Back to Journals » Infection and Drug Resistance » Volume 12
Whole-genome characterization and resistance-associated substitutions in a new HCV genotype 1 subtype
Authors von Massow G, Garcia-Cehic D, Gregori J ![]() , Rodriguez-Frias F
, Rodriguez-Frias F ![]() , Macià MD, Escarda A, Esteban JI, Quer J
, Macià MD, Escarda A, Esteban JI, Quer J ![]()
Received 21 November 2018
Accepted for publication 7 March 2019
Published 24 April 2019 Volume 2019:12 Pages 947—955
DOI https://doi.org/10.2147/IDR.S195441
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Suresh Antony
Georg von Massow,1 Damir Garcia-Cehic,1,2 Josep Gregori,1–3 Francisco Rodriguez-Frias,2,4 María Dolores Macià,5 Ana Escarda,6 Juan Ignacio Esteban,1–2,7 Josep Quer1–2,7
1Liver Unit, Liver Diseases – Viral Hepatitis, Vall d’Hebron Institut of Research (VHIR) – Hospital Universitari Vall d’Hebron (HUVH), Barcelona, Spain; 2Centro de Investigación Biomédica en Red de Enfermedades Hepáticas y Digestivas (CIBERehd), Instituto de Salud Carlos III, Madrid, Spain; 3Roche Diagnostics S.L., Sant Cugat del Vallès, Barcelona, Spain; 4Biochemistry and Microbiology Department, VHIR-HUVH, Barcelona, Spain; 5Unidad de Microbiología Molecular, Servicio de Microbiología, Instituto de Investigación Sanitaria de les Illes Balears (IdISBa), Hospital Universitario Son Espases, Mallorca, Spain; 6Digestive Department, Hospital Universitario Son Espases, Mallorca, Spain; 7Medicine Department, Universitat Autònoma de Barcelona, Barcelona, Spain
Abstract: Hepatitis C virus (HCV) is a highly variable infectious agent, classified into 8 genotypes and 86 subtypes. Our laboratory has implemented an in-house developed high-resolution HCV subtyping method based on next-generation sequencing (NGS) for error-free classification of the virus using phylogenetic analysis and analysis of genetic distances in sequences from patient samples compared to reference sequences. During routine diagnostic, a sample from an Equatorial Guinea patient could not be classified into any of the existing subtypes. The whole genome was analyzed to confirm that the new isolate could be classified as a new HCV subtype. In addition, naturally occurring resistance-associated substitutions (RAS) were analyzed by NGS. Whole-genome analysis based on p-distances suggests that the sample belongs to a new HCV genotype 1 subtype. Several RAS in the NS3 (S122T, D168E and I170V) and NS5A protein (Q(1b)24K, R(1b)30Q and Y93L+Y93F) were found, which could limit the use of some inhibitors for treating this subtype. RAS studies of new subtypes are of great interest for tailoring treatment, as no data on treatment efficacy are reported. In our case, the patient has not yet been treated, and the RAS report will be used to design the most effective treatment.
Keywords: subtype, direct-acting antivirals, HCV, genotype 1
Background
Hepatitis C virus (HCV) is a blood-borne virus affecting an estimated 71 million people worldwide resulting in a global prevalence of 1%-3%.1
HCV is a single-stranded, positive-sense RNA virus containing approximately 9600 nucleotides and one open reading frame. Eight genotypes and 86 subtypes have been reported according to the International Committee on Taxonomy of Viruses (ICTV).2 A genotype is assigned when its genetic distance to all other genotypes is >30%, whereas subtypes are defined as having a genetic distance of >15% to other subtypes of the same genotype.3 In a single patient, HCV can be present as a complex mixture of viral variants having small genetic differences (1–3%), known as a quasispecies.4 Because of this, HCV has high genetic heterogeneity, which has important implications for the diagnosis and treatment of infected patients, and has impaired the development of an effective vaccine. However, with the recent advances in HCV therapy, direct-acting antiviral (DAA) treatment in chronically infected patients leads to a sustained virologic response (SVR) rate greater than 90%. However, drug resistance-associated substitutions (RAS) can emerge during this therapy and lead to treatment failure in 2%–10% of patients.5 In addition, RAS have been detected in some HCV subtypes as natural occurrences.6 Therefore, as HCV genotype and subtype in an infected patient may have an impact on the outcome of the treatment received, the therapy should be adjusted to these characteristics of the virus for it to be effective.7–9 In this scenario, accurate identification of HCV genotype and subtype is of paramount importance for proper patient management, especially in countries where pan-genotypic drugs are not available. Additionally, the degree of liver fibrosis and previous exposure to DAA and/or interferon, together with the infecting genotype, are of pivotal importance for the treatment strategy (including the duration of therapy).
