Back to Journals » Clinical Interventions in Aging » Volume 14
Vitamin E and Alzheimer’s disease: what do we know so far?
Authors Browne D, McGuinness B ![]() , Woodside JV, McKay GJ
, Woodside JV, McKay GJ ![]()
Received 10 May 2019
Accepted for publication 4 July 2019
Published 18 July 2019 Volume 2019:14 Pages 1303—1317
DOI https://doi.org/10.2147/CIA.S186760
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Richard Walker
Declan Browne, Bernadette McGuinness, Jayne V Woodside, Gareth J McKay
Centre for Public Health, Queen’s University Belfast, Belfast, UK
Abstract: Vitamin E has been proposed as a potential clinical intervention for Alzheimer’s disease (AD) given the plausibility of its various biological functions in influencing the neurodegenerative processes associated with the condition. The tocopherol and tocotrienol isoforms of vitamin E have multiple properties including potent antioxidant and anti-inflammatory characteristics, in addition to influences on immune function, cellular signalling and lowering cholesterol. Several of these roles offer a theoretical rationale for providing benefit for the treatment of AD-associated pathology. Diminished circulating concentrations of vitamin E have been demonstrated in individuals with AD. Reduced plasma levels have furthermore been associated with an increased risk of AD development while intake, particularly from dietary sources, may limit or reduce the rate of disease progression. This benefit may be linked to synergistic actions between vitamin E isoforms and other micronutrients. Nevertheless, randomised trials have found limited and inconsistent evidence of vitamin E supplementation as an effective clinical intervention. Thus, despite a strong rationale in support of a beneficial role for vitamin E for the treatment of AD, the evidence remains inconclusive. Several factors may partly explain this discrepancy and represent the difficulties of translating complex laboratory evidence and dietary interactions into clinical interventions. Methodological design limitations of existing randomised trials and restrictions to supplementation with a single vitamin E isoform may also limit the influence of effect. Moreover, several factors influence individual responsiveness to vitamin E intake and recent findings suggest variation in the underlying genetic architecture attenuates vitamin E biological availability and activity which likely contributes to the variation in clinical responsiveness and the failure of randomised trials to date. Importantly, the clinical safety of vitamin E remains controversial and warrants further investigation.
Keywords: vitamin E, Alzheimer’s disease, tocopherols, tocotrienols, antioxidants
Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder that accounts for up to 80% of dementia cases.1 It is clinically characterised by the insidious onset of episodic memory impairment that evolves over time and is associated with subsequent decline in other cognitive domains that diminish functional ability.2 Its histopathological hallmarks include neurofibrillary tangles (NFTs), amyloid plaques and loss of neuronal synapses in the brain.3 AD represents a major global disease burden with an estimated 50 million people currently living with dementia, a figure expected to increase threefold by 2050 with associated global economic costs expected to double to US$2 trillion by 2030.4,5 As such, prevention and treatment interventions for AD are paramount, given an estimated 9.2 million deaths could be prevented by 2050 if AD onset was delayed by one year.6
It is widely accepted that AD pathology begins decades before the appearance of clinical manifestations; changes may be present up to 30 years before the onset of symptoms.7 Advances in neuro-imaging modalities and the ongoing development of biomarkers from cerebrospinal fluid (CSF) offer some aid to predicting the development of AD in those with mild cognitive impairment (MCI) when combined with validated clinical tests.8
However, existing therapeutic options are largely limited to delayed disease progression and ease of symptom burden, albeit without modification of disease-course.9 This has resulted in greater focus on the development of alternative interventions to delay or prevent onset that have included dietary and antioxidant measures.10 Vitamin E has been studied extensively, primarily due to its potent antioxidant properties and the biological plausibility of its potential role in combatting the pathological processes of AD. However, its use as an effective clinical intervention remains controversial.
This review will assess the current evidence for the role of vitamin E as a treatment option in the context of AD. A literature search of Medline, a major article database, was conducted using the keywords “Alzheimer Disease” AND “Vitamin E” OR “tocopherols” OR “tocotrienols”. The keywords were searched in all possible combinations. Original journal articles that were written in English and published prior to 1st March 2019 were retrieved. All studies incorporating cell, animal and human evidence were included in addition to review articles to achieve a comprehensive search of the topic and to retrieve the maximum number of articles possible. A total of 341 articles were retrieved.
Pathogenesis of AD
Several theories have proposed the onset and progression of AD as a corollary of the uncertainty of the pathogenic mechanisms that lead to disease and the likely overlapping contributions they make to the phenotype observed. Since the characteristic “plaques” and “tangles” of AD were first reported by Alois Alzheimer in 1907, the role of amyloid and tau protein deposits have remained central to AD pathogenesis.11 The amyloid cascade hypothesis postulates that excessive accumulation of senile plaques, composed of amyloid-beta (Aβ) protein, directly induces the clinical manifestations of AD through neurodegeneration mediated by inflammation, immunological mechanisms and the effects of free radical species.12 Aβ is a beta-sheet protein derived from the amyloid beta precursor protein (AβPP) molecule through the activity of β and γ-secretase.13 Importantly, expression of the ApoE4 allele reduces elimination of Aβ and is associated with increased AD risk.14
Similarly, the accumulation of intra-cellular NFTs have been implicated in AD pathology.15 NFTs consist of hyper-phosphorylated tau-protein, an important component of the neuronal cytoskeleton. Significantly, an increased quantity of NFTs is inversely associated with cognitive impairment and is more closely correlated with dementia severity than Aβ plaques.16 Furthermore, hypercholesterolaemia has been associated with AD pathology through its association with the amyloid pathway, although its precise role remains uncertain.17 Laboratory studies have shown higher cholesterol levels are associated with increased proteolysis of AβPP and Aβ production through secretase enzyme activity.18 Dysfunctional cholesterol metabolism has also been associated with its accumulation in the brains of AD patients.19 However, the clinical application of this remains controversial as evidence from randomised trials indicates that cholesterol-reducing statins have no effect on validated measures of cognition.20 Despite the obvious implications of the amyloid cascade theory, several concerns have questioned the assumption of direct causality, including the failure of therapeutic interventions targeting the amyloid pathway to provide clinical benefit21 and an extensive body of evidence suggests the pathological processes of AD are complex and multifactorial.22
The mitochondrial cascade hypothesis of AD pathogenesis postulated that genetic variation influences the impact of age-related mitochondrial changes which upon reaching a threshold value initiate a pathological cascade, including the amyloid pathway.23 In addition, critical mitochondrial dysfunction may precipitate other cellular and molecular changes associated with AD including synaptic degeneration, production of free radical species and neuro-inflammation.24,25 The processes of neuro-inflammation have been reported as an early event in AD, perhaps occurring before the appearance of Aβ deposits.26 In vitro studies have demonstrated elevation of multiple interleukins (ILs), tumour necrosis factor-α (TNF-α) and granulocyte macrophage colony stimulating factor (GM-CFS) in AD murine models early in the disease process.27 Additionally, elevated levels of inflammatory cytokines have been detected in the brain and CSF of AD patients.27
