")
Back to Journals » Clinical Ophthalmology » Volume 14
Voretigene Neparvovec in Retinal Diseases: A Review of the Current Clinical Evidence
Authors Gao J , Hussain RM, Weng CY
Received 23 September 2020
Accepted for publication 21 October 2020
Published 13 November 2020 Volume 2020:14 Pages 3855—3869
DOI https://doi.org/10.2147/OPTH.S231804
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Jie Gao,1 Rehan M Hussain,2 Christina Y Weng1
1Department of Ophthalmology, Baylor College of Medicine, Houston, TX, USA; 2Retina Associates, Elmhurst, IL, USA
Correspondence: Christina Y Weng
Baylor College of Medicine, Alkek Eye Center, 1977 Butler Blvd, Houston, TX 77030, USA
Email [email protected]
Abstract: Subretinal gene therapy trials began with the discovery of RPE65 variants and their association with Leber congenital amaurosis. The RPE65 protein is critical for the normal functioning of the visual phototransduction cascade. RPE65 gene knockout animal models were developed and showed similar diseased phenotypes to their human counterparts. Proof of concept studies were carried out in these animal models using subretinal RPE65 gene replacement therapy, resulting in improvements in various visual function markers including electroretinograms, pupillary light responses, and object avoidance behaviors. Positive results in animal models led to Phase 1 human studies using adeno-associated viral vectors. Results in these initial human studies also showed positive impact on visual function and acceptable safety. A landmark Phase 3 study was then conducted by Spark Therapeutics using a dose of 1.5 x1011 vector genomes after dose-escalation studies confirmed its efficacy and safety. Multi-luminance mobility testing was used to measure the primary efficacy endpoint due to its excellent reliability in detecting the progression of inherited retinal diseases. After the study met its primary endpoint, the Food and Drug Administration approved voretigene neparvovec (Luxturna®) for use in RPE65-associated inherited retinal diseases.
Keywords: gene therapy, inherited retinal diseases, Leber congenital amaurosis, Luxturna, RPE65, voretigene neparvovec, retinitis pigmentosa, retina
Introduction
Inherited retinal diseases (IRDs) are a heterogenous group of disorders characterized by varying degrees of functional vision loss and associated retinal degenerative changes. For the collective 270 gene mutations that have been identified in association with clinically diagnosed IRDs, the incidence is approximately 1 in 2000.1,2 In most IRDs, the visual loss occurs early and can be profound, resulting in significant disability to the patient. The primary site of degeneration usually involves the photoreceptor and retinal pigment epithelium (RPE) complex. IRDs can be classified as either stationary, such as in congenital stationary night blindness (CSNB), or progressive, such as in retinitis pigmentosa (RP).1 Leber congenital amaurosis (LCA) is one of the most severe types of progressive IRDs, presenting with significant functional vision decline within the first year of life.3,4 This review will provide a brief overview on the RPE65 gene mutation-related dystrophies and focus on the clinical evidence that led to the approval of voretigene neparvovec-rzyl, the first FDA (Food and Drug Administration)-approved gene replacement therapy in the United States and in the European Union.5,6
The RPE65 Gene
Gene Function
The RPE65 gene encodes for a 65 kDa protein located primarily on the smooth endoplasmic reticulum of RPE cells.7 Electroretinograms (ERGs) of biallelic knockout (RPE65 -/-) mice demonstrated diminished or absent waveforms similar to what is seen in humans. Dark-adapted ERGs tended to be worse than light-adapted or flicker ERGs suggesting that rod function is more severely impacted than cone function. In the photoreceptors of these eyes, no detectable rhodopsin was found. In the RPE cells, there was an absence of 11-cis-retinol and an overaccumulation of all-trans-retinol.8 In initial in vitro studies, human cells were transfected with the RPE65 gene along with a LRAT coenzyme gene, followed by exposure to all-trans-retinol. This resulted in decreased all-trans-retinol levels and a dramatic increase in 11-cis-retinol levels, suggesting that the RPE65 protein plays a direct enzymatic role in the isomerization of 11-trans-retinol to 11-cis-retinol.9,10
Other studies suggest that a non-enzymatic role for the RPE65 protein may exist. One study noted that heterozygous (RPE65 +/–) mice have a much larger drop in rod pigment recycling than would be expected compared to wild type (RPE65 +/+) mice if the RPE65 protein was acting solely as an enzyme. This study also showed that when RPE65 -/- mice were given oral 9-cis-retinal supplementation, a small amount of 11-cis-retinal could be detected. The author suggested that other proteins may be involved in the visual cycle pathway that eventually produces 11-cis-retinal. They concluded that the RPE65 protein could either have a dual function as both an isomerase enzyme and a structural protein or as an organizer protein involved in the distribution of retinyl esters within the RPE cells.11 Currently, the exact function of the RPE65 protein is still debated, but what is known is that 11-cis-retinol is converted to 11-cis-retinal which is required by the photoreceptor outer segments in order to combine with opsin and produce the visual pigment that is responsible for detecting light and initiating the phototransduction cascade.7
Mutant RPE65 Gene Phenotypes
Biallelic RPE65 gene mutations can manifest clinically as Leber congenital amaurosis type 2 (LCA2) or as rare types of retinitis pigmentosa. The incidence of LCA is estimated to be around 2 to 3 per 100,000, and this condition accounts for 5% of all IRDs diagnosed. LCA2 is due to a mutation in the RPE65 gene mapped to chromosome 1p31. Phenotypically, patients present in infancy with absent fixation, eye wandering, nyctalopia, nystagmus, and progressive vision loss.3,12–17 Fundus examination may initially be normal, but ERG will show severely diminished or absent waveforms consistent with severe rod and cone dysfunction.18 Over time, the dilated exam may show signs of degeneration presenting as retinal vascular attenuation, optic disc atrophy, peripheral pigmentary spicules, nummular pigmentation, peripheral yellow spots, or para-arteriolar pigmentary changes.12 While over 100 mutations have been identified in RP, only 2% of autosomal recessive RP patients have mutations in the RPE65 gene. Clinical presentation may vary, but most patients will present in early childhood with nyctalopia and peripheral vision loss that eventually progresses to central vision loss. On exam, diffuse pigmentary changes, arteriolar attenuation, and optic nerve pallor can be seen (Figure 1). A choroideremia-like picture has also been reported with an autosomal dominant type of RP associated with the RPE65 gene mutation. On ERG, a reduction in a- and b-wave amplitudes is noted in the earlier stages of the disease, but these waveforms may completely extinguish in later stages of the disease.19
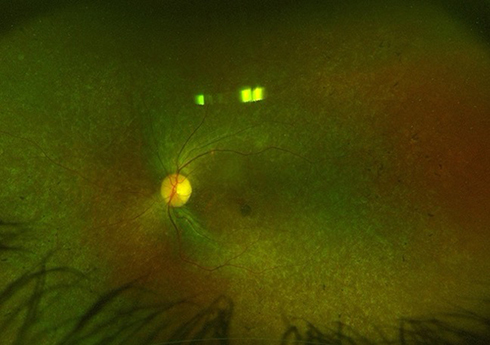 |
Figure 1 Fundus photograph of the left eye of a 33-year-old female with biallelic RPE65 gene mutation demonstrating diffuse pigmentary changes, vascular attenuation, and temporal disc pallor. |
One study of over 200 patients diagnosed with either isolated RP, autosomal recessive RP, or LCA showed that 2% of the RP patients and 16% of the LCA patients had mutations in the RPE65 gene. This suggests phenotypic heterogeneity within RPE65 gene mutants. The authors clinically differentiated between RP and LCA by stating that LCA patients were either born blind or developed severe vision loss within the first few months of life while RP patients showed good central vision within the first decade of life with eventual worsening afterwards.20 This phenotypic heterogeneity could be explained by the variability in mutant protein function depending on the exact location of the mutation within the gene. Given the potential variability in phenotypic presentation for the same gene mutation and the increased accessibility of genetic testing, there has been a general shift from diagnosing IRDs by their phenotypic presentation towards identifying IRDs by their genotypic profile.
