Back to Journals » Cancer Management and Research » Volume 11
Whole exome sequencing of multiple meningiomas with varying histopathological presentation in one patient revealed distinctive somatic mutation burden and independent clonal origins
Authors Sheng HS, Shen F ![]() , Zhang N, Yu LS, Lu XQ, Zhang Z, Fang HY, Zhou LL
, Zhang N, Yu LS, Lu XQ, Zhang Z, Fang HY, Zhou LL ![]() , Lin J
, Lin J
Received 21 January 2019
Accepted for publication 21 March 2019
Published 6 May 2019 Volume 2019:11 Pages 4085—4095
DOI https://doi.org/10.2147/CMAR.S202394
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Kenan Onel
Han-Song Sheng,1 Fang Shen,2 Nu Zhang,1 Li-Sheng Yu,1 Xiang-Qi Lu,1 Zhe Zhang,1,3 Huang-Yi Fang,1,3 Ling-Li Zhou,4 Jian Lin1
1Department of Neurosurgery, Second Affiliated Hospital of Wenzhou Medical University, Wenzhou, People’s Republic of China; 2Department of Orthopedic Surgery‘s Spine Division, The Affiliated Hospital of Medical School of Ningbo University, Ningbo, People’s Republic of China; 3School of the 2nd Clinical Medical Sciences, Wenzhou Medical University, Wenzhou, People’s Republic of China; 4Department of Pathology, Second Affiliated Hospital of Wenzhou Medical University, Wenzhou, People’s Republic of China
Background: Although meningiomas are common intracranial tumors, multiple meningiomas (MMs) are rare entities in patients without neurofibromatosis type 2. Previous studies suggest most sporadic MMs are of monoclone in origin.
Objective: To elucidate the clonal relationship between two sporadic meningiomas from the same patient by using the next-generation sequencing (NGS) platform.
Methods: Two MMs, located frontally and parietally on the right side, were surgically removed from a 52-year-old male. Pathological examinations and whole exome sequencing were performed on tumor samples, followed by Sanger sequencing validation.
Results: MMs were diagnosed as secretory and fibrous subtypes, respectively, on histology (WHO grade I) and tumor DNA exhibited distinctive somatic mutation patterns. Specifically, the secretory subtype carried more single nucleotide variant while the fibrous subtype had much higher copy number variation. Besides, the two tumors demonstrated different mutation profiles in predisposing genes and known driver mutations. For example, the secretory subtype had missense mutations in TRAF7 and KLF4, while the fibrous subtype had frameshift deletion of NF2 gene in addition to copy number loss of NF2 and SMARCB1, genetic events that have already been associated with the development of meningiomas. Significantly mutated gene analysis revealed novel mutations of LOC729159 in the secretory subtype and RPGRIP1L and DPP6 in the fibrous subtype. Sanger sequencing validated important point mutations in TRAF7 (c.1678G>A, p.G560S), KLF4 (c.1225A>C, p.K409Q) and CDH11 (c.169T>G, p.W57G).
Conclusion: Our data suggest the two meningiomas might develop independently in this patient and molecular subtyping by NGS is a valuable supplement to conventional pathology. Further study is needed to ascertain whether these novel genetic events are tumorigenic or simply passenger mutations, as well as their clinical implications
Keywords: multiple meningiomas, whole exome sequencing, secretory meningioma, fibrous meningioma, TRAF7, NF2
Introduction
Meningioma is the most common intracranial tumor developed from the arachnoid cells of the meninges. Although meningioma can affect both genders, its incidence is much higher among females than males. The majority of meningiomas are benign, and histologically, they can be classified into various subtypes, such as meningothelial, fibrous, psammomatous, microcystic, and secretory meningiomas. Atypical and malignant meningiomas are rare (~5%) and have a slight male dominance.1
Various genetic abnormalities have been associated with the development of meningiomas.2 Historically, loss of chromosome 22 and focal chromosomal deletion in 1p, 6q, 14q, and 18q were first observed. Later, deletion and inactivation of neurofibromin 2 (NF2) gene, a tumor suppressor gene on chromosome 22q12, has been identified as the most common genetic event in patients with both sporadic meningiomas and neurofibromatosis type 2.3 The advent of the next-generation sequencing (NGS) technology further revealed important genetic aberrations in the development of meningiomas not associated with neurofibromatosis type 2 (NF2) abnormality,4–9 such as TNF receptor-associated factor 7 (TRAF7), Krupplelike factor 4 (KLF4), v-akt murine thymoma viral oncogene homolog 1 (AKT1), phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA), smoothened, frizzled family receptor (SMO) and SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily b, member 1 (SMARCB1).
It is common for patients with NF2 to develop multiple meningiomas (MMs). However, the incidence of sporadic MMs in patients without a history of NF2 is low.10 It is much less common to find these sporadic MMs bearing different histological features in the same patient.11 The etiology of MM is still being debated because of two hypotheses for the development of MMs, ie, monoclonal or independent.12 The monoclonal hypothesis suggests that MMs originate from a certain neoplastic transformed clone that subsequently spreads along the meninges to form multi-foci.13,14 This hypothesis is supported by the observations that most sporadic MMs presented the same histological features.15 However, it is also possible for MMs that evolve independently and are driven by different key genetic events, such as the cases of MMs showing different histological types or grades.12 Nevertheless, most of the studies on the etiology of MM were performed based on histological and cytogenetic evidence. To our knowledge, the authors reported here the first comprehensive genomic profiling with whole exome sequencing (WES) on two meningiomas of different histological types in the same patient. Our results revealed distinctive somatic mutation burdens in the two tumors and supported the independent clonal origins hypotheses for the current case.