While it is likely each of these mechanisms contribute to AD pathogenesis, oxidative stress (OS) represents a common underlying theme. The role of OS in disease is characterised by the generation of reactive oxygen species (ROS) through the metabolism of oxygen within the mitochondria that manifests as structural and functional alterations in various biomolecules.28 Multiple indicators of OS are significantly elevated in AD with detectable oxidative effects on lipids, proteins, nucleic acids and sugars.29,30 Importantly, cerebral glucose metabolism is reduced early in the AD process and may be accompanied by metabolic dysregulation and increased production of ROS.31
Several factors render the brain particularly vulnerable to the effects of ROS including its high oxygen demand and consumption, the proportion of polyunsaturated fats in neural tissues and the relative scarcity of endogenous antioxidants to address this high metabolic demand.32 Interestingly, AD-associated mitochondrial defects beyond the central nervous system represent characteristic features of a systemic disease.33 This is supported by factors such as diabetes mellitus, obesity and physical inactivity as potential risk factors for AD development.34 In addition, strong associations have been reported between vascular risk factors and AD development, with Aβ protein deposition found within vessels early in the disease course.35 As such, there is a strong rationale in support of systemic antioxidant therapy as a preventative or therapeutic intervention for AD with particular support for vitamin E in accordance with its biological activities.36,37
Basis of vitamin E as a clinical intervention in AD
Biological properties of vitamin E
Vitamin E is a collective term that describes a family of eight naturally-occurring homologues with potent antioxidant properties. The group is composed of four tocopherols and four tocotrienols, each of which has an α, β, γ and δ isoform. All eight congeners are differentially distributed within food sources such as vegetable oils, grains and various nuts and seeds38 and α-tocopherol is the primary isoform normally found within vitamin E supplements.39 The recommended dietary allowance for α-tocopherol is currently 15 mg/day in adults with a recommended upper intake level of 1000 mg/day for supplemental vitamin E as the highest dose unlikely to result in haemorrhage - however, high-doses (>1000 mg/day) have been used in a number of studies of vitamin E supplementation to date.32
Plasma levels of vitamin E are dependent upon the absorption, distribution and excretion rates of each isoform. All eight homologues have lipophilic properties and are absorbed from the intestine following ingestion in micelles formed by pancreatobiliary secretions.32 The plasma half-life of α-tocopherol is estimated at 20 hrs, which is considerably longer than that of other isoforms, particularly the tocotrienol congeners.40 This is significant in that α-tocopherol is therefore the predominant isoform found in tissues whereas the other congeners are metabolised and more quickly removed.41 Additionally, important interactions have been previously reported between various isoforms including antagonistic interactions between plasma α and γ-tocopherol.42
Biological functions of vitamin E
Vitamin E has a broad range of biological functions that vary according to the relevant isoform. The tocopherol and tocotrienol sub-groups possess varying properties and functions linked with the level of chemical saturation in their molecular structures with tocopherols having phytyl side-chains, while tocotrienols possess three carbon-carbon double bonds.32 However, as a collective group, the potent antioxidant capabilities of vitamin E are well known and each of the eight tocopherol and tocotrienol congeners are considered free-radical scavengers.43 The antioxidant capacity results from the presence of a hydroxyl group on the aromatic ring of tochocromanols that quenches free radicals through hydrogen atom donation.44
The various vitamin E isoforms enact a key role in the protection of cell membranes, rich in highly unsaturated fatty acids, from oxidative damage.45 Several studies have shown that different isoforms are differentially located within the cell membrane and that this may influence their biological activity in the lipid membrane.46,47 In vivo studies have reported that the antioxidant activity of α-tocopherol is superior to other tocopherol congeners, followed in potency by the β, γ and δ isoforms, respectively.48 While the effectiveness of α-tocopherol has also been reported through in vitro evidence, it has been suggested that its relative laboratory efficacy may be dependent upon experimental conditions.49 Tocotrienols may exhibit more potent antioxidant activities than tocopherols due to their shorter side-chains enabling easier incorporation into the cell membrane and the presence of their unsaturated side-chain.50 However, it has been suggested that α-tocopherol retains a superior in vivo role in neuroprotection due to its relatively greater bioavailability and preferential retention by tissues.51 Similar to tocopherols, the δ-tocotrienol isoform demonstrates reduced antioxidant potency compared to the other tocotrienol congeners.48
Importantly, the scope of vitamin E activity extends beyond its antioxidant capabilities and includes other neuro-protective, anti-inflammatory and cholesterol-reducing properties, in addition to influencing gene expression and potentially ensuing AD pathology.50
Several studies have demonstrated the beneficial effects of vitamin E supplementation on various markers of inflammatory stress, cellular signalling and immune function in humans and its influence on AD-associated pathology.52,53 Additionally, studies in murine AD models have identified associations between vitamin E deficiency and increased expression of genes associated with AD progression including those involved in the regulation of apoptosis, neuro-transmission and Aβ metabolism.54 Similarly, vitamin E has been shown to confer a protective effect against hyper-phosphorylated tau protein.55 The enzyme-inhibiting activity of various tocopherol and tocotrienol isoforms also incorporates several AD-associated enzymes, including cyclo-oxygenases (COX), which contribute to neuro-inflammation and OS.56 The activity of both sub-groups have also been associated with reduced Aβ production through inhibiting secretase enzyme activity.39
Comparison of tocopherol and tocotrienol isomers
Tocopherols are a necessary constituent of physiological neuronal activity with high levels of the vitamin E transfer protein (α-tocopherol transfer protein, α-TPP) found in the cerebellum.57 Its importance is emphasised by a loss-of-function mutation within the α-TPP gene that results in a rare condition known as ataxia with vitamin E deficiency (AVED)58 and several studies have also reported potential benefits of α-tocopherol in other neurodegenerative disorders: for example, increasing duration (years) of vitamin E supplement use has been inversely correlated with amyotrophic lateral sclerosis (ALS) rates while a meta-analysis found a protective influence from moderate-high intake of vitamin E in participants with regards to their risk of developing Parkinson’s disease.59,60
In contrast, research on tocotrienols has been limited, although not all are believed to be essential for normal physiological function and there is no evidence to date for any genetic mutations that alter tocotrienol metabolism and result in clinically significant sequelae.61 However, tocotrienols are an important vitamin E sub-group with biological functions that differ from tocopherols, with stronger antioxidant and anti-inflammatory effects according to some measures.17 Tocotrienols influence the mevalonate pathway by suppressing hydroxyl-methyl-glutaryl co-enzyme A (HMG-CoA) reductase resulting in cholesterol-lowering and anti-inflammatory properties that influence AD pathology.62 These findings have been reported for several tocotrienol isoforms including the δ congener which suppresses the action of HMG-CoA reductase at the transcriptional level.63,64 However, combined supplementation with α-tocopherol has been shown to attenuate these inhibitory effects with a dose-dependent relationship in hamster models.65 This study furthermore found that α-tocopherol supplementation alone may act as a stimulant to HMG-CoA activity and therefore demonstrate hypercholesterolaemic activity.65 Such interactions highlight the importance of a thorough understanding of the interactions between isoforms and the appropriate selection of congeners in interventional studies.