Gene Therapy and the Development of Voretigene Neparvovec
Immune Privilege and the Subretinal Space
The eye is an excellent site for gene therapy because of its immune privileged status and its compartmentalized structure that allows for targeted delivery of medication to intraocular cells. Immune privilege was originally described by a phenomenon known as anterior chamber-associated immune deviation (ACAID) where foreign antigens presented into the anterior chamber (AC) were observed to produce less of an immune response than expected. It was hypothesized that the AC had the ability to upregulate the expression of suppressive regulatory T cells towards foreign antigens.21 This same concept was tested in the subretinal space by injecting P815 mastocytoma cells and ovalbumin acting as a soluble antigen into mice. Immune response was tested by seeing if a delayed-type hypersensitivity (DTH) reaction would occur after reinoculation of the P815 cells or ovalbumin behind the mice’s ears. The ear challenge confirmed the lack of an antigen-specific DTH reaction.22 Experiments like this proved that the subretinal space also exhibited immune deviation likely due to the presence of a blood-retinal barrier and local production of regulatory suppressor T cells. This unique immune property opened the pathway to exploring the use of viral vectors within the subretinal space.
Viral Vector Development
Successful gene replacement relies on the ability of a vector to safely insert itself into the targeted cell followed by implantation of the packaged genetic material. This must then lead to further translation of the gene into functional proteins without causing insertional mutagenesis, oncogenesis, or an exorbitant host immune response.23 One of the most commonly used vectors for gene therapy is the adeno-associated viral (AAV) vector. AAVs are small (25 nm) non-enveloped single-stranded DNA viruses belonging to the Parvoviridae family. Because they require helper viruses to replicate, they are considered non-pathogenic. Another advantage is that AAV vector DNA remains episomal and does not integrate into host DNA thereby decreasing the risk for oncogenesis or mutagenesis. Disadvantages include a small packing capacity (can only hold up to 4.5 kB of complementary DNA), and the inability of DNA expression to perpetuate in dividing cells.24 There are over 100 AAV variants with varying degrees of tropism for different human tissue types. Serotypes 1 through 9 all have affinity for ocular tissue and efficiently transduce RPE cells. AAV is fairly ubiquitous within humans with over 70% of the population showing positive antibodies (Ab) to AAV1 and AAV2 antigens.25 Many factors play a role in determining the efficiency of the DNA expression, including the type of promoter used, the addition of enhancer sequences, and vector capsid components.26,27 Capsid proteins can be modified to improve transduction, cell targeting, and counter pre-existing immunity.
The AAV2 vector used in the RPE65 gene therapy trials transduces mainly RPE cells and, to a lesser degree, photoreceptors. This transduction and transgenic gene expression occur by 2 to 4 weeks following subretinal inoculation.28 This property of the AAV2 vector to preferentially target RPE cells coupled with the fact that the RPE65 gene is mainly expressed in RPE cells and also small enough to be packaged into the AAV2 vector all contributed to the success observed in later animal and human trials.
Pre-Clinical Animal Studies
Unlike many other IRDs, RPE65-associated LCA had the advantage of being established in small and large animal models which provided a platform for forthcoming gene replacement therapy experiments. RPE65 -/- mice and dogs were the first animal models tested.29,30 In 2001, the first report of a subretinal gene therapy trial in a canine model was released. The study used an AAV vector to deliver functional RPE65 genes into RPE65 -/- dogs. Both cone and rod ERGs improved after subretinal injections, but not after an intravitreal injection. Pupillary light response (PLR), object avoidance, and visual evoked potential also improved. Enucleation of one of the eyes that received a subretinal injection confirmed the expression of transgenic wild RPE65 gene by PCR testing.31 A follow-up study showed that improvements were stable at 3 to 5 years without significant inflammation or diminished ERG responses.32 Additional mice studies also showed improvements in both cone and rod ERG waveforms. Furthermore, structural improvements were seen with respect to a decrease in lipid inclusions within the RPE cell, although this did not appear to halt photoreceptor degeneration.33
Further dose escalation trials were done in mice, dogs, and eventually monkeys to establish a potentially safe and efficacious starting dose for human trials. An initial study in dogs evaluated a wide range of vector genomes (vg) from 1.5 x108 vg to 4.5 x1012 vg. Improvements in ERG amplitudes were noted in most dogs at doses of 1.5 x1011 vg or higher. Transient ocular inflammation and variable mild serum Ab responses to the AAV antigen were noted in some animals, but no major systemic sequalae were noted. Later studies in macaque monkeys showed that dose levels of 1.5 x1012 vg and 4.5 x1012 vg were well-tolerated without evidence of retinal toxicity.34 These studies helped pave the way for subsequent human trials.
Pre-Treatment Considerations in Humans
Prior to undergoing surgery for gene replacement therapy, the integrity of the photoreceptor layer must be evaluated. The formal indication for voretigene neparvovec use requires the presence of “viable retinal cells”. Visual function loss in RPE65-associated IRD is comprised of two components: a visual cycle blockade that is reversible with gene therapy and a cellular degenerative process which is unlikely to be reversible in its end stages. Experiments in older mice showed that the therapeutic responders on average had 4 rows of photoreceptor nuclei in histological sectioning compared to therapeutic failures which only had 1 row.35 Because of the implausibility of actual retinal tissue sampling, optical coherence tomography (OCT) has been used as a surrogate marker for photoreceptor health (Figure 2). Even so, OCT studies on human RPE65 gene mutants have not shown a strong correlation between the degree of visual function loss or age and OCT characteristics such as outer nuclear layer (ONL) thickness. Therefore, deciding whether a patient’s photoreceptor layer has sufficient viable cells to successfully respond to gene therapy is left to the discretion of the treating provider.35,36
 |
Figure 2 Optical coherence tomography images of the right (A) and left (B) macula of a 6-year-old boy with biallelic RPE65 gene mutation. Notice the diffuse loss of the ellipsoid zone except for a small subfoveal island in both eyes. Also note the marked atrophy of the outer nuclear layer (arrows) which slightly increases in thickness in the parafoveal and foveal regions. |
Phase I/II Human Studies
In 2008, the results from the first human gene therapy trials were published from 3 separate groups. Each group recruited 3 patients (age 16–24 years) and gave unilateral subretinal injections of the RPE65 gene carried by AAV2 vectors to the worse-seeing eye. Despite differences in baseline visual acuity (VA), treatment dosages (eg, 1.5 x1010 vg, 5.96 x1010 vg, 1 x1011 vg), regulatory elements (eg, promoters, enhancers), surgical techniques (eg, gas displacement of subretinal bleb in one study) and injection locations (eg, 6 fovea-involving, 3 extramacular) between the studies, improvements in various visual function markers were observed in all three studies. No serious systemic immune-related side effects were seen.37–39 These encouraging safety and efficacy results spurred further dose-escalation and sequential fellow eye treatment studies.