A high-resolution HCV subtyping (HRCS) method based on deep sequencing has been implemented in our routine clinical laboratory.10 This method involves phylogenetic analysis and genetic distance analysis of the new template compared to the confirmed HCV subtypes.11,12 Because of the large number of reads obtained with this method (>2,000), new subtypes can be detected, as well as mixed infections in patients with more than 1 genotype or subtype.
Objectives
A genotype 1 sample from an untreated patient from Equatorial Guinea could not be classified during routine genotype-subtype analysis at the diagnostic laboratory. The aim of this study was to characterize the sample through complete analysis of the HCV genome sequence and investigation of baseline RAS.
Methods
The original serum sample was obtained from an HCV-infected woman from Equatorial Guinea diagnosed at the Molecular Microbiology Department of the Instituto de Investigación Sanitaria de les Illes Balears (IdISBa), Hospital Universitario Son Espases (Spain) in May 2017. Patient signed an informed consent to have the case details published. As this study has no legal implications for the institution, special approval from it was not required to publish the case details. As it was impossible to classify HCV subtype of this patient during the routine diagnostic procedure and because HCV sequencing for further classification is a requirement during the diagnostic and treatment procedure as the data sheet supports, a sample taken in June 2018 was delivered to our laboratory on dry ice for characterization using an HRCS technique, recently adapted to the MiSeq platform. HRCS is based on studying a short, highly variable region from the NS5B gene flanked with highly conserved primers.10
In this study, we used next-generation sequencing (NGS) to subtype HCV and characterize the RAS profile of the proteins NS3, NS5A and NS5B, targeted by DAA inhibitors. Sanger sequencing was used to sequence the whole genome.
The complete HCV genome was obtained by nested RT-PCR with 3 external primer pairs and 11 overlapping internal primer pairs (Table S1). The first externals (1, 2) and the internals (1, 2, 3, 5, 6, 7) were the primers reported by Ordeig et al in 2016.11 The remaining external and internal primer pairs were newly designed, using the closest subtype sequences (1l, 1h, 1c and 1e), identified during the subtyping test performed by HRCS after 200 bootstrapping cycles. All primers were numbered according to isolate H77 (accession number AF009606).13 The external fragments were amplified using the Transcriptor One Step RT-PCR Kit (#04655885001 Sigma, Roche), and for the internals, the FastStart High Fidelity PCR System, dNTPack (#04738292001 Sigma, Roche), adapting the various PCR programs to the lengths and temperatures of each specific primer pair. Due to the high variability of the region corresponding to internal fragment number 11, a random priming (#11034731001 Sigma, Roche) PCR was necessary to perform between the RT-PCR and the nested PCR with the specific primers, to obtain enough DNA for analysis. All amplifications were checked in 1.5% agarose gel to ensure that there was no contamination, and are purified in columns using the QIAquick PCR Purification Kit (#28106 QIAGEN). All fragments were Sanger-sequenced, and were assembled, verified and edited using the GeneDoc 2.7.000 (2006) software.