AD and vitamin E: human studies
Plasma, serum and CSF concentrations of vitamin E in AD
A large number of case-control studies have previously evaluated vitamin E levels in the plasma, serum and CSF of patients with AD (Table 1). An early study reported reduced vitamin E levels in a small number of AD patients compared with cognitively intact controls.83 Many studies have since replicated these findings and have been summarised in a 2014 meta-analysis.84 This study reviewed the plasma status of several micro-nutrients in AD and reported diminished vitamin E levels in AD patients compared with cognitively intact controls and concluded this was not as a consequence of patient malnourishment.84 Similar findings were reported in the sensitivity analysis of a 2017 case-control study that found no correlation between AD severity and plasma vitamin E levels, suggesting diminished plasma antioxidant status may result from early disease pathology rather than as a consequence of reduced vitamin E intake.77
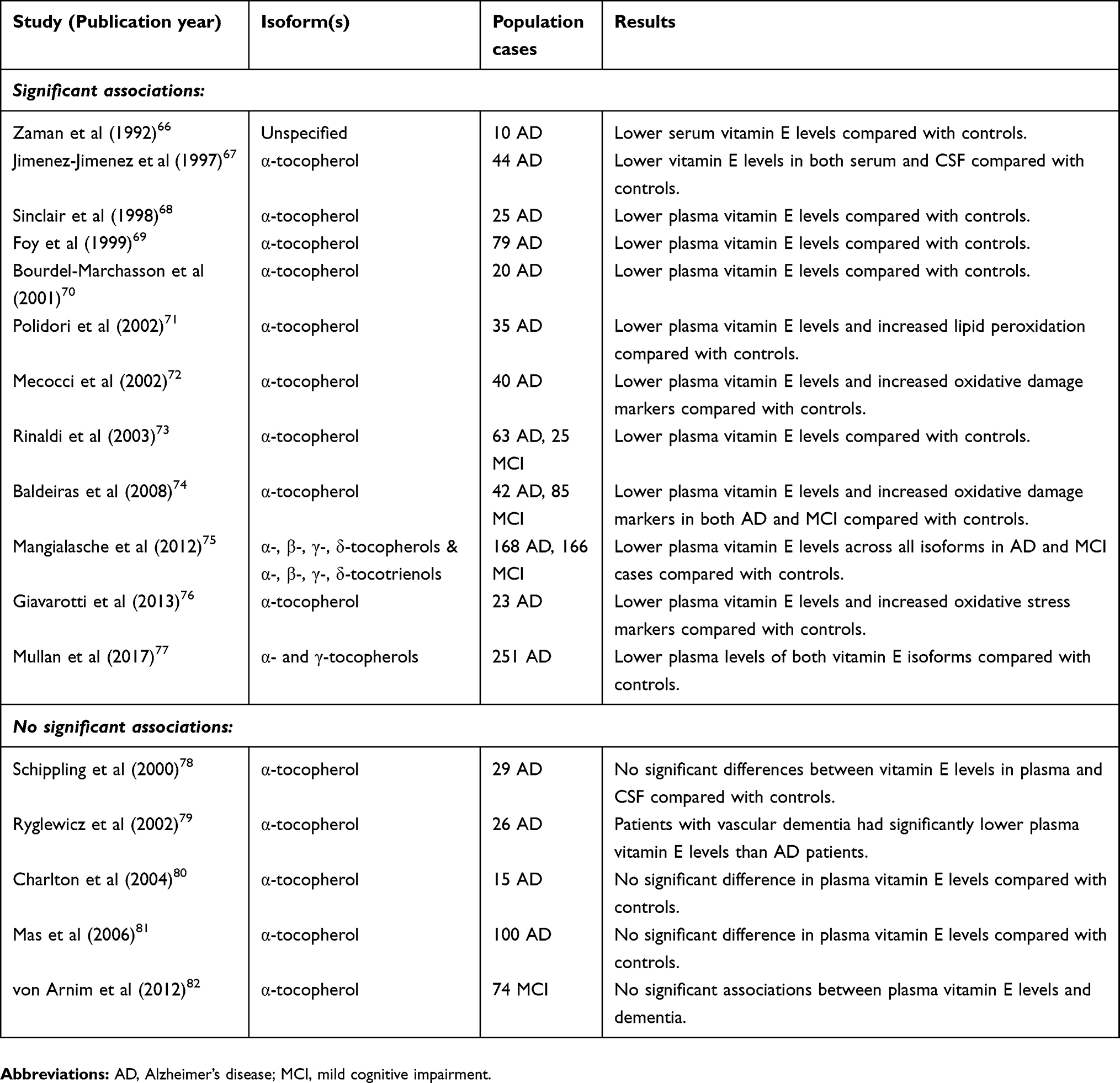 |
Table 1 Cross-sectional studies investigating vitamin E levels in AD patients |
The most recent meta-analysis conducted by Mullan and colleagues (2018) evaluated 51 studies comparing the plasma nutrient status in AD participants compared to cognitively intact controls.85 This study reported that vitamin E was the most extensively studied dietary plasma antioxidant and concluded that vitamin E levels are 11% lower in AD patients compared to cognitively normal subjects85 corroborating the findings from a previous smaller meta-analysis of 17 studies.86
In addition to diminished plasma vitamin E levels in AD, a meta-analysis of 116 studies reported significantly reduced levels in the central nervous system of participants with AD and suggested nutrient status of the brain parallels that of the systemic circulation.87 One cross-sectional study was notable for its measurement of tocotrienol isoforms: 521 subjects were recruited including 168 AD, 166 MCI cases and 187 cognitively intact controls.75 The study reported significantly diminished plasma vitamin E levels for each isoform in those with AD compared to controls and low tocopherol and tocotrienol levels were associated with increased risk of both MCI and AD.75
Despite a substantial body of evidence in support of reduced vitamin E levels in AD, a relatively small number of case-control studies have reported no significant differences in plasma levels compared with cognitively normal controls (Table 1). Although these studies were of limited sample size, they were supported by findings from a larger Mendelian randomised study which included data from two genome-wide association studies of vitamin E (n=7,781) and AD cases and controls (17,007 AD cases and 37,154 controls) that found no association between circulating vitamin E levels and AD.88
