Back to Journals » Therapeutics and Clinical Risk Management » Volume 14
What is known about deferasirox chelation therapy in pediatric HSCT recipients: two case reports of metabolic acidosis
Authors Fucile C, Mattioli F ![]() , Marini V, Gregori M, Sonzogni A, Martelli A
, Marini V, Gregori M, Sonzogni A, Martelli A ![]() , Maximova N
, Maximova N ![]()
Received 17 April 2018
Accepted for publication 24 June 2018
Published 7 September 2018 Volume 2018:14 Pages 1649—1655
DOI https://doi.org/10.2147/TCRM.S170761
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Carmen Fucile,1 Francesca Mattioli,1 Valeria Marini,1 Massimo Gregori,2 Aurelio Sonzogni,3 Antonietta Martelli,1 Natalia Maximova4
1Pharmacology and Toxicology Unit, University of Genoa, Genoa, Italy; 2Department of Pediatric Radiology, Institute for Maternal and Child Health – IRCCS Burlo Garofalo, Trieste, Italy; 3Department of Pathology, Ospedale Beato Papa Giovanni XIII, Bergamo, Italy; 4Bone Marrow Transplant Unit, Institute for Maternal and Child Health – IRCCS Burlo Garofalo, Trieste, Italy
Abstract: To date, in pediatric field, various hematological malignancies are increasingly treated with allogeneic hematopoietic stem cell transplantation (allo-HSCT). Iron overload and systemic siderosis often occur in this particular cohort of patients and are associated with poor prognosis. We describe herein the case of two allo-HSCT patients, on treatment with deferasirox; they showed histopathological elements compatible with venoocclusive disease or vanishing bile duct syndrome in ductopenic evolution before deferasirox started. The first patient developed drug-induced liver damage with metabolic acidosis and the second one a liver impairment with Fanconi syndrome. After withdrawing deferasirox treatment, both patients showed improvement. Measurements of drug plasma concentrations were performed by HPLC assay. The reduction and consequent disappearance of symptoms after the suspension of deferasirox substantiate its role in inducing hepatic damage, probably enabling the diagnosis of drug-induced liver damage. But the difficulties in diagnosing drug-related toxicity must be underlined, especially in compromised subjects. For these reasons, in patients requiring iron-chelating therapy, close and careful drug therapeutic monitoring is strongly recommended.
Keywords: deferasirox, allogeneic hematopoietic stem cell transplantation, allo-HSCT, pediatric, ductopenia, therapeutic drug monitoring, adverse events
Introduction
Allogeneic hematopoietic stem cell transplantation (allo-HSCT) is an increasingly used treatment modality for a wide variety of hematological malignancies and benign disease in the pediatric population. Iron overload (IO) has been associated with poor prognosis in patients undergoing allo-HSCT.1–5 Systemic siderosis in this cohort of patients is multifactorial and is not exclusively due to intense transfusion regimen.6 Intensive cytotoxic therapy before HSCT destroys both bone marrow and neoplastic cells, releasing internal iron contents from cells and increasing free iron concentrations.7 High-intensity myeloablative conditioning prior to HSCT can damage hepatic cells, which releases cellular iron pools and increases further iron load.8
Liver dysfunction (LD) is a common problem in these patients, both before and after transplantation. Almost half of pediatric HSCT patients have laboratory evidence of LD before transplantation, and 74% to 85.5% have post-transplantation transaminase elevations.9,10 Many causes of LD are well known, such as graft-versus-host disease (GVHD), drug hepatotoxicity, viral hepatitis, sepsis, venoocclusive disease (VOD), and disease recurrence. Nevertheless, a lesser proportion of liver complications are still classified as of undetermined cause.
Deferasirox (DFX), an oral iron chelator, is widely used for the treatment of patients with IO. Excessive iron accumulation often results in tissue damage and organ failure; it has been shown that pre-HSCT chelation with DFX leads to a rapid decrease in the labile plasma iron, improving survival. However, there are only few studies regarding the effects of chelation therapy in the post-transplant period in a pediatric setting or about the safety and tolerability of DFX in pediatric patients who developed LD after allo-HSCT.2,11
The aim of our study was to describe the adverse events (AEs) observed in two allo-HSCT patients treated with DFX. We have analyzed the development of symptoms before and after therapy discontinuation and the possible association with DFX.