Twenty-two complete reference genomes, corresponding to all the confirmed genotype 1 (G1) subtypes (1a, 1b, 1c, 1d, 1e, 1g, 1h, 1i, 1j, 1k, 1l, 1m, 1n) and 8 previously unassigned isolates, were used to classify the sequence under study;2 phylogenetic trees and a heatmap were constructed. The sequence was analyzed using a sliding window procedure to investigate possible viral recombination of different subtypes. The scripts in R language used to obtain the figures are provided The scripts in R language used to obtain the figures and provided elsewhere.11,14 The R libraries included Biostrings, ape, stringr, RColorBrewer and psych. MUSCLE was used for the required multiple alignments.15 To characterize any naturally occurring baseline RAS in the putative new subtype taking into account that any isolate is composed by a population of sequences,4 four new internal primers pairs were designed, corresponding to the three DAA-targeted regions of the HCV genome (1 NS3, 1 NS5A and 2 NS5B); using this time the isolate's full genome sequence, obtained with Sanger ssequencing, was used as reference (Table S1). To amplify these new fragments, we used the products of the external RT-PCRs obtained previously on the FastStart High Fidelity PCR System, dNTPack (#04738292001 Sigma, Roche), adapting the thermocycler programs to the characteristics of each different primer pair. Again all fragments were checked and purified on 1.5% agarose gel, and were deep-sequenced using the MiSeq Reagent Kit v3 (600 cycle) (#MS-102–3003, Illumina) according to a previously described method.16 Briefly, each nested-PCR purified product was pooled and normalized to 4nM, and appropriate volumes of each pool were added to the final library, which was quantified by LightCycler480 (Kapa Library Quantification Kit, KapaBiosystems, Roche, Pleasanton, CA, USA). The last dilution of the library was prepared and mixed with an internal DNA control (PhiX control V3, Illumina, San Diego, CA) before sequencing on the MiSeq platform using the MiSeq Reagent Kit V3 (Illumina, San Diego, CA). Data management includes a bioinformatics haplotype-centric procedure to exclude full reads that did not meet minimum quality requirements.16 The final steps included demultiplexing by specific primers to obtain a fasta file by region. Reads were then collapsed into haplotypes with corresponding frequencies. Haplotypes were aligned with the reference sequence, and haplotypes containing more than two indeterminations, three gaps, or 99 differences were also discarded. Accepted indeterminations and gaps were repaired as per the contents of the dominant haplotype. The yield of this process was above 90% in all cases. Then, reads were translated to amino acids, and the intersection between forward and reverse haplotypes with abundances not below 0.2% was performed. The yield of this process has been found in the range of 45%–60%. Based on the controls, all amino acid variants by site or haplotype at 1% or above are reported. The global yield is 15%–30% of raw reads.
Results
HCV subtyping of a short fragment of the NS5B gene using a high-resolution method based on deep sequencing (HRCS) revealed that the all reads belonged to the same subtype and that this subtype was included in the genotype 1 cluster (Figure S1). However, it could not be classified into any of the genotype 1 subtypes, with subtype 1h being the closest. The next step was to design primers to sequence and characterize the whole genome.
Phylogenetic analysis and pairwise comparison of full-length genomes
The full sequence (9182nt) deposited in the GenBank database with accession number “MH921830” includes the almost complete coding region and a partial sequence from 5ʹ UTR.
Phylogenetic analysis of the complete new genome with the confirmed subtypes of all 8 genotypes showed that our sequence clustered with G1 (Figure 1). Furthermore, the new genome did not cluster with any of the confirmed G1 subtypes when compared to the confirmed subtype reference sequences (Figure 2).