Prospective studies of vitamin E concentrations and subsequent AD risk
Several prospective cohort studies have also investigated plasma vitamin E levels and the subsequent risk of developing AD (Table 2). In 2010, a prospective study assessed plasma levels of all eight vitamin E isoforms in 232 subjects aged at least 80 years from the Kungsholmen Project with a six-year follow-up.89 Individuals with total plasma tocopherols, tocotrienols or vitamin E in the highest tertile had a reduced risk of incident AD compared with participants in the lowest tertile. The authors suggested that any neuro-protective effects may result from the combination of vitamin E isoforms rather than specifically to any individual congener.89
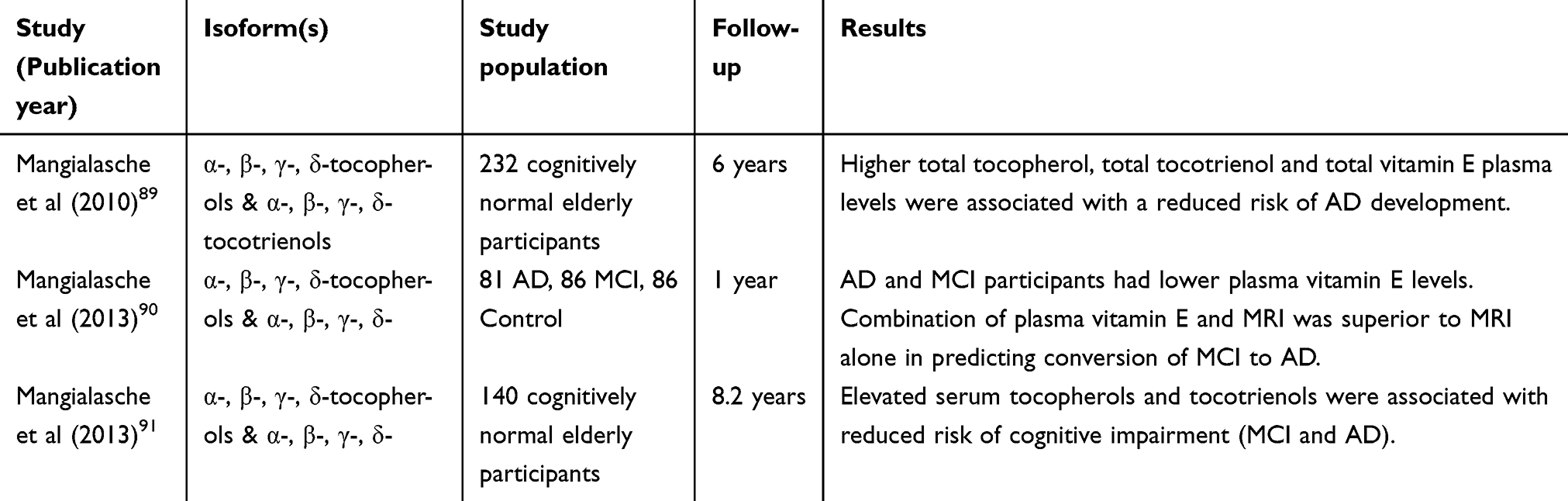 |
Table 2 Studies investigating cross-sectional levels of vitamin E and subsequent risk of AD development |
Another prospective study evaluated vitamin E status and magnetic resonance imaging (MRI) with regard to MCI conversion to AD.90 Data from 253 participants including 81 AD, 86 MCI cases and 86 cognitively intact controls revealed lower absolute values for all vitamin E isomers in both the AD and MCI subjects compared to controls. The authors suggested the combination of vitamin E measures and MRI scanning was superior to imaging alone in predicting MCI conversion to AD, with 95% sensitivity after one-year follow-up.90
Similar findings were reported from the Cardiovascular Risk Factor, Aging and Dementia (CAIDE) study,91 which analysed data from 140 cognitively intact participants with a follow-up time of 8 years. Elevated values of both tocopherol and tocotrienol isoforms were associated with a reduced risk of “cognitive impairment”, defined as the development of either MCI or AD.91
Evaluation of vitamin E intake and AD risk
Associations between vitamin E intake through supplementary or dietary sources and the risk of developing AD have also been extensively investigated (Table 3). A prospective study investigated vitamin E supplementation and incident AD in a population of 633 individuals with a mean 4.3-year follow-up and reported that none of the 27 subjects taking vitamin E supplements developed AD in contrast to the predicted incidence of 3.9.92 Similarly, the prospective Cache County Study reported that individuals taking vitamin E supplements in addition to multivitamins containing vitamin C had reduced AD risk.93 However, the study found no significant benefit from use of vitamin E supplements alone.93 A more recent study published in 2017 reported that vitamin E supplementation was associated with decreased risk of cognitive decline in a cohort of 560 AD patients from the Canadian Study of Health and Aging although, no significant association was detected between vitamin E intake and AD risk specifically.94
