Back to Journals » Infection and Drug Resistance » Volume 11
Which methods are appropriate for the detection of Staphylococcus argenteus and is it worthwhile to distinguish S. argenteus from S. aureus?
Authors Kaden R ![]() , Engstrand L, Rautelin H
, Engstrand L, Rautelin H ![]() , Johansson C
, Johansson C
Received 6 July 2018
Accepted for publication 11 October 2018
Published 21 November 2018 Volume 2018:11 Pages 2335—2344
DOI https://doi.org/10.2147/IDR.S179390
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Sahil Khanna
René Kaden,1 Lars Engstrand,2 Hilpi Rautelin,1 Cecilia Johansson1
1Department of Medical Sciences, Clinical Microbiology, Uppsala University, Uppsala, Sweden; 2Department of Microbiology, Tumor and Cell Biology, Karolinska Institute, Solna, Sweden
Purpose: To further analyze a clinical isolate originally identified as methicillin-resistant Staphylococcus aureus (MRSA) using whole-genome sequencing and comparative genomics.
Materials and methods: Classical diagnostic methods such as cultivation, biochemical tests, and PCR were supplemented with whole-genome sequencing and comparative genomics, to identify the isolate.
Results: The isolate was phenotypically similar to MRSA. However, the presence of the nuc gene could not be confirmed using PCR, while it was positive for the mecA gene. Whole-genome sequencing correctly identified the isolate as Staphylococcus argenteus. The isolate possessed several resistance genes, such as mecA, blaZ (β-lactam antibiotics) and dfrG (trimethoprim). The nuc gene differed from that of MRSA. Six phylogenetic distinct clusters were identified by average nucleotide identity (ANI) analysis of all available S. argenteus whole-genome sequences. Our isolate, RK308, clustered with those isolated in Europe and Asia.
Conclusion: Due to the invasive potential, the multi-drug resistance and the similarity to MRSA, S. argenteus should be included in the MRSA screening. Due to the divergent genome compared to MRSA, new PCR approaches have to be developed to avoid an unnoticed spreading of S. argenteus.
Keywords: Staphylococcus argenteus, Staphylococcus aureus, MRSA, whole-genome sequencing, clinical diagnostics
Introduction
The first outbreaks of methicillin-resistant Staphylococcus aureus (MRSA) were reported from European hospitals in the early 1960s.1 Gradually, MRSA turned out to be one of the main causes of nosocomial infections worldwide but also community-acquired infections are reported frequently.
S. aureus clonal complex 75 (CC75) was described as the distinct species Staphylococcus argenteus in 2015.2 S. argenteus usually have the same virulence factors3 and antibiotic resistance genes as S. aureus. The lack of genes for staphyloxanthin production first led to the conclusion that S. argenteus might be less virulent4 since this carotenoid pigment protects against oxidative stress and impairs neutrophil killing of S. aureus.5 This hypothesis could then be refuted through studies showing comparable morbidity and health care-associated infection rates for both species6,7 and furthermore, a recent study reported even increased mortality rates compared to S. aureus.8
Due to the close relationship between S. aureus and S. argenteus, it is difficult to distinguish the species with common diagnostic methods,9 as both species have identical 16S rRNA genes10,11 and also harbor thermostable nuclease genes, such as nuc. Positive mecA assays commonly lead to initial characterization of S. argenteus as MRSA.12
Here, we report how complementation of the classical diagnostic methods for S. aureus and MRSA screening with whole-genome sequencing correctly identified S. argenteus and also provided clues on virulence and antibiotic resistance.
Materials and methods
Bacterial isolates
The bacterial isolate RK308 was originally isolated from a clinical sample sent to the Clinical Microbiology Laboratory at Uppsala University Hospital and was thereafter anonymously analyzed in this study. The type strain of the species S. aureus 1800T and the MRSA CCUG 35601 reference strain were also included for comparison.
Phenotypical analyses
The original clinical sample was selectively enriched overnight at 37°C in MRSA broth (Iso-Sensitest broth, Oxoid, Basingstoke, United Kingdom) with 4 mg/L cefoxitin (Sigma-Aldrich, Steinheim, Germany) and 60 mg/L aztreonam (Bristol-Myers Squibb, Solna, Sweden). A broth volume of 5 µL was transferred to blood and S. aureus ID (SAID) agar plates (bioMérieux, La Balme Les Grottes, France) with a cefoxitin disc (30 µg; Oxoid, Basingstoke, United Kingdom) placed on the agar surface. Bacterial colonies that grew into the antibiotic zone that represented the chromogenic effect of SAID agar, and that had a phenotypical occurrence like that of S. aureus were chosen for further analysis. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF, Bruker Daltonik GmbH, Leipzig, Germany) was used to determine the bacterial species.13 Separate colonies were transferred to analysis plates, and 1 µL HCCA (α-Cyano-4-hydroxycinnamic acid) matrix was added to each sample spot to ionize peptides, smaller proteins, triacylglycerols and many other compounds in the initial phase of the analysis. Bruker’s default settings were used for analysis of the obtained spectra. The StaphAurex test (Remel, Lenexa, USA) was applied to confirm suspected S. aureus colonies. Furthermore, heat-inactivated colonies (100°C, 15 minutes) were placed on DNase Agar (Thermo Fisher Scientific, Basingstoke, United Kingdom) plates for confirming extracellular heat-stable DNase activity. The ID32 Staph system (bioMérieux, SA, Marcy l’Etoile, France), commonly used for phenotypical differentiation of 27 staphylococcal species,14 was also applied.