Case reports
Case 1
The first patient was a 4-year-old male child (whose mother was an Ethiopian) affected by acute lymphoblastic leukemia who underwent allo-HSCT after total body irradiation (TBI)-based myeloablative conditioning. GVHD prophylaxis was performed with tacrolimus only. Prophylaxis of liver complications was performed with ursodeoxycholic acid (UDCA), 30 mg/kg/day, from the first day of conditioning. During the pre-transplant work-up, the child was subjected to abdominal magnetic resonance evaluation of abdominal organ iron concentration. He was found to have severe IO with an iron concentration value higher than 200 μmol/g (normal value: <40 μmol/g) in three of the four organs evaluated (ie, in spleen, bone, and liver, except pancreas), and the child underwent chelation therapy with deferoxamine (30 mg/kg/day, continuous intravenous infusion) during the entire stay in the Transplant Unit.
The post-transplantation period was complicated by a severe VOD, with a complete inversion of the portal flow and an important ascite (Figure 1). The severity of the clinical picture was further complicated by a hyperacute GVHD, characterized by hypertransaminasemia with aspartate aminotransferase and an alanine aminotransferase value higher than 800 U/L (normal values: <38 U/L and <25 U/L, respectively). The portal flow returned to normal after 10 days of defibrotide treatment. Hepatic GVHD resolved completely after three doses of infliximab.
 | Figure 1 Case 1: color Doppler sonographic evaluation. Complete inversion of the portal venous flow. |
After ascites resolution, a liver biopsy was performed and a histological evaluation showed the presence of central ischemic-hemorrhagic necrosis suggestive of VOD and a high degree of IO. Another histological feature was ductopenia (absence of interlobular bile ducts in portal tracts) suggestive of liver GVHD (Figure 2).
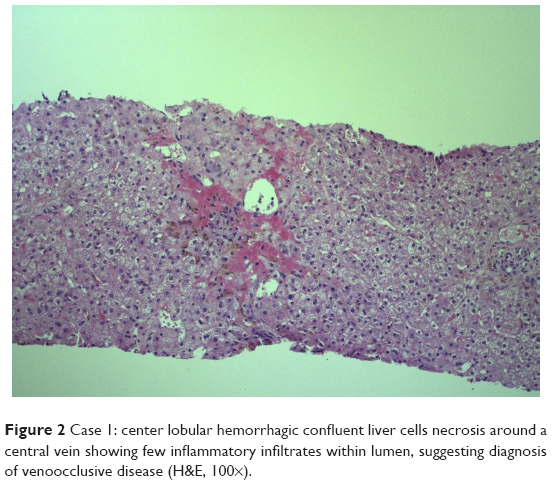 | Figure 2 Case 1: center lobular hemorrhagic confluent liver cells necrosis around a central vein showing few inflammatory infiltrates within lumen, suggesting diagnosis of venoocclusive disease (H&E, 100×). |
The patient underwent a second MRI-based iron load evaluation before discharge from our Transplant Unit and showed a general worsening of iron accumulation. All four abdominal organs evaluated had an abnormal iron concentration. The liver, spleen, and bone showed a higher degree of siderosis compared to pre-transplant values. Ferritin values stabilized around 3,000 ng/mL (normal range: 20–300 ng/mL).
Therefore, the patient was treated with DFX at a dosage of 375 mg/day (17.6 mg/kg). One month later, he was readmitted to our Unit with a marked deterioration of clinical conditions. He experienced fatigue, weakness, nausea, vomiting, abdominal pain, and anorexia that had begun one week before. Laboratory investigations on admission showed a normal white blood cell count with mild neutropenia, a normal platelets count, and hemoglobin concentration. His liver and renal function was mildly deranged. Furthermore, he was found to have a metabolic acidosis.