Methods
Patient recruitment
A 52-year-old male presented with headaches for 2 months was recruited from Second Affiliated Hospital of Wenzhou Medical University. Physical and neurological examinations were performed to confirm whether the patient has pathological signs. Surgery was performed in one stage for resection of both tumors which then were sent for pathological examinations. The patient received detailed information on the study and provided his written informed consent prior to inclusion in the study and has given written informed consent for the publication of information about his case. The present study complied with the Declaration of Helsinki and the experimental protocols and informed consents were approved by the Hospital Institutional Ethics Committee.
DNA extraction and whole-exome sequencing
DNA from the two meningioma tissues and the corresponding patient’s peripheral lymphocytes (control) were isolated by standard methods, as described before.16 The quality of isolated genomic DNA was verified by using 1% agarose gel electrophoresis and Qubit® DNA Assay Kit in Qubit® 2.0 Fluorometer (Life Technologies, CA, USA).
For the WES protocol, libraries were prepared for each sample with an Agilent SureSelect Human All Exon kit (Agilent Technologies, Santa Clara, CA, USA) following manufacturer’s recommendations and index codes were added to each sample. A total amount of 0.6 μg genomic DNA per sample was used as input material for the DNA sample preparation. Libraries products were purified using AMPure XP system (Beckman Coulter, Beverly, MA, USA) and quantified using the Agilent high sensitivity DNA assay on the Agilent Bioanalyzer 2100 system. The clustering of the index-coded samples was performed on a cBot Cluster Generation System using Hiseq PE Cluster Kit (Illumina) according to the manufacturer’s instructions. After cluster generation, the DNA libraries were sequenced on Illumina Hiseq platform and 150 bp paired-end reads were generated.
Variant calling and bioinformatics analysis
FASTQ files of exomes obtained from the three samples (one from peripheral blood and two different tumors from the same patient) were first examined by QC steps to generate high-quality clean data. Valid sequencing data were then mapped to the reference human genome (UCSC hg19) by Burrows-Wheeler Aligner (BWA) software17 to get the original mapping results stored in BAM format. Then, SAMtools,18 Picard (
Sanger sequencing validation
The primers used for CDH11 amplification were as follows: Forward, 5’‑CACAGCCATGCCTTTGCC‑3’ and reverse, 5’‑GCCTTACCCTGCCCACAA‑3’. The primers used for KLF4 amplification were as follows: Forward, 5’‑CACCCCACCTTCTTCACCC‑3’ and reverse, 5’‑CTGGGAAGTCAAGGAGGCAC‑3’. The primers used for TRAF7 amplification were as follows: Forward, 5’‑CATCTGCCCTGTTCCTACCTTCG‑3’ and reverse, 5’‑GGCCTTACGTGGATGAGGTTCTC‑3’. PCR was conducted with 40 cycles of denaturation (95°C for 25 s), annealing (56°C for 25 s) and extension (72°C for 40 s). PCR was performed in a LineGene 9600 Plus thermal cycler (BIOER, Hangzhou Bioer Technology Co. Ltd., China), using deoxynucleotides (Takara Bio, Inc., Japan) and Tris-borate ethylenediaminetetraacetic acid (Takara Bio, Inc.) as a buffer. Amplified DNA fragments were recovered from a low melting temperature agarose gel, purified with a Magnetic Beads Genomic DNA Extraction Kit (MSi100-DNA, Enriching Biotechnology Ltd, China) and subjected to direct sequencing analysis using an automated ABI-3730 Sequencer (Applied Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA, USA).
Results
Clinical characteristics of the patient
A 52-year-old male has presented with headaches for 2 months. Physical and neurological examination revealed normal sensation and muscle strength, normal reflexes and negative pathological signs. There was no evidence of neurofibromatosis either. Preoperative magnetic resonance imaging (MRI) scan showed a right frontal tumor with strong homogenous contrast enhancement, marked perifocal edema, and significant midline shift (Figure1A-C). Another well-demarcated and homogenously enhancing tumor was also found at the right parietal convexity, but with minimal perifocal edema (Figure1E-G). Surgery was performed in one stage and both tumors were successfully totally resected and sent for pathological examinations. The postoperative course was uneventful, and his symptoms improved after surgery. He was discharged in one week postoperatively and follow-up MRI scan 8 months later confirmed the complete removal of both tumors (Figure 1D, H).
 | Figure 1 Magnetic resonance imaging (MRI) images of the two sporadic multiple meningiomas in a 52-year-old male patient. (A-C) Preoperative MRI showed a right frontal tumor with strong homogenous contrast enhancement, marked perifocal edema, and significant midline shift. (E-G) Preoperative MRI showed another well-demarcated and homogenously enhancing tumor at the right parietal convexity, but with minimal perifocal edema. (D, H) Follow-up MRI scan performed 8 months later confirmed the complete removal of both tumors. |
Histopathological examination
Histopathological examination of right frontal meningioma revealed the tumor cells were in a form of syncytium-like appearance, with unclear boundaries between the cells. The nucleus was oval with fine chromatin and some of the nucleus was transparent in the middle. Cells were arranged in large lobulated structures or formed a small swirling structure. Some tumor cells were partially epithelial-differentiated and the intraepithelial microgland contained eosinophilic substances (Figure 2A). Immunohistochemistry (IHC) showed CK (+), Vimentin (+++), EMA (sporadic +), CEA (+), S-100 (−), Ki-67 (~1%), CD34 (−), ER (−) and PR (+). Histopathological examination of the right parietal meningioma showed spindle-shaped tumor cells, parallel or bundled in a cross-shaped arrangement. There was formation of swirling structure in focal areas, with calcification scattered in a small amount of mature adipose tissues. The shapes of nucleus were long spindle, fatty spindle, or oval, with fine chromatin and some nucleus were transparent (Figure 2B). IHC showed Vimentin (+++), Ki-67 (~1%), CK (−), and EMA (+). The right frontal meningioma was pathologically diagnosed as secretory meningioma (WHO grade I) whereas the right parietal one was fibrous meningioma (WHO grade I).