PCR analyses
The DNA from one pure colony was extracted using the Amplicor Respiratory Specimen Preparation Kit (Roche Diagnostics, Mannheim, Germany). The presence or absence of the thermostable nuclease gene (nuc) and the mecA gene was tested by PCR with the primers 5´-TCA GCA AAT GCA TCA CAA ACA G-3´ and 5´-CGT AAA TGC ACT TGC TTC AGG-3´ specific for the nuc gene and 5´-GGG ATC ATA GCG TCA TTA TTC-3´ and 5´-AAC GAT TGT GAC ACG ATA GCC-3´ specific for the mecA gene.15 MRSA broth without bacteria was used as the negative control while the positive control was the MRSA strain CCUG 35601. PCR was performed using a GeneAmp PCR System 9700 (Applied Biosystems, Houston, USA) with initial denaturation at 95°C for 15 minutes and 35 cycles at 94°C for 30 seconds, 55°C for 30 seconds and 72°C for 30 seconds.
Whole genome sequencing
Pure colonies from blood agar plates were transferred to MRSA broth (Oxoid) containing cefoxitin (4 mg/L) and aztreonam (60 mg/L) and incubated overnight at 37°C. DNA extraction was performed from 400 µL of broth with MagNA Pure Compact Nucleic Acid Isolation Kit I (Roche, Mannheim, Germany) according to the manufacturer’s protocol version 12. The libraries for WGS were prepared with a Nextera XT sample preparation kit (Illumina, San Diego, USA). An Illumina HiSeq platform with a 2 × 100 paired end run was used for WGS. The single reads were assembled to contigs with Velvet.16 The paired reads and merging contigs were assembled by Geneious version 8.1.5.17 The average nucleotide identity (ANI), which is based on base by base comparison, to closely related taxa was calculated using the Gegenees software version 2.2.1 with a threshold of 20%.18 The ANI-based phylogenetic tree was constructed in Geneious. The PathoFinder 1.1 database of the Center for Genomic Epidemiology Denmark19 was used to predict the pathogenicity.
Ethical considerations
The isolate studied was obtained as part of the routine hospital laboratory procedure in the identification of infectious agents for the patient. Patient data were kept anonymous for analysis in the research project. As the study only focused on a bacterial isolate, neither written informed consent nor ethical approval was necessary for the study, according to the Swedish act concerning the ethical review of research involving humans, Etikprövningslagen (2003:460).
Results
Identification of RK308 with classical methods
The analyzed isolate, RK308 grew on blood agar plates like S. aureus with beta hemolysis but as whitish colonies, similar to MRSA CCUG 35601 (Figure 1A). On SAID agar plates, the RK308 colonies were clear blue while S. aureus was green (Figure 1). The DNase reaction, as well as the StaphAurex test, were positive for RK308 (Table 1). The analyzed isolate was identified as S. aureus with MALDI-TOF with a score value of 1.916 in the corresponding database (Bruker). ID32 Staph analysis also resulted in determination as S. aureus according to the ID32 Staph manual. To determine if the isolate was an MRSA, a multiplex PCR for amplification of a nuc gene fragment and a mecA gene fragment was applied. The PCR gave a negative signal for nuc but a positive signal for mecA (Table 1), ruling out other nuc-negative Staphylococci.