Primary disease recurrence was excluded. Prednisone at a dosage of 1 mg/kg was added to tacrolimus as systemic GVHD was suspected. To correct acidosis, the patient was hydrated by administering electrolyte solution enriched with bicarbonate.
Measurement, by high performance liquid chromatography (HPLC) assay, previously described and validated,12,13 of DFX plasma concentration confirmed a moderate increase of the drug plasmatic level (Ctrough =27 μM; normal Ctrough value <20 μM; CDFX/DDFX ratio =0.56). Tacrolimus therapeutic plasma concentrations were maintained at about 10 ng/mL.
He continued to demonstrate mild liver and renal dysfunction during the following month, while metabolic acidosis progressively worsened. The child underwent another liver biopsy that showed a histological feature suggestive of drug-induced liver injury (DILI) (Figure 3). After 79 days of chelation treatment, DFX was immediately stopped. His biochemical parameters normalized rapidly after discontinuation of DFX and the bicarbonate supplement was stopped two weeks later (see Table 1).
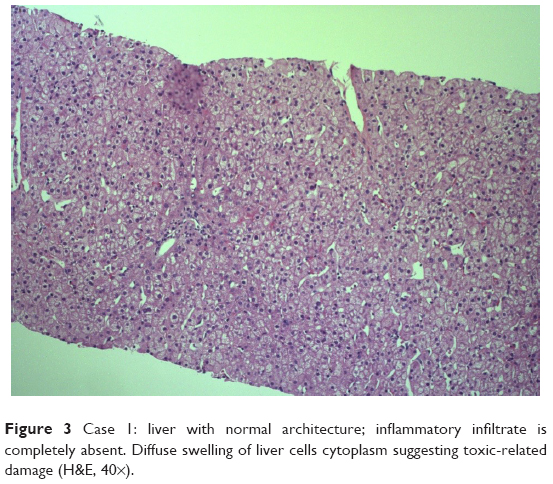 | Figure 3 Case 1: liver with normal architecture; inflammatory infiltrate is completely absent. Diffuse swelling of liver cells cytoplasm suggesting toxic-related damage (H&E, 40×). |
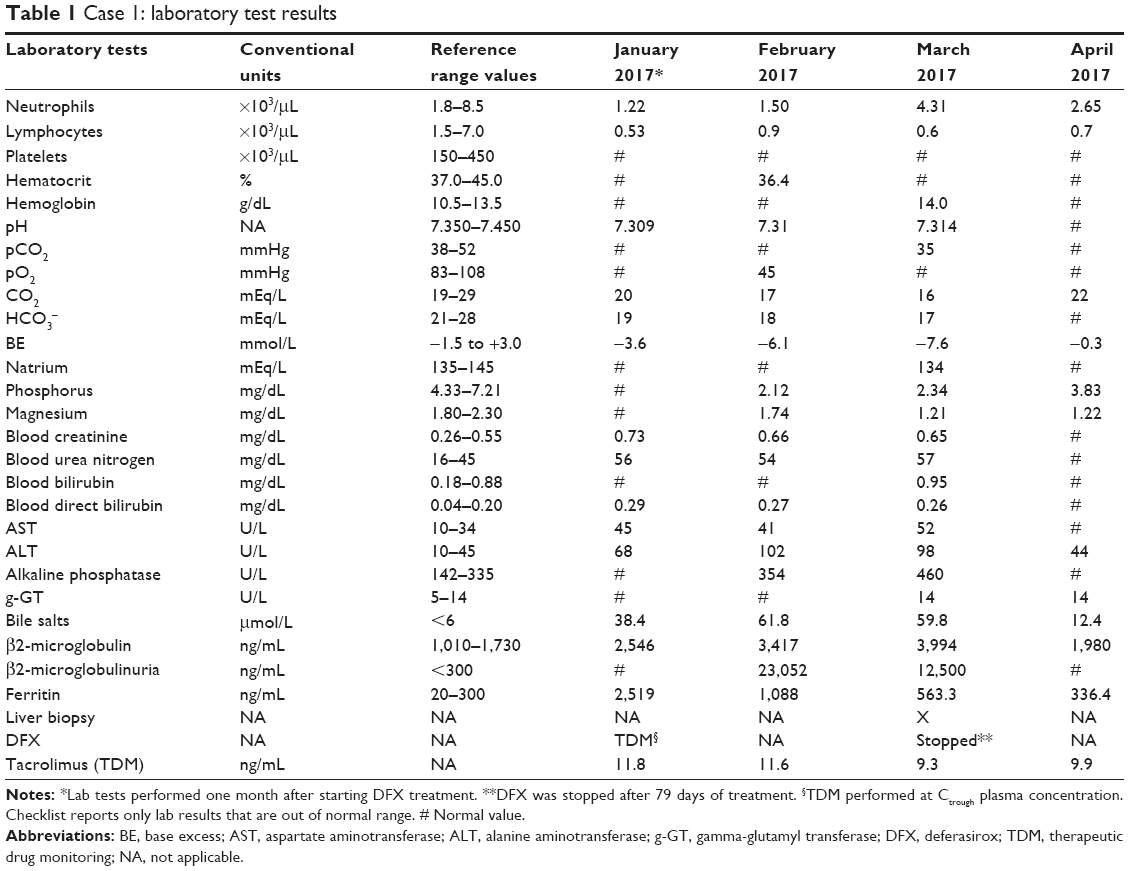 | Table 1 Case 1: laboratory test results |
Case 2
The second clinical case was a 16-year-old male affected by X-linked lymphoproliferative syndrome. The diagnosis was performed at the beginning of January 2012, after a severe Epstein–Barr virus infection complicated by hepatic and respiratory failure, and erythrophagocytic histiocytosis affecting the brain.
In April 2012, he underwent a segmentectomy of the upper lung lobe, with a histological diagnosis of lymphomatoid granulomatosis. Then, he was treated with rituximab, vincristine, and dexamethasone.
In April 2013, after a busulfan-based myeloablative conditioning, he underwent a matched unrelated donor-HSCT. GVHD prophylaxis was performed with tacrolimus and mycophenolate mofetil (MMF). The post-transplant period was uneventful and MMF was stopped at day +30.