Maguire et al reported the first dose-escalation study in 2009 involving 12 patients (age 8–44 years) subdivided into low (1 x1010 vg), medium (4.8 x1010 vg) and high (1.5 x1011 vg) dose treatment groups. Again, unilateral treatments were given to the worse-seeing eye. VA gains were seen in 7 patients (3 low, 3 medium, and 1 high dose) with no association to age or dosage received. Goldmann visual field (GVF) improved in all 12 and was associated with subretinal injection location as well as the amount of quantifiable healthy outer retina. Five of 7 patients improved in dark-adapted full-field sensitivity threshold (FST) testing, but no improvements were noted on ERG, unlike what was observed in animal models. No systemic toxicity or adverse immunologic responses were seen despite transient elevations in Ab against AAV2 vector antigens in the tears and blood of some participants.40 A follow-up study showed that the improvements were stable out to 3 years. Analysis of patients with sustained VA gains and improved fixation on microperimetry suggested that the improvements were likely due to decreases in nystagmus which was observed in both treated and untreated eyes.41 A functional MRI (fMRI) study on 10 of the 12 patients showed improved signaling in the occipital lobe corresponding to the subretinal treatment location.42
Additional dose-escalation trials followed. Jacobson et al showed similar improvements in dark-adapted FST in all 15 subjects studied. Blue light stimulus was used for FST to show that the improvements were largely rod-mediated. About half the patients improved on mobility testing in low light levels when using the treated eye, but not with the fellow control eye.43 Three patients were recruited for a long-term follow-up study. Surprisingly, results showed that the dark-adapted retinal sensitivity maps initially expanded in size and peaked in the first 1 to 3 years after treatment, but then slowly contracted in subsequent years. ONL thickness scans showed gradual but persistent declines from baseline to years 4 to 6. This could be explained by either initial photoreceptor rescue secondary to restoration of the visual cycle from gene therapy with subsequent decline in transgenic expression over time; or an RPE65 protein-independent progressive photoreceptor degeneration; or a combination of both.44 Bainbridge et al employed the highest dosing regimens (1 x1011 vg, 1 x1012 vg). This study of 12 patients (age 6–23 years) showed similar initial improvements in dark-adapted perimetry, microperimetry, and navigation, especially in the higher dose group. However, the higher dose group also showed a higher incidence of intraocular inflammation with 1 patient developing focal pigmentary changes in the macula resulting in a 15 letter VA loss. Again, no correlation was seen between treatment success and age. In fact, the older patients (age 17–23 years) had greater initial improvements. Similar to earlier studies, improvements waned beyond the first year post-treatment causing focus to shift from increasing the vg dosage to optimizing the efficacy of the vector construct.45 More recently, Weleber et al published results at 2 years showing at least 1 visual function improvement in 9 out of 12 patients (age 6–39 years) using medium-dose regimens (1.8 x1011 vg, 6 x1011 vg) which were sustained at 5 years. While the effects were not dose-dependent, they did find that younger patients (age 6–11 years) tended to have greater improvements. The visual function improvements corroborated the previously observed correlation between dampening of nystagmus and improvement in foveal fixation, explaining why both treated and untreated eyes improved. Of note, results could have been skewed because of very poor baseline vision in the older group (age 28–39) with the majority of these patients having VA of counting fingers or worse along with severely reduced baseline visual fields (VF).46,47
Gene therapy retreatments to the contralateral eye of previously-treated canines and non-human primates showed that sequential treatments were safe. Re-exposure to subretinal gene therapy in the fellow eye did not incite an exaggerated immune response or a decrease in RPE65 protein expression. Early human contralateral eye studies demonstrated efficacy through improvements in FST, obstacle avoidance, and bilateral cortical activation on fMRI.48 Results from a more recent study evaluating sequential contralateral treatments showed FST improvements for both rod- and cone-directed stimuli, although the rod response appeared more robust. Improvements in cortical activation on fMRI and navigational performance in progressively lower light levels were also noted. Previous AAV2 serum Ab positivity did not diminish the visual function gains.49 Given the positive results from these dose-escalation and contralateral redosing studies (Table 1), Spark Therapeutics proceeded to sponsor a phase 3 clinical trial.
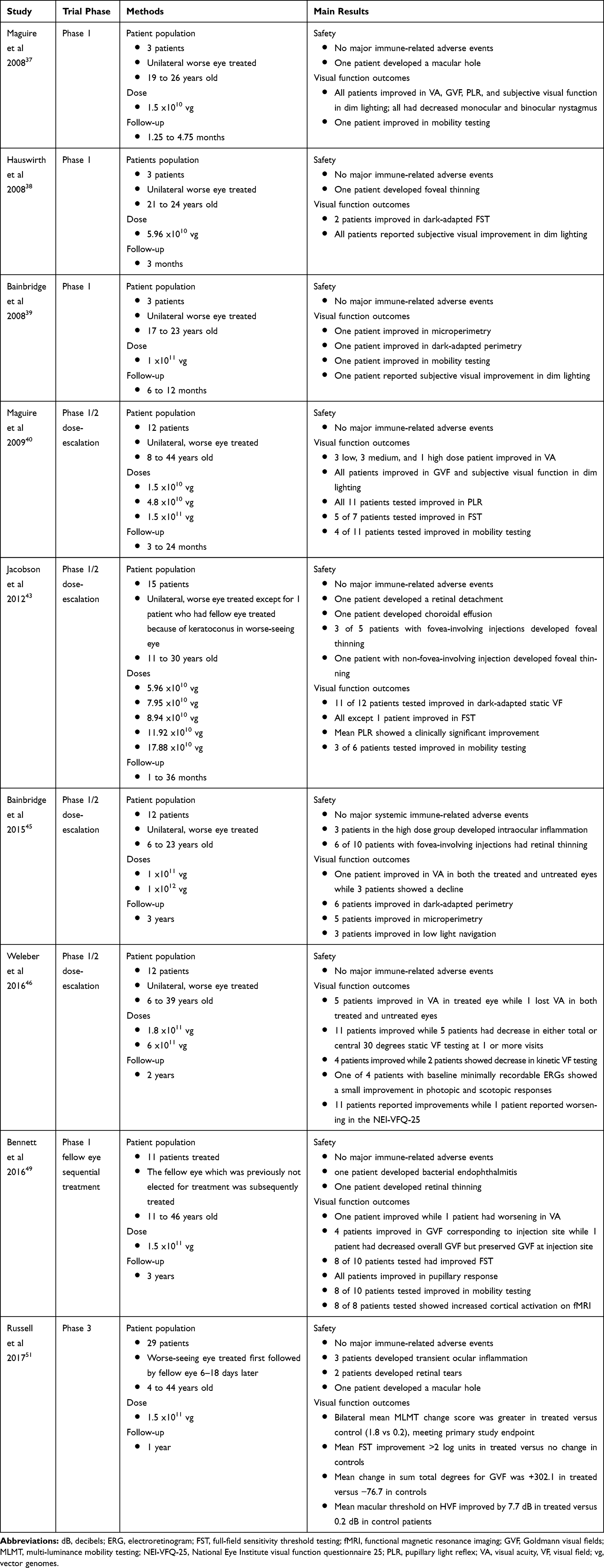 |
Table 1 Summary of the Major Human Clinical Trials Evaluating RPE65 Gene Therapy |
Novel Primary Endpoint in IRD Studies
Before initiating a phase 3 human gene therapy trial, an appropriate primary efficacy endpoint needed to be established. Because affected patients typically have very poor baseline VA, using VA as the primary outcome measure is not ideal. Since rod dysfunction is more prominent in the early stages of RPE65-associated dystrophy, it is reasonable to assume that navigational abilities in lower light settings will initially be more significantly affected. Chung et al designed a multi-luminance mobility test (MLMT) in an effort to create a single quantifiable test that could be used to assess visual function changes in patients with rod-dominant IRDs. The MLMT involved navigating an obstacle course at 7 different light levels resembling real-world environments. The lowest light level was 1 lux which equates to a moonless night, and the highest was 400 lux which mimics the illumination of a well-lit office space. A score was given for passing the course with higher points being awarded for passing at lower light levels. The highest possible score was 6 points for successfully navigating the course at 1 lux, and the point values decreased at each increasing light level with 0 points being awarded for passing at 400 lux. Accuracy and time scores were combined to determine a passing or failing score. The patients were allowed to attempt the course at progressively lower light levels until either failure occurred at any light level or success occurred at 1 lux. A MLMT change score was defined as the difference in the MLMT score at 1 year compared to the baseline MLMT score.50
The initial MLMT validation studies were done by comparing normal controls to patients with various IRDs including RP, LCA, choroideremia, Stargardt disease, and Usher syndrome. Children as young as 4 years old were able to complete the obstacle course. Passing or failing grades were given after review of the MLMT course videos by multiple masked graders and showed an inter-grader reliability of 97.9%. Based on the time and accuracy scores, MLMT had an excellent ability to distinguish between normal vision patients and patients with visual impairment from IRDs. MLMT also showed the ability to detect disease progression over 1 year in the IRD patient populations. This was especially apparent in the LCA and RP cohorts. Due to these results, the MLMT score was used as the primary outcome measure in the RPE65 gene replacement therapy phase 3 human trials.50
Landmark Phase III Human Study
The phase 3 clinical trial enrolled 31 patients between the ages of 4 to 44 years old and randomized them 2:1 to intervention versus control. Individuals with a confirmed biallelic RPE65 gene mutation could be included if they were gene therapy- or oral retinoid therapy-naïve, had best-corrected VA (BCVA) ≤20/60 (Snellen), had <20 degrees of VF in any meridian, and could perform the MLMT protocol using both eyes. Viable retinal cells had to be present as determined by a combination of fundus photography, clinical examination, and OCT retinal thickness map >100 microns in the posterior pole. The primary outcome measured was the change in bilateral MLMT performance at 1 year compared to baseline. Video recordings of the MLMT were graded at an independent reading center by two separate well-trained masked graders. The human RPE65 cDNA carried within an AAV2 viral vector, named voretigene neparvovec-rzyl (VN), included a modified Kozak sequence at the translation site and hybrid chicken beta-actin promoter linked to a cytomegalovirus enhancer to optimize transgenic gene expression within RPE cells.51 A dose of 1.5 x1011 vg was chosen due to success and safety seen in prior studies.40,48,49 Patients were temporarily immunosuppressed using pre-operative systemic prednisone 3 days prior to subretinal injections. Both eyes were treated with a 1 to 2 week delay between first and fellow eye treatments. One patient in each group left after consent but before intervention, resulting in a final intention-to-treat analysis of 20 patients in the treatment group and 9 in the control group. The control group was permitted to crossover to the treatment group at the one-year timepoint.51
