Back to Journals » Infection and Drug Resistance » Volume 12
Whole genome analysis reveals new insights into the molecular characteristics of Clostridioides difficile NAP1/BI/027/ST1 clinical isolates in the People’s Republic of China
Authors Lv T, Chen Y, Guo L, Xu Q, Gu S, Shen P, Quan J, Fang Y ![]() , Chen L, Gui Q, Ye G, Li L
, Chen L, Gui Q, Ye G, Li L ![]()
Received 28 January 2019
Accepted for publication 8 May 2019
Published 1 July 2019 Volume 2019:12 Pages 1783—1794
DOI https://doi.org/10.2147/IDR.S203238
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Suresh Antony
Tao Lv,1,* Yunbo Chen1,* Lihua Guo,1 Qiaomai Xu,1 Silan Gu,1 Ping Shen,1 Jiazheng Quan,1 Yunhui Fang,1 Lifeng Chen,2 Qiaodi Gui,3 Guangyong Ye,4 Lanjuan Li1
1State Key Laboratory for Diagnosis and Treatment of Infectious Diseases, Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases, The First Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, Zhejiang, People’s Republic of China; 2Medical Engineering Department, The First Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, Zhejiang, People’s Republic of China; 3Department of Clinical Laboratory, Shaanxi Provincial People’s Hospital, Xi’an, Shaanxi, People’s Republic of China; 4Women’s Hospital, School of Medicine, Zhejiang University, Hangzhou, Zhejiang, People’s Republic of China
*These authors contributed equally to this work
Background: The epidemic new strain NAP1/BI/027/ST-1 of Clostridioides difficile (C. difficile) causes more severe coliti and a higher mortality rate than historical strains. However, C. difficile NAP1/BI/027/ST-1 (C. difficile RT027) infections have been rarely reported in Asia, particularly in China.
Purpose: The objective of this study was to strengthen the understanding of the molecular characterizations of C. difficile RT027 in China.
Patients and methods: Two C. difficile NAP1/BI/027/ST-1 were detected from two patients, and no additional isolates were found. Whole genome sequencing (WGS) was used to characterize two C. difficile RT027 isolates and control strain CD6 (from Hong Kong), and comparative genomic analysis was performed to compare genomic differences between seven isolates from Mainland China, CD6, and 10 isolates from North America and Europe.
Results: The comparative genomic analysis revealed that isolates obtained from Mainlan China were outside of the two epidemic lineages, FQR1 and FQR2, and might have decreased virulence and transmissibility for outbreak. Furthermore, unique SNP mutations were detected in isolates obtained from Mainland China, which may affect the biological function of C. difficile.
Conclusion: We speculate that C. difficile RT027 isolates in Mainland China may have different features, compared to those in North America and Europe.
Keywords: People’s Republic of China, multilocus sequence typing, Clostridioides difficile infection, whole genome sequencing
Introduction
Clostridioides difficile is widely recognized as an important diarrheal pathogen in North America and Europe. The mechanisms by which C. difficile causes severe disease in one individual, but silently colonizes another, are multifactorial but primarily associated with the use of antibiotics and other factors involving immune status, age, microbiota composition, and C. difficile strain types.1 A hypervirulent strain of C. difficile, designated as NAP1/BI/027 or ST-1, caused more severe colitis and higher mortality than other types in North America and Europe since 2003.2 Phylogeographic analysis of the whole genome sequencing data identified two major genetic lineages (FQR1 and FQR2) in C. difficile RT027, with independently acquired, identical mutations that convey high-level fluoroquinolone resistance (FQR), and traced their distinct patterns of global spread. The FQR1 lineage emerged in North America in 2001 and spread to South Korea and Switzerland, whereas the FQR2 lineage originated in 2003 in North America, but subsequently spread to the United Kingdom, continental Europe and Australia.3
The phylogenetic analysis has demonstrated that C. difficile is a genetically diverse lineage.4 Many different typing or fingerprinting methods have been applied to type and characterize C. difficile strains during an outbreak or infection, including PCR ribotyping and pulsed field gel electrophoresis (PFGE), among which PCR ribotyping has evolved as the standard typing method in Europe.5 Multilocus sequence typing (MLST) has gradually become popular in the molecular typing of multiple loci, which characterizes allelic variations of housekeeping genes, and assigns each particular allelic profile to a sequence type (ST).6
The use of whole-genome sequencing (WGS) has provided evidence for a higher degree of C. difficile strain diversity than previously acknowledged.7 WGS can be used to compare single nucleotide variants (SNVs) between isolates across the nonrepetitive core genome, which accounts for 80% of the 4.3 million-base pair C. difficile strain 630 reference genome.8–10 WGS also offers additional benefits, including the reconstruction of long-term evolutionary histories and the in silico determination of virulence factors and antimicrobial resistance.11
C. difficile RT027 first appeared in Mainland China in 2014, and subsequently caused sporadic cases, including a small healthcare-associated outbreak.12–14 However, it has not replaced the other epidemic clones, such as ST54 (RT012), as the predominant clone across regions or the country.15–17 This was quite different from North America and Europe, where hypervirulent C. difficile RT027 displaced endemic strains after it occurred. For example, RT027 has disseminated with a clear shift throughout Europe from UK to the Eastern Europe.18,19 On the other hand, some countries have successfully controlled its spread and decreased its prevalence after taking measures.20 The molecular epidemiology of C. difficile RT027 isolates from Mainland China may be different from other regions of the world. In the present study, we identified two clinical isolates of C. difficile, which were confirmed as C. difficile RT 027 by PCR ribotyping, NAP1 by PFGE and ST-1 by MLST and characterized by WGS. Then, comparative genomic analysis was performed to compare genomic differences between seven isolates from Mainland China and ten isolates from North America and Europe. This study will further our understanding of the molecular characterizations of C. difficile NAP1/BI/027/ST-1 in China.
