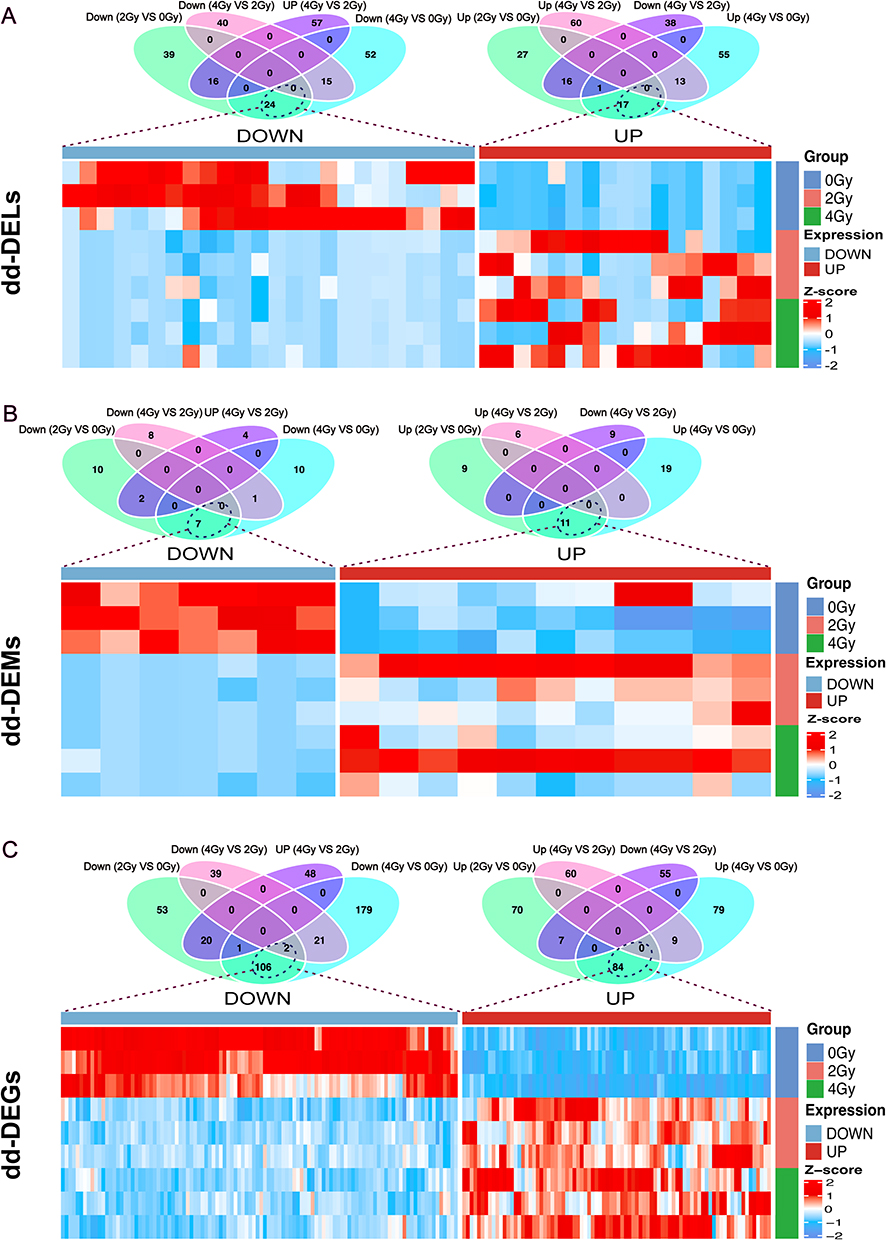 |
Figure 5 Identification of dose-dependent differentially expressed RNAs (dd-DERs). (A) Venn diagrams and heatmap of the dose-dependent differentially expressed lncRNAs (dd-DELs). (B) The Venn diagrams and heatmap of the dose-dependent differentially expressed miRNAs (dd-DEMs). (C) The Venn diagrams and heatmap of the dose-dependent differentially expressed mRNAs (dd-DEGs). |
Investigation of the Potential Functions and Pathways
We performed Gene Ontology-Biological Pathway (GO-BP) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses to identify the potential biological functions and pathways associated with dd-DEGs in irradiated cells. According to the GO-BP analysis, the top five enriched BP terms included various metabolic processes, including hormone metabolism, cellular glucuronidation, uronic acid metabolism, glucuronate metabolism, and xenobiotic metabolism (Figure 6). The KEGG pathway analysis revealed the top five terms, including pentose and glucuronate interconversions, retinol metabolism, drug metabolism - cytochrome P450, bile secretion, and porphyrin metabolism (Figure 6). The inner circle in this figure represents the terms, while the outer circle represents the genes enriched in each term. Among the genes presented in the outer circle, nine members of the UGT1A family, including UGT1A1, UGT1A3, UGT1A4, UGT1A5, UGT1A6, UGT1A7, UGT1A8, UGT1A9, and UGT1A10, have roles in a variety of metabolic pathways. The above results suggest that dd-DEGs, especially the members of the UGT1A family, primarily exert their functions by regulating metabolic pathways, thereby providing insights into the effects of radiation on protein functions.