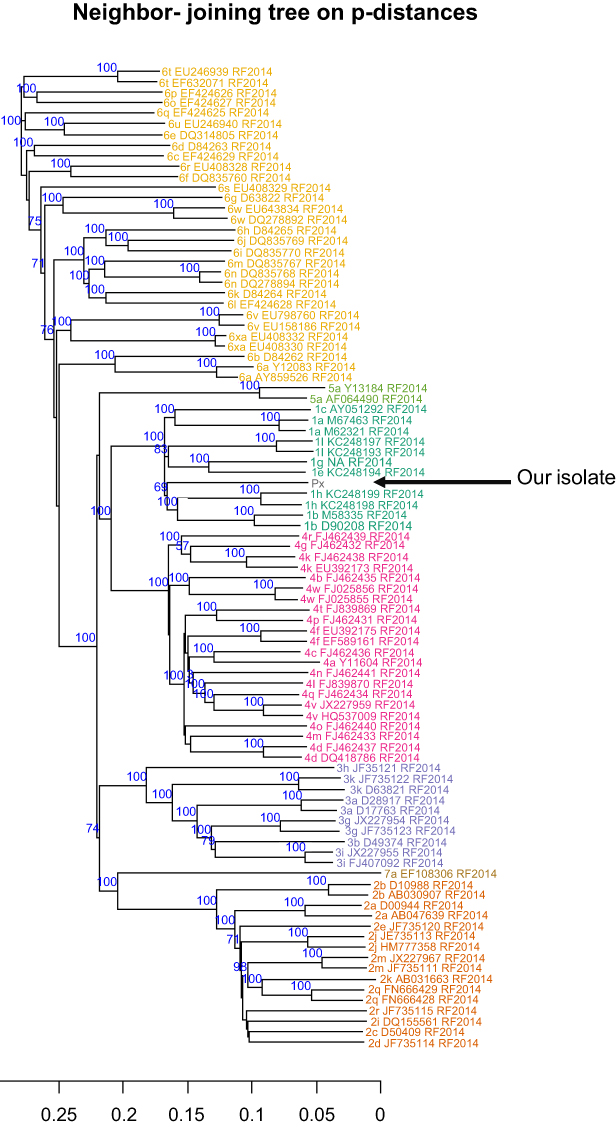 | Figure 1 Phylogenetic tree of the entire sequences, including reference from accepted genotypes2 (shown in different colors) and our sequence (Px). Trees were generated using neighbor-joining based on the p-distances. Only those bootstrap-values greater than 60 are shown. |
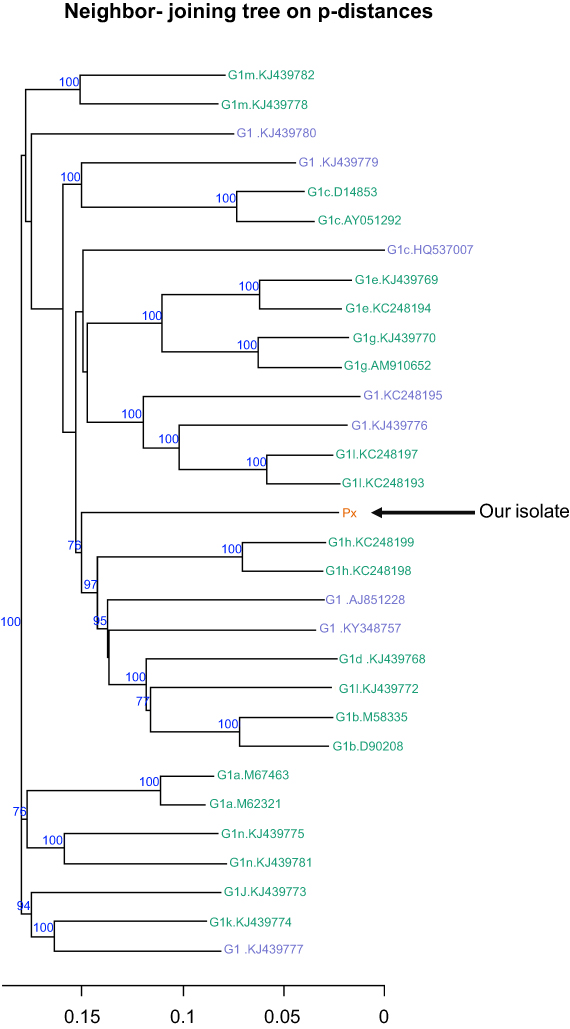 | Figure 2 Phylogenetic tree of entire genomes from all 13 subtypes of genotype 1, and including 8 unassigned isolates (green) and our Px sequence (orange). The tree was generated using neighbor-joining based on p-distances. Only those bootstrap-values greater than 60 are shown. |
The heatmap on the matrix of p-distances further demonstrated that our sequence did not cluster with any confirmed subtype or unassigned isolate (Figure 3). The shortest genetic p-distance measured (0.2447) was to a still unassigned isolate “KY348757”,11 whereas the shortest p-distance to a confirmed subtype was 0.2495 with subtype 1k (Table S2). Therefore, our sequence could not be classified into any of the existing G1 subtypes and could not be grouped with any other unassigned isolate.