 | Figure 2 Histopathological examinations of the two sporadic multiple meningiomas. (A) The right frontal tumor cells showed syncytium-like appearance with unclear boundaries. Cells were arranged in large lobulated structures. Some cells were partially epithelial-differentiated and the intraepithelial microgland contained eosinophilic substances. Pathological diagnosis was secretory meningioma (WHO grade I). (B) The right parietal tumor showed spindle cells with bland nuclei arranged in storiform pattern, with calcification in some adipose tissues. Pathological diagnosis was fibrous meningioma (WHO grade I). |
Genetic analysis of the two meningiomas
Given the different subtypes in both meningiomas, we tried to examine the genetic differences and the clonal relationship between two sporadic meningiomas. We applied WES on the two meningiomas to investigate all variants annotated with in-house pipeline. Somatic mutation analysis revealed, in the secretory subtype, there was a total of 1,343 SNVs (59 in the CoDing Sequence (CDS)) and 57 InDels (3 in the CDS) in a total of 1,085 genes (Figure 3A). Whereas 1034 SNVs (28 in the CDS) and 32 InDels (3 in the CDS) in a total of 798 genes were identified in the fibrous subtype (see Tables S1 and S2). In addition, we found that there were 128 common genes with somatic mutations between two subtypes, whereas there were only 37 common somatic mutations (SNVs and InDels) in these common genes (Figure 3A). Focusing on the exons, there were no common genes with somatic mutations in exons of the whole genome between two subtypes (Figure 3B). Furthermore, we also performed CNV analysis that revealed only 13 gain counts in the secretory subtype, while there were 109 gain counts and 10 loss counts, including copy number loss of NF2 and SMARCB1, in the fibrous subtype (see Table S3).
 | Figure 3 Distinctive somatic mutation patterns for single nucleotide variants (SNVs) and InDels in the two sporadic multiple meningiomas. (A) The number of genes with somatic mutations including SNVs and InDels between two subtypes. The box showed the number of SNVs and InDels in the common genes between the two subtypes. (B) All the genes with somatic mutations in exons of the whole genome of the two subtypes. |
The two tumors also demonstrated different mutation profiles in terms of predisposing gene mutation and known driver gene mutation. For predisposing gene mutation analysis, the secretory subtype had the highest mutation burden. Besides, as compared with the control, it contained missense mutations in TRAF7, KLF4, and CDH11, in-frame deletion in ELN, a nonsense mutation in PDE4DIP, as well as mutations in the splice sites of NUP214, KDM6A, and ZMYM2. In contrast, the fibrous subtype had a missense mutation in PDE4DIP, in-frame deletion in ZRSR2, frame-shift deletion in NF2, and splice site mutation of ZMYM2 (Figure 4A). Known driver gene mutation analysis indicated the missense mutations of ABCB1, AHNAK, CDH11, TRAF7 and H3F3A, and frame-shift deletion of KMT2D in the secretory subtype. Whereas the fibrous subtype had missense mutations of GMPS and frame-shift deletion of NF2 (Figure 4B). Significantly mutated genes analysis showed missense mutations of LOC729159 in the secretory subtype and RPGRIP1L and DPP6 in the fibrous subtype (Figure 4C). A whole-genome view of the two samples confirmed a higher number of SNV and InDel events in secretory subtype while a more prominent CNV abnormality in the fibrous subtype (Figure 5). Subsequent validation by Sanger sequencing confirmed nonsynonymous mutations in KLF4 (c.1225A>C, p.K409Q), CDH11 (c.169T>G, p.W57G) and TRAF7 (c.1678G>A, p.G560S) in secretory subtype (Figure 6).