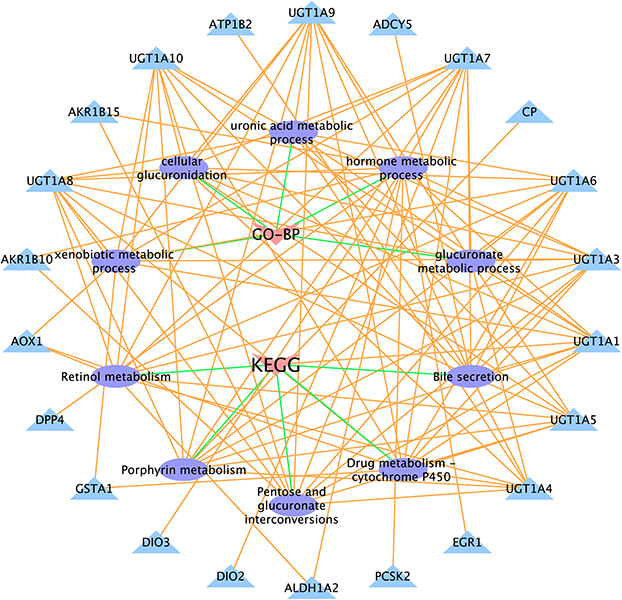 |
Figure 6 Gene Ontology-Biological Process and Kyoto Encyclopedia of Genes and Genomes pathway enrichment analyses based on dose-dependent differentially expressed genes (dd-DEGs). |
Construction of Competing Endogenous RNA (ceRNA) Network
The relationships among dd-DELs, dd-DEMs and dd-DEGs were investigated by constructing a ceRNA network to explore the reciprocal regulatory interactions. We able to identify a total of 113 co-expressed dd-DELs/dd-DEMs exhibiting a strong negative correlation (correlation coefficient < −0.7, P-value < 0.05) (Figure 7A). Further online predictions indicated interactions for 141 dd-DELs/dd-DEMs (Figure 7A). Additionally, correlation among 770 co-expressed dd-DEMs/dd-DEGs were observed, and online predictions suggested binding for 31,639 dd-DEMs/dd-DEGs (Figure 7B). We constructed a ceRNA network through the intersection of the co-expression and online prediction sets (Figure 7C). This network comprised 10 lncRNAs, 5 miRNAs, and 55 mRNAs. The heatmap was generated to visualize the expression levels of all RNAs extracted from the network (Figure 7D). These RNA molecules are believed to be involved in RIEI through the competitive mechanism of ceRNA, where they compete to regulate the expression of target genes. The ceRNA network offers valuable insights into the regulatory interactions between diverse RNA types and their potential roles in RIEI. The RNA relationships involved in constructing the ceRNA network are shown in Supplementary Material 4.
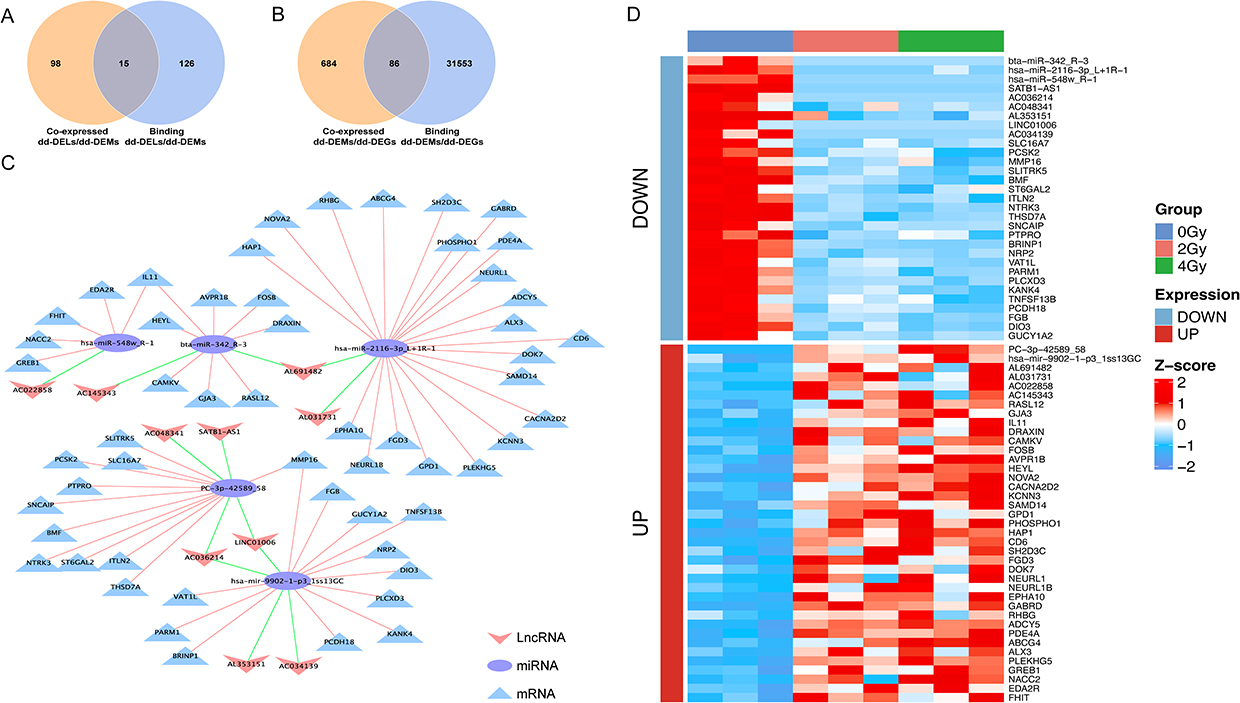 |
Figure 7 Competing endogenous RNA (ceRNA) network composed of lncRNA, miRNA, and mRNA. (A) Venn diagram illustrating the co-expression dose-dependent differentially expressed lncRNAs/ dose-dependent differentially expressed miRNAs/ (dd-DELs/dd-DEMs) and binding dd-DELs/dd-DEMs. (B) Venn diagram depicting the co-expression dd-DEMs/ dose-dependent differentially expressed genes (dd-DEMs/dd-DEGs) and binding dd-DEMs/dd-DEGs. (C) CeRNA network diagram constructed based on filtered dd-DELs, dd-DEMs, and dd-DEGs. lncRNA, miRNA, and mRNA are represented by “V” shape, circle, and triangle, respectively. (D) Heatmap of RNA expression in the ceRNA network. |