Methods
Ethics
The present study is a retrospective study and ethical approval was given by the Medical Ethics Committee of The First Affiliated Hospital, School of Medicine, Zhejiang University (Reference Number: 2018–1020). None of the test results were used to alter individual care.
Study setting and general study design
This epidemiological study was conducted at the First Affiliated Hospital, School of Medicine, Zhejiang University, which is a 2,500-bed tertiary teaching hospital in Hangzhou, Zhejiang, China. Since September 2009, diagnosis of C. difficile infection (CDI) was carried out according to the clinician requirements using C. difficile culture and toxin gene tests.
Diarrhea was defined as three or more loose stools within 24 hours. Inpatients with diarrhea, whose stool samples were positive for both C. difficile culture and toxin gene tests and without evidence of another cause of diarrhea, were diagnosed as CDI. HA-CDI (Hospital-associated C. difficile infection) was defined as a patient with CDI symptoms onset more than 48 hours after admission to a healthcare-facility or with onset of symptoms in the community within 12 weeks following discharge from a health-care facility.21 CDI outbreak was defined as >2 isolates of the same genotype detected less than 7 days apart in one hospital either with onset of symptoms on the same ward, or accompanied by an increased CDI monthly incidence within the hospital.
Isolation and detection of C. difficile toxins
Stool samples were cultured on cycloserine-cefoxitin-fructose agar (CCFA) in an atmosphere composed of 80% N2, 10% H2 and 10% CO2 at 37°C for 48 hours. The colonies were identified through the typical morphology and odor of C. difficile, and were further confirmed by Matrix-Assisted Laser Desorption-Ionization mass spectrometry (MALDI-MS, FlexControl 3.3-Microflex). Bacterial cultures from both stool specimens were negative for common diarrhea pathogens, including Salmonellaspp., Shigellaspp., Vibriospp., Staphylococcus aureus and Escherichia coli.
Genomic DNA was isolated from the two isolates using a QIAamp DNA Mini Kit (Qiagen NV, Hilden, Germany). The presence of tcdA and tcdB genes in two isolates was detected by PCR, as previously described.22 The presence of binary toxin genes cdtA, and cdtB were detected, as described by Stubbs et al.23
A C. difficile NAP1/BI/027 strain, designated as CD6, which was kindly provided by Dr K. Y. Yuen (Department of Microbiology, Queen Mary Hospital, Hong Kong) was used as the positive control.24
Capillary gel electrophoresis (CGE)-based PCR ribotyping and pulsed-field gel electrophoresis typing
The PCR-ribotyping of two isolates targeted the intergenic spacer region of C. difficile between the 16S and 23S rRNA genes.25 The PCR products were analyzed by CGE. Strain CD6 was used as an internal control for PCR ribotyping. PFGE was performed, as previously described.26
Multilocus sequence typing
MLST was performed by amplifying and sequencing seven housekeeping genes (adk, atpA, dxr, glyA, recA, sodA and tpi), according to previously described protocols.27 MLST sequences were uploaded to the PubMLST database (http://pubmlst.org/cdifficile/) to determine the individual ST from the allelic combination.
Genome sequencing, assembly and annotation
The sequencing-quality genomic DNA of the three strains was extracted using a DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany), according to manufacturer’s instructions, and quantitated by Qubit 2.0. Whole-genome sequencing was performed on the Illumina HiSeq 2,500 System (Illumina, San Diego, CA, USA). Before performing the assembly, the sequencing reads were filtered to remove reads (<Q20) with low-quality base calls or reads that were similar to Illumina adapters. Subsequently, the raw reads of C. difficile RT027 strains were trimmed and mapped to the genome sequence of the C. difficile RT027 reference strain R20291 using BWA-SW to detect the genomic single nucleotide polymorphisms (SNPs) using a Genome Analysis Toolkit (GATK).28,29 Finally, the genomes were submitted to the Rapid Annotation using Subsystem Technology (RAST) servers (http://rast.nmpdr.org/) for annotations.30
Phylogenetic and comparative genomic analysis
In order to perform the comparative genomic analysis with previously published genome data, sequencing reads from representative C. difficile RT027 strains were downloaded from the National Center for Biotechnology Information (NCBI) genome database. As shown in Table 1, five strains reported by Jia et al14 were the first to cause a ward transmission in a hospital in Mainland China, whereas ten strains from the report of He et al3 were the node at the base of the star-like topology, and were associated with severe outbreaks in North America and Europe. SNPs were used to reconstruct the phylogenetic tree and maximum-likelihood tree by RAxML. Meanwhile, the heat-map reconstruction and analysis were performed using Roary Matrix for further comparative analysis.