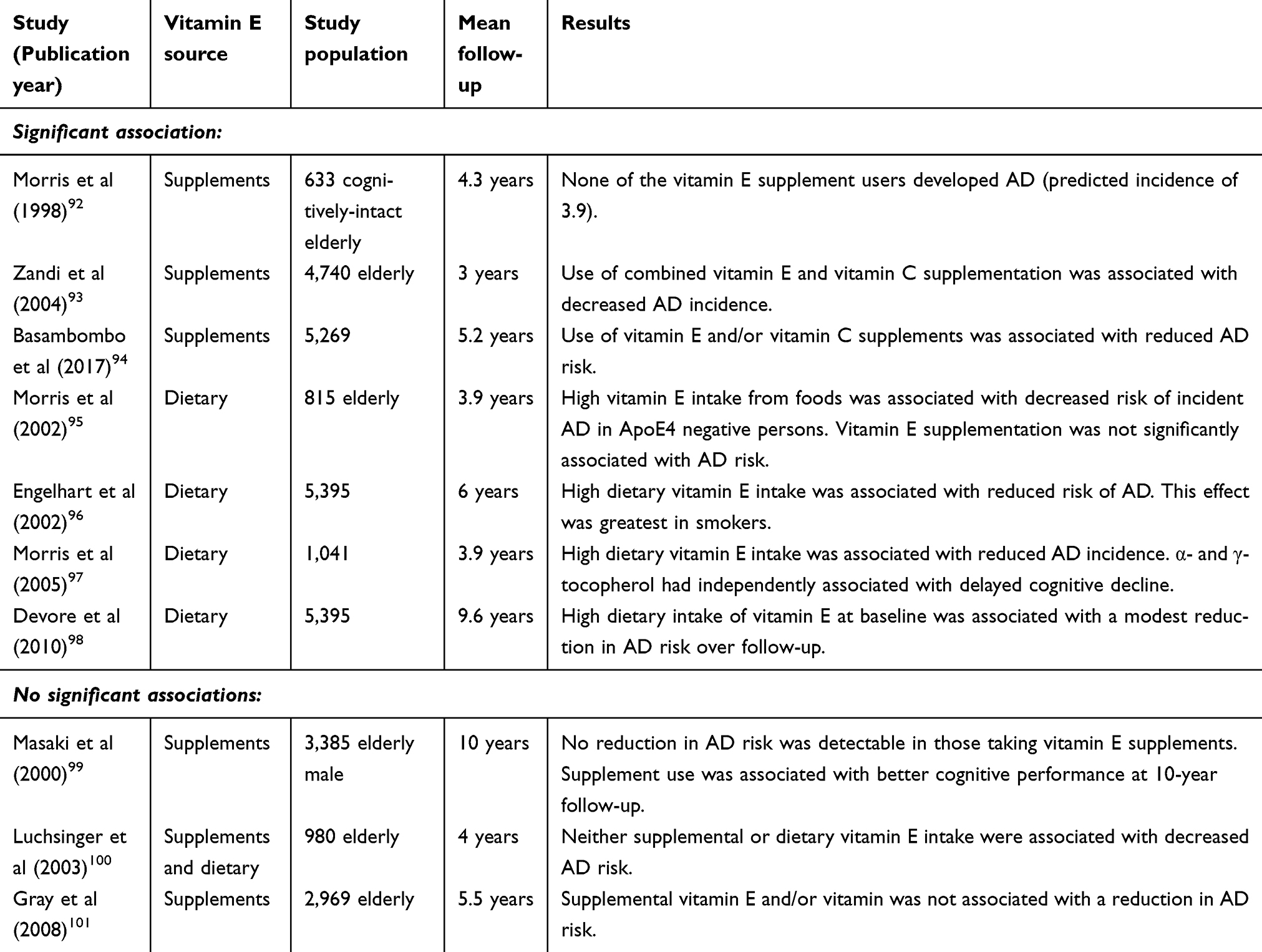 |
Table 3 Epidemiological studies investigating associations between vitamin E intake and risk of AD |
Several prospective studies of vitamin E dietary intake also reported beneficial effects associated with reduced AD risk. One study evaluated 815 cognitively normal elderly individuals with a mean follow-up time of 3.9-years and found that increased vitamin E intake from dietary sources was associated with a lower risk of developing AD, although the benefit was limited to those not carrying the ApoE4 risk allele.95 Interestingly, this study suggested that vitamin E supplementation from non-dietary sources was not significantly associated with diminished AD risk.95 Similar findings in a study of 5,395 participants reported that high dietary intake of both vitamin E and vitamin C was associated with a reduced risk of developing AD.96 This effect was greatest among smokers and was independent of the ApoE4 risk allele.96 Furthermore, Morris and colleagues found that subjects with higher dietary intake of vitamin E had a lower incidence of AD and that plasma α- and γ-tocopherol levels were independently associated with AD risk.97 Finally, the prospective population based Rotterdam Study included 365 participants with AD and identified a modest reduction in risk over the longer term in participants from the highest tertile of dietary vitamin E intake compared with those in the lowest tertile, independent of any supplement use and other potential confounders.98
However, several studies failed to detect associations between either dietary intake or vitamin E supplementation and AD. The Honolulu-Asia Aging Study analysed data from 3,385 men who reported taking vitamin E and vitamin C supplements at baseline in addition to measuring AD prevalence ten-years later.99 Although the study identified a protective effect of vitamin E with vascular dementia, no association with AD risk was noted.99 The analysis of 980 individuals within the Washington Heights-Inwood Columbia Aging Project concluded that neither supplementary nor dietary intake of vitamin E, alone or in combination, significantly attenuated AD risk.100 A prospective study of 2,969 individuals followed-up over a mean 5.5 years found no association between AD risk and the use of vitamin E supplements, with or without vitamin C.101
While these prospective studies provide limited evidence for the benefits of vitamin E supplementation, they nevertheless suggest that a high intake from dietary sources may confer some benefit in reducing the risk of developing AD compared to those with lower intake.