Validation of Competing Endogenous RNA Network Through RT-qPCR
The accuracy of whole transcriptome sequencing was validated by selecting several lncRNAs, miRNAs, and mRNAs exhibiting high connectivity in the network for validation through reverse transcription-quantitative polymerase chain reaction (RT-qPCR). The RT-qPCR findings demonstrated an upregulation of IL11 mRNA expression with increasing radiation dose, while NRP2, SLC16A7, and KANK4 were downregulated. Moreover, the expression levels of lncRNAs (AC048341, AL353151) decreased in response to escalating radiation dose. Conversely, miRNA (hsa-mir-9902-1-p3_1ss13GC) exhibited enhanced expression, whereas the miRNA (hsa-miR-2116-3p_L+1R-1) level decreased (Figure 8). Significantly, the experimental RT-qPCR results aligned with the sequencing data, partially confirming the accuracy of our sequencing outcomes. These findings provide valuable insights into the potential mechanisms of radiation-induced damage in esophageal epithelial cells.
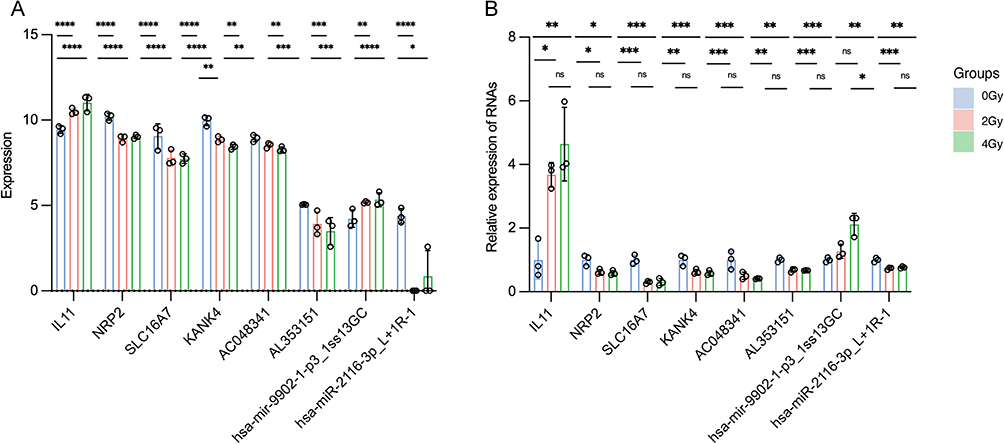 |
Figure 8 Validation of multiple dose-dependent differentially expressed RNAs (dd-DERs). (A) Summary of the validation of whole transcriptome results. (B) Validation of expression levels of multiple RNAs in the network by real time-quantitative polymerase chain reaction. *P-value < 0.05; ** P-value < 0.01; *** P-value < 0.001; **** P-value < 0.0001. Data are presented as mean ± standard error (n = 3). |
Discussion
In our study, we employed whole transcriptome sequencing technology to investigate the differential expression of mRNA, lncRNA, circRNAs, and miRNA in human esophageal epithelial cells following exposure to different radiation doses. Using dose-dependent genes as an entry point, we identified key biological processes and pathways closely associated with radiation-induced esophageal epithelial cell injury. Through correlation analysis and sequence-based prediction, we successfully identified a potential ceRNA network of dose-dependent differentially expressed mRNAs, lncRNAs, and miRNAs.