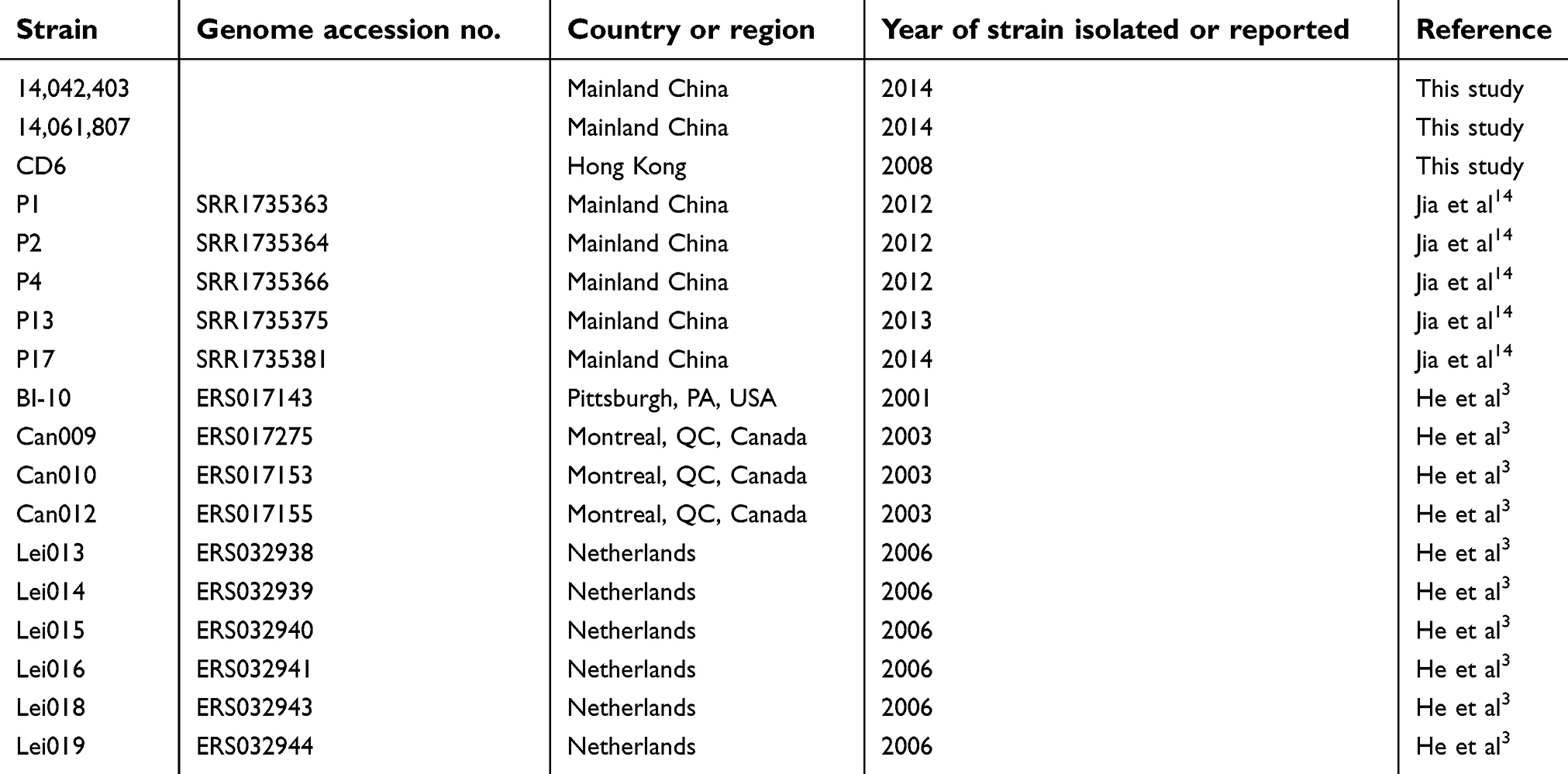 |
Table 1 Information about the isolates in this study |
Antimicrobial susceptibility
Antimicrobial susceptibility testing was performed on Brucella agar plates containing 1 mg/L of vitamin K, 5 mg/L of hemin and 5% sheep red blood cells with the eight antimicrobial agents: metronidazole, vancomycin, clindamycin, erythromycin, linezolid, moxifloxacin, levofloxacin, and rifampicin. The minimum inhibitory concentration (MIC) breakpoints used were 8 mg/L for erythromycin, clindamycin, and the fluoroquinolones, and 32 mg/L for metronidazole, in accordance with Clinical Laboratory Stsandards Institute (CLSI) interpretative categories approved for anaerobic bacteria.31 For vancomycin, linezolid and rifampicin, in which the breakpoints were not available in CLSI documents, >2 mg/L, >4 mg/L and >32 mg/L were used as the breakpoints, respectively, according to the European Committee on Antimicrobial Susceptibility Testing (EUCAST) (http://www.eucast.org/clinical _breakpoints/). C. difficile ATCC 70,057 was used as a control.
Results
Epidemiological analysis and characterization of C. difficile isolates
In total, 421 (8.1%) non-duplicate toxigenic C. difficile isolates were identified from 5,171 patients suffering from diarrhea (parts data has been published) during the period of study (by the end of August 31, 2017).32 We have identified 33 STs among these isolates. The most prevalent STs were ST-54, ST-35, ST-37, and ST-3, followed by ST-2, ST-81, and ST-8. However, STs with binary toxin genes, such as ST-11, ST-5, and ST-201, were not common.32
All positive strains of C. difficile isolated from stool samples were retained in our laboratory, from which two clinical strains were tested positive for both toxin A and toxin B, and binary toxin genes by PCR assay, which were further confirmed as ST-1 by MLST. Compared to CD6, these two isolates were assigned as C. difficile RT027 by PCR ribotyping and NAP1 type by PFGE (Figure 1A and B).
 |
Figure 1 (A) PCR ribotyping of Clostridioides difficile (C. difficile) PCR ribotype 027 reference strains (A-01) and two clinical isolates (A-02 and B-03) identified in the present study. (B) Pulsed-field gel electrophoresis of C. difficile PCR ribotype 027 reference strains (A-01) and two clinical isolates (A-02 and B-03). Marker: Xba 1 was used for the reference marker, Salmonella H9812. Line A-01=CD06; Line A-02=14,042,403; Line B-03=14,061,807. |
C. difficile strain 14,042,403 was isolated from the stool sample from a 71-year-old man with diarrhea during the hospitalization. The patient was treated with antibiotics for intra-abdominal infection before he was diagnosed as CDI. C. difficile strain 14,061,807 was isolated from an 83-year-old hospitalized man in the same hospital. This patient was also treated with different antibiotics for pulmonary infection and continued to suffer from persistent fever with persistent diarrhea.