One-year results met the primary efficacy endpoint. The mean bilateral MLMT score change showed a statistically significant improvement by 1.8 light levels in the VN group versus only 0.2 light levels in the controls (P = 0.0013). Improvements were seen as soon as 30 days post-treatment and were stable at 1 year. Mean unilateral MLMT score change was very similar to the bilateral mean MLMT score change. Thirteen out of 20 patients (65%) in the VN group were able to pass the MLMT at the lowest luminance level (1 lux) at 1 year as opposed to none in the control group. Mean FST improved by over 2 log units in the VN group compared to none in the control group (P = 0.0004). The 18 out of 20 patients who improved in MLMT change scores were also the ones who improved in FST. Mean sum total degrees of GVF nearly doubled in the treatment arm while it decreased in the control arm. There was a trend towards improved VA, but this was not clinically significant (P = 0.17). At 1 year, there were no major AAV2 vector-related adverse events or systemic side effects. Most common ocular side effects appeared to be surgery-related and included elevated intraocular pressure (20%), cataracts (15%), ocular inflammation (10%), retinal tears (10%), macular holes or degeneration (5%), and epiretinal membranes (5%). Of note, one treated patient experienced a large decrease in post-operative VA, and was also the only patient who did not show improvement in MLMT performance (Table 1). The overall positive results seen in the phase 3 human trial led to the FDA approval of VN for the treatment of RPE65-associated IRDs.51,52
Long-Term Phase 1 and 3 Follow-Up
Maguire et al reported long-term follow-up data on 20 patients from the original intervention (OI) group and 9 in the control-crossover (CC) group from the phase 3 trial along with 8 bilaterally-treated patients from a phase 1 contralateral eye retreatment study.53 Updated MLMT change scores were reported at 1 year for the CC group, 2 years for the OI group, and 4 years for the phase 1 patients with gains of 2.1, 1.9, and 2.4, respectively. Improvements in FST were again highly correlated to gains in MLMT change scores. Results were stable at 1 to 4 years with 89% of patients who improved on FST being able to successfully pass the MLMT at the lowest light level (1 lux). VA again showed no statistically significant improvements at 1 to 4 years, although no significant decline was seen either. VF testing was too variable in the phase 1 patient group to allow for statistical comparison, likely due to the poorer baseline VA. GVF for the phase 3 patients were reported using the III4e stimuli. The CC group showed a 49% increase at 1 year compared to a 16% loss in the prior year before crossing over. The OI group had a 99% increase at 2 years compared to baseline. No gene therapy-related immunologic systemic adverse effects were observed. All ocular side effects were attributed to the expected risks from vitrectomy surgery.53 Subsequent follow-up at 3 years for the CC cohort and 4 years for the OI cohort showed mean MLMT change scores of 2.4 and 1.7 light levels, respectively. Within the OI cohort, 4 out of 20 patients had a decrease of 1 lux which was still above their baselines; one patient had a gain of 1 lux. The authors suggested that amblyopia is unlikely to be a major deterrent to gene therapy, but acknowledged that there may be other ongoing degenerative processes that may limit outcomes after gene therapy.54 Ongoing long-term follow-up studies will provide additional information in the years to come.55
Post-Approval Use of Voretigene Neparvovec-rzyl
Approval and Launch
Positive results from the phase 3 human trial led to the FDA approval of voretigene neparvovec-rzyl (Luxturna®) on December 19, 2017 for the treatment of patients with confirmed biallelic RPE65 mutation-associated retinal dystrophy, marking the first-ever FDA-approved gene therapy. Included in the indication is the condition that the patient must have viable retinal cells remaining, as determined by the treating physician.52 On November 23, 2018, approval was granted for use in all 28 member states of the European Union as well as Iceland, Liechtenstein, and Norway.5 Spark Therapeutics decided to roll out the product at a limited number of Ocular Gene Therapy Treatment Centers around the country. These sites were selected by the company based on specialized scientific or clinical experience with VN, the ability to properly store and prepare VN, the ability to coordinate and manage patient logistics with the Spark Therapeutics team, and formal training of pharmacy and surgical specialists on proper VN handling. Currently, there are ten sites in the United States which have been approved: Bascom Palmer Eye Institute, Baylor College of Medicine, Casey Eye Center, Children’s Hospital of Philadelphia, Cincinnati Children’s Hospital, Kellogg Eye Center, Massachusetts Eye and Ear, Scheie Eye Institute, University of Iowa Hospitals and Clinics, and The Vision Center at Children’s Hospital Los Angeles.56
Dosage and Safety
The medication supplied to treatment centers includes a 2-mL vial containing 0.5 mL of the medication at a concentration of 5 x1012 vg/mL and two 2-mL vials of diluent. The initial concentration needs to be diluted 1:10 prior to subretinal administration to create a final recommended concentration of 1.5 x1011 vg in 0.3 mL of solution. Three days prior to surgery, it is recommended that patients start a 7-day course of systemic corticosteroids equivalent to 1 mg/kg/day of oral prednisone (max 40 mg/day) followed by a taper over 10 days. It is recommended that the contralateral eye be treated within a close time interval, but with a minimum delay of 6 days between eyes. Pediatric use for children under 1 year of age is not recommended because of the active retinal cell proliferation occurring during this age which can result in possible dilution and loss of efficacy.6
Results from Phase 1 and 3 studies from 41 patients (81 eyes) showed no significant immune-related or systemic adverse events related to initial and repeat exposure to VN through 4 years. Transient AAV2 vector antigen and Ab may rarely be found in tears or blood without evidence of immunologic sequalae. Most ocular side effects were minor and consistent with what is expected after vitrectomy surgery including cataracts (19%), conjunctival hyperemia (11%), increased intraocular pressure (10%), and retinal tears (5%). More serious ocular events were rare and included foveal thinning (2%), endophthalmitis (1%), and retinal detachment (1%). Most reports of foveal thinning suggested that this was a result of the surgically-induced retinal detachment rather than medication-induced retinal toxicity or progressive retinal degeneration. No clinical safety data exists for females who are pregnant or lactating or for the geriatric population.6,49,51,53
Preparation and Surgical Technique
VN must be prepared and diluted within 4 hours of surgery. To prepare the medication, 0.3 mL of concentrated VN and 2.7 mL of diluent are injected into a 10-mL glass vial and inverted gently a few times to allow for even mixing. Then, two 1-mL syringes are used to draw out 2 aliquots of 0.8 mL of the diluted VN mixture (one syringe serves as a backup) and set aside at room temperature for surgery. At the time of surgery, the patient is prepped and draped in usual aseptic manner; most patients are treated under general anesthesia, especially if they are of childhood age. One of the 1-mL syringes containing the prepared VN mixture is connected to polyvinyl chloride extension tubing and attached to a 41-gauge subretinal injection cannula (Figure 3). The extension tubing should not exceed a length of 15.2 cm and an inner diameter of 1.4 mm to avoid excessive “dead space” volume during priming.6 The syringe is primed by depressing the plunger until a few droplets of the medication is seen at the tip of the injection cannula. Some surgeons will choose to express the solution until 0.3 mL is left in the syringe to ensure that the intended delivery volume is preset while others elect to self-gauge the 0.3 mL volume at time of injection in case extra volume is needed.