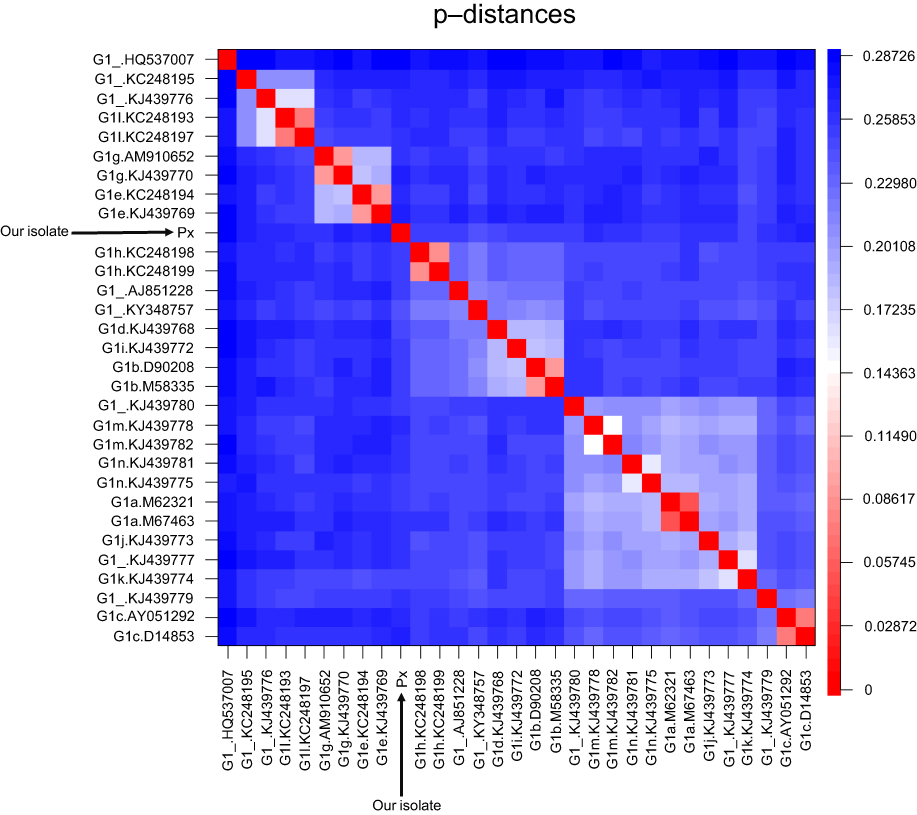 | Figure 3 Heatmap visualizing the p-distances between all G1 reference sequences and the new isolate (Px). The scale on the right indicates the p-distance according to color, with red representing a short and blue a long p-distance. The clusters shown in light colors indicate the grouping of subtypes based on their similarity. The new isolate did not cluster with any subtype. |
Recombination: sliding window analysis
Sliding window analysis was conducted to exclude the possibility of viral recombination between HCV genotype 1 subtypes (Figure 4). The p-distance of our sequence to the nearest G1 subtype at any given position was higher than the minimum p-distance between all subtypes.