 | Figure 1 (A) S. argenteus RK308, S. aureus 1800 and MRSA CCUG 35601 on blood agar plates. (B) S. aureus 1800 (green) and S. argenteus RK308 (blue) on SAID agar plate with cefoxitin disc (30 µg). Abbreviations: MRSA, methicillin-resistant Staphylococcus aureus; SAID, S. aureus ID. |
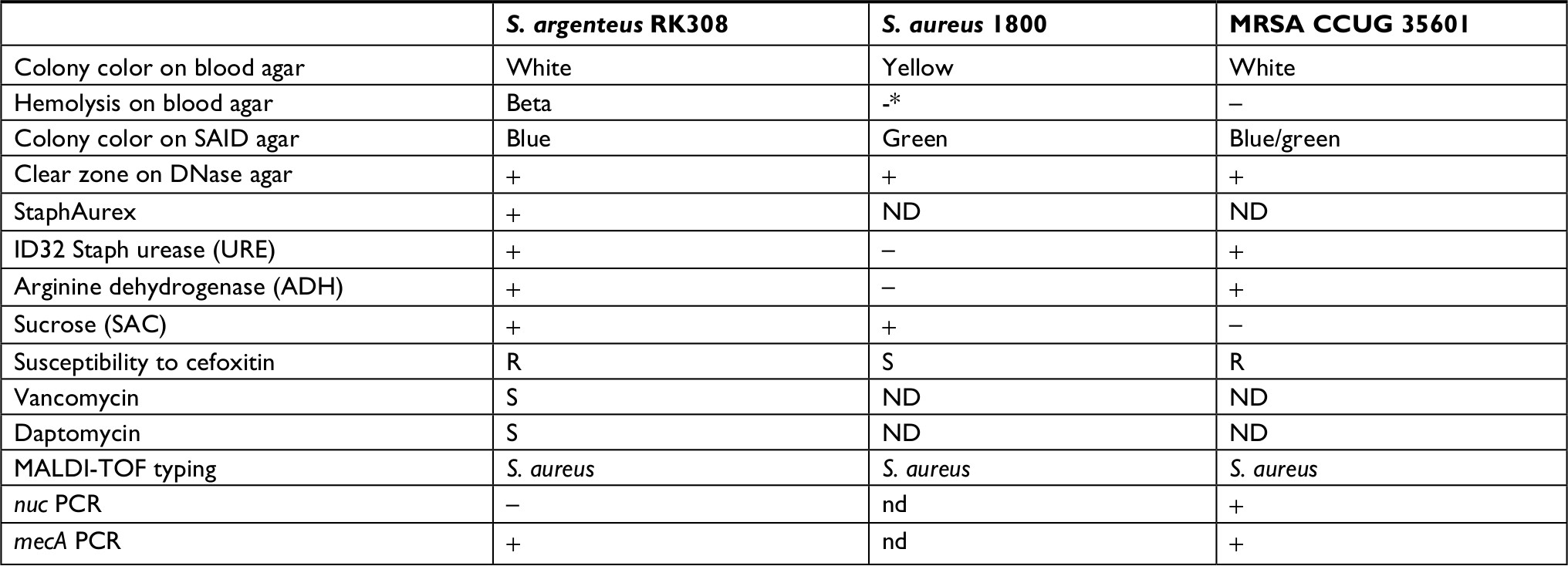 | Table 1 Summary of phenotypical characterization of S. argenteus RK308, S. aureus 1800 and MRSA CCUG 35601 Notes: +, positive result; -, negative result; R, resistant; S, sensitive; ND, not determined. *, unusual since many S. aureus strains are hemolysin positive Abbreviations: MALDI-TOF, Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry; MRSA, methicillin-resistant Staphylococcus aureus; SAID, S. aureus ID. |
Genomic characterization
The RK308 isolate was whole-genome sequenced. Gap closing was not performed for the RK308 genome since it is not applicable to clinical diagnostic approaches and not essential for the current analysis.20 The final assemblies are available from the NCBI database (BioProject number PRJNA310972, GenBank/EMBL/DDBJ accession number LSFQ00000000).
The S. aureus strain Newman (NCBI accession AP009351), which is a representative strain of S. aureus,21 shares 93% identity of the whole genome with S. aureus subspecies anaerobius. These strains showed 74% and 73% identical bases with RK308 in ANI-analysis, which is based on base by base comparison, respectively, while the whole genomes of S. argenteus MSHR1132T and RK308 had 97.2% identical bases.
All available S. argenteus whole genome sequences (n=116, May 2018, Table S1) were compared using ANI-analysis. The diversity of the analyzed genomes ranged from 0% to 4.2% (data not shown). Phylogenetic analysis identified six clusters (I-VI, Figure 2), where RK308 clustered with strains mainly isolated in Asia (cluster VI), although its closest relatives seemed to be from Europe (Figure 2).