General genomic features
After filtering to remove low quality and adapter contamination reads, 1,656,804, 5,048,333 and 5,248,180 paired reads were produced for CD6, 14,042,403 and 14,061,807 strains. Following the assembly, the average genome coverage was approximately 290× and the genome assembly produced 80,110,116 contigs, respectively, with an average GC content between 29.2% and 32.8%. The features of these three strains, including the assembly stats and gene content, are shown in Table S1.
Phylogenetic analyses
In order to further understand the phylogenetic relationship between the two isolates in the present study, paired-end DNA sequencing reads were mapped to the reference genomes of R20291. The two strains were highly similar with a difference of only four SNPs between them. Among these four different SNPs as shown in Figure 2A, two were in the non-coding regions, and two were in the coding regions (conserved hypothetical protein and ABC transporter ATP-binding protein). The phylogenetic tree revealed that these two strains were closely clustered into the same predominant evolutionary branch, which is similar to the other isolates obtained from Mainland China. However, the maximum-likelihood tree revealed different group patterns, in which two isolates were separated into the same clade, but were far away from the other strains (Figure 2B).
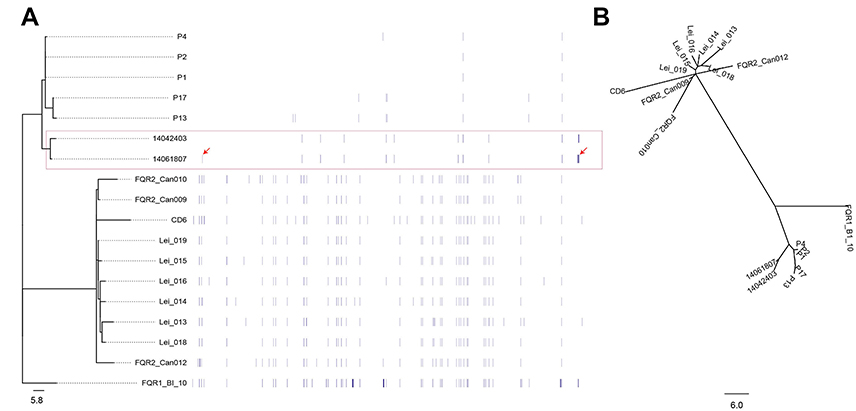 |
Figure 2 Patterns of molecular variation in 18 strains of C. difficile RT027, including the three isolates in this study, the other five strains from the People's Republic of China and the ten from North America and Europe. (A) Annotation by RAST revealed several SNPs, which were significantly different between the strains obtained from Mainland China and North America and Europe. The symbol arrow indicates the four different SNPs between two isolates in this study. (B) Maximum-likelihood phylogenetic analysis of based on core genome SNPs which were grouped into three different clades. The previously described strains of fluoroquinolone-resistant C. difficile RT027 were clustered into two clades (FQR1 and FQR 2), whereas the isolates from Mainland China were classified into a third clade. |
In order to investigate the difference and relationship between C. difficile RT027 strains isolated from Mainland China and those obtained from North America and Europe, all WGS data of available C. difficile RT027 from Mainland China (by the end of October 2017) and ten isolates from North America and Europe were downloaded to align with the genome of the C. difficile RT027 strain R20291. A total of 628 SNPs were identified and grouped into three different clades (Figure 2B). The previously described strains of fluoroquinolone-resistant C. difficile RT027 were clustered into two clades (FQR1 and FQR 2), whereas the isolates from Mainland China were classified into a third clade.3 However, CD6, the strain isolated from Hong Kong, was grouped into the FQR2 lineage. Annotation by RAST revealed several SNPs, which were significantly different between the strains obtained from Mainland China and North America and Europe. These were located at significant protein-coding sites, including the conserved hypothetical protein, putative membrane protein, putative exported protein and two-component response regulator (Table S2). In addition, a total of 3,944 genes were identified (Figure 3), including 3,397 common core genes without soft core genes, 295 different shell genes, and 252 different cloud genes. The annotation for identification of the predicted function of proteins coded by different genes revealed that 12 kinds of proteins were only encoded by unique genes in the seven strains obtained from Mainland China, including excisionase and transposase from transposon Tn916 (Table S3). Bifunctional AAC/APH, which belongs to the aminoglycoside phosphotransferase family, and ribosomal-protein-alanine N-acetyltransferase, which acetylates the N-terminal alanine residues of specific ribosomal proteins, were the unique proteins in Chinese C. difficile RT027 isolates.
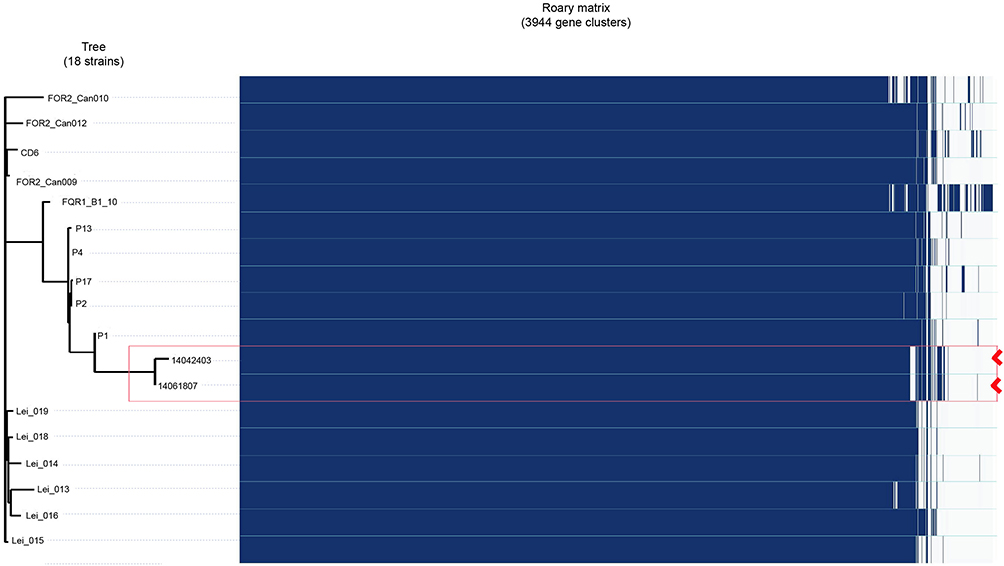 |
Figure 3 Heatmap reconstruction of 18 strains resulting from SNPs. A total of 3,944 genes were identified, including 3,397 common core genes without soft core genes, 295 different shell genes, and 252 different cloud genes. Two isolates in this study are labeled with point brackets. |
Antimicrobial susceptibility
The antibiotic susceptibility patterns of C. difficile isolates are presented in Table 2. Strain 14,042,403 and 14,061,807 were resistant to most of the tested antibiotics, including fluoroquinolones with high MICs, but susceptible to metronidazole, vancomycin and linezolid.