Randomised clinical trials of vitamin E supplementation
Several randomised trials have investigated the efficacy of vitamin E as a potential therapeutic intervention for AD (Table 4). The first of these was published over 20 years ago and was a double-blind, randomised multi-centre clinical trial which compared the effectiveness of selegiline (a selective monoamine oxidase inhibitor) and α-tocopherol (2000 IU/day), individually or in combination, with a placebo in 341 subjects with moderate AD for 2 years.102 Several outcome measures were considered including time-to-death, institutionalisation, functional ability and dementia severity. The study concluded that vitamin E and selegiline slow the progression of moderate AD both independently and as a combination therapy compared with placebo but with no additive benefit of the combined regimen.102
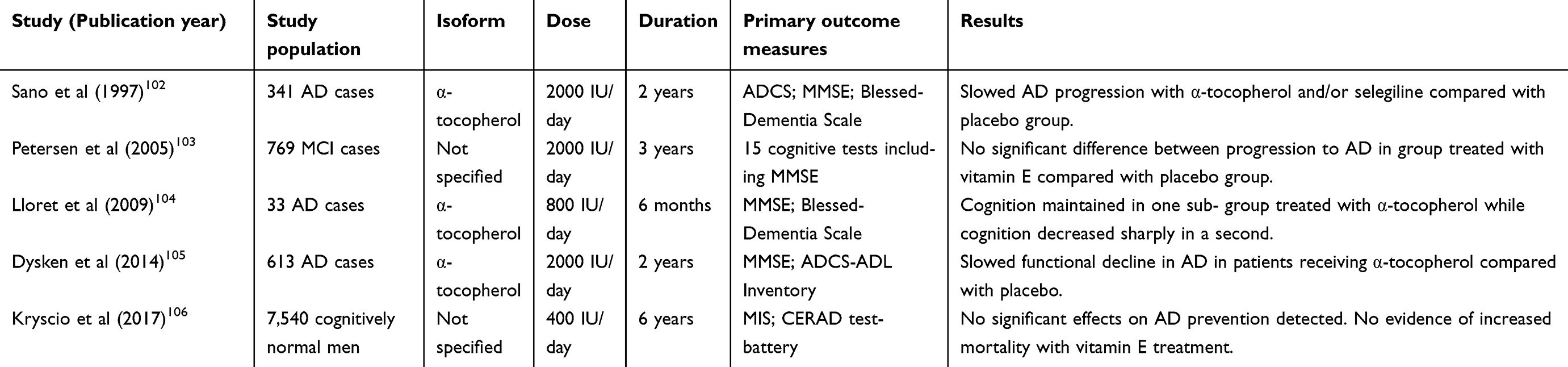 |
Table 4 Randomised trials investigating vitamin E supplementation as a treatment for AD |
A larger double-blind study was undertaken several years later and compared the effects of 2000 IU/day of vitamin E, donepezil or placebo daily for 3 years in 769 subjects with the amnestic subtype of MCI.103 Although the authors did not specify the vitamin E isoform used in supplementation, no significant difference in the rate of conversion to AD was found at any point during follow-up in the group receiving vitamin E and only minimal effects on secondary measures of cognition were detected compared with those receiving placebo.103
In a small trial, 57 AD participants were randomised (33 completed the study) to receive either vitamin E (800 IU/day) or placebo for six-months and markers of OS and cognitive function were assessed.104 Among those randomised to receive vitamin E, two sub-groups were identified: (1) those who had lower measures of OS and retained their cognitive function and (2) those with no significant changes in OS levels who demonstrated marked cognitive decline throughout the six months of the study.104 This cognitive decline was greater in the latter sub-group compared with the group of participants who received placebo.
Another double-blind, randomised clinical trial (The TEAM-AD VA Cooperative Randomised Trial) investigated supplementation of α-tocopherol (2000 IU/day) and/or memantine compared to placebo in 613 patients with mild-to-moderate AD.105 The study reported that α-tocopherol supplementation alone resulted in slower functional decline compared with the placebo group.105 Unexpectedly, the combination of α-tocopherol and memantine demonstrated less benefit than α-tocopherol supplementation alone, although a convincing rationale for this was lacking. Furthermore, evaluation of the α-tocopherol safety data suggested no significant increase in mortality in contrast to an earlier meta-analysis.105,107
More recently, the double-blind PREADViSE study (2017) evaluated the effects of low-dose vitamin E (400 IU/d, unspecified isoform) and/or selenium versus placebo in 7,540 cognitively intact elderly men.106 The study found that neither vitamin E nor selenium, individually or in combination, offered any benefit in delaying the onset of AD.106 Of note, Naeini and colleagues investigated the effects of combined vitamin E and vitamin C supplementation for 1 year versus placebo in elderly individuals with MCI in a double-blind, randomised trial.108 Although supplementation offered some improvement in selected measures of oxidative stress, there were no detectable benefits in cognition.108 A further small trial investigating the effects of supplementation with vitamin E and other nutrients similarly found reductions in measures of OS with treatment but no derived clinical benefit.109
Failure of vitamin E as a clinical intervention so far
Since Harman first postulated the “free radical theory of ageing” in 1956, the potential implications for providing “chemical means of prolonging effective life” has received significant attention.110 Nevertheless, evidence in support of vitamin E providing clinical benefit against AD remains inconsistent and inconclusive. Despite substantial evidence of increased OS in AD aetiology and reduced circulating vitamin E levels, the findings from clinical trials investigating vitamin E as an intervention have yet to match the expectation.
This translational discrepancy is not uncommon among studies investigating the role of OS in disease and its implication in pathogenesis and potential therapeutic approaches.111 Indeed, several studies have shown that antioxidant supplementation is ineffective or possibly even harmful.112 In the context of vitamin E and AD, the failure of clinical trials may be due to general weaknesses in the studies investigating OS or limitations of studies specific to this area. A major and controversial limitation includes determination of the most appropriate OS markers.113,114 Consequently, it has been suggested that disease specific proteins or combinations of markers in large-scale panels should be considered for monitoring therapeutic response and predicting outcomes.115 In the specific context of AD, the use of myeloperoxidase (MPO), trans-4-hydroxy-2-nonenal (HNE) or several advanced glycation end products (AGEs) resulting from glycoxidation have been suggested as potentially useful OS markers.115
However, several factors may reflect the translational difficulties from laboratory to clinical evidence in the existing vitamin E studies in AD. While increased OS and reduced plasma vitamin E levels have been associated with AD, a weakness of many vitamin E supplementation trials has been their failure to measure antioxidant and nutritional levels at baseline. As such, an unknown proportion of participants may not have had sufficiently depleted vitamin E levels upon study entry and therefore the likelihood of observing any beneficial effects of vitamin E on primary outcome measures in these individuals may have been reduced.22 The requirement of low baseline levels for supplementation to cause an efficacious increase in plasma levels has been highlighted in non-vitamin E studies and suggests that supplementation is beneficial only in the setting of deficient or insufficient nutrient status.116
In addition, differences in study design between randomised trials may explain, in part, the inconsistent findings to date. For example, a study by Lloret and colleagues consisted of a relatively small number of participants with a lower vitamin E dose and shorter duration while in the study by Sano et al there were large differences in baseline Mini-Mental State Examinations between the placebo and vitamin E groups.102,104 It also remains unclear whether the duration of intervention in these studies is sufficient for the detection of clinical effects. A 2017 Cochrane review concluded that only the study by Dysken and colleagues (which found that that vitamin E slows functional decline in AD) was of moderate quality and that future trials were likely to counter its findings of a lack of support for the beneficial effects of vitamin E in AD.117
Importantly, a major limitation of trials to date may have been the choice of intervention. While two studies (Petersen et al and Kryscio et al) failed to specify which vitamin E isoform was used, the remaining studies have focused solely on α-tocopherol. There is support that supplementation with a single vitamin E isoform (aside from questions of dose and duration) is a less than optimal approach. Firstly, there is sufficient evidence for the biological activity of the other vitamin E isoforms to warrant their investigation in randomised trials.17,61 There have been no trials to date investigating tocotrienol supplements. Secondly, the administration of high-doses of α-tocopherol alone may inhibit the absorption of other tocopherol and tocotrienol isoforms, leading to a damaging biochemical imbalance rather than clinical benefit.22