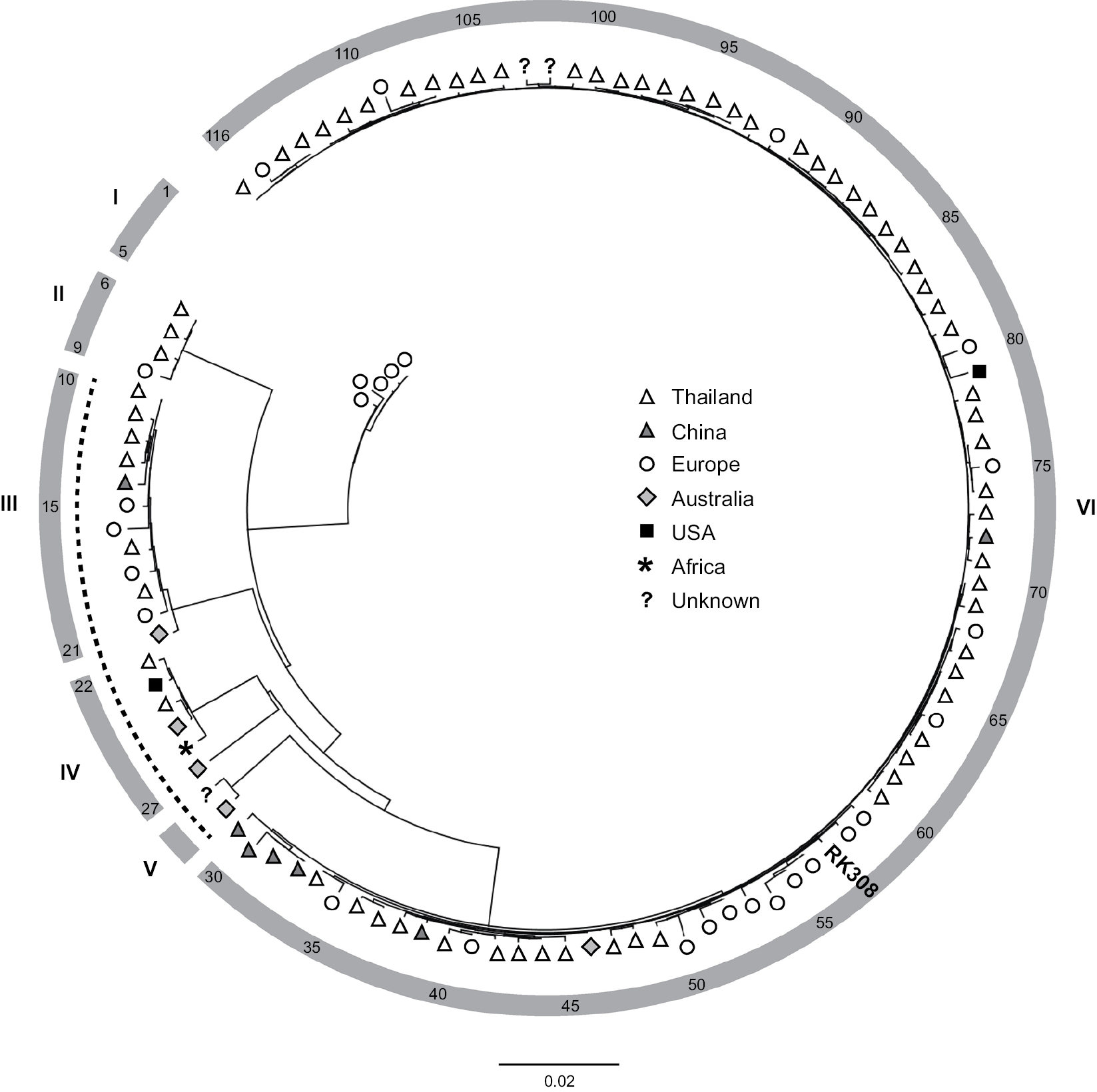 | Figure 2 Phylogenetic ANI-based analysis of S. argenteus whole-genome sequences. Notes: Symbols indicate geographical location of isolation. The position of isolate RK308 is indicated (#57). Numbers in outer circle refer to isolate numbers from Table S1. Roman numerals correspond to identified clusters I-VI. The dotted line indicates PVL-negative isolates. Abbreviation: PVL, Panton-Valentine leucocidins. |
The spa gene was extracted from the whole genome. The spa-type was t6675 (299-25-17-17-16-16-16). However, the spa gene contained 3 variations in the binding site of the forward primer compared to S. aureus. Furthermore, a low frequent G → A variation was observed in the spa gene.
Both thermonucleases of strain Newman were found in RK308. For one of them, amino acid alignment showed high variation in the N-terminal part of the protein (Figure 3A), while the other variant was very similar (Figure 3B).
 | Figure 3 Amino acid alignment of two heat-stable nucleases of S. aureus Newman and S. argenteus RK308 (A and B). Notes: Black: identical amino acids; Gray: similar amino acids; White: different amino acid; line: gaps. |
All existing 16S rRNA gene sequences of the whole genome were blasted22 to ensure that there was no contamination within the culturing and sequencing procedure. All sequences were determined as S. aureus with an identity of 99%.