We identified a ceRNA regulatory network comprising 10 lncRNAs, 5 miRNAs, and 55 mRNAs. In irradiated cells, the lncRNAs LINC01006 and SATB1-AS1 were significantly downregulated. Although some identified lncRNAs have not been previously reported to act through endogenous competition in RIEI, their roles in other diseases have been implicated. LINC01006, in particular, has been shown to influence cancer progression through endogenous competition across various cancers. In hepatocellular carcinoma (HCC), LINC01006 suppresses the development of tumors via the LINC01006/miR-194-5p/CADM1 axis.30 Additionally, it regulates HCC cell proliferation, migration, and invasion through the LINC01006/miR-433-3p/CBX3 axis.31 In another example, the downregulation of SATB1-AS1 has been linked to enhanced migration and invasion of laryngeal squamous cell carcinoma cells through the SATB1-AS1/miR-1299/SAV1/CCNG2/SH3KBP1/JADE1/HIPK2 axis.32 The most significantly upregulated and downregulated mRNAs in the network are, respectively, RHBG and PLCXD3. Research shows that RHBG can initiate intracellular signaling cascades that ultimately activate NFκB,33 with nuclear factor-κB activation contributing to inflammation, such as radiotherapy-induced mucositis.34 Downregulated PLCXD3 is associated with impaired insulin signaling and biosynthesis, and reduced glucose sensing in pancreatic β cells.35 The miRNAs involved in this endogenous competition network are variants of mature miRNAs available in the database, and their specific effects in the regulatory functions of cells or tissues remain unclear. Nonetheless, certain mechanisms associated with some miRNAs in the database have been confirmed. For example, Wu et al described a positive feedback loop in breast cancer cells (LINC01433/miR-2116-3p/MYC), whereas LINC01433 promotes cell progression by regulating the miR-2116-3p/MYC axis.36 While these RNAs play a role in disease progression through endogenous competition, their roles in RIEI require further investigation.
The GSEA analysis identified the channel activity activation, including the enrichment of potassium ion and monovalent ion channel activities. It also revealed the activation of immune responses and the suppression of cell mitosis. Earlier studies have documented that ion channels are potentially promising targets for modulating diverse cellular functions. Furthermore, specific ion channels on the cell membrane are accompanied by intracellular counterparts, each serving distinct roles within various metabolic pathways.37 Our phenotypic results revealed a gradual increase in the rate of cell apoptosis with escalating radiation dose in agreement with elevated levels of ROS. Notably, mitochondria serve as the main source of ROS production and enhanced ROS levels can trigger the release of cytochrome c from mitochondria through mitochondrial pathways, ultimately culminating in cell apoptosis.38 Experimental evidence supports the hypothesis that ion channels play a potential regulatory role at the mitochondrial level during apoptosis. Among these channels, potassium (K+) channels are widely present in organisms and facilitate the transport of K+ across biological membranes. Several events can be triggered through the regulation of K+ channels, including K+ flux, such as mitochondrial membrane hyperpolarization, significantly impacting the generation of mitochondrial ROS.39–41 Voltage-gated potassium channels (Kv) constitute the largest and most diverse family of potassium channels, divided into 12 families (Kv1-12).42 The Kv1.3 channel, a member of the Kv channels, belongs to the Shaker family.43 It was initially discovered in the plasma membrane (PM),44 where it is involved in proliferation and cell volume regulation. Studies have shown that MitoKv1.3 participates in apoptotic cell death.45 In cells lacking Kv1.3, the expression of mitochondria-targeted Kv1.3 is sufficient to restore these cells’ response to several apoptotic stimuli.45 MitoKv1.3 has been identified as a target of the pro-apoptotic protein Bax, a member of the Bcl-2 family.45,46 The physical interaction between Bax and mitoKv1.3 occurs only upon the induction of apoptosis. Similar to Kv1.3, Kv1.1 and Kv1.5 also exhibit dual localization in the inner mitochondrial membrane (IMM) and PM, and can interact with Bax under apoptotic stimuli, sensitizing cells to death signals.47,48 Radiation can induce DNA damage in cells, leading to several cellular responses. These include the removing and repairing DNA damage, restoring DNA double-strand continuity, activating DNA damage checkpoints to halt cell cycle progression for repair, and preventing the propagation of damaged or incompletely replicated chromosomes. Additionally, cells may elicit transcriptional responses that could induce beneficial changes or apoptosis to eliminate severely damaged or dysfunctional cells.49 Our GSEA results indicate the activation of mitotic inhibition and DNA damage checkpoints within the cell. Furthermore, our enrichment analysis revealed a suppression of nucleotide excision repair (NER) and base excision repair (BER). Nucleotide excision repair removes large DNA lesions caused by radiation, chemicals, or adding proteins to DNA. The damaged bases are excised by “excision nucleases”, the multi-subunit enzyme system that creates double incisions in the damaged strand to remove the lesion.50–53 In humans, excision repair involves six repair factors (RPA, XPA, XPC, TFIIH, XPG, and XPF•ERCC1) composed of 15 polypeptides.54–56 Although our transcriptome data could detect expression changes in several repair factors, these changes were not statistically significant according to the defined threshold.