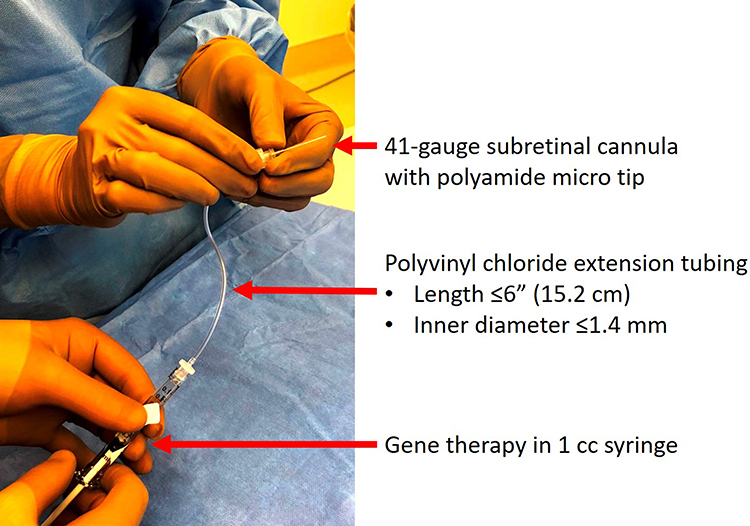 |
Figure 3 Image illustrating the set-up of the injection apparatus. A 1-mL syringe containing the prepared voretigene neparvovec is connected to extension tubing and a 41-gauge subretinal injection cannula. |
A standard 3-port pars plana vitrectomy is performed along with the induction of a posterior hyaloid detachment. Some surgeons elect to use triamcinolone acetonide for hyaloidal staining while others do not as its interaction with VN is unknown.57 Once the vitreous has been removed, the injection site is inspected. It is recommended to select an area near the superior arcade that is at least 2 mm away from the foveal center and avoids retinal vessels or obvious pathology. Areas of dense atrophy or pigmentary changes should be avoided as these areas may be excessively adherent to the underlying RPE rendering subretinal bleb creation and propagation more difficult and traumatic.6,58 The tip of the injection cannula is trimmed, and sometimes beveled, to prevent kinking during cannula insertion and to facilitate entrance into the subretinal space.59 Once the subretinal cannula is in contact with the retina, slow and steady injection of drug initiates bleb creation (Figure 4). Of note, some surgeons will first create a pre-bleb with balanced salt solution or air, but how this affects the concentration of gene therapy is not well-understood.57 Microscope-integrated OCT may be used to confirm that the bleb has been created within the correct tissue plane (Figure 5). The remaining VN solution is injected into the bleb until the entire 0.3 mL is delivered. A foot pedal-assisted injection method has also been described using infusion pressures around 12–16 psi; this eliminates the need for a surgical assistant.58,60 Care should be used to avoid over-stretching the bleb, especially if the fovea is involved, as an iatrogenic full-thickness macular hole can be created. Another injection site may be used if over-stretching of the first bleb is of concern.59 To stabilize bleb localization and possibly mitigate reflux of viral vector into the vitreous cavity, an air-fluid exchange is performed with care to avoid aspirating directly over the injection site. Instruments are removed and all sclerotomies are sutured. Post-operatively, patients are advised to remain supine for the next 24 hours so that the medication can diffuse over the macula. Usual post-vitrectomy precautions, eye drops, and follow-up visits are recommended.57
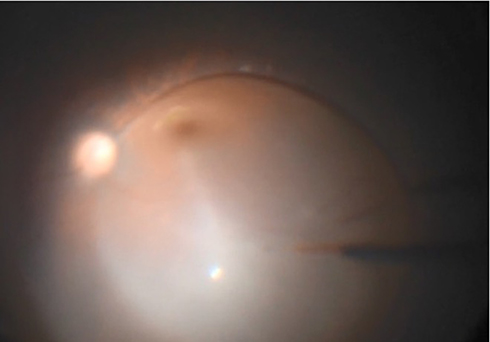 |
Figure 4 Intraoperative photograph illustrating a fovea-involving subretinal bleb created by injecting 0.3 mL of voretigene neparvovec. |
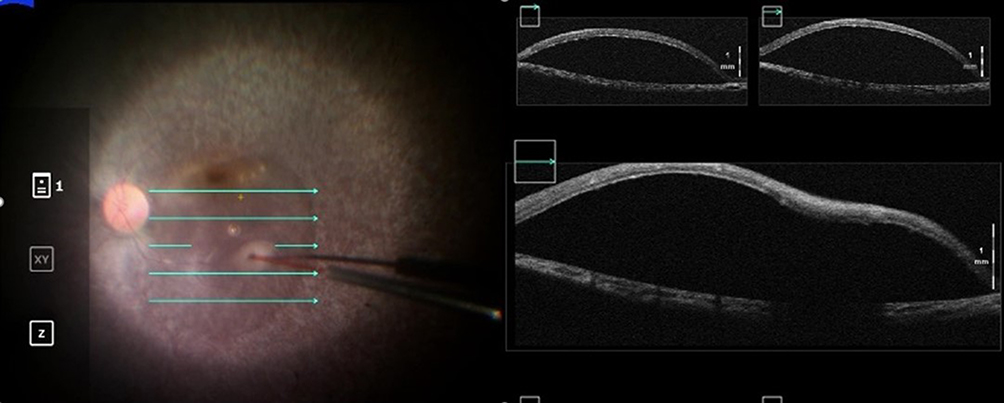 |
Figure 5 Intraoperative optical coherence tomography imaging can be used to guide the formation of a subretinal bleb of voretigene neparvovec and confirm its proper localization in the subretinal space. |
Cost-Effectiveness
The voretigene neparvovec-rzyl drug itself costs $425,000 per eye. When accounting for facility and surgical fees, the total cost is even higher.61 Some have raised concerns regarding the cost of this therapy relative to the actual benefits received.62 Since the 1990s, the incremental cost-effectiveness ratio (ICER) has been established as a metric for evaluating cost-effectiveness of medical therapies. ICER is calculated by dividing the cost of therapy by the quality-adjusted life years (QALY). QALY is defined as the life expectancy multiplied by the quality of life on a scale of 0 to 1 (1 equates to perfect health while 0 equates to death). ICER is reported as cost per QALY. An ICER range of $50,000 to $150,000 United States dollars (USD), which equates to approximately 45,000 to 137,000 euros, has been generally considered as the threshold range for cost-efficiency such that anything at or below this range is considered to be cost-effective.63,64
Johnson et al used VA and VF data from a retrospective natural history study of RPE65-associated IRD patients and compared their progression to that of patients from the gene therapy trials. Direct medical costs from visual impairment were estimated based on a neovascular age-related macular degeneration (nAMD) study population of 200 patients with an adjustment for inflation. Indirect costs were approximated using national surveys evaluating productivity loss, caregiver burden, and government program costs for patients with IRD. A standard 3% annual discount for costs and benefits was included. Assuming gene therapy benefits were completely lost after year 3, the study estimated an ICER of $380,185 and $237,140 per QALY excluding and including the burdens of indirect costs, respectively. This exceeds the standard range accepted as cost-effective. However, the study calculated that if at least 8.8% (including indirect costs) or 43.3% (excluding indirect costs) of the long-term treatment effects persisted after year 3, then VN therapy would fall below the $150,000 per QALY threshold to be considered cost-effective.61 Another group using slightly different assumptions and estimates also found VN to be cost-ineffective. Their study assumed a 10-year duration of treatment effect followed by a 10-year waning period. The resulting ICER value accounting for average total lifetime direct medical costs without gene therapy was $643,813 per QALY and $480,130 per QALY when nonmedical and indirect costs were also accounted for.65 Unlike the prior two studies, a United Kingdom study assumed a 40-year duration of treatment effect, and did find VN to be cost-effective. Costs calculations include VN acquisition price, administration, testing, monitoring, and adverse events. The study found a final ICER of 95,072 euros per QALY.66