Presence of virulence and antibiotic resistance genes
Resistance genes for β-lactams (mecA), penicillins (blaZ) and trimethoprim (dfrG) were detected in the whole genome of RK308. The mecA gene had three-point mutations located outside of the first open reading frame but was otherwise completely functional in vitro. There was no variation within the mecA primer binding sites of the primers listed above. Further analysis of the RK308 staphylococcal cassette chromosome mec (SCCmec) showed that it contained a class A mec gene complex, harboring the mecA, mecR1, and mecI genes and a type 2 (A2B2) ccr gene complex, resulting in a SCCmec type II according to the nomenclature.23 Furthermore, the RK308 SCCmec had almost identical nucleotide sequence to that of MRSA USA300.
The mprF gene, involved in daptomycin resistance, was also detected in the whole genome but did not have any of the previously reported amino acid substitutions involved in resistance24 and RK308 did not show resistance to daptomycin in in vitro susceptibility testing (data not shown). Neither did RK308 show vancomycin resistance in vitro, and nor were vancomycin resistance genes found in silico.
RK308 was predicted as a human pathogen with a probability of 95.3% and with 100 matched pathogenic families according to the database of the Center for Genomic Epidemiology Denmark.19 Further in silico analyses revealed the presence of the Panton-Valentine leucocidins (PVL, lukF-PV, and lukS-PV) in RK308 and in 83% (96/116) of all the isolates included here. Interestingly, all PVL-negative isolates (isolates #10–29, Table S1), including the S. argenteus type strain MSHR1132, were found in clusters III, IV and V (Figure 2), while all isolates in clusters I, II and VI contained PVL. Other toxin genes, such as gamma-hemolysin (hlg) and exfoliative toxin A (eta) were also found in RK308, while the toxic shock syndrome toxin gene (tsst) and enterotoxins sea-see were not found in RK308.
Discussion
Despite the fact that S. aureus is known to have a clonal population structure,25 a genetic divergence of 26% of the whole genome has been determined. Regarding the mutation frequency that was estimated to an average of 9.2 mutations per year,26 S. argenteus and S. aureus have divided about 41, 000 years ago from the common ancestors.
The species determination of S. argenteus is challenging with classical methods,9 and here RK308 was incorrectly identified as S. aureus. The cultivation on SAID agar plates on which the isolate grew with a deviant gray color raised doubts about the correct species identification. The isolate was therefore further analysed with an MRSA-specific multiplex PCR. The mecA gene was detected with PCR while the nuc gene (thermostable nuclease), which is a common marker for S. aureus, gave a negative result. However, two thermostable nuclease genes were found in the genome where the one for which diagnostic primers are used showed many variations between S. aureus strain Newman and S. argenteus RK308, possibly explaining the negative PCR. The positive reaction on DNase plates might, therefore, be due to redundant functions of the two thermostable nuclease genes, as previously reported.27
The variations in the binding site of the forward primer in the spa gene may also lead to difficulties in conventional PCR for spa typing.
With the results of WGS, the isolate RK308 could be assigned to the species S. argenteus. While S. aureus is a common species and an important cause of both nosocomial and community-acquired infections, S. argenteus occurs rarely but worldwide (Table S1). However, the real distribution and prevalence of S. argenteus are still unclear. Due to the difficulties in diagnostics, a high number of incorrectly diagnosed S. aureus / MRSA can be suspected if the methods are not appropriate to distinguish between S. aureus and S. argenteus. Thaipadungpanit et al6 reported 4.1% of 246 molecularly typed S. aureus isolates to be S. argenteus. However, the patients might have received the appropriate treatment even if the species identification has been wrong. The results from the in vitro susceptibility testing here showed that RK308 was susceptible to both vancomycin and daptomycin, which would offer good options for treatment. A worse scenario is the application of diagnostic methods specific for S. aureus, like the multiplex PCR described above, which might lead to the unrecognition and further unnoticed spread of S. argenteus. The PCR method described above15 is used by national and reference laboratories worldwide and is considered to be robust and reliable, and a recommended method for detecting MRSA. However, the presence of the complete SCCmec cassette in RK308 might open up new options for screening and detection of S. argenteus in the future.
On the basis of the present results, it seems obvious that if both species need to be detected simultaneously, alternative methods have to be applied. There is no consensus, neither national nor international, whether S. argenteus should be included in or excluded from diagnostics of mecA-positive bacteria.28 S. argenteus was previously denoted S. aureus CC75 and all detected MRSA CC75-cases were conclusively clinically treated as MRSA. Conclusively, the updated method for detection of MRSA at the laboratory of the Uppsala University Hospital now also includes S. argenteus and will do so until there is a clear definition in the Communicable Disease Act. Correct identification and separation of the two species will be important for epidemiological studies but might have an even larger impact on clinical practice and treatments as new studies point toward significant mortality rates for S. argenteus combined with sustained susceptibility to several antibiotics.7,8
Although the presence of several resistance genes and the high prevalence of PVL would suggest that S. argenteus is able to cause infections similar to those of S. aureus, the effect of RK308 on human cells remains to be tested.