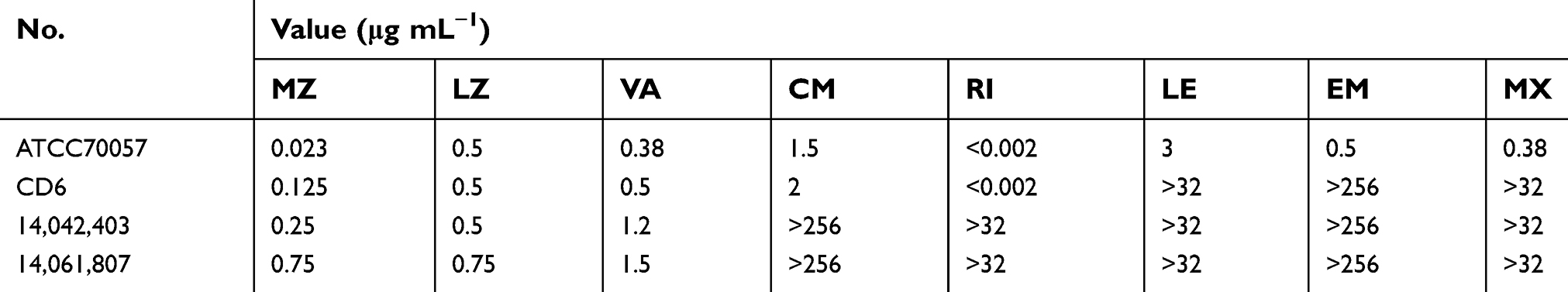 |
Table 2 Antimicrobial susceptibility of the four C. difficile isolates |
Discussion
In 2014, two strains of C. difficile NAP1/BI/027/ST-1 were successfully isolated from a tertiary hospital in East China. To the best of our knowledge, this is the first report of C. difficile NAP1/BI/027/ST-1 infection in this region.
C. difficile RT027 infection is not common in Mainland China, even in almost all regions of Asia. In the present study, we report two cases infected by C. difficile RT027, of which one patient recovered and another patient died. Although the two C. difficile RT027 strains were highly similar, with only differences in four SNPs, the genotypic characteristic demonstrates that the two isolates were not derived from the same clone, according to the previously estimated evolutionary rate of 0.74 SNP per year for C. difficile, indicating no direct transmission of C. difficile between patients hospitalized at the same time, although both cases had a history of sharing the same ward (Gastroenterology Department) for 2 days.7 It has been reported that exposure to fluoroquinolones was an independent risk factor for the spread of C. difficile RT027, while the consumption of fluoroquinolones in the People's Republic of China was similar to that in Europe and North America.33,34 The two isolates were multi-drug resistant strains and also resistant to fluoroquinolones. All these evidences demonstrate the occurrence of C. difficile RT027 strains in this region, but there were no more isolates of C. difficile RT027 in the subsequent surveillance, which has been done since 2009.32 The review of relevant reports on C. difficile RT027 revealed no evidence to support the predominance of hypervirulent C. difficile RT027 strains in Mainland China, which was quite different from Europe or North America, where the prevalence of C. difficile RT027 emerged after it occurred.35 This may be due to the fact that the lack of adequate laboratory diagnostic capacity and high-quality and multiple-center surveillance system for CDI, and the low submission rate of samples in the People's Republic of China. However, in this study, we tried to explain the difference by comparative genomic analysis.
Two distinct epidemic lineages of C. difficile RT027, FQR1 or FQR2, emerged in North America within a relatively short period after acquiring the same fluoroquinolone resistance-conferring mutations.3 The present results confirm that for all C. difficile isolates detected from Mainland China, all of them were outside of both FQR lineages, which might have decreased virulence and transmissibility for outbreak. In the study reported by He et al3 that the isolates outside of the both lineages represent the pre-epidemic C. difficile RT027 genetic background, from which FQR epidemic lineages emerge may support our hypothesis. Although the two isolates in this study were both resistant to levofloxacin and moxifloxacin while they had mutations in DNA gyrase subunit A gene gyrA (Asp87→Tyr), this mutation was different from an identical mutation (Thr82→Ile) in another C. difficile RT027.36 A future study is needed to know if these mutations are hotspots within the so-called fluoroquinolone resistance determining region.