We identified a total of 55 dd-DEGs, with dose changes categorized as 4 Gy ≥ 2 Gy > 0 Gy or 4 Gy ≤ 2 Gy < 0 Gy. Enrichment analysis of these dd-DEGs revealed that several pathways related to glucuronic acid metabolism were enriched, and the UDP-glycosyltransferases (UGT) superfamily genes were found to be involved in multiple metabolic pathways. UGTs are crucial enzymes involved in biotransformation, regulating the levels and distribution of many endogenous signaling molecules and the metabolism of a broad range of endogenous as well as exogenous compounds. In mammals, the UGT superfamily includes the UGT1, UGT2, UGT3, and UGT8. UGTs serve as rate-limiting enzymes in the glucuronic acid pathway, and alterations in their activity can impact gluconeogenesis and lipid metabolism. UGTs conjugate various lipophilic molecules to sugars, which include galactose, xylose, acetylglucosamine, glucuronic acid, and glucose, thereby converting these molecules into water-soluble substrates, including carcinogens, drugs, steroids, lipids, fatty acids, and bile acids.57 UGTs expression and their enzymatic activity are influenced by various factors and regulated through multiple mechanisms of epigenetic modifications (eg, DNA methylation and histone modifications), transcriptional regulation, post-transcriptional regulation (eg, microRNAs), and post-translational modifications (eg, structural and functional changes and protein-protein interactions). Methylation of the transcription factor HNF1A in colon cells negatively regulates UGT1A1.58,59 UGT expression is also influenced by histone modifications, which together with DNA methylation, regulate UGT1A1 expression.60 MicroRNAs can directly target UGT mRNAs to modulate their expression and indirectly affect UGT expression through the inhibition of transcription factors.61–63 Additionally, the interaction between UGT1A9 and bisphenol leads to intracellular calcium overload through the activation of the calcium signaling pathway, a finding also supported by our analysis. This overload induces mitochondrial stress, disrupts mitochondrial homeostasis, and promotes bisphenol-induced cell death.64 We hypothesize that members of the UGT superfamily may significantly contribute to radiation-induced esophageal epithelial cell injury, warranting further investigational studies.
Conclusion
In conclusion, our study provides significant insights into the effects of ionizing radiation on the entire transcriptome of human esophageal epithelial cells. We identified several differentially expressed RNAs (DD-RNAs) at the 48-hour time point following ionizing radiation and used these RNAs to construct an associated ceRNA network. However, the ceRNA network is a dynamic regulatory process. Our study focused on a single time point, which limits the ability to fully capture the temporal changes within the ceRNA network. Additionally, the internal regulatory relationships require further validation. Therefore, future experimental studies will aim to identify key factors and regulatory relationships that contribute to early damage and explore potential therapeutic strategies to mitigate radiation-induced damage to esophageal epithelial cells through the competitive regulatory interactions between RNAs in the ceRNA network. These findings offer potential avenues for radiation protection of the entire esophagus.
Abbreviations
ceRNA, competitive endogenous, RNA; RIEI, radiation-induced esophageal injury; DERs, differentially expressed RNAs; DECs, differentially expressed circRNAs; DELs, differentially expressed lncRNAs, DEMs, differentially expressed miRNAs; DEGs, differentially expressed mRNAs; dd-DELs, dd-DERs, dose-dependent differentially expressed RNAs; dose-dependent differentially expressed lncRNAs; dd-DEMs, dose-dependent differentially expressed miRNAs; dd-DEGs, dose-dependent differentially expressed mRNAs; dd-DECs, dose-dependent differentially expressed circRNAs; PCA, principal component analysis; GSEA, gene set enrichment analysis; GO, Gene Ontology; MF, Molecular Function; BP, Biological Process; CC, Cellular Component; KEGG, Kyoto Encyclopedia of Genes and Genomes; RT-qPCR, reverse transcription quantitative polymerase chain reaction.
Data Sharing Statement
The RNA-seq raw data from this study have been submitted to Sequence Read Archive (SRA) database (https://www.ncbi.nlm.nih.gov/sra). The SRA accession number was PRJNA1101704.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (82473568, 82103773), the Young Talent Project of China National Nuclear Corporation (CNNC202379, CNNC202275), Foundation of Department of Science and Technology of Sichuan Provincial (2024NSFSC0764, 2023ZYD0084, 2023NSFSC0680, 2023JDRC0108), the Joint Fund of Chengdu Medical College and the Second Affiliated Hospital of Chengdu Medical College (2022LHFSZYB-01), Natural Science Foundation of Chengdu Medical College (CYZ19-30), and the Foundation of Key Laboratory of Radiation Physics and Technology of the Ministry of Education (2023SCURPT03).