Less is also known about the S. argenteus transmission ways, but several hosts and sources of isolation have been described. According to the metadata belonging to the deposited sequences in the NCBI database,22 the majority of S. argenteus isolates have been isolated from humans but also from various animals, such as gorilla and bats,29,30 and from environmental samples of unknown origin. Although globally distributed, most of the S. argenteus isolates originate from Asia. RK308 clustered among other European and Asian isolates of clonal complex 225031 in the phylogenetic tree.
Conclusion
The differentiation between methicillin-resistant S. argenteus and MRSA is challenging. In this study, all clinical diagnostic methods failed to identify S. argenteus correctly while identification to the species level was obtained by WGS. The S. argenteus isolates studied here, RK308, possessed several virulences and resistance genes usually detected in MRSA, which suggests that S. argenteus could be as virulent as MRSA and should not be left unnoticed. To prevent future spread, appropriate diagnostic methods should be used.
Acknowledgments
The authors thank Hugo Wefer and Cecilia Svensson for fruitful discussions.
Disclosure
The authors report no conflicts of interest in this work.
References
Chambers HF. The changing epidemiology of Staphylococcus aureus? Emerg Infect Dis. 2001;7(2):178–182. | ||
Tong SY, Schaumburg F, Ellington MJ, et al. Novel staphylococcal species that form part of a Staphylococcus aureus-related complex: the non-pigmented Staphylococcus argenteus sp. nov. and the non-human primate-associated Staphylococcus schweitzeri sp. nov. Int J Syst Evol Microbiol. 2015;65(Pt 1):15–22. | ||
Zhang DF, Zhi XY, Zhang J, et al. Preliminary comparative genomics revealed pathogenic potential and international spread of Staphylococcus argenteus. BMC Genomics. 2017;18(1):808. | ||
Tong SY, Sharma-Kuinkel BK, Thaden JT, et al. Virulence of endemic nonpigmented northern Australian Staphylococcus aureus clone (clonal complex 75, S. argenteus) is not augmented by staphyloxanthin. J Infect Dis. 2013;208(3):520–527. | ||
Liu GY, Essex A, Buchanan JT, et al. Staphylococcus aureus golden pigment impairs neutrophil killing and promotes virulence through its antioxidant activity. J Exp Med. 2005;202(2):209–215. | ||
Thaipadungpanit J, Amornchai P, Nickerson EK, et al. Clinical and molecular epidemiology of Staphylococcus argenteus infections in Thailand. J Clin Microbiol. 2015;53(3):1005–1008. | ||
Chantratita N, Wikraiphat C, Tandhavanant S, et al. Comparison of community-onset Staphylococcus argenteus and Staphylococcus aureus sepsis in Thailand: a prospective multicentre observational study. Clin Microbiol Infect. 2016;22(5):458.e11–458.e19. | ||
Chen SY, Lee H, Wang XM, et al. High mortality impact of Staphylococcus argenteus on patients with community-onset staphylococcal bacteraemia. Int J Antimicrob Agents. Epub 2018 Aug 24. | ||
Tunsjø HS, Kalyanasundaram S, Charnock C, Leegaard TM, Moen AEF. Challenges in the identification of methicillin-resistant Staphylococcus argenteus by routine diagnostics. APMIS. 2018;126(6):533–537. | ||
Ng JW, Holt DC, Lilliebridge RA, et al. Phylogenetically distinct Staphylococcus aureus lineage prevalent among indigenous communities in northern Australia. J Clin Microbiol. 2009;47(7):2295–2300. | ||