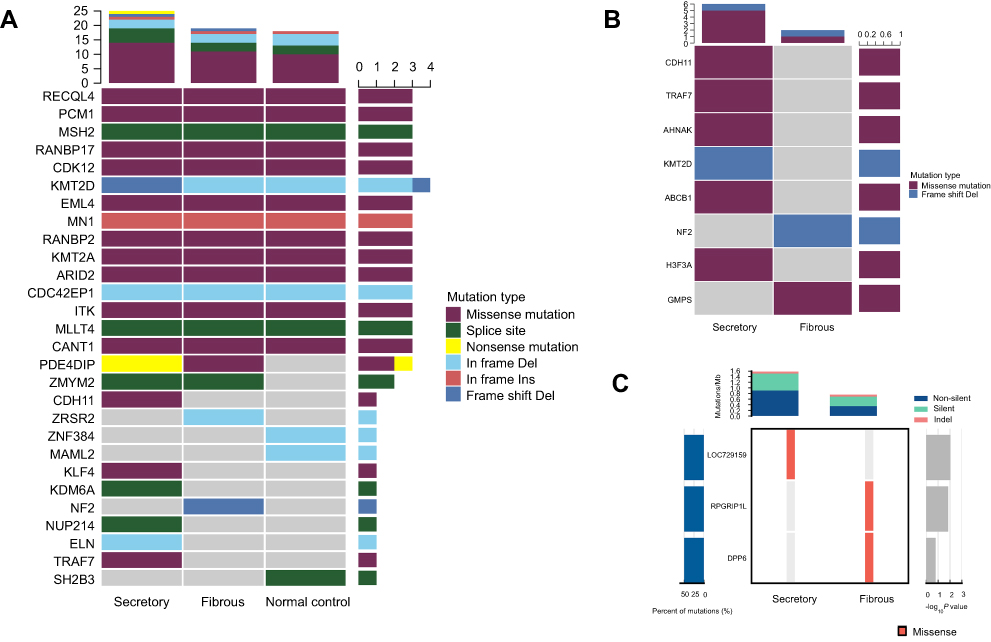 | Figure 4 Whole exome sequencing revealed distinctive mutational patterns in the two sporadic multiple meningiomas of different histological features. (A) Predisposing gene mutation analysis showed the secretory subtype had the highest mutation burden and contained missense mutations in TRAF7, KLF4, and CDH11. In contrast, the fibrous subtype had a frame-shift deletion in NF2. (B) Known driver gene mutation analysis indicated the missense mutations of CDH11, TRAF7, and H3F3A in the secretory subtype. Whereas the fibrous subtype had a frame-shift deletion of NF2. (C) Significantly mutated genes analysis showed missense mutations of LOC729159 in the secretory subtype and RPGRIP1L and DPP6 in the fibrous subtype. |
 | Figure 5 The somatic mutation landscapes of the two histologically different multiple meningiomas presented in Circos plots. Chromosome ideograms are shown around the outer ring and are oriented pter–qter in a clockwise direction with centromeres indicated in red. Other tracks contain somatic alterations (from outside to inside): sequencing coverage; green dots represent the density of validated somatic insertions and deletions (InDel) and single nucleotide variants (SNV); somatic copy number variation (CNV) events are represented as loss (blue dots), gain (red dots) or normal (green dots). (A) Secretory subtype featured with a higher number of SNV and InDel events. (B) Fibrous subtype had a more prominent CNV abnormality. |
 | Figure 6 Sanger sequencing validation of point mutations in the secretory subtype meningioma. (A) Sanger sequencing confirmed a c.1225A>C mutation was observed in the KLF4 gene, resulting in p.K409Q. (B) Sanger sequencing confirmed a c.169T>G mutation was observed in the CDH11 gene, resulting in p.W57G. (C) Sanger sequencing confirmed a c.1678G>A mutation was observed in the TRAF7 gene, resulting in p.G560S. |
Discussion
The definition of MM is generally accepted as two or more than two spatially separated meningiomas that have developed from at least two distinct brain regions. Although MMs can occur in patients with predisposing conditions such as NF2, hereditary SWI/SNF complex deficiency syndromes,22 Cowden syndrome,23 Turner syndrome24 and Rubinstein-Taybi syndrome,25 sporadic MMs with no family history are not commonly reported in the literature. Besides, most of these sporadic MMs are of the same histological subtypes13,14,26–28 and sporadic MMs bearing different histological features or grades in the same patient are much less common. Huang et al15 evaluated 456 patients with an intracranial meningioma and found 8.6% of them had more than one meningioma. Compared with solitary meningiomas, MMs have their own clinical features such as female preponderance and stronger PR expression. Koh et al12 reported a case of MMs with both malignant and benign histological features and proposed MMs originated from multicentric neoplastic foci. Similarly, Liu et al11 showed the coexistence of fibrous meningioma (WHO grade I) and atypical meningioma (WHO grade II) in the same patient. More recently, Tsermoulas et al29 reviewed a total of 133 consecutive patients with MMs for over 25 years and 18 patients had surgical removal of more than one meningioma. Among these patients, 4 had tumors of different grades and 6 patients had meningiomas of different histological types.
Despite the lack of knowledge on the incidence of meningiomas of different histological subtypes in the same patient, there is a keen interest in revealing the etiology of MMs. Since the majority of surgically removed MMs showed the same histopathological features, it is likely they might derive from one single clone that disseminates through the subarachnoid space during the long history of this benign condition. Another hypothesis, which is supported by the observations of MMs presenting different pathological features, is that tumors can develop independently at multiple foci through different neoplastic transition mechanisms. Previous investigations have mainly focused on the molecular mechanisms underpinning the pathogenesis of solitary meningiomas by different techniques including cytogenetic analysis, gene expression arrays and NGS, and revealed a wealth of information on genetic abnormalities,3,30 the most notable of which is the NF2 gene. However, not much is yet known about the genetic basis of those solitary MMs. Shen et al31 applied comparative genomic hybridization arrays and showed a distinct pattern between MMs from patients with familial predisposition and those sporadic cases. Dewan et al32 performed in-depth genomic profiling of two cranial meningiomas (WHO grade I and grade II) with WES and spectral karyotyping from a patient predisposed with NF2. They found, in addition to second NF2 copy inactivation, both tumors had a low somatic burden. However, the grade II tumor exhibited a high level of genomic instability and mutations of ADAMTSL3 and CAPN5, which might explain its more aggressive biological behaviors. Recently, Torres-Martı´n et al33 performed WES on four meningioma samples from a patient with sporadic MM. They identified three common mutational events (NF2, FAM109B, and TPRXL) for all tumors as well as unique mutations for each individual tumor. Therefore, they proposed a monoclonal origin for their particular case. That patient underwent multiple surgeries for progressive tumor growth, including a dorsal meningioma (1975), an olfactory sulcus meningioma (1987) and multiple neoplastic nodules from the frontoparietal convexity and falx (1990), over a period of 15 years. Although all tumors were diagnosed histologically as transitional meningioma, the authors did not specify which four tumor tissues, among the multiple samples they had collected, had been sequenced.