Author Contributions
All authors have made significant contributions to the work reported, whether in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas. They participated in drafting, revising, or critically reviewing the article; gave final approval of the version to be published; agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Werner-Wasik M, Yorke E, Deasy J, Nam J, Marks LB. Radiation dose-volume effects in the esophagus. Int J Radiat Oncol Biol Phys. 2010;76(3 Suppl):S86–93. doi:10.1016/j.ijrobp.2009.05.070
2. Simone CB. Thoracic radiation normal tissue injury. Semin Radiat Oncol. 2017;27(4):370–377. doi:10.1016/j.semradonc.2017.04.009
3. Yu Y, Guan H, Dong Y, Xing L, Li X. Advances in dosimetry and biological predictors of radiation-induced esophagitis. Onco Targets Ther. 2016;9:597–603. doi:10.2147/OTT.S97019
4. Syllaios A, Vailas M, Tolia M, et al. Radiation-induced esophageal cancer: investigating the pathogenesis, management, and prognosis. Medicina. 2022;58(7):949. doi:10.3390/medicina58070949
5. Da P, S S, O C, et al. Predicting esophagitis after chemoradiation therapy for non-small cell lung cancer: an individual patient data meta-analysis. Int J Radiat Oncol Biol Phys. 2013;87(4). doi:10.1016/j.ijrobp.2013.07.029
6. Li Y, Lin J, Xiao J, et al. Therapeutic effects of co-venenum bufonis oral liquid on radiation-induced esophagitis in rats. Exp Anim. 2020;69(3):354–362. doi:10.1538/expanim.19-0142
7. Jakubczyk K, Dec K, Kałduńska J, Kawczuga D, Kochman J, Janda K. Reactive oxygen species - sources, functions, oxidative damage. Pol Merkur Lekarski. 2020;48(284):124–127.
8. Yakovlev VA. Role of nitric oxide in the radiation-induced bystander effect. Redox Biol. 2015;6:396–400. doi:10.1016/j.redox.2015.08.018
9. Khodamoradi E, Hoseini-Ghahfarokhi M, Amini P, et al. Targets for protection and mitigation of radiation injury. Cell mol Life Sci. 2020;77(16):3129–3159. doi:10.1007/s00018-020-03479-x
10. Djordjević VB. Free radicals in cell biology. Int Rev Cytol. 2004;237:57–89. doi:10.1016/S0074-7696(04)37002-6
11. Kitamura H, Tanigawa T, Kuzumoto T, et al. Interferon-α exerts proinflammatory properties in experimental radiation-induced esophagitis: possible involvement of plasmacytoid dendritic cells. Life Sci. 2022;289:120215. doi:10.1016/j.lfs.2021.120215
12. Usami S, Motoyama S, Koyota S, et al. Regenerating gene I regulates interleukin-6 production in squamous esophageal cancer cells. Biochem Biophys Res Commun. 2010;392(1):4–8. doi:10.1016/j.bbrc.2009.12.129
13. Epperly MW, Gretton JA, DeFilippi SJ, et al. Modulation of radiation-induced cytokine elevation associated with esophagitis and esophageal stricture by manganese superoxide dismutase-plasmid/liposome (SOD2-PL) gene therapy. Radiat Res. 2001;155(1 Pt 1):2–14. doi:10.1667/0033-7587(2001)155[0002:morice2.0.co;2
14. Yao J, Zhang J, Wang J, et al. Transcriptome profiling unveils a critical role of IL-17 signaling-mediated inflammation in radiation-induced esophageal injury in rats. Dose Response. 2022;20(2):15593258221104609. doi:10.1177/15593258221104609
15. Tu W, Feng Y, Lai Q, et al. Metabolic profiling implicates a critical role of cyclooxygenase-2-mediated arachidonic acid metabolism in radiation-induced esophageal injury in rats. Radiat Res. 2022;197(5):480–490. doi:10.1667/RADE-20-00240.1
16. Xi T, Zhou Y, Ma S, et al. Construction of a potential long noncoding RNA prognostic model involved competitive endogenous RNA for patients with gastric cancer. Medicine. 2024;103(24):e38458. doi:10.1097/MD.0000000000038458
17. Kohansal M, Alghanimi YK, Banoon SR, et al. CircRNA-associated ceRNA regulatory networks as emerging mechanisms governing the development and biophysiopathology of epilepsy. CNS Neurosci Ther. 2024;30(4):e14735. doi:10.1111/cns.14735
18. Asadi MR, Abed S, Kouchakali G, et al. Competing endogenous RNA (ceRNA) networks in Parkinson’s disease: a systematic review. Front Cell Neurosci. 2023;17:1044634. doi:10.3389/fncel.2023.1044634
19. Zhang Y, Qian L, Liu Y, Liu Y, Yu W, Zhao Y. CircRNA-ceRNA network revealing the potential regulatory roles of CircRNA in alzheimer’s disease involved the cGMP-PKG signal pathway. Front Mol Neurosci. 2021;14:665788. doi:10.3389/fnmol.2021.665788
20. Li X, An Y, Wang Q, Han X. The new ceRNA crosstalk between mRNAs and miRNAs in intervertebral disc degeneration. Front Cell Dev Biol. 2022;10:1083983. doi:10.3389/fcell.2022.1083983