Ruimy R, Armand-Lefevre L, Barbier F, et al. Comparisons between geographically diverse samples of carried Staphylococcus aureus. J Bacteriol. 2009;191(18):5577–5583. | ||
Tång Hallbäck E, Karami N, Adlerberth I, et al. Methicillin-resistant Staphylococcus argenteus misidentified as methicillin-resistant Staphylococcus aureus emerging in western Sweden. J Med Microbiol. Epub 2018 May 17. | ||
Seng P, Drancourt M, Gouriet F, et al. Ongoing revolution in bacteriology: routine identification of bacteria by matrix-assisted laser desorption ionization time-of-flight mass spectrometry. Clin Infect Dis. 2009;49(4):543–551. | ||
Chesneau O, Aubert S, Morvan A, Guesdon JL, El Solh N. Usefulness of the ID32 staph system and a method based on rRNA gene restriction site polymorphism analysis for species and subspecies identification of staphylococcal clinical isolates. J Clin Microbiol. 1992;30(9):2346–2352. | ||
Poulsen AB, Skov R, Pallesen LV. Detection of methicillin resistance in coagulase-negative staphylococci and in staphylococci directly from simulated blood cultures using the EVIGENE MRSA Detection Kit. J Antimicrob Chemother. 2003;51(2):419–421. | ||
Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18(5):821–829. | ||
Kearse M, Moir R, Wilson A, et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28(12):1647–1649. | ||
Ågren J, Sundström A, Håfström T, Segerman B. Gegenees: fragmented alignment of multiple genomes for determining phylogenomic distances and genetic signatures unique for specified target groups. PLoS One. 2012;7(6):e39107. | ||
Cosentino S, Voldby Larsen M, Møller Aarestrup F, Lund O. PathogenFinder – distinguishing friend from foe using bacterial whole genome sequence data. PLoS One. 2013;8(10):e77302. | ||
Greub G, Kebbi-Beghdadi C, Bertelli C, et al. High throughput sequencing and proteomics to identify immunogenic proteins of a new pathogen: the dirty genome approach. PLoS One. 2009;4(12):e8423. | ||
Baba T, Bae T, Schneewind O, Takeuchi F, Hiramatsu K. Genome sequence of Staphylococcus aureus strain Newman and comparative analysis of staphylococcal genomes: polymorphism and evolution of two major pathogenicity islands. J Bacteriol. 2008;190(1):300–310. | ||
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410. | ||
International Working Group on the Classification of Staphylococcal Cassette Chromosome Elements (IWG-SCC). Classification of staphylococcal cassette chromosome mec (SCCmec): guidelines for reporting novel SCCmec elements. Antimicrob Agents Chemother. 2009;53(12):4961–4967. | ||
Bayer AS, Mishra NN, Chen L, Kreiswirth BN, Rubio A, Yang SJ. Frequency and Distribution of Single-Nucleotide Polymorphisms within mprF in Methicillin-Resistant Staphylococcus aureus Clinical Isolates and Their Role in Cross-Resistance to Daptomycin and Host Defense Antimicrobial Peptides. Antimicrob Agents Chemother. 2015;59(8):4930–4937. | ||
Feil EJ, Cooper JE, Grundmann H, et al. How clonal is Staphylococcus aureus? J Bacteriol. 2003;185(11):3307–3316. | ||
Harris SR, Feil EJ, Holden MT, et al. Evolution of MRSA during hospital transmission and intercontinental spread. Science. 2010;327(5964):469–474. | ||
Tang J, Zhou R, Shi X, Kang M, Wang H, Chen H. Two thermostable nucleases coexisted in Staphylococcus aureus: evidence from mutagenesis and in vitro expression. FEMS Microbiol Lett. 2008;284(2):176–183. | ||