A major limitation in all these studies is the use of VA and VF as the primary outcomes representing benefit from therapy. These two markers have been found to be extremely variable and are unlikely to be accurate reflections of the benefits gained from gene therapy in RPE65-associated IRDs. MLMT or FST may be better metrics for visual function gains, but have not been used because no cost-association data exists for either. Other limitations include the unknown true durability of VN therapy, variability in assumptions incorporated into the costs, and the visual function results being extrapolated from small sample sizes due to the rarity of the disease.
Spark-Sponsored Patient Assistance
Spark Therapeutics has enacted several strategies to improve patient access to voretigene neparvovec treatment in light of prohibitory high costs. One includes a unique outcomes-based rebate model which shares the cost responsibilities with the payer if outcomes fail to meet predetermined thresholds. These thresholds will be based on short-term (30–90 days) and long-term (30 months) FST testing scores. The second involves bypassing the typical billing model known as “buy and bill” in which the treating facility first purchases a drug and later initiates a bill to the payer for reimbursement. Instead, Spark Therapeutics offers a different contracting model where the company enters into a direct agreement with commercial payers for the purchase of VN which may cap patient out-of-pocket expenses at an in-network limit. The third involves a proposal to be reviewed by the Centers for Medicare and Medicaid Services for a long-term payment model that would allow the costs of therapy to be paid over several years rather than as a single lumpsum payment upfront. This also increases the possibility of receiving company rebates linked to clinical outcomes. If this proposal is rejected, the company will propose that its distributors be permitted to independently make alternative payment options available to the payer which may include installments or financing options. The company has also developed a team known as the Spark Therapeutic Generation Patient Services which assists commercially insured patients in navigating the insurance process. This includes support for travel and lodging costs related to treatment as well as other out-of-pocket treatment-related costs.67–69
Conclusion
Over the last decade, gene therapy research has made significant progress, and new mutations are being discovered at a rapid pace.70 The success of VN in human clinical trials has paved the way for investigational studies targeting other genetic mutations associated with a variety of IRDs. Currently, there are dozens of ongoing gene therapy trials at both preclinical and clinical stages for diseases such as achromatopsia, X-linked retinoschisis, X-linked retinitis pigmentosa, choroideremia, and even non-IRDs such as neovascular age-related macular degeneration and diabetic macular edema. In addition to addressing the mutations themselves, scientists are also evaluating factors that contribute to the pathogenesis of IRDs. For example, oxidative stress may induce alternative gene expression pathways, contributing to progression of certain IRDs.71,72 As our knowledge about gene therapy and IRDs expands, so does the likelihood of having more therapeutic options to treat once-untreatable blinding diseases.
Disclosure
Dr Rehan M Hussain reports personal fees from Alimera Sciences, outside the submitted work. Dr Christina Y Weng reports personal fees from Alcon, Inc., Alimera Sciences, Inc., Allergan/AbbVie, Dutch Ophthalmic Research Center, Novartis, Regeneron, and REGENXBIO, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Kutluer M, Huang L, Marigo V. Targeting molecular pathways for the treatment of inherited retinal degeneration. Neural Regen Res. 2020;15(10):1784–1791. doi:10.4103/1673-5374.280303
2. RetNet. Summaries of genes and loci causing retinal diseases. 2-14-2020. [cited May 5, 2020]. Available from: https://sph.uth.edu/retnet/sum-dis.htm.
3. Hanein S, Perrault I, Gerber S, et al. Leber congenital amaurosis: comprehensive survey of the genetic heterogeneity, refinement of the clinical definition, and genotype-phenotype correlations as a strategy for molecular diagnosis. Hum Mutat. 2004;23(4):306–317. doi:10.1002/humu.20010
4. Gu S-M, Thompson DA, Srikumari CR, et al. Mutations in RPE65 cause autosomal recessive childhood–onset severe retinal dystrophy. Nat Genet. 1997;17(2):194–197. doi:10.1038/ng1097-194
5. Novartis announces landmark EU approval for one-time gene therapy Luxturna® to restore vision in people with rare inherited retinal disease. November 23, 2018. [cited May 5, 2020]. Available from: https://novartis.gcs-web.com/Novartis-announces-landmark-EU-approval-for-one-time-gene-therapy-Luxturna-to-restore-vision-in-people-with-rare-inherited-retinal-disease.
6. LUXTURNA (voretigene neparvovec-rzyl) US full prescribing information. 2017. [cited May 5, 2020]. Available from: https://sparktx.com/LUXTURNA_US_Prescribing_Information.pdf.