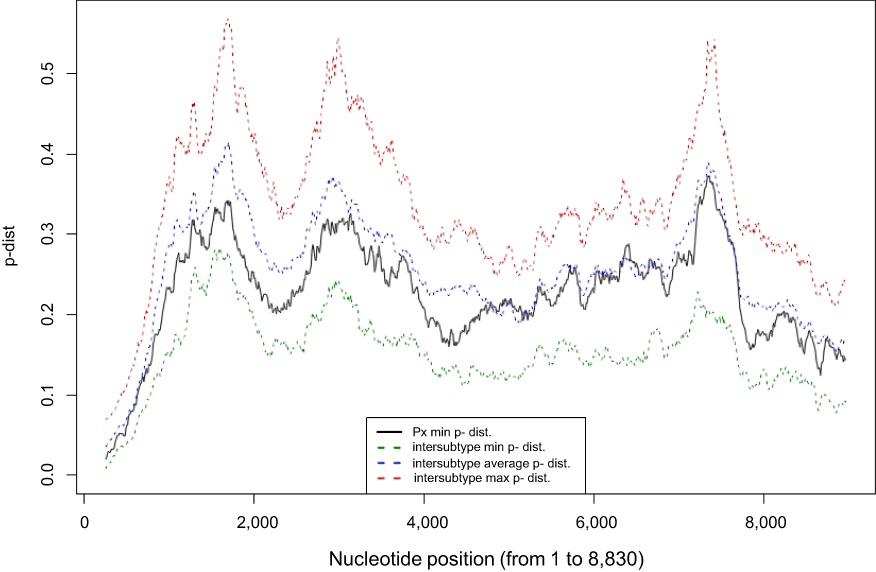 | Figure 4 Visual representation of the sliding window analysis (500-bp, by 10-bp steps). The minimum p-distance between the new isolate (Px) and any reference sequence of accepted subtypes for each 500-bp window is shown in black. The green dotted line represents the minimum p-distance between the closest sequences belonging to the various subtypes. The blue dotted line indicates the average p-distance of all accepted G1 subtypes for each window. The red dotted line represents the maximum p-distance between the farthest sequences belonging to the accepted subtypes for each window. |
Resistance-associated substitutions (RAS)
To characterize the viral genome, RAS were analyzed for the NS3, NS5A and NS5B proteins by deep sequencing using the Illumina MiSeq sequencing platform, to identify resistances to various DAAs between our isolate and the wild-type replicons 1a and 1b, used as references (Table 1). RAS S122T, D168E and I170V were found in NS3 in almost all genomes reported after NGS, and the substitutions Y93F/L and R30Q in NS5A. Interestingly, variability was found in the Y93 residue of NS5A, in which 72% of viral genomes were carrying amino acid 93L (leucine) and 28% were carrying 93F (phenylalanine). The amino acid composition of our isolate in the main RAS positions in the NS3 protein was 80Q, 122T, 155R, 156A, 168E and 170V; and in the NS5A protein 24K, 28L, 30Q, 58P, 62Q and 93L/F (Table 1). Later, we discuss the scope of each mutation and the combination of them in the same genome. No RAS were detected in the NS5B protein.
 | Table 1 Mean fold change in resistance compared with wild type, found in G1a and G1b (in brackets)7,8,9,18 |
Discussion
This report describes and characterizes the full-length genome of a putative new HCV subtype identified during routine HCV classification of a serum sample from an Equatorial Guinea patient. Commercial methods were unable to genotype the viral isolate and it was then subjected to high-resolution HCV subtyping, a methodology based on NGS10 adapted to the MiSeq platform, and phylogenetic analysis of a short region of the NS5B gene, confirming that the isolate could be member of a new subtype.