In April 2014, one year later, the patient underwent a liver biopsy for a mild but steady alteration of liver functional tests (LFT). The histological report highlighted portal tracts characterized by minimal fibrosis and marginal biliary ductular proliferation; numeric reduction of the interlobular bile ducts present in one of the eight portal tracts of the sample; Kupffer cells hyperplasia; and reticuloendothelial siderosis (2–3 degree). Widespread structural anomalies of the portal venous vessels appear numerically increased, with dilated lumen, sometimes herniated in the perivascular parenchyma (Figure 4).
 | Figure 4 Case 2: widespread structural anomalies of the portal venous vessels with dilated lumen, sometimes herniated in the perivascular parenchyma (CD34 immunoreaction, 40×). |
The histological picture was compatible with chronic GVHD associated with vascular lesions such as hepatoportal sclerosis.
The MMF was again added to tacrolimus therapy and was suspended in May 2015, one year later. In June 2015, after a complete normalization of the LFT, tacrolimus was also stopped. The patient underwent an MRI-based iron-load evaluation that demonstrated moderate liver siderosis (LIC 140 μmol/g). Ferritin also stabilized at values above 2,000 ng/mL. At the end of June 2015, chelation therapy was started with DFX (1,500 mg/day; 26.4 mg/kg). The only concomitant drug was UDCA.
In July 2015, the patient returned to the observation of clinicians due to aggravating asthenia, which had started about 20 days earlier and became more complicated in the previous days, with symptoms such as severe abdominal pain, persistent vomiting, and anorexia, in particular after the intake of DFX.
A physical examination showed poor general conditions, weight loss of 8 kg in the past 2 weeks, pallor, signs of dehydration, polypnea without dyspnea, tachycardia, and palpable liver at 2 cm from the costal arch. No other abnormalities were found.