To the best of our knowledge, our current study is the first to adopt WES on the genetic profiling of two histologically different primary sporadic MMs in the same patient. Our results showed distinctive genetic features underlying them and thus we support an independent clonal origin in the current case. For example, secretory subtype featured with a higher frequency of SNV and InDel events while the fibrous subtype had a more prominent CNV abnormality. Our results also identified some key mutations that have been associated with the development of meningiomas. Specifically, mutations in TRAF7, KLF4, and CDH11 for the secretory subtype and deletion of NF2, copy number loss of NF2 and SMARCB1 for the fibrous subtype. There are several comprehensive review articles on the germline and somatic mutations underlying the pathogenesis meningiomas1,34,35 and a detailed analysis of those known gene mutations and their complex pathway interactions are beyond the scope of the current paper. However, we would discuss here briefly on important mutant genes that have been confirmed by Sanger sequencing. For example, we identified a c.1225A>C (p.K409Q) in KLF4, which is consistent with previous reports.36–38 KLF4 belongs to a family of DNA-binding transcriptional regulators and were suggested to be involved in some cancers as potential tumor suppressor, despite the fact that KLF4 K409Q mutation being specific for meningioma.39,40 In contrast, the TRAF7 c.1678G>A (p.G560S) mutation in our secretory subtype is relatively novel compared with previous reports (N520S, G536S, K615E, R641C and R641H).30 TRAF7 depletion by RNA interference has been shown to result in resistance to TNFα cytotoxicity and TRAF7 downregulation was observed in breast cancer expression and was proposed to contribute to p53 accumulation.41,42 Nevertheless, similar to KLF4, TRAF7 mutation is highly specific for meningioma. This concomitant TRAF7/KLF4 mutation pattern was seen in about 8% of meningiomas and provided both diagnostic (secretory subtype) and prognostic value (better prognosis compared with NF2 type).30 Besides, there might be potential benefits by targeting either KLF4, TRAF7 or both, though further research is needed.
Moreover, these authors suggest the establishment of a classification system on meningiomas by combing clinical feature, histology, and genetic mutation to better refine personalized treatments. Yuzawa et al30 classified meningiomas into seven genotypes: NF2, TRAF7/KLF4, TRAF7/AKT1, SMO, “Others,” “Complex,” and “None”. They also found these genotypes were related to clinical features such as tumor location, and histological types and grades. Interestingly, both the fibrous subtype and the secretory subtype in our case matched the proposed NF2 genotype and TRAF7/KLF4 genotype. Another recent study further proposed a classification and grading system for meningioma based on DNA methylation pattern.43 They have shown the six methylation classes are better at predicting recurrence and prognosis than the WHO classification system. Specifically, the methylation class (MC) ben-1 is featured by NF2 mutation, 22q deletion with predominant histology of fibroblastic, transitional and atypical. While the MC ben-2 is featured by TRAF7, KLF4, SMO, and AKT mutations, balanced chromosome and secretory, transitional and meningothelial histology. Accordingly, the two meningiomas of our current case matched the MC ben-1 and MC ben-2 subclass. Therefore, our sequencing results support these molecular classification systems. In 2016 WHO classification of the tumors of the central nervous system,44 molecular classification has already been applied in the diagnosis of gliomas, as their genetic makeup can significantly influence patients’ prognosis and choice of treatments. Therefore, it is also promising to incorporate molecular classification of meningiomas into existent WHO histological classification system.
Conclusion
Genetic profiling with NGS is a valuable supplement to conventional pathological diagnosis in answering the questions regarding the clonal origin of MMs. Further study is needed to ascertain whether these genetic abnormalities are tumorigenic or simply passenger mutations, as well as their clinical implications. Current efforts on genetic profiling of meningiomas, together with further investigation on the associations between genetic abnormalities, histology, and patients’ prognosis, will help to establish a more integrate diagnosis on meningiomas including MMs.
Ethical approval and consent to participate
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Ethical approval was obtained from the institutional ethics committee.
Consent for publication
The patient has given his informed consent to the publication of his data.
Data sharing statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Acknowledgments
This study was funded by Zhejiang Province Science and Technology Program (Grant No.: 2016C33213) and Ningbo Natural Science Foundation (Grant No.: 2018A610256).