7. Thompson DA, Gal A. Vitamin A metabolism in the retinal pigment epithelium: genes, mutations, and diseases. Prog Retin Eye Res. 2003;22(5):683–703. doi:10.1016/S1350-9462(03)00051-X
8. Redmond TM, Yu S, Lee E, et al. Rpe65 is necessary for production of 11-cis-vitamin A in the retinal visual cycle. Nat Genet. 1998;20(4):344–351. doi:10.1038/3813
9. Redmond TM, Poliakov E, Yu S, Tsai JY, Lu Z, Gentleman S. Mutation of key residues of RPE65 abolishes its enzymatic role as isomerohydrolase in the visual cycle. Proc Natl Acad Sci USA. 2005;102(38):13658–13663. doi:10.1073/pnas.0504167102
10. Moiseyev G, Chen Y, Takahashi Y, Wu BX, Ma JX. RPE65 is the isomerohydrolase in the retinoid visual cycle. Proc Natl Acad Sci USA. 2005;102(35):12413–12418. doi:10.1073/pnas.0503460102
11. Van Hooser JP, Aleman TS, He YG, et al. Rapid restoration of visual pigment and function with oral retinoid in a mouse model of childhood blindness. Proc Natl Acad Sci USA. 2000;97(15):8623–8628. doi:10.1073/pnas.150236297
12. Den Hollander AI, Roepman R, Koenekoop RK, Cremers FP. Leber congenital amaurosis: genes, proteins and disease mechanisms. Prog Retin Eye Res. 2008;27(4):391–419. doi:10.1016/j.preteyeres.2008.05.003
13. Wiszniewski W, Lewis RA, Stockton DW, et al. Potential involvement of more than one locus in trait manifestation for individuals with leber congenital amaurosis. Hum Genet. 2011;129(3):319–327. doi:10.1007/s00439-010-0928-y
14. Marlhens F, Bareil C, Griffoin JM, et al. Mutations in RPE65 cause leber’s congenital amaurosis. Nat Genet. 1997;17(2):139–141. doi:10.1038/ng1097-139
15. Chung DC, Traboulsi EI. Leber congenital amaurosis: clinical correlations with genotypes, gene therapy trials update, and future directions. J AAPOS. 2009;13(6):587–592. doi:10.1016/j.jaapos.2009.10.004
16. Perrault I, Rozet JM, Gerber S, et al. Leber congenital amaurosis. Mol Genet Metab. 1999;68(2):200–208. doi:10.1006/mgme.1999.2906
17. Leber T. Uber retinitis pigmentosa and angeborene amaurose. Graefes Arch Clin Exp Ophthalmol. 1869;15:13–20. doi:10.1007/BF02721213
18. Astuti GD, Bertelsen M, Preising MN, et al. Comprehensive genotyping reveals RPE65 as the most frequently mutated gene in Leber congenital amaurosis in Denmark. Eur J Hum Genet. 2016;24(7):1071–1079. doi:10.1038/ejhg.2015.241
19. Jauregui R, Park KS, Tsang SH. Two-year progression analysis of RPE65 autosomal dominant retinitis pigmentosa. Ophthalmic Genet. 2018;39(4):544–549. doi:10.1080/13816810.2018.1484929
20. Morimura H, Fishman GA, Grover SA, Fulton AB, Berson EL, Dryja TP. Mutations in the RPE65 gene in patients with autosomal recessive retinitis pigmentosa or leber congenital amaurosis. Proc Natl Acad Sci USA. 1998;95(6):3088–3093. doi:10.1073/pnas.95.6.3088
21. Wilbanks GA, Streilein JW. Characterization of suppressor cells in anterior chamber-associated immune deviation (ACAID) induced by soluble antigen. Evidence of two functionally and phenotypically distinct T-suppressor cell populations. Immunology. 1990;71(3):383–389.
22. Wenkel H, Streilein JW. Analysis of immune deviation elicited by antigens injected into the subretinal space. Invest Ophthalmol Vis Sci. 1998;39(10):1823–1834.
23. Han Z, Conley SM, Naash MI. AAV and compacted DNA nanoparticles for the treatment of retinal disorders: challenges and future prospects. Invest Ophthalmol Vis Sci. 2011;52(6):3051–3059. doi:10.1167/iovs.10-6916
24. Rodrigues GA, Shalaev E, Karami TK, Cunningham J, Slater NKH, Rivers HM. Pharmaceutical development of AAV-based gene therapy products for the eye. Pharm Res. 2018;36(2):29. doi:10.1007/s11095-018-2554-7
25. Nidetz NF, Mcgee MC, Tse LV, et al. Adeno-associated viral vector-mediated immune responses: understanding barriers to gene delivery. Pharmacol Ther. 2020;207:107453. doi:10.1016/j.pharmthera.2019.107453
26. Khani SC, Pawlyk BS, Bulgakov OV, et al. AAV-mediated expression targeting of rod and cone photoreceptors with a human rhodopsin kinase promoter. Invest Ophthalmol Vis Sci. 2007;48(9):3954–3961.
27. Li Q, Timmers AM, Guy J, Pang J, Hauswirth WW. Cone-specific expression using a human red opsin promoter in recombinant AAV. Vision Res. 2008;48(3):332–338. doi:10.1016/j.visres.2007.07.026
28. Auricchio A, Kobinger G, Anand V, et al. Exchange of surface proteins impacts on viral vector cellular specificity and transduction characteristics: the retina as a model. Hum Mol Genet. 2001;10(26):3075–3081. doi:10.1093/hmg/10.26.3075
29. Redmond TM, Hamel CP. Genetic analysis of RPE65: from human disease to mouse model. Meth Enzymol. 2000;316:705–724.
30. Veske A, Nilsson SE, Narfström K, Gal A. Retinal dystrophy of Swedish briard/briard-beagle dogs is due to a 4-bp deletion in RPE65. Genomics. 1999;57(1):57–61. doi:10.1006/geno.1999.5754
31. Acland GM, Aguirre GD, Ray J, et al. Gene therapy restores vision in a canine model of childhood blindness. Nat Genet. 2001;28(1):92–95. doi:10.1038/ng0501-92
32. Acland GM, Aguirre GD, Bennett J, et al. Long-term restoration of rod and cone vision by single dose rAAV-mediated gene transfer to the retina in a canine model of childhood blindness. Mol Ther. 2005;12(6):1072–1082. doi:10.1016/j.ymthe.2005.08.008
33. Lai CM, Yu MJ, Brankov M, et al. Recombinant adeno-associated virus type 2-mediated gene delivery into the knockout mouse eye results in limited rescue. Genet Vaccines Ther. 2004;2:3. doi:10.1186/1479-0556-2-3
34. Jacobson SG, Boye SL, Aleman TS, et al. Safety in nonhuman primates of ocular AAV2-RPE65, a candidate treatment for blindness in leber congenital amaurosis. Hum Gene Ther. 2006;17(8):845–858. doi:10.1089/hum.2006.17.845
35. Jacobson SG, Aleman TS, Cideciyan AV, et al. Identifying photoreceptors in blind eyes caused by RPE65 mutations: prerequisite for human gene therapy success. Proc Natl Acad Sci USA. 2005;102(17):6177–6182. doi:10.1073/pnas.0500646102
36. Jacobson SG, Cideciyan AV, Aleman TS, et al. Photoreceptor layer topography in children with leber congenital amaurosis caused by RPE65 mutations. Invest Ophthalmol Vis Sci. 2008;49(10):4573–4577. doi:10.1167/iovs.08-2121
37. Maguire AM, Simonelli F, Pierce EA, et al. Safety and efficacy of gene transfer for leber’s congenital amaurosis. N Engl J Med. 2008;358(21):2240–2248. doi:10.1056/NEJMoa0802315
38. Hauswirth WW, Aleman TS, Kaushal S, et al. Treatment of leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno-associated virus gene vector: short-term results of a Phase I trial. Hum Gene Ther. 2008;19(10):979–990. doi:10.1089/hum.2008.107
39. Bainbridge JW, Smith AJ, Barker SS, et al. Effect of gene therapy on visual function in leber’s congenital amaurosis. N Engl J Med. 2008;358(21):2231–2239. doi:10.1056/NEJMoa0802268
40. Maguire AM, High KA, Auricchio A, et al. Age-dependent effects of RPE65 gene therapy for leber’s congenital amaurosis: a phase 1 dose-escalation trial. Lancet. 2009;374(9701):1597–1605. doi:10.1016/S0140-6736(09)61836-5
41. Testa F, Maguire AM, Rossi S, et al. Three-year follow-up after unilateral subretinal delivery of adeno-associated virus in patients with leber congenital amaurosis type 2. Ophthalmology. 2013;120(6):1283–1291. doi:10.1016/j.ophtha.2012.11.048