Furthermore, existing epidemiological studies suggest that dietary sources of vitamin E are more effective in reducing the risk of developing AD than supplementation alone. It is therefore likely that this benefit can be attributed to synergistic interactions which are obscured in trials that investigate supplementation with only a single isoform.118 It is worth noting that dietary vitamin E consumption is more likely to reflect long-term intake than supplement use which may offer partial explanation as to why diet-based sources seem more effective than supplementation in reducing associated AD risk.119
There is also evidence to suggest that the combination of vitamin intake is influential. Vitamin C plays an important role in the reduction of vitamin E after it has been oxidised by free radicals and therefore in maintaining its antioxidant capabilities in tissues.120,121 It is possible that α-tocopherol radicals can themselves induce lipid peroxidation in the context of inadequate co-antioxidant (including vitamin C) levels, particularly in settings of increased oxidative stress or where α-tocopherol levels have been increased alone.122,123 The possibility of α-tocopherol exhibiting pro-oxidant activity under such circumstances is supported by a small randomised trial which found increased plasma oxidant activity in patients receiving vitamin E supplementation compared with placebo.124 It may therefore be important to consider the effects of supplementation with other vitamins in combination with vitamin E to maximise antioxidant efficacy. This is supported by studies that have shown that vitamin E contributes only a relatively small proportion to overall serum antioxidant capacity.105,125
Consideration of the complex bioavailability of vitamin E is important as it is influenced by several factors. The intestinal absorption of vitamin E can vary significantly depending on the food source and its composition of tocopherol or tocotrienol isomers and other nutrients.126 It has been shown that various dietary compounds, including sterols, can reduce intestinal absorption of vitamin E.127 Furthermore, there is large variation between individuals in the availability of high-density lipoproteins (HDLs) which are necessary for vitamin E integration into the central nervous system.128 There is evidence to suggest this may explain the apparently higher maximum plasma levels of vitamin E in women compared to men.129
Several other variables, including age, smoking status and obesity, have also been associated with variation in vitamin E bioavailability, with reduced plasma levels reported in those over 80 years of age although this may be partially attributable to comorbid illness and diminished food intake.130 Low α-tocopherol levels have also been demonstrated in smokers, while obesity has been inversely correlated with plasma α-tocopherol levels131,132 although potential confounding through variation in dietary patterns and nutrient intake may exist.131 Such variations may have important consequences for determining which individuals are likely to benefit from vitamin E intervention. This concept is augmented by the recent identification of 28 genetic polymorphisms that have been shown to influence vitamin E absorption, metabolism and bioavailability.133,134 These genetic variants have been proposed as a rationale that explains the substantial individual variability of vitamin E bioavailability and responsiveness of randomised trial participants.133 Improved understanding of the genetic architecture that underpins vitamin E bioavailability and bioactivity will enable personalised and more effective recommendations of vitamin E intake.135 Such factors may also clarify responder status in vitamin E supplementation studies and warrants further consideration.104
While vitamin E is an essential micronutrient and has been internationally incorporated into many guidelines for dietary intake, its safety as a clinical intervention remains controversial.136 Significantly, it has been reported to have a modifying effect on platelet function and may therefore theoretically increase the risk of clinically significant bleeding.137 One meta-analysis found that supplementation with low-dose vitamin E increased the risk of haemorrhagic stroke amongst study participants.138 These effects may be important in the setting of individuals taking other anticoagulant or antiplatelet agents including aspirin. Increased risk of prostate cancer has also been linked with vitamin E supplementation.139 Similar concerns have been raised by several meta-analyses which concluded that vitamin E supplementation may lead to increased overall mortality.107,112 However, the conclusions of such meta-analyses have been questioned and different analytical approaches have produced contradictory results.140,141 Therefore, the potential adverse effects of vitamin E remain an important clinical consideration and should be explored in future studies.
Several reviews that provide an overview to various aspects of the relationship between vitamin E and AD already exist.39,61 However, this narrative review is broader in scope with a flexible structure which has allowed the authors to be more exploratory and current in their approach that considers the use of vitamin E as a potential therapeutic for the treatment of AD. This review reflects certain points based on the authors’ experience and uses a specified strategy that details how the literature was searched (keywords), time limits of searches, and bibliographic databases accessed. The methodological approach described provides a reference point in time from which future narrative reviews may focus on new literature, thereby limiting redundancy.
Conclusions
In spite of a strong rationale for the role of vitamin E as an effective intervention for AD, the existing clinical evidence remains inconclusive. This review has presented findings from cross-sectional studies that reported significantly lower plasma and CSF levels of vitamin E in those with AD. Additionally, reduced plasma vitamin E status has been associated with increased future risk of developing AD. Epidemiological studies have offered mixed results with regards to vitamin E supplementation but have suggested that intake of high levels of vitamin E from dietary sources may be beneficial. However, clinical trials to date have investigated only the α-tocopherol isoform and have several limitations including failure to measure antioxidant and nutritional levels of participants at baseline. Therefore, there is insufficient evidence to accept or reject the premise that vitamin E is an effective clinical intervention for delaying or preventing the onset of AD and further research is necessary. Importantly, investigation of the underlying genetic architecture with regard to responder status to vitamin E supplementation is warranted, given it is a likely significant contributor to the failure of clinical trials to date.
Acknowledgments
No sources of funding were used to assist in the conduct of this study
Author contributions
All authors contributed toward data analysis, drafting and revising the paper, gave final approval of the version to be published and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Ashraf GM, Chibber S, Zaidi SK, et al. Recent updates on the association between Alzheimer’s disease and vascular dementia. J Med Chem. 2016;12(3):226–237. doi:10.2174/1573406411666151030111820
2. Scheltens P, Blennow K, Breteler MBM, et al. Alzheimer’s disease. Lancet. 2016;388(10043):505–517. doi:10.1016/S0140-6736(15)01124-1
3. Perl DP. Neuropathology of Alzheimer’s disease. Mt Sinai J Med. 2010;77(1):32–42. doi:10.1002/msj.20157
4. Prince M, Wimo A, Guerchet M, Ali GC, Wu YT, Prina N Alzheimer’s Disease International. World Alzheimer Report; Alzheimers Disease International: London, UK; 2015.