Author contributions
All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Preusser M, Brastianos PK, Mawrin C. Advances in meningioma genetics: novel therapeutic opportunities. Nat Rev Neurol. 2018;14(2):106–115. doi:10.1038/nrneurol.2017.168
2. Bi WL, Zhang M, Wu WW, Mei Y, Dunn IF. Meningioma genomics: diagnostic, prognostic, and therapeutic applications. Front Surg. 2016;3:40. doi:10.3389/fsurg.2016.00040
3. Bi WL, Mei Y, Agarwalla PK, Beroukhim R, Dunn IF. Genomic and epigenomic landscape in meningioma. Neurosurg Clin N Am. 2016;27(2):167–179. doi:10.1016/j.nec.2015.11.009
4. Clark VE, Erson-Omay EZ, Serin A, et al. Genomic analysis of non-NF2 meningiomas reveals mutations in TRAF7, KLF4, AKT1, and SMO. Science. 2013;339(6123):1077–1080. doi:10.1126/science.1233009
5. Abedalthagafi M, Bi WL, Aizer AA, et al. Oncogenic PI3K mutations are as common as AKT1 and SMO mutations in meningioma. Neuro Oncol. 2016;18(5):649–655. doi:10.1093/neuonc/nov316
6. Brastianos PK, Horowitz PM, Santagata S, et al. Genomic sequencing of meningiomas identifies oncogenic SMO and AKT1 mutations. Nat Genet. 2013;45(3):285–289. doi:10.1038/ng.2526
7. Wu J, Kong M, Bi Q. Identification of a novel germline SMARCB1 nonsense mutation in a family manifesting both schwannomatosis and unilateral vestibular schwannoma. J Neurooncol. 2015;125(2):439–441. doi:10.1007/s11060-015-1918-7
8. van den Munckhof P, Christiaans I, Kenter SB, Baas F, Hulsebos TJ. Germline SMARCB1 mutation predisposes to multiple meningiomas and schwannomas with preferential location of cranial meningiomas at the falx cerebri. Neurogenetics. 2012;13(1):1–7. doi:10.1007/s10048-011-0300-y
9. Christiaans I, Kenter SB, Brink HC, et al. Germline SMARCB1 mutation and somatic NF2 mutations in familial multiple meningiomas. J Med Genet. 2011;48(2):93–97. doi:10.1136/jmg.2010.082420
10. Kerr K, Qualmann K, Esquenazi Y, Hagan J, Kim DH. Familial syndromes involving meningiomas provide mechanistic insight into sporadic disease. Neurosurgery. 2018. doi:10.1093/neuros/nyy121
11. Liu Y, Song DP, Wang T. Meningiomas with different histological grade in the same patient: case report. Medicine (Baltimore). 2017;96(50):e9086. doi:10.1097/MD.0000000000009086
12. Koh YC, Yoo H, Whang GC, Kwon OK, Park HI. Multiple meningiomas of different pathological features: case report. J Clin Neurosci. 2001;8(Suppl 1):40–43. doi:10.1054/jocn.2001.0875
13. Zhu JJ, Maruyama T, Jacoby LB, et al. Clonal analysis of a case of multiple meningiomas using multiple molecular genetic approaches: pathology case report. Neurosurgery. 1999;45(2):409–416.
14. von Deimling A, Kraus JA, Stangl AP, et al. Evidence for subarachnoid spread in the development of multiple meningiomas. Brain Pathol. 1995;5(1):11–14.
15. Huang H, Buhl R, Hugo HH, Mehdorn HM. Clinical and histological features of multiple meningiomas compared with solitary meningiomas. Neurol Res. 2005;27(3):324–332. doi:10.1179/016164105X39932
16. Robbe P, Popitsch N, Knight SJL, et al. Clinical whole-genome sequencing from routine formalin-fixed, paraffin-embedded specimens: pilot study for the 100,000 genomes project. Genet Med. 2018. doi:10.1038/gim.2017.241
17. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi:10.1093/bioinformatics/btp324
18. Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi:10.1093/bioinformatics/btp352
19. DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491–498. doi:10.1038/ng.806
20. Cibulskis K, Lawrence MS, Carter SL, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31(3):213–219. doi:10.1038/nbt.2514
21. Saunders CT, Wong WS, Swamy S, Becq J, Murray LJ, Cheetham RK. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics. 2012;28(14):1811–1817. doi:10.1093/bioinformatics/bts271
22. Agaimy A, Foulkes WD. Hereditary SWI/SNF complex deficiency syndromes. Semin Diagn Pathol. 2018;35(3):193–198. doi:10.1053/j.semdp.2018.01.002
23. Pain M, Darbinyan A, Fowkes M, Shrivastava R. Multiple meningiomas in a patient with cowden syndrome. J Neurol Surg Rep. 2016;77(3):e128–133. doi:10.1055/s-0036-1584265
24. Amelot A, Lemaistre G, Cornu P, Kalamarides M, Peyre M. Multiple meningiomas in patients with turner syndrome. Acta Neurochir (Wien). 2015;157(4):621–623. doi:10.1007/s00701-015-2360-5
25. Verstegen MJ, van den Munckhof P, Troost D, Bouma GJ. Multiple meningiomas in a patient with Rubinstein-Taybi syndrome. Case report. J Neurosurg. 2005;102(1):167–168. doi:10.3171/jns.2005.102.1.0167
26. Neuss M, Westphal M, Hansel M, Herrmann HD. Clinical and laboratory findings in patients with multiple meningiomas. Br J Neurosurg. 1988;2(2):249–256.
27. Borovich B, Doron Y, Braun J, et al. The incidence of multiple meningiomas – do solitary meningiomas exist? Acta Neurochir (Wien). 1988;90(1–2):15–22.