42. Ashtari M, Zhang H, Cook PA, et al. Plasticity of the human visual system after retinal gene therapy in patients with leber’s congenital amaurosis. Sci Transl Med. 2015;7(296):296ra110. doi:10.1126/scitranslmed.aaa8791
43. Jacobson SG, Cideciyan AV, Ratnakaram R, et al. Gene therapy for leber congenital amaurosis caused by RPE65 mutations: safety and efficacy in 15 children and adults followed up to 3 years. Arch Ophthalmol. 2012;130(1):9–24. doi:10.1001/archophthalmol.2011.298
44. Jacobson SG, Cideciyan AV, Roman AJ, et al. Improvement and decline in vision with gene therapy in childhood blindness. N Engl J Med. 2015;372(20):1920–1926. doi:10.1056/NEJMoa1412965
45. Bainbridge JW, Mehat MS, Sundaram V, et al. Long-term effect of gene therapy on leber’s congenital amaurosis. N Engl J Med. 2015;372(20):1887–1897. doi:10.1056/NEJMoa1414221
46. Weleber RG, Pennesi ME, Wilson DJ, et al. Results at 2 years after gene therapy for RPE65-deficient leber congenital amaurosis and severe early-childhood-onset retinal dystrophy. Ophthalmology. 2016;123(7):1606–1620. doi:10.1016/j.ophtha.2016.03.003
47. Pennesi ME, Weleber RG, Yang P, et al. Results at 5 years after gene therapy for RPE65-deficient retinal dystrophy. Hum Gene Ther. 2018;29(12):1428–1437. doi:10.1089/hum.2018.014
48. Bennett J, Ashtari M, Wellman J, et al. AAV2 gene therapy re-administration in three adults with congenital blindness. Sci Transl Med. 2012;4(120):120ra15. doi:10.1126/scitranslmed.3002865
49. Bennett J, Wellman J, Marshall KA, et al. Safety and durability of effect of contralateral-eye administration of AAV2 gene therapy in patients with childhood-onset blindness caused by RPE65 mutations: a follow-on phase 1 trial. Lancet. 2016;388(10045):661–672. doi:10.1016/S0140-6736(16)30371-3
50. Chung DC, Mccague S, Yu ZF, et al. Novel mobility test to assess functional vision in patients with inherited retinal dystrophies. Clin Experiment Ophthalmol. 2018;46(3):247–259. doi:10.1111/ceo.13022
51. Russell S, Bennett J, Wellman JA, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomized, controlled, open-label, phase 3 trial. Lancet. 2017;390(10097):849–860. doi:10.1016/S0140-6736(17)31868-8
52. LUXTURNATM (voretigene neparvovec-rzyl) December 19, 2017 approval letter. 2017. [cited May 5, 2020]. Available from: https://www.fda.gov/downloads/BiologicsBloodVaccines/CellularGeneTherapyProducts/ApprovedProducts/UCM589690.pdf.
53. Maguire AM, Russell S, Wellman JA, et al. Efficacy, safety, and durability of voretigene neparvovec-rzyl in RPE65 mutation-associated inherited retinal dystrophy: results of phase 1 and 3 trials. Ophthalmology. 2019;126(9):1273–1285. doi:10.1016/j.ophtha.2019.06.017
54. Drack AV, Bennett J, Russell S, et al. How long does gene therapy last? 4-year follow up of phase 3 voretigene neparvovec trial in RPE65-associated LCA/inherited retinal disease. JAAPOS. 2019;23(4):e7.
55. A Patient Registry Study for Patients Treated With Voretigene Neparvovec. [cited May 5, 2020]. Available from: https://clinicaltrials.gov/ct2/show/NCT03597399.
56. Treatment Centers. HCP support services | spark Therapeutics. [cited May 5, 2020]. Available from: http://mysparkgeneration.com/hcp-support.html#TreatmentCenters.
57. Weng CY Making history exploring luxturna gene therapy and its surgical delivery. New Retinal Physician. September, 2018. [cited May 5, 2020]. Available from: https://www.retinalphysician.com/supplements/2018/september-2018/new-retinal-physician/making-history.
58. Davis JL. The blunt end: surgical challenges of gene therapy for inherited retinal diseases. Am J Ophthalmol. 2018;196:xxv–xxix. doi:10.1016/j.ajo.2018.08.038
59. Hussain RM, Tran KD, Maguire AM, Berrocal AM. Subretinal injection of voretigene neparvovec-rzyl in a patient with RPE65-associated leber’s congenital amaurosis. Ophthalmic Surg Lasers Imaging Retina. 2019;50(10):661–663. doi:10.3928/23258160-20191009-01
60. Xue K, Groppe M, Salvetti AP, Maclaren RE. Technique of retinal gene therapy: delivery of viral vector into the subretinal space. Eye (Lond). 2017;31(9):1308–1316.
61. Johnson S, Buessing M, O’connell T, Pitluck S, Ciulla TA. Cost-effectiveness of voretigene neparvovec-rzyl vs standard care for RPE65-mediated inherited retinal disease. JAMA Ophthalmol. 2019;137(10):1115–1123. doi:10.1001/jamaophthalmol.2019.2512
62. Darrow JJ. Luxturna: FDA documents reveal the value of a costly gene therapy. Drug Discov Today. 2019;24(4):949–954. doi:10.1016/j.drudis.2019.01.019
63. Grosse SD. Assessing cost-effectiveness in healthcare: history of the $50,000 per QALY threshold. Expert Rev Pharmacoecon Outcomes Res. 2008;8(2):165–178. doi:10.1586/14737167.8.2.165
64. Neumann PJ, Cohen JT, Weinstein MC. Updating cost-effectiveness–the curious resilience of the $50,000-per-QALY threshold. N Engl J Med. 2014;371(9):796–797. doi:10.1056/NEJMp1405158
65. Zimmermann M, Lubinga SJ, Banken R, et al. Cost utility of voretigene neparvovec for biallelic RPE65-mediated inherited retinal disease. Value Health. 2019;22(2):161–167. doi:10.1016/j.jval.2018.09.2841
66. Viriato D, Bennett N, Sidhu R, et al. An economic evaluation of voretigene neparvovec for the treatment of biallelic RPE65-mediated inherited retinal dystrophies in the UK. Adv Ther. 2020;37(3):1233–1247. doi:10.1007/s12325-020-01243-y
67. Spark Therapeutics Announces First-of-their-kind Programs to Improve Patient Access to LUXTURNA™ (voretigene neparvovec-rzyl), a One-time Gene Therapy Treatment. [cited May 5, 2020]. Available from: https://sparktx.com/press_releases/spark-therapeutics-announces-first-of-their-kind-programs-to-improve-patient-access-to-luxturna-voretigene-neparvovec-rzyl-a-one-time-gene-therapy-treatment/.
68. Spark Therapeutic Generation Patient Services Enrollment Form. [cited May 5, 2020]. Available from: https://www.mysparkgeneration.com/pdf/LUX2692_LUXTURNA_Enrollment_Form_P-RPE65-US-76000_12-9-19_Editable.pdf.
69. Luxturna voretigene neparvovec-rzyl for subretinal injection Statement of Medical Necessity. [cited May 5, 2020]. Available from: https://mysparkgeneration.com/pdf/LUX2693_Luxturna_SMN_Form_FIN_Editable.pdf.
70. Scimone C, Donato L, Esposito T, Rinaldi C, D’Angelo R, Sidoti A. A novel RLBP1 gene geographical area-related mutation present in a young patient with retinitis punctata albescens. Hum Genomics. 2017;11(1):18. doi:10.1186/s40246-017-0114-6
71. Donato L, D’Angelo R, Alibrandi S, Rinaldi C, Sidoti A, Scimone C. Effects of A2E-induced oxidative stress on retinal epithelial cells: new insights on differential gene response and retinal dystrophies. Antioxidants (Basel). 2020;9(4):307. doi:10.3390/antiox9040307
72. Donato L, Scimone C, Alibrandi S, et al. Discovery of GLO1 new related genes and pathways by RNA-seq on A2E-stressed retinal epithelial cells could improve knowledge on retinitis pigmentosa. Antioxidants (Basel). 2020;9(5):416. doi:10.3390/antiox9050416
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.