Laboratory tests showed a severe impairment of liver and renal functions; the patient was found to have metabolic acidosis (see Table 2). Urinalysis showed abnormal proteinuria (192.9 mg/24 hours), albuminuria (25.89 mg/24 hours), β2-microglobulinuria (>50,000 ng/mL), and glycosuria (113 mg/24 hours), suggesting proximal tubular dysfunction. The patient was hydrated and corrected for acidosis with bicarbonates. As DFX liver toxicity associated with Fanconi syndrome was suspected, therapeutic drug monitoring of DFX was performed. It showed a minimum plasma concentration (Ctrough) of 207 μM (CDFX/DDFX ratio =2.93).
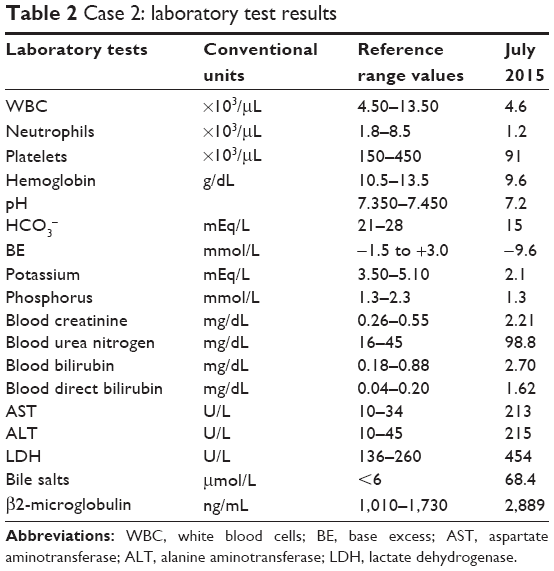 | Table 2 Case 2: laboratory test results |
DFX was immediately discontinued. Four days later, fatigue and gastrointestinal symptoms cleared and the patient gained 3 kg of weight. One month later, LFT and laboratory findings associated with Fanconi syndrome were normal. DFX was not rechallenged to the patient and no recurrence of LD or Fanconi syndrome was evident for more than 2 years.
Results
DFX is an oral iron-chelating agent used to treat chronic IO. It binds two iron molecules with high affinity and specificity, thus forming a stable complex with each iron (Fe3+) atom.14 DFX is licensed for use as first-line therapy for chronic blood transfusion-related IO in patients aged 2 years and older.15
Although its use represents a great improvement in terms of patients’ quality of life, there have been reports of several AEs in both adult and pediatric patients.
Mild AEs are the most frequently reported in pediatric and adult patients with transfusion-dependent anemias, for example, transient gastrointestinal issues in 15%, skin rashes in 11%, and mild, dose-dependent increases in serum creatinine in 38% of patients.15 Uncommon, but potentially severe, AEs are renal failure, gastrointestinal bleeding, hypersensitivity reactions, and bone marrow suppression mostly in patients with pre-existing hematologic disorders.16
Few data are available on tolerability and safety of DFX in allo-HSCT recipients. Jaekel et al have documented drug-related AEs in 71.1% patients.3
As of December 2017, EudraVigilance (the European database of suspected adverse drug reaction reports) had identified 1,679 individual cases of suspected adverse drug reactions for DFX in the European Economic Area and 11,570 in the non-European Economic Area.17 Independent of age and Economic Area, the number of individual cases of reported suspected hepatobiliary reactions for DFX was 920 (29 cases of DILI, 2 cases of VOD, and 9 cases of bile duct obstruction or stenosis) and renal and urinary disorders was 1,774. EudraVigilance reports 101 individual cases of suspected metabolic acidosis, 62 cases of acquired Fanconi syndrome, 17 cases of acidosis, and 4 cases of hyperchloremic acidosis. Several single cases of clinically apparent liver injury, which was often severe and occasionally fatal, arising during DFX therapy have been reported. The onset of acute liver injury occurred in a period ranging from a few days to eight weeks after beginning DFX, and the injury was usually hepatocellular. Immunoallergic and autoimmune features were absent. Once DFX was stopped, recovery was usually rapid but some cases were affected by progressive liver injury and hepatic failure. DFX is sold with a health warning regarding hepatotoxicity and a recommendation for regular monitoring of serum bilirubin and aminotransferase levels.18
We have described herein cases of two allo-HSCT patients who were treated with DFX; they showed histopathological elements compatible with VOD or vanishing bile duct syndrome in ductopenic evolution before DFX started. The first patient developed DILI with metabolic acidosis and the second one a liver impairment with Fanconi syndrome. After withdrawing DFX treatment, both patients improved.