The present study revealed that several unique SNPs of C. difficile RT027 strains isolated in Mainland China were located at significant protein-coding sites that might affect the biological function of C. difficile. Hundreds of two-component response regulatory systems that allow organisms to sense and respond to environmental changes have now been found in bacterial genomes.37 The sporulation program is one of two-component systems, and capsular polysaccharides play important roles in the mechanism for C. difficile to efficiently persist in the environment and transmit the disease.38 SNP alterations in C. difficile strains from Mainland China change codons from ACT (threonine) to GCT (alanine) in amino acid sequences of the two-component response regulator involved in the sporulation program, and cause proline (CCG) to be replaced by leucine (CTG) in the capsular polysaccharide biosynthesis protein, respectively. However, although the mutation of particular protein-coding sites can affect protein function in two-component systems and capsular polysaccharides, it remains to be determined whether SNP mutations that result in amino acid substitutions are associated with the epidemiological features of C. difficile RT027 in the People's Republic of China.
Several special proteins have been detected in C. difficile isolates from Mainland China, including excisionase and transposase from transposon Tn916. Tn916 contains genes that encode a tyrosine recombinase and an excisionase, which are responsible for the integration and excision of an element.39 Acting as one of the most widespread conjugative transposons, Tn916 is responsible for the dissemination of resistance genes, can be transferred from a donor cell to a recipient, and can regulate the resistance gene by transcriptional attenuation.40,41 Further studies are needed to understand the molecular mechanism of SNP mutations in protein-coding sites that affect the features of C. difficile RT027 in the People's Republic of China.
The present study has several limitations. WGS results revealed significant genomic differences in C. difficile RT027 between Mainland China and North America and Europe, and the effects of SNP mutations on pathogenesis and transmission remain largely unclear. Secondly, only two C. difficile RT027 strains were identified and it is not enough data to draw conclusions about differences in bacterial strains on the ability of C. dfficile to cause epidemic disease in the People's Republic of China. However, we collected the whole genome data of RT027 reported in China, and combined with our data to analyze the difference between RT027 in the People's Republic of China and those in North America and Europe. Thirdly, we didn’t confirm the source of the C. difficile RT027 due to no environmental samples taking and no more C. difficile RT027 being identified.
In conclusion, two isolates of C. difficile NAP1/BI/027/ST-1 were identified from two patients, and no outbreak occurred. We speculate that the isolates of C. difficile RT027 isolated from Mainland China may have lower levels of transmissibility when compared to those in North America and Europe. The comparative genomic analysis revealed that C. difficile RT027 isolates from Mainland China were outside of the two epidemic lineages, FQR1 and FQR2, and might also have different epidemiological features. Unique SNP mutations were detected in isolates from Mainland China, leading to the probable alterations of the biological functions of C. difficile RT027. The results of the present study strengthen the understanding of the molecular characterizations of C. difficile RT027 in the People's Republic of China.
Availability of data and materials
The Whole Genome Sequencing of C. difficile isolates 14061807, CD6 and 14042403 have been deposited in GenBank under the following accession number: PNER00000000, PNES00000000, PNET00000000, respectively.
Acknowledgments
The authors wish to thank Dr KY Yuen and WC Yam of the Department of Microbiology Queen Mary Hospital Faculty of Medicine, University of Hong Kong, who provided the PCR ribotype027 strain of C. difficile, CD6. This study was partially funded by grants from the National Key Research and Development Program of China (2017YFC1200203), the Mega-projects of Science research of China (2018ZX10712001-005) and the Zhejiang Provincial Natural Science Foundation of China under Grant No. LY18H040001.
Author contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Zhou Y, Burnham CA, Hink T, et al. Phenotypic and genotypic analysis of Clostridium difficile isolates: a single-center study. J Clin Microbiol. 2014;52(12):4260–4266. doi:10.1128/JCM.02115-14
2. Loo VG, Poirier L, Miller MA, et al. A predominantly clonal multi-institutional outbreak of Clostridium difficile-associated diarrhea with high morbidity and mortality. N Engl J Med. 2005;353(23):2442–2449. doi:10.1056/NEJMoa051639
3. He M, Miyajima F, Roberts P, et al. Emergence and global spread of epidemic healthcare-associated Clostridium difficile. Nature Genet. 2013;45(1):109–113. doi:10.1038/ng.2478
4. He M, Sebaihia M, Lawley TD, et al. Evolutionary dynamics of Clostridium difficile over short and long time scales. Proc Natl Acad Sci U S A. 2010;107(16):7527–7532. doi:10.1073/pnas.0914322107
5. Brazier JS. Typing of Clostridium difficile. Clin Microbiol Infect. 2001;7(8):428–431.
6. Munoz M, Rios-Chaparro DI, Patarroyo MA, Ramirez JD. Determining Clostridium difficile intra-taxa diversity by mining multilocus sequence typing databases. BMC Microbiol. 2017;17(1):62. doi:10.1186/s12866-017-0969-7
7. Eyre DW, Cule ML, Wilson DJ, et al. Diverse sources of C. difficile infection identified on whole-genome sequencing. N Engl J Med. 2013;369(13):1195–1205. doi:10.1056/NEJMoa1216064
8. Didelot X, Eyre DW, Cule M, et al. Microevolutionary analysis of Clostridium difficile genomes to investigate transmission. Genome Biol. 2012;13(12):R118. doi:10.1186/gb-2012-13-12-r118
9. Sebaihia M, Wren BW, Mullany P, et al. The multidrug-resistant human pathogen Clostridium difficile has a highly mobile, mosaic genome. Nat Genet. 2006;38(7):779–786. doi:10.1038/ng1830
10. Eyre DW, Golubchik T, Gordon NC, et al. A pilot study of rapid benchtop sequencing of Staphylococcus aureus and Clostridium difficile for outbreak detection and surveillance. BMJ Open. 2012;2:3. doi:10.1136/bmjopen-2012-001124
11. Didelot X, Bowden R, Wilson DJ, Peto TEA, Crook DW. Transforming clinical microbiology with bacterial genome sequencing. Nature Rev Genet. 2012;13(9):601–612. doi:10.1038/nrg3226
12. Wang P, Zhou Y, Wang Z, et al. Identification of Clostridium difficile ribotype 027 for the first time in Mainland China. Infect Control Hosp Epidemiol. 2014;35(1):95–98. doi:10.1086/674405
13. Cheng JW, Xiao M, Kudinha T, et al. The first two Clostridium difficile ribotype 027/ST1 isolates identified in Beijing, China – an emerging problem or a neglected threat? Sci Rep. 2016;6:18834.