28. Larson JJ, Tew JM
29. Tsermoulas G, Turel MK, Wilcox JT, et al. Management of multiple meningiomas. J Neurosurg. 2018;128(5):1403–1409. doi:10.3171/2017.2.JNS162608
30. Yuzawa S, Nishihara H, Tanaka S. Genetic landscape of meningioma. Brain Tumor Pathol. 2016;33(4):237–247. doi:10.1007/s10014-016-0271-7
31. Shen Y, Nunes F, Stemmer-Rachamimov A, et al. Genomic profiling distinguishes familial multiple and sporadic multiple meningiomas. BMC Med Genomics. 2009;2:42. doi:10.1186/1755-8794-2-42
32. Dewan R, Pemov A, Dutra AS, et al. First insight into the somatic mutation burden of neurofibromatosis type 2-associated grade I and grade II meningiomas: a case report comprehensive genomic study of two cranial meningiomas with vastly different clinical presentation. BMC Cancer. 2017;17(1):127. doi:10.1186/s12885-017-3127-6
33. Torres-Martin M, Kusak ME, Isla A, et al. Whole exome sequencing in a case of sporadic multiple meningioma reveals shared NF2, FAM109B, and TPRXL mutations, together with unique SMARCB1 alterations in a subset of tumor nodules. Cancer Genet. 2015;208(6):327–332. doi:10.1016/j.cancergen.2015.03.012
34. Smith MJ. Germline and somatic mutations in meningiomas. Cancer Genet. 2015;208(4):107–114. doi:10.1016/j.cancergen.2015.02.003
35. Galani V, Lampri E, Varouktsi A, Alexiou G, Mitselou A, Kyritsis AP. Genetic and epigenetic alterations in meningiomas. Clin Neurol Neurosurg. 2017;158:119–125. doi:10.1016/j.clineuro.2017.05.002
36. Marrero-Rodriguez D, la Cruz HA, Taniguchi-Ponciano K, et al. Kruppel like factors family expression in cervical cancer cells. Arch Med Res. 2017;48(4):314–322. doi:10.1016/j.arcmed.2017.06.011
37. Farrugia MK, Sharma SB, Lin CC, et al. Regulation of anti-apoptotic signaling by Kruppel-like factors 4 and 5 mediates lapatinib resistance in breast cancer. Cell Death Dis. 2015;6:e1699. doi:10.1038/cddis.2015.65