According to both direct and indirect evidence, the main pathway of the DFX metabolism is through hepatic glucuronidation to metabolites M3 (acyl glucuronide) and M6 (2-O-glucuronide) and it is eliminated by biliary excretion mainly through the feces (78.5%–86.9% over 7 days).19,20
We hypothesized that the DFX elimination pathway may not work optimally in these two patients; the biliary tract injury, leading to the reduction of the metabolism and biliary elimination of the drugs, could be considered the main cause of DFX overload in our patients, also confirmed by high DFX plasmatic concentrations (26 μM and 207 μM, respectively). Therefore, the excess of free DFX molecules cannot be effectively eliminated and it accumulates in blood and in organs, such as the liver and kidney, leading to a worsening of the liver damage (DILI) and metabolic acidosis as observed in our first patient (Pt 1) and the development of Fanconi syndrome in the second patient (Pt 2).
The mechanism of occurrence of liver injury during DFX therapy is still unclear. Hepatocellular injury may be caused due to the intrinsic toxicity of DFX which determined direct damage to the hepatocytes, associated with elevated serum transaminase levels, and a worsening outcome in patients with pre-existing liver disease as a result of IO.18 Patients affected by hepatic dysfunction often have acid-base disorders and tend to be more susceptible to the development of metabolic acidosis.21 Moreover, in agreement with ScP and the EudraVigilance database, “… there have been post-marketing reports of metabolic acidosis occurring during treatment with deferasirox. Acid-base balance should be monitored as clinically indicated in these populations”.
The data in the literature also show DFX renal toxicity in adult and pediatric patients, with an increase in plasma creatinine and onset of proteinuria in about 35% of adult patients and in 19%–50% of pediatric patients.22 As widely reported, several drugs show a direct toxicity to the renal proximal tubule and can cause Fanconi syndrome; among them, DFX has also been recently reported to be associated with Fanconi syndrome.23,24 By increasing hemodynamic iron removal, DFX causes elevated iron absorption in organs, mitochondrial dysfunction, and vacuolization of proximal tubular epithelial cells together with a diminished excretion of hydrogen ions (H +), which is consistent with Fanconi syndrome. All of these could lead to the development of acidosis.25
Our patients did not experience plasma electrolyte disturbance prior to treatment with DFX during the routine follow-up. In our two patients, the first signs of acidosis appeared only after starting DFX – in the first patient, laboratory findings were compatible with mild renal dysfunction and in the second patient, laboratory findings were compatible with Fanconi syndrome. Hence, DFX was discontinued in both patients, and acidosis was corrected.
It is reasonable to think that the development of acidosis in both cases is correlated with the overload of DFX and its potential toxicity, but we believe that, in the first patient, there was also a clear hepatic histopathological feature, as revealed by the biopsy, that contributed to the onset of the acidosis itself that developed more slowly over time (29 days of therapy). According to our hypothesis, the second patient by contrast rapidly developed acidosis (Fanconi syndrome) following a greater direct renal toxic effect of the drug associated with hepatic damage, considering also the brevity of treatment before the onset of the clinical condition (12 days of therapy) and the high plasma concentration detected (279 μM).
In our patients, other potential causes of metabolic acidosis were ruled out, although the nephrotoxic effects of tacrolimus are also a major concern; coadministration of DFX with tacrolimus could have increased the risk of metabolic acidosis.26
The Naranjo adverse drug reaction scale27 was used to assess the probability that DFX caused acidosis. Based on this scale, there is a “probable” (Naranjo score =5) association between DFX and the development of acidosis in both patients.
In line with the literature, our clinical cases do not seem to be the first to highlight the onset of metabolic acidosis due to the use of DFX also in a pediatric setting.23,26,28,29 The reduction and consequent disappearance of symptoms after the suspension of DFX substantiate its role in inducing hepatic damage, probably enabling the diagnosis of DILI. The clinical suspicion of drug toxicity was also supported by the measurement of DFX plasma concentrations.