Bogestam K, Vondracek M, Karlsson M, Fang H, Giske CG. Introduction of a hydrolysis probe PCR assay for high-throughput screening of methicillin-resistant Staphylococcus aureus with the ability to include or exclude detection of Staphylococcus argenteus. PLoS One. 2018;13(2):e0192782. | ||
Olatimehin A, Shittu AO, Onwugamba FC, Mellmann A, Becker K, Schaumburg F. Staphylococcus aureus Complex in the Straw-Colored Fruit Bat (Eidolon helvum) in Nigeria. Front Microbiol. 2018;9:162. | ||
Schuster D, Rickmeyer J, Gajdiss M, et al. Differentiation of Staphylococcus argenteus (formerly: Staphylococcus aureus clonal complex 75) by mass spectrometry from S. aureus using the first strain isolated from a wild African great ape. Int J Med Microbiol. 2017;307(1):57–63. | ||
Hansen TA, Bartels MD, Høgh SV, et al. Whole Genome Sequencing of Danish Staphylococcus argenteus Reveals a Genetically Diverse Collection with Clear Separation from Staphylococcus aureus. Front Microbiol. 2017;8:1512. |
Supplementary material
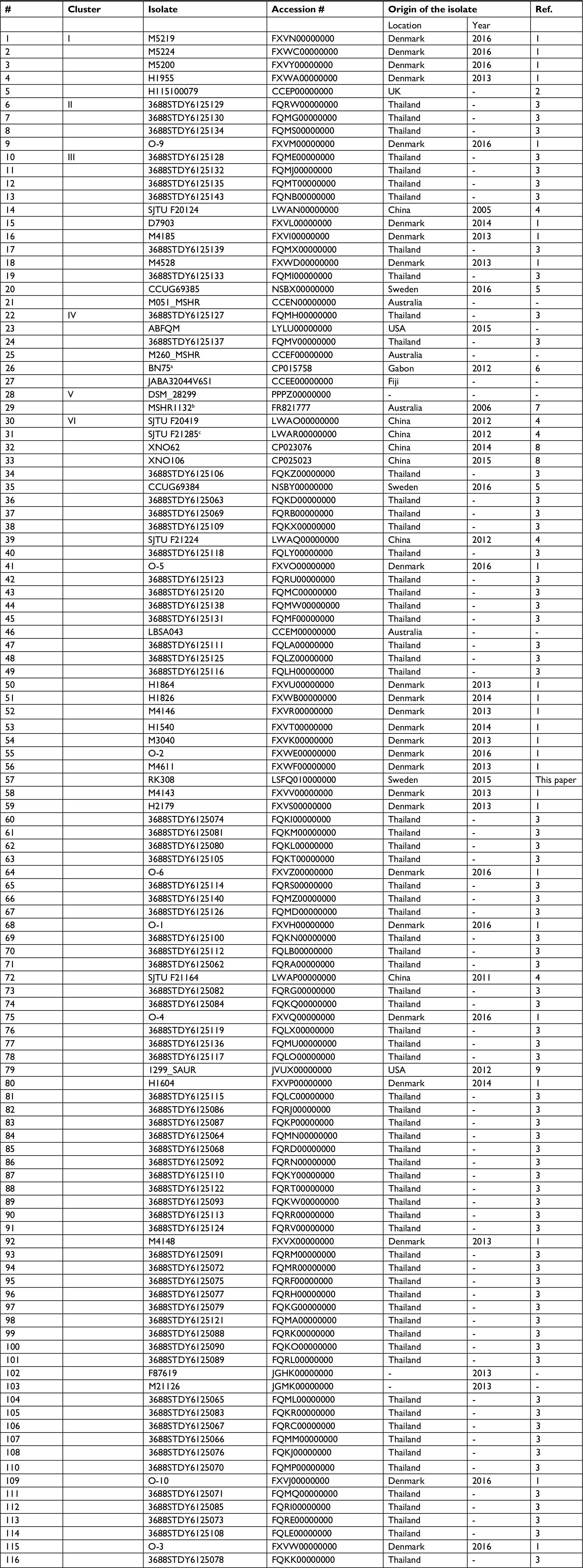 | Table S1 S. argenteus genomes used in in silico analyses Note: aOrigin: Gorilla, bType strain of S. argenteus, cOrigin: Pig, -; unknown |
References
Hansen TA, Bartels MD, Hogh SV, et al. Whole Genome Sequencing of Danish Staphylococcus argenteus Reveals a Genetically Diverse Collection with Clear Separation from Staphylococcus aureus. Front Microbiol. 2017;8:1512. | ||
Tong SY, Schaumburg F, Ellington MJ, et al. Novel staphylococcal species that form part of a Staphylococcus aureus-related complex: the non-pigmented Staphylococcus argenteus sp. nov. and the non-human primate-associated Staphylococcus schweitzeri sp. nov. Int J Syst Evol Microbiol. 2015;65(Pt 1):15–22. | ||
Thaipadungpanit J, Amornchai P, Nickerson EK, et al. Clinical and molecular epidemiology of Staphylococcus argenteus infections in Thailand. J Clin Microbiol. 2015;53(3):1005–1008. | ||
Zhang DF, Zhi XY, Zhang J, et al. Preliminary comparative genomics revealed pathogenic potential and international spread of Staphylococcus argenteus. BMC Genomics. 2017;18(1):808. | ||
Tang Hallback E, Karami N, Adlerberth I, et al. Methicillin-resistant Staphylococcus argenteus misidentified as methicillin-resistant Staphylococcus aureus emerging in western Sweden. J Med Microbiol. Epub 2018 May 17. | ||
Schuster D, Rickmeyer J, Gajdiss M, et al. Differentiation of Staphylococcus argenteus (formerly: Staphylococcus aureus clonal complex 75) by mass spectrometry from S. aureus using the first strain isolated from a wild African great ape. Int J Med Microbiol. 2017;307(1):57–63. | ||
Holt DC, Holden MT, Tong SY, et al. A very early-branching Staphylococcus aureus lineage lacking the carotenoid pigment staphyloxanthin. Genome Biol Evol. 2011;3:881–895. | ||
Jiang B, You B, Tan L, et al. Clinical Staphylococcus argenteus Develops to Small Colony Variants to Promote Persistent Infection. Front Microbiol. 2018;9:1347. | ||
Roach DJ, Burton JN, Lee C, et al. A Year of Infection in the Intensive Care Unit: Prospective Whole Genome Sequencing of Bacterial Clinical Isolates Reveals Cryptic Transmissions and Novel Microbiota. PLoS Genet. 2015;11(7):e1005413. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.