38. Tetreault MP, Yang Y, Katz JP. Kruppel-like factors in cancer. Nat Rev Cancer. 2013;13(10):701–713. doi:10.1038/nrc3582
39. Zhao W, Hisamuddin IM, Nandan MO, Babbin BA, Lamb NE, Yang VW. Identification of Kruppel-like factor 4 as a potential tumor suppressor gene in colorectal cancer. Oncogene. 2004;23(2):395–402. doi:10.1038/sj.onc.1207067
40. Zammarchi F, Morelli M, Menicagli M, et al. KLF4 is a novel candidate tumor suppressor gene in pancreatic ductal carcinoma. Am J Pathol. 2011;178(1):361–372. doi:10.1016/j.ajpath.2010.11.021
41. Scudiero I, Zotti T, Ferravante A, et al. Tumor necrosis factor (TNF) receptor-associated factor 7 is required for TNFalpha-induced Jun NH2-terminal kinase activation and promotes cell death by regulating polyubiquitination and lysosomal degradation of c-FLIP protein. J Biol Chem. 2012;287(8):6053–6061. doi:10.1074/jbc.M111.300137
42. Wang L, Wang L, Zhang S, et al. Downregulation of ubiquitin E3 ligase TNF receptor-associated factor 7 leads to stabilization of p53 in breast cancer. Oncol Rep. 2013;29(1):283–287. doi:10.3892/or.2012.2121
43. Sahm F, Schrimpf D, Stichel D, et al. DNA methylation-based classification and grading system for meningioma: a multicentre, retrospective analysis. Lancet Oncol. 2017;18(5):682–694. doi:10.1016/S1470-2045(17)30155-9
44. Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131(6):803–820. doi:10.1007/s00401-016-1545-1
Supplementary materials
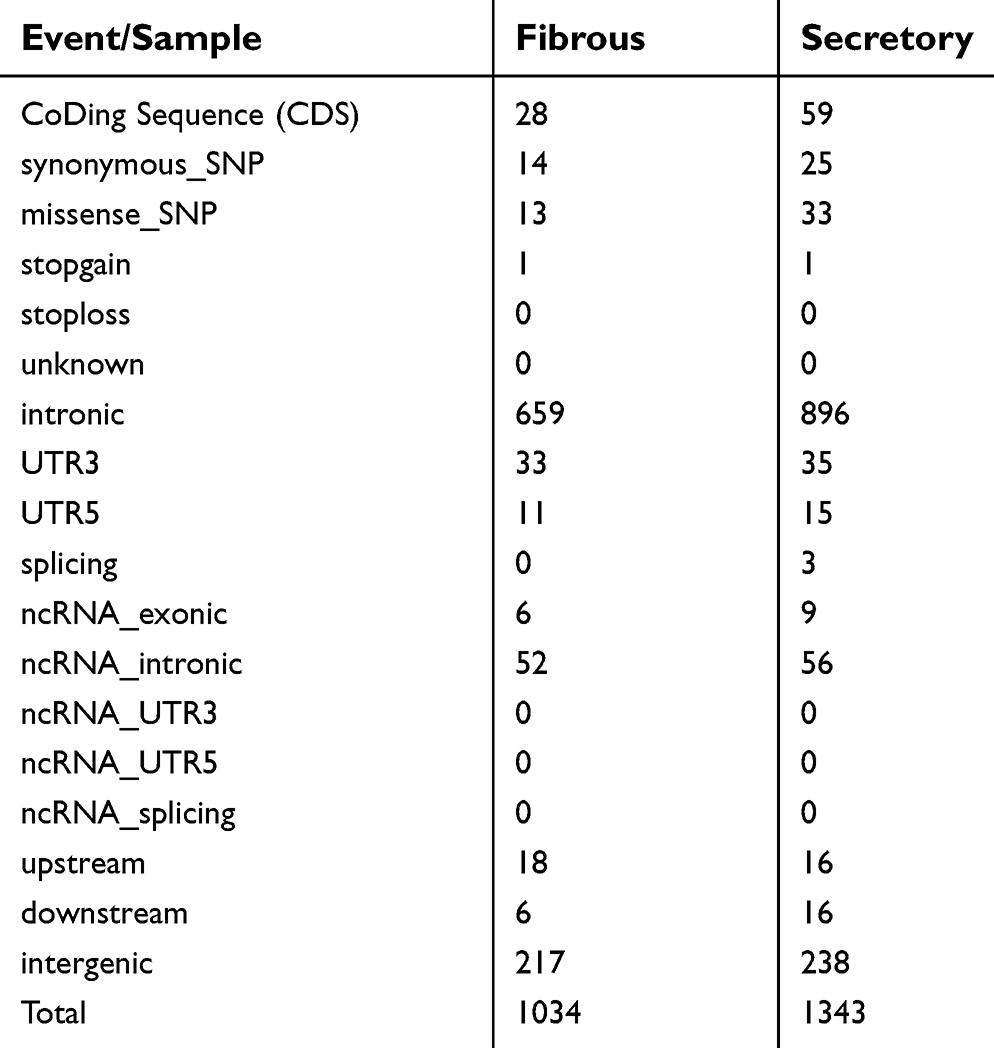 | Table S1 The number of somatic single nucleotide variant (SNV) in different tumor samples |
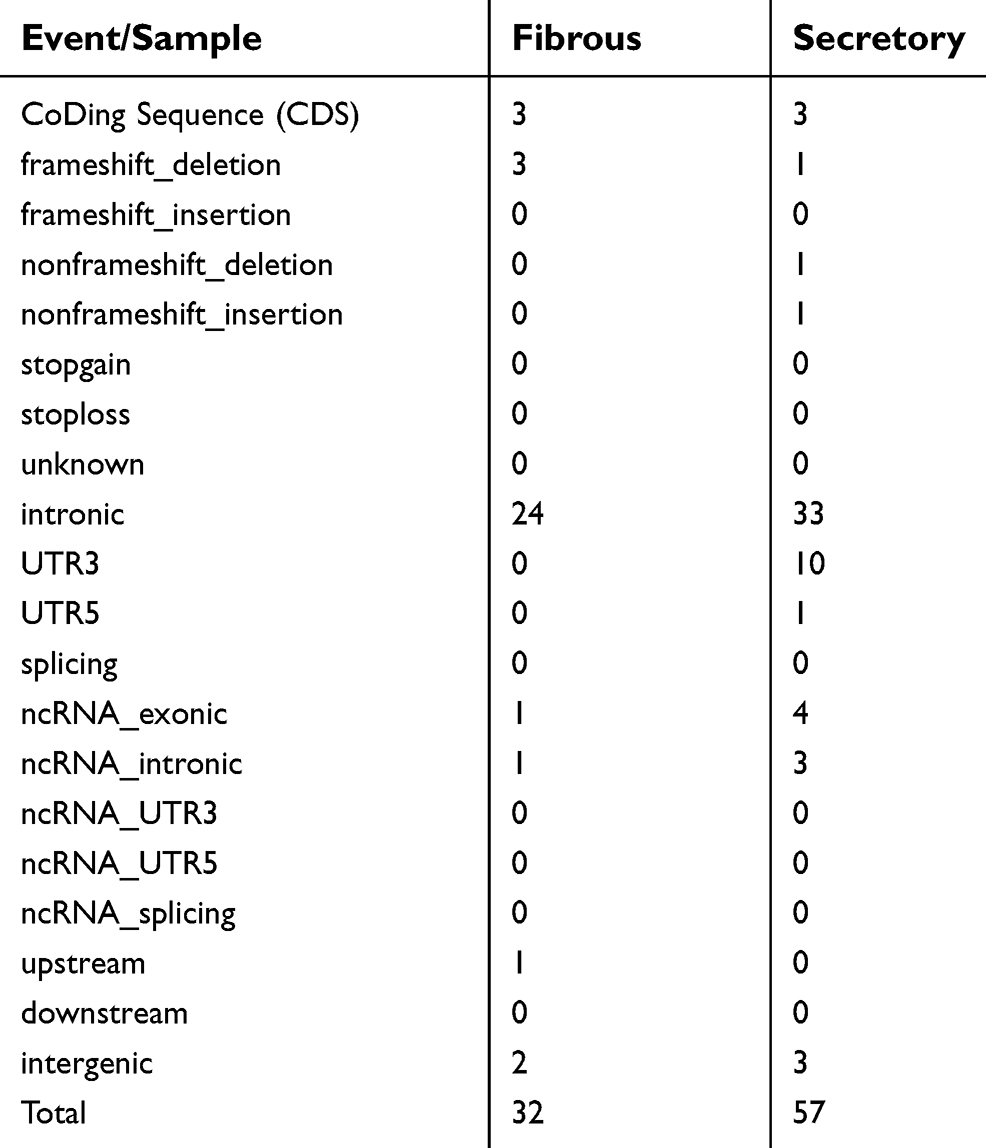 | Table S2 The number of somatic insertion and deletion (InDel) in different tumor samples |
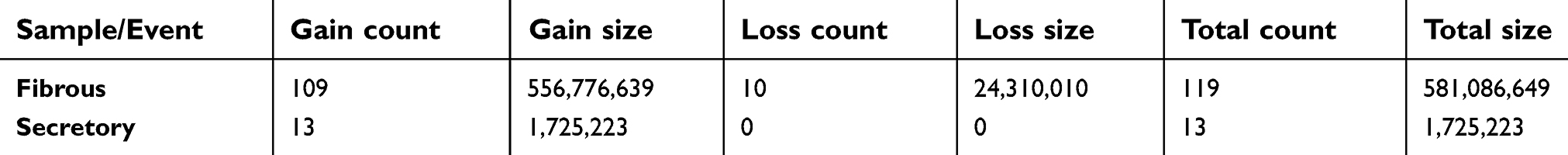 | Table S3 The number of somatic copy number variation (CNV) in different tumor samples |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.