The complete genome sequence was then obtained after PCR and Sanger sequencing and uploaded to GenBank (MH921830). Through phylogenetic analysis, genetic p-distance calculations by pairs, and heat-mapping, we found that our sequence (Px) had a genetic distance greater that 15% to the closest confirmed reference sequence, which belonged to G1k (Table S2). Interestingly, the closest sequence was a subtype-unassigned sequence (KY348757), also isolated from an Equatorial Guinean patient. This suggests that G1 has a long-lasting presence in that country, which has facilitated divergence of this genotype into new, unclassified subtypes. Additionally, sliding window analysis provided evidence that the sequence did not result from recombination of different G1 subtypes. However, it will remain as an unclassified lineage until a related sequence is discovered, in keeping with the recommendations for a unified nomenclature system.2
Deep sequencing analysis was used to trace the RAS profile of the targeted NS3, NS5A, and NS5B proteins, to identify with maximum reliability minor mutations, exactly report the frequency at which each mutation was present in the viral population, and determine whether two mutations were combined in the same viral genome, particularly minor mutations. Interestingly, all genomes isolated from this new isolate were carrying the NS3 mutations S122T and D168E.17 S122T has been found to be a polymorphism of genotypes 4 and 6, but not genotype 1. S122T and D168E have been associated with resistance to antivirals, especially in subtypes 1a and 1b, and D168E additionally in subtypes 2a, 2b, 4a and 6,7,18 suggesting that this subtype maybe basically resistant to NS3 inhibitors.7,8,9,18 NS3-D168E is especially relevant, as in subtypes 1a and 1b, it can make the virus resistant to almost all NS3 inhibitors, including vaniprevir (used for genotype 1 in many countries) and voxilaprevir (NS3 inhibitor present in the pan-genotypic triple salvage treatment with velpatasvir and sofosbuvir).19 The new subtype carries the amino acid 170V which is considered wild type for many subtypes, but we cannot exclude it as a RAS because it has been described for subtype 1a.18 Variability was found in the Y93 residue of NS5A in our new subtype (Y93F/L). Y93F is reported to confer resistance in G1a to ledipasvir and ombitasvir, and Y93L to ombitasvir and velpatasvir in cell cultures as well as in treatment-failing patients.18 Lysine (K) and glutamine (Q) in residues 24 and 30, respectively, have been found to be wild type in G1a17 However, in genotype 1b, Q24K has been reported to confer resistance to daclatasvir and elbasvir, and R30Q to velpatasvir.18 Cell culture studies are needed to determine whether these NS5A mutations (93F/L and 30Q) and some of the NS3 mutations (170V) can be considered RAS for the new subtype. The level of resistance to an inhibitor depends on several conditions: the genetic barrier to resistance of the HCV region and the drug, type of RAS, freguency at which the RAS is present in the viral isolate, and presence of RAS combinations in the same viral particle, all of which can significantly increase resistance to some DAA treatments. In the study of combinations of RAS residues in the same genome, none of the combinations found could be associated with a fold-change increase in resistance to the inhibitors.18
Taking all this information together, the RAS report suggests that our naïve patient should be treated with a combination of an NS5B inhibitor such as sofosbuvir, an NS3 inhibitor such as glecaprevir or voxilaprevir, and/or the NS5A inhibitor pibrentasvir. As any treatment should include a combination of two or even three inhibitors with or without ribavirin, our patient could be treated combining glecaprevir+pibrentasvir(Maviret)+RBV or using voxilaprevir+velpatasvir+sofosbuvir+RBV. In the latter option, velpatasvir (which is included in the Vosevi commercial treatment) will probably not work, but the combination of voxilaprevir plus sofosbuvir with ribavirin should be effective. Inclusion of ribavirin is recommended, if possible, due to the presence of several baseline RAS.
In summary, high HCV variability among genotype 1 subtypes has been observed in Equatorial Guinean patients. In general, HCV Isolates that cannot be assigned by means of commercially available techniques should be analyzed using a high-throughput technology, with the special recommendation to use NGS and phylogenetic analysis for the NS5B region, as mixed infections and minority mutants can be evidenced with these techniques, Further characterization of the whole genome is interesting for epidemiological reasons and study of RAS mutations using a high-throughput technique is useful to identify whether the new subtype is naturally harboring resistance-associated mutations, which will be of help to prescribe the most effective antiviral treatment.
Acknowledgments
This study was supported by the Spanish Ministry of Health, Consumption and Social Welfare grant name “Plan Estratégico Nacional contra la Hepatitis C”. This study was funded by Instituto de Salud Carlos III grants, PI15/00856, PI15/00829 and PI16/00337, and cofinanced by CIBERehd (Consorcio Centro de Investigación en Red de Enfermedades Hepaticas y Digestivas), which is funded by Instituto de Salud Carlos III and Centro para el Desarrollo Tecnológico Industrial-CDTI from the Spanish Ministry of Economy and Business, grant number, IDI-20151125.