14. Jia H, Du P, Yang H, et al. Nosocomial transmission of Clostridium difficile ribotype 027 in a Chinese hospital, 2012-2014, traced by whole genome sequencing. BMC Genomics. 2016;17:405. doi:10.1186/s12864-016-3328-4
15. Wang R, Suo L, Chen HX, Song LJ, Shen YY, Luo YP. Molecular epidemiology and antimicrobial susceptibility of Clostridium difficile isolated from the Chinese People’s Liberation Army General Hospital in China. Int J Infect Dis. 2018;67:86–91. doi:10.1016/j.ijid.2017.07.010
16. Gao Q, Wu S, Huang H, et al. Toxin profiles, PCR ribotypes and resistance patterns of Clostridium difficile: a multicentre study in China, 2012-2013. Int J Antimicrob Agents. 2016;48(6):736–739. doi:10.1016/j.ijantimicag.2016.09.009
17. Chen YB, Gu SL, Shen P, et al. Molecular epidemiology and antimicrobial susceptibility of Clostridium difficile isolated from hospitals during a 4-year period in China. J Med Microbiol. 2018;67(1):52–59. doi:10.1099/jmm.0.000646
18. Bauer MP, Notermans DW, van Benthem BH, et al. Clostridium difficile infection in Europe: a hospital-based survey. Lancet. 2011;377(9759):63–73. doi:10.1016/S0140-6736(10)61266-4
19. Davies KA, Ashwin H, Longshaw CM, et al. Diversity of Clostridium difficile PCR ribotypes in Europe: results from the European, multicentre, prospective, biannual, point-prevalence study of Clostridium difficile infection in hospitalised patients with diarrhoea (EUCLID), 2012 and 2013. Euro Surveill. 2016;21(29):30294. doi:10.2807/1560-7917.ES.2016.21.29.30294
20. Wilcox MH, Shetty N, Fawley WN, et al. Changing epidemiology of Clostridium difficile infection following the introduction of a national ribotyping-based surveillance scheme in England. Clin Infect Dis. 2012;55(8):1056–1063. doi:10.1093/cid/cis614
21. McDonald LC, Coignard B, Dubberke E, Song X, Horan T, Kutty PK. Recommendations for surveillance of Clostridium difficile-associated disease. Infect Control Hosp Epidemiol. 2007;28(2):140–145. doi:10.1086/511798
22. Kato H, Kato N, Watanabe K, et al. Identification of toxin A-negative, toxin B-positive Clostridium difficile by PCR. J Clin Microbiol. 1998;36(8):2178–2182.
23. Stubbs S, Rupnik M, Gibert M, Brazier J, Duerden B, Popoff M. Production of actin-specific ADP-ribosyltransferase (binary toxin) by strains of Clostridium difficile. FEMS Microbiol Lett. 2000;186(2):307–312. doi:10.1111/j.1574-6968.2000.tb09122.x
24. Cheng VC, Yam WC, Chan JF, To KK, Ho PL, Yuen KY. Clostridium difficile ribotype 027 arrives in Hong Kong. Int J Antimicrob Agents. 2009;34(5):492–493. doi:10.1016/j.ijantimicag.2009.04.004
25. Indra A, Huhulescu S, Schneeweis M, et al. Characterization of Clostridium difficile isolates using capillary gel electrophoresis-based PCR ribotyping. J Med Microbiol. 2008;57(Pt 11):1377–1382. doi:10.1099/jmm.0.47714-0
26. Gal M, Northey G, Brazier JS. A modified pulsed-field gel electrophoresis (PFGE) protocol for subtyping previously non-PFGE typeable isolates of Clostridium difficile polymerase chain reaction ribotype 001. J Hosp Infect. 2005;61(3):231–236. doi:10.1016/j.jhin.2005.01.017
27. Griffiths D, Fawley W, Kachrimanidou M, et al. Multilocus sequence typing of Clostridium difficile. J Clin Microbiol. 2010;48(3):770–778. doi:10.1128/JCM.01796-09
28. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi:10.1093/bioinformatics/btp324
29. McKenna A, Hanna M, Banks E, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–1303. doi:10.1101/gr.107524.110
30. Aziz RK, Bartels D, Best AA, et al. The RAST server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75. doi:10.1186/1471-2164-9-75
31. Clinical and Laboratory Standards Institute (CLSI). M11-A8. Methods for Antimicrobial Susceptibility Testing of Anaerobic Bacteria; Approved Standard-Eighth Edition. Wayne, PA: Clinical and Laboratory Standards Institute; 2012.