21. Lang Y, Zhang J, Yuan Z. Construction and dissection of the ceRNA‑ceRNA network reveals critical modules in depression. Mol Med Rep. 2019;19(5):3411–3420. doi:10.3892/mmr.2019.10009
22. Wu X, Sui Z, Zhang H, Wang Y, Yu Z. Integrated analysis of lncRNA-mediated ceRNA network in lung adenocarcinoma. Front Oncol. 2020;10:554759. doi:10.3389/fonc.2020.554759
23. Xiao Y, Tang J, Yang D, et al. Long noncoding RNA LIPH-4 promotes esophageal squamous cell carcinoma progression by regulating the miR-216b/IGF2BP2 axis. Biomark Res. 2022;10(1):60. doi:10.1186/s40364-022-00408-x
24. Sun Z, Li J, Lin M, Zhang S, Luo J, Tang Y. An RNA-seq-based expression profiling of radiation-induced esophageal injury in a rat model. Dose Response. 2019;17(2):1559325819843373. doi:10.1177/1559325819843373
25. Luo J, Zhang C, Zhan Q, et al. Profiling circRNA and miRNA of radiation-induced esophageal injury in a rat model. Sci Rep. 2018;8(1):14605. doi:10.1038/s41598-018-33038-1
26. Wu F, Zhang X, Zhang S, et al. Construction of an immune-related lncRNA-miRNA-mRNA regulatory network in radiation-induced esophageal injury in rats. Int Immunopharmacol. 2023;122:110606. doi:10.1016/j.intimp.2023.110606
27. Liu M, Sun Z, Tang Y, Zhang S, Luo J. The regulation of exosome-mediated miR-132-3p/miR-132-3p-UUU on radiation-induced esophageal injury. Radiat Res. 2023;200(2):151–161. doi:10.1667/RADE-22-00070.1
28. Sun Z, Zhang J, Zeng F, et al. Differentially expressed mRNAs and potential mechanisms of radiation-induced TUT4-/- esophageal cell injury. Dose Response. 2022;20(4):15593258221136810. doi:10.1177/15593258221136810
29. Jafarzadeh N, Mani-Varnosfaderani A, Gilany K, Eynali S, Ghaznavi H, Shakeri-Zadeh A. The molecular cues for the biological effects of ionizing radiation dose and post-irradiation time on human breast cancer SKBR3 cell line: a Raman spectroscopy study. J Photochem Photobiol B. 2018;180:1–8. doi:10.1016/j.jphotobiol.2018.01.014
30. Sun Z, Zhao L, Wang S, Wang H. Knockdown of long non-coding RNA LINC01006 represses the development of hepatocellular carcinoma by modulating the miR-194-5p/CADM1 axis. Ann Hepatol. 2022;27 Suppl 1:100571. doi:10.1016/j.aohep.2021.100571
31. Song Y, Wang S, Cheng X. LINC01006 regulates the proliferation, migration and invasion of hepatocellular carcinoma cells through regulating miR-433-3p/CBX3 axis. Ann Hepatol. 2021;25:100343. doi:10.1016/j.aohep.2021.100343
32. Lyu K, Li Y, Xu Y, et al. Using RNA sequencing to identify a putative lncRNA-associated ceRNA network in laryngeal squamous cell carcinoma. RNA Biol. 2020;17(7):977–989. doi:10.1080/15476286.2020.1741282
33. Mishra S, Welch N, Singh SS, et al. Ammonia transporter RhBG initiates downstream signaling and functional responses by activating NFκB. Proc Natl Acad Sci U S A. 2024;121(31):e2314760121. doi:10.1073/pnas.2314760121
34. Abeesh P, Guruvayoorappan C. NF-κB as a potential target for the treatment and prevention of mucositis. Curr Pharm Biotechnol. 2023;24(13):1613–1622. doi:10.2174/1389201024666230331121328
35. Aljaibeji H, Mukhopadhyay D, Mohammed AK, et al. Reduced expression of PLCXD3 associates with disruption of glucose sensing and insulin signaling in pancreatic β-cells. Front Endocrinol (Lausanne). 2019;10:735. doi:10.3389/fendo.2019.00735
36. Wu M, Wu W, Ding J, Yang J. LINC01433/miR-2116-3p/MYC feedback loop promotes cell proliferation, migration, and the epithelial-mesenchymal transition in breast cancer. Cancer Biother Radiopharm. 2019;34(6):388–397. doi:10.1089/cbr.2019.2772
37. Checchetto V, Azzolini M, Peruzzo R, Capitanio P, Leanza L. Mitochondrial potassium channels in cell death. Biochem Biophys Res Commun. 2018;500(1):51–58. doi:10.1016/j.bbrc.2017.06.095
38. Chandra J, Samali A, Orrenius S. Triggering and modulation of apoptosis by oxidative stress. Free Radic Biol Med. 2000;29(3–4):323–333. doi:10.1016/s0891-5849(00)00302-6
39. Kadenbach B. Intrinsic and extrinsic uncoupling of oxidative phosphorylation. Biochim Biophys Acta. 2003;1604(2):77–94. doi:10.1016/s0005-2728(03)00027-6
40. Korshunov SS, Skulachev VP, Starkov AA. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997;416(1):15–18. doi:10.1016/s0014-5793(97)01159-9
41. Skulachev VP. Uncoupling: new approaches to an old problem of bioenergetics. Biochim Biophys Acta. 1998;1363(2):100–124. doi:10.1016/s0005-2728(97)00091-1