Conclusion
One of the greatest difficulties when dealing with iron chelation therapy is tailoring the dose of chelators to the individual patient. This has to be done according to the patient’s clinical condition and considering available pharmacokinetic data. Indeed, it is known that DFX pK parameters show a wide intra- and interindividual variability that strongly supports the adoption of therapeutic drug monitoring protocols. To date, DFX pharmacokinetic data in pediatric allo-HSCT patients is still lacking. In this particular cohort of patients, various factors can affect DFX kinetics, thereby exposing the patient to a non-negligible risk of adverse reactions. Increasingly, clinical practice requires a complex assessment of the interaction between the metabolic effects of the drug and individual susceptibility through pharmacokinetic approaches.
Based on our previously study in pediatric HSCT patients, we suggest a DFX starting dose of 10 mg/kg/day with close monitoring of DFX plasma concentrations (normal Ctrough value <20 μM), in order to reduce the incidence of AEs and the discontinuation of therapy. Moreover, monitoring DFX plasma concentrations over time may be useful in restoring adequate exposure to the drug following any changes in the clinical picture of patients which can lead to a variation in drug dosage.
The difficulties in diagnosing drug-related toxicity must be underlined, especially in compromised subjects. For these reasons, in patients requiring iron-chelating therapy, close and careful drug therapeutic monitoring is strongly recommended.
Acknowledgments
The authors would like to thank the participating patients. Written informed consent has been provided by the patients to have the case details and any accompanying images published. The authors would like to thank Dr Claire Marie O’Neil for her English language support. The authors have not presented or posted this study or data anywhere and have solely submitted it to this journal. The study did not receive any source of funding from a sponsor.
Author contributions
All authors contributed toward data analysis, drafting and critically revising the paper, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Reddy K, Eng J, Carlson C, Ginsberg J, Fish J. Impact of transfusional support for childhood cancer – peering behind the iron curtain: (Poster 119). Pediatr Blood Cancer. 2011;56:908. | ||
Inati A, Kahale M, Sbeiti N, et al. One-year results from a prospective randomized trial comparing phlebotomy with deferasirox for the treatment of iron overload in pediatric patients with thalassemia major following curative stem cell transplantation. Pediatr Blood Cancer. 2017;64(1):188–196. | ||
Jaekel N, Lieder K, Albrecht S, et al. Efficacy and safety of deferasirox in non-thalassemic patients with elevated ferritin levels after allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant. 2016;51(1):89–95. | ||
Sivgin S, Baldane S, Akyol G, et al. The oral iron chelator deferasirox might improve survival in allogeneic hematopoietic cell transplant (alloHSCT) recipients with transfusional iron overload. Transfus Apher Sci. 2013;49(2):295–301. | ||
Sivgin S, Eser B, Bahcebasi S, et al. Efficacy and safety of oral deferasirox treatment in the posttransplant period for patients who have undergone allogeneic hematopoietic stem cell transplantation (alloHSCT). Ann Hematol. 2012;91(5):743–749. | ||
Maximova N, Gregori M, Boz G, et al. MRI-based evaluation of multiorgan iron overload is a predictor of adverse outcomes in pediatric patients undergoing allogeneic hematopoietic stem cell transplantation. Oncotarget. 2017;8(45):79650–79661. | ||
Evens AM, Mehta J, Gordon LI. Rust and corrosion in hematopoietic stem cell transplantation: the problem of iron and oxidative stress. Bone Marrow Transplant. 2004;34(7):561–571. | ||