32. Xu Q, Chen Y, Gu S, et al. Hospital-acquired Clostridium difficile infection in Mainland China: a seven-year (2009-2016) retrospective study in a large university hospital. Sci Rep. 2017;7(1):9645. doi:10.1038/s41598-017-09961-0
33. Drudy D, Kyne L, O’Mahony R, Fanning S. gyrA mutations in fluoroquinolone-resistant Clostridium difficile PCR-027. Emerg Infect Dis. 2007;13(3):504–505. doi:10.3201/eid1303.060771
34. Van Boeckel TP, Gandra S, Ashok A, et al. Global antibiotic consumption 2000 to 2010: an analysis of national pharmaceutical sales data. Lancet Infect Dis. 2014;14(8):742–750. doi:10.1016/S1473-3099(14)70780-7
35. Stabler RA, He M, Dawson L, et al. Comparative genome and phenotypic analysis of Clostridium difficile 027 strains provides insight into the evolution of a hypervirulent bacterium. Genome Biol. 2009;10(9):R102. doi:10.1186/gb-2009-10-9-r102
36. Spigaglia P, Carattoli A, Barbanti F, Mastrantonio P. Detection of gyrA and gyrB mutations in Clostridium difficile isolates by real-time PCR. Mol Cell Probes. 2010;24(2):61–67. doi:10.1016/j.mcp.2009.10.002
37. Stock AM, Robinson VL, Goudreau PN. Two-component signal transduction. Annu Rev Biochem. 2000;69:183–215. doi:10.1146/annurev.biochem.69.1.183
38. Dembek M, Barquist L, Boinett CJ, et al. High-throughput analysis of gene essentiality and sporulation in Clostridium difficile. mBio. 2015;6(2):e02383. doi:10.1128/mBio.02383-14
39. Mullany P, Allan E, Roberts AP. Mobile genetic elements in Clostridium difficile and their role in genome function. Res Microbiol. 2015;166(4):361–367. doi:10.1016/j.resmic.2014.12.005
40. Su YA, He P, Clewell DB. Characterization of the tet(M) determinant of Tn916: evidence for regulation by transcription attenuation. Antimicrob Agents Chemother. 1992;36(4):769–778. doi:10.1128/aac.36.4.769
41. Roberts AP, Mullany P. A modular master on the move: the Tn916 family of mobile genetic elements. Trends Microbiol. 2009;17(6):251–258. doi:10.1016/j.tim.2009.03.002
Supplementary materials
 |
Table S1 Genomic features of the three strains |
 |
Table S2 SNPs located within the protein-coding genes |
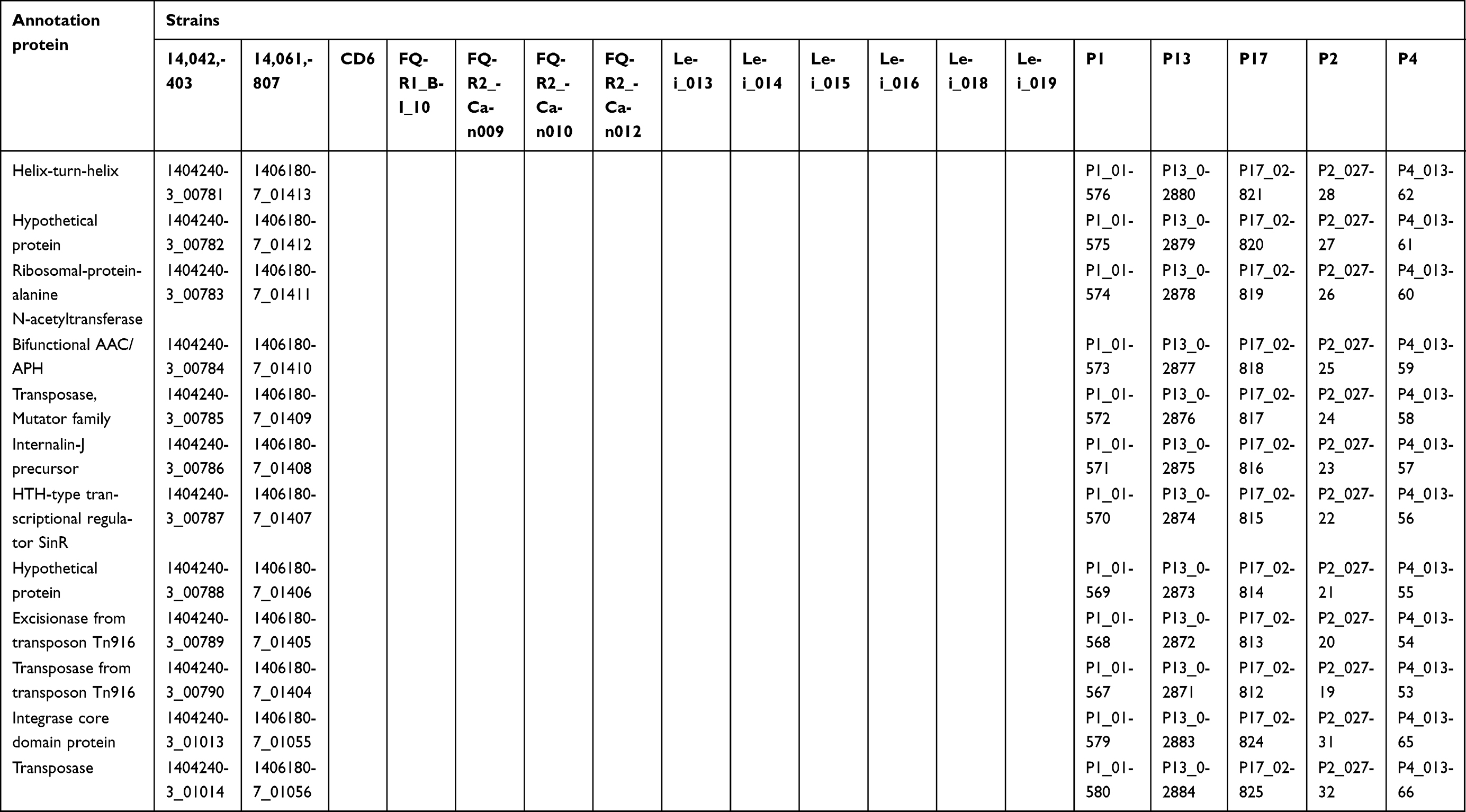 |
Table S3 Annotation proteins for the unique genes of C. difficile in mainland China |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.