42. Gutman GA, Chandy KG, Grissmer S, et al. International Union of Pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol Rev. 2005;57(4):473–508. doi:10.1124/pr.57.4.10
43. Yellen G. The voltage-gated potassium channels and their relatives. Nature. 2002;419(6902):35–42. doi:10.1038/nature00978
44. DeCoursey TE, Chandy KG, Gupta S, Cahalan MD. Voltage-gated K+ channels in human T lymphocytes: a role in mitogenesis? Nature. 1984;307(5950):465–468. doi:10.1038/307465a0
45. Szabó I, Bock J, Grassmé H, et al. Mitochondrial potassium channel Kv1.3 mediates Bax-induced apoptosis in lymphocytes. Proc Natl Acad Sci U S A. 2008;105(39):14861–14866. doi:10.1073/pnas.0804236105
46. Szabò I, Soddemann M, Leanza L, Zoratti M, Gulbins E. Single-point mutations of a lysine residue change function of Bax and Bcl-xL expressed in Bax- and Bak-less mouse embryonic fibroblasts: novel insights into the molecular mechanisms of Bax-induced apoptosis. Cell Death Differ. 2011;18(3):427–438. doi:10.1038/cdd.2010.112
47. Leanza L, Biasutto L, Managò A, Gulbins E, Zoratti M, Szabò I. Intracellular ion channels and cancer. Front Physiol. 2013;4:227. doi:10.3389/fphys.2013.00227
48. Leanza L, Zoratti M, Gulbins E, Szabò I. Induction of apoptosis in macrophages via Kv1.3 and Kv1.5 potassium channels. Curr Med Chem. 2012;19(31):5394–5404. doi:10.2174/092986712803833281
49. Sancar A, Lindsey-Boltz LA, Unsal-Kaçmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85. doi:10.1146/annurev.biochem.73.011303.073723
50. Sancar A. DNA excision repair. Annu Rev Biochem. 1996;65:43–81. doi:10.1146/annurev.bi.65.070196.000355
51. Petit C, Sancar A. Nucleotide excision repair: from E. coli to man. Biochimie. 1999;81(1–2):15–25. doi:10.1016/s0300-9084(99)80034-0
52. Wood RD. Nucleotide excision repair in mammalian cells. J Biol Chem. 1997;272(38):23465–23468. doi:10.1074/jbc.272.38.23465
53. Wood RD. DNA damage recognition during nucleotide excision repair in mammalian cells. Biochimie. 1999;81(1–2):39–44. doi:10.1016/s0300-9084(99)80036-4
54. Mu D, Park CH, Matsunaga T, Hsu DS, Reardon JT, Sancar A. Reconstitution of human DNA repair excision nuclease in a highly defined system. J Biol Chem. 1995;270(6):2415–2418. doi:10.1074/jbc.270.6.2415
55. Mu D, Hsu DS, Sancar A. Reaction mechanism of human DNA repair excision nuclease. J Biol Chem. 1996;271(14):8285–8294. doi:10.1074/jbc.271.14.8285
56. Evans E, Moggs JG, Hwang JR, Egly JM, Wood RD. Mechanism of open complex and dual incision formation by human nucleotide excision repair factors. EMBO J. 1997;16(21):6559–6573. doi:10.1093/emboj/16.21.6559
57. Liu W, Li J, Zhao R, Lu Y, Huang P. The Uridine diphosphate (UDP)-glycosyltransferases (UGTs) superfamily: the role in tumor cell metabolism. Front Oncol. 2022;12:1088458. doi:10.3389/fonc.2022.1088458
58. Yasar U, Greenblatt DJ, Guillemette C, Court MH. Evidence for regulation of UDP-glucuronosyltransferase (UGT) 1A1 protein expression and activity via DNA methylation in healthy human livers. J Pharm Pharmacol. 2013;65(6):874–883. doi:10.1111/jphp.12053
59. Bélanger AS, Tojcic J, Harvey M, Guillemette C. Regulation of UGT1A1 and HNF1 transcription factor gene expression by DNA methylation in colon cancer cells. BMC Mol Biol. 2010;11:9. doi:10.1186/1471-2199-11-9
60. Luo W, Karpf AR, Deeb KK, et al. Epigenetic regulation of vitamin D 24-hydroxylase/CYP24A1 in human prostate cancer. Cancer Res. 2010;70(14):5953–5962. doi:10.1158/0008-5472.CAN-10-0617
61. Dluzen DF, Sun D, Salzberg AC, et al. Regulation of UDP-glucuronosyltransferase 1A1 expression and activity by microRNA 491-3p. J Pharmacol Exp Ther. 2014;348(3):465–477. doi:10.1124/jpet.113.210658
62. Tatsumi N, Tokumitsu S, Nakano M, Fukami T, Nakajima M. miR-141-3p commonly regulates human UGT1A isoforms via different mechanisms. Drug Metab Pharmacokinet. 2018;33(4):203–210. doi:10.1016/j.dmpk.2018.05.002
63. Papageorgiou I, Freytsis M, Court MH. Transcriptome association analysis identifies miR-375 as a major determinant of variable Acetaminophen glucuronidation by human liver. Biochem Pharmacol. 2016;117:78–87. doi:10.1016/j.bcp.2016.08.014
64. Tian M, Xia P, Gou X, Yan L, Yu H, Zhang X. CRISPR screen identified that UGT1A9 was required for bisphenols-induced mitochondria dyshomeostasis. Environ Res. 2022;205:112427. doi:10.1016/j.envres.2021.112427
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.