Gordon LI, Brown SG, Tallman MS, et al. Sequential changes in serum iron and ferritin in patients undergoing high-dose chemotherapy and radiation with autologous bone marrow transplantation: possible implications for treatment related toxicity. Free Radic Biol Med. 1995;18(3):383–389. | ||
Subbarao G, Haut PR, Johnson CS, Gowan D, Molleston JP. Incidence, etiology, and risk factors for liver dysfunction in children following hematopoietic stem cell transplantation. Pediatr Transplant. 2006;10:682–689. | ||
Locasciulli A, Testa M, Valsecchi MG, et al. Morbidity and mortality due to liver disease in children undergoing allogeneic bone marrow transplantation: a 10-year prospective study. Blood. 1997;90(9):3799–3805. | ||
Maximova N, Gregori M, Simeone R, et al. Safety and tolerability of deferasirox in pediatric hematopoietic stem cell transplant recipients: one facility’s five years’ experience of chelation treatment. Oncotarget. 2017;8(38):63177–63186. | ||
Rouan MC, Marfil F, Mangoni P, Séchaud R, Humbert H, Maurer G. Determination of a new oral iron chelator, ICL670, and its iron complex in plasma by high-performance liquid chromatography and ultraviolet detection. J Chromatogr B Biomed Sci Appl. 2001;755(1–2):203–213. | ||
Mattioli F, Puntoni M, Marini V, et al. Determination of deferasirox plasma concentrations: do gender, physical and genetic differences affect chelation efficacy? Eur J Haematol. 2015;94:310–317. | ||
Cappellini MD, Taher A. Long-term experience with deferasirox (ICL670), a once-daily oral iron chelator, in the treatment of transfusional iron overload. Expert Opin Pharmacother. 2008;9(13):2391–2402. | ||
EXJADE® (deferasirox) SmPC. Novartis Pharmaceuticals; 2013. Available from: http://www.ema.europa.eu/ema/. Accessed July 20, 2018. | ||
Cappellini MD, Porter J, El-Beshlawy A, et al. Tailoring iron chelation by iron intake and serum ferritin: the prospective EPIC study of deferasirox in 1744 patients with transfusion-dependent anemias. Haematologica. 2010;95(4):557–566. | ||
The European Medicines Agency. EudraVigilance – European Database of suspected adverse drug reaction reports. Available from: http://www.adrreports.eu/index.html. Accessed December 20, 2017. | ||
National Institutes of Health, US Department of Health and Human Services. LIVERTOX. Available from: https://livertox.nlm.nih.gov. Accessed December 22, 2017. | ||
Waldmeier F, Bruin GJ, Glaenzel U, et al. Pharmacokinetics, metabolism, and disposition of deferasirox in beta-thalassemic patients with transfusion-dependent iron overload who are at pharmacokinetic steady state. Drug Metab Dispos. 2010;38(5):808–816. | ||
Tanaka C. Clinical pharmacology of deferasirox. Clin Pharmacokinet. 2014;53(8):679–694. | ||
Scheiner B, Lindner G, Reiberger T, et al. Acid-base disorders in liver disease. J Hepatol. 2017;67(5):1062–1073. | ||
Dubourg L, Laurain C, Ranchin B, et al. Deferasirox-induced renal impairment in children: an increasing concern for pediatricians. Pediatr Nephrol. 2012;27(11):2115–2122. | ||
Hall AM, Bass P, Unwin RJ. Drug-induced renal Fanconi syndrome. QJM. 2014;107(4):261–269. | ||
Baum M. Renal Fanconi syndrome secondary to deferasirox: where there is smoke there is fire. J Pediatr Hematol Oncol. 2010;32(7):525–526. | ||
Pham AQ, Xu LH, Moe OW. Drug-induced metabolic acidosis. F1000Res. 2015;4:4. | ||
Naesens M, Kuypers DR, Sarwal M. Calcineurin inhibitor nephrotoxicity. Clin J Am Soc Nephrol. 2009;4(2):481–508. | ||
Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30(2):239–245. | ||
Dell’Orto VG, Bianchetti MG, Brazzola P. Hyperchloraemic metabolic acidosis induced by the iron chelator deferasirox: a case report and review of the literature. J Clin Pharm Ther. 2013;38(6):526–527. | ||
Marano M, Bottaro G, Goffredo B, et al. Deferasirox-induced serious adverse reaction in a pediatric patient: pharmacokinetic and pharmacogenetic analysis. Eur J Clin Pharmacol. 2016;72(2):247–248. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.