The authors thank Celine Cavallo for English language support and helpful editing suggestions.
Disclosure
The funder “Roche Diagnostics S.L.” provided support in the form of salaries for one author (Josep Gregori), but the company did not have any additional role in the study design, data collection and analysis, decision to publish or preparation of the manuscript. The other authors report no conflicts of interest in this work.
References
1.
2. Smith D. HCV classification. A web resource to manage the classifcation and genotype and subtype assignements of hepatits virus; 2018. Available from:
3. Smith DB, Bukh J, Kuiken C, et al. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: updated criteria and assignment web resource. Hepatology. 2014;59:318–327. doi:10.1002/hep.26744
4. Martell M, Esteban JI, Quer J, et al. Hepatitis C virus (HCV) circulates as a population of different but closely related genomes: quasispecies nature of HCV genome distribution. J Virol. 1992;66:3225–3229.
5. Vermehren J, Sarrazin C. The role of resistance in HCV treatment. Best Pract Res Clin Gastroenterol. 2012;26:487–503. doi:10.1016/j.bpg.2012.09.011
6. Eltahla AA, Leung P, Pirozyan MR, et al. Dynamic evolution of hepatitis C virus resistance-associated substitutions in the absence of antiviral treatment. Sci Rep. 2017;7:41719. doi:10.1038/srep41719
7.
8. Lontok E, Harrington P, Howe A, et al. Hepatitis C virus drug resistance-associated substitutions: state of the art summary. Hepatology. 2015;62:1623–1632. doi:10.1002/hep.27934
9. Sarrazin C. The importance of resistance to direct antiviral drugs in HCV infection in clinical practice. J Hepatol. 2016;64:486–504. doi:10.1016/j.jhep.2015.09.011
10. Quer J, Gregori J, Rodriguez-Frias F, et al. High-resolution hepatitis C virus subtyping using NS5B deep sequencing and phylogeny, an alternative to current methods. J Clin Microbiol. 2015;53:219–226. doi:10.1128/JCM.02093-14
11. Ordeig L, Garcia-Cehic D, Gregori J, et al. New hepatitis C virus genotype 1 subtype naturally harbouring resistance-associated mutations to NS5A inhibitors. J Gen Virol. 2018;99:97–102. doi:10.1099/jgv.0.000996
12. Rodriguez-Frias F, Nieto-Aponte L, Gregori J, et al. High HCV subtype heterogeneity in a chronically infected general population revealed by high-resolution hepatitis C virus subtyping. Clin Microbiol Infect. 2017;23:775e1–775e6. doi:10.1016/j.cmi.2017.02.007
13. Kuiken C, Combet C, Bukh J, et al. A comprehensive system for consistent numbering of HCV sequences, proteins and epitopes. Hepatology. 2006;44:1355–1361. doi:10.1002/hep.21377
14.
15. Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113. doi:10.1186/1471-2105-5-113
16. Perales C, Chen Q, Soria ME, et al. Baseline hepatitis C virus resistance-associated substitutions present at frequencies lower than 15% may be clinically significant. Infect Drug Res. 2018;11:2207–2210.
17. Palanisamy N, Kalaghatgi P, Akaberi D, et al. Worldwide prevalence of baseline resistance-associated polymorphisms and resistance mutations in HCV against current direct-acting antivirals. Antivir Ther. 2018;23:485–493. doi:10.3851/IMP3237
18. Sorbo MC, Cento V, Di Maio V, et al. Hepatitis C virus drug resistance associated substitutions and their clinical relevance: update 2018. Drug Resist Updat. 2018;37:17–39. doi:10.1016/j.drup.2018.01.004
19. Lawitz E, Yang JC, Stamm LM, et al. Characterization of HCV resistance from a 3-day monotherapy study of voxilaprevir, a novel pangenotypic NS3/4A protease inhibitor. Antivir Ther. 2018;23:325–334. doi:10.3851/IMP3202
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.