5. Wimo A, Jonsson L, Bond J, Prince M, Winblad B. The worldwide economic impact of dementia 2010. Alzheimers Dement. 2013;9(1):1. doi:10.1016/j.jalz.2012.11.006
6. Brookmeyer R, Johnson E, Ziegler-Graham K, Arrighi HM. Forecasting the global burden of Alzheimer’s disease. Alzheimers Dement. 2007;3(3):186–191. doi:10.1016/j.jalz.2007.04.381
7. Niemantsverdriet E, Valckx S, Bierke M and Engelborghs S. Alzheimer’s disease and CSF biomarkers: clinical indications and rationale use. Acta Neurol Belg. 2017;117(3):591–602. doi:10.1007/s13760-017-0816-5
8. Cui Y, Liu B, Luo S, et al. Alzheimer’s disease neuroimaging initiative. Identification of conversion from mild cognitive impairment to Alzheimer’s disease using multivariate predictors. PLoS One. 2011;6(7):e21896. doi:10.1371/journal.pone.0021896
9. Kumar A, Singh A. A review on Alzheimer’s disease pathophysiology and its management: an update. Pharmacol Rep. 2015;67(2):195–203.
10. Ghezzi L, Scarpini E, Galimberti D. Disease-modifying drugs in Alzheimer’s disease. Drug Des Devel Ther. 2013;7:1471.
11. Alzheimer A, Stelzmann RA, Schnitzlein HN, Murtagh FR. An English translation of Alzheimer’s 1907 paper, “Uber eine eigenartige Erkankung der Hirnrinde”. Clin Anat. 1995;8(6):429–431. doi:10.1002/ca.980080612
12. Cummings JL, Cole G. Alzheimer disease. JAMA. 2002;287(18):2335–2338. doi:10.1001/jama.287.18.2335
13. Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down Syndrome. Proc Natl Acad Sci USA. 1985;82(12):4245–4249. doi:10.1073/pnas.82.12.4245
14. Castellano JM, Kim J, Stewart FR, et al. Human apoE isoforms differentially regulate brain amyloid- β peptide clearance. Sci Transl Med. 2011;3:89ra57. doi:10.1126/scitranslmed.3002156
15. Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Ann Rev Neurosci. 2001;24(1):1121–1159. doi:10.1146/annurev.neuro.24.1.1121
16. Giannakopoulos P, Herrmann FR, Bussière T, et al. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s Disease. Neurology. 2003;60(9):1495–1500. doi:10.1212/01.wnl.0000063311.58879.01
17. Xia W, Mo H. Potential of tocotrienols in the prevention and therapy of Alzheimer’s disease. J Nutr Biochem. 2016;31:1–9. doi:10.1016/j.jnutbio.2015.10.011
18. Yao ZX, Papadopoulos. Function of β-amyloid in cholesterol transport: a lead to neurotoxicity. Faseb J. 2002;16(12):1677–1679. doi:10.1096/fj.02-0285fje
19. Cutler RG, Kelly J, Storie K, et al. Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc Natl Acad Sci USA. 2004;101(7):2070–2075. doi:10.1073/pnas.0305799101
20. McGuinness B, Craig D, Bullock R, Passmore P. Statins for the prevention of dementia. Cochrane Databse Syst Rev. 2016;(1):CD003160. doi:10.1002/14651858.CD003160.pub3
21. Karran E, Mercken M, De Strooper B. The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov. 2011;10(9):698. doi:10.1038/nrd3505
22. Mecocci P, Boccardi V, Cecchetti R, et al. A long journey into aging, brain aging, and Alzheimer’s disease following the oxidative stress tracks. J Alzheimers Dis. 2018;62(3):1319–1335. doi:10.3233/JAD-170732
23. Swerdlow RH, Khan SM. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med Hypotheses. 2004;63(1):8–20. doi:10.1016/j.mehy.2003.12.045
24. Swerdlow RH. Alzheimer’s disease pathologic cascades: who comes first, what drives what. Neurotox Res. 2012;22(3):182–194. doi:10.1007/s12640-011-9272-9
25. Swerdlow RH. Brain aging, Alzheimer’s disease, and mitochondria. Biochim Biophys Acta. 2011;1812(12):1630–1639. doi:10.1016/j.bbadis.2011.08.012
26. Kitazawa M, Oddo S, Yamasaki TR, Green KN, LaFerla FM. Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer’s disease. J Neurosci. 2005;25(39):8843–8853. doi:10.1523/JNEUROSCI.2868-05.2005
27. Heppner FL. Ransohoff RM and Becher B. Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci. 2015;16(6):358. doi:10.1038/nrn3880
28. Zuo L, Zhou T, Pannell BK, Ziegler AC, Best TM. Biological and physiological role of reactive oxygen species–the good, the bad and the ugly. Acta Physiol. 2015;214(3):329–348. doi:10.1111/apha.2015.214.issue-3
29. Nunomura A, Perry G, Aliey G, et al. Oxidative damage is the earliest event in Alzheimer disease. J Neuropath Exp Neurol. 2001;60(8):759–767. doi:10.1093/jnen/60.8.759
30. Gustaw-Rothenberg K, Kowalczuk K, Stryjecka-Zimmer M. Lipids’ peroxidation markers in Alzheimer’s disease and vascular dementia. Geriatr Gerontol. 2010;10(2):161–166.
31. Mosconi L, Pupi A, De Leon MJ. Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer’s disease. Ann NY Acad Sci. 2008;1147(1):180–195. doi:10.1196/annals.1427.007
32. Boccardi V, Baroni M, Mangialasche F, Mecocci P. Vitamin E family: role in the pathogenesis and treatment of Alzheimer’s disease. Alzheimers Dement. 2016;2(3):182–191.
33. Curti D, Rognoni F, Gasparini L, et al. Oxidative metabolism in fibroblasts derived from sporadic Alzheimer’s disease (AD) patients. Neurosci Lett. 2008;1147(1):180–195.
34. Nelson ME, Reieski WJ, Blair SN, et al. Physical activity and public health in older adults: recommendation from the American College of Sports Medicine and the American Heart Association. Circulation. 2007;116(9):1094. doi:10.1161/CIRCULATIONAHA.107.185650
35. Jack C. The vascular hypothesis of Alzheimer’s disease: bench to bedside and beyond. Neurodegener Dis. 2010;7(1–3):116–121. doi:10.1159/000285520
36. Feng Y, Wang X. Antioxidant therapies for Alzheimer’s disease. Oxid Med Cell Longev. 2012;2012:472932. doi:10.1155/2012/472932
37. Grundman M. Vitamin E and Alzheimer disease: the basis for additional clinical trials. Am J Clin Nutr. 2000;71(2):630S–636S. doi:10.1093/ajcn/71.2.630s
38. Bieri JG. Sources and consumption of antioxidants in the diet. J Am Oil Chem Soc. 1984;61(12):1917–1918. doi:10.1007/BF02540831
39. Grimm M, Mett J, Hartmann T. The impact of vitamin E and other fat-soluble vitamins on Alzheimer’s disease. Int J Mol Sci. 2016;17(11):1785. doi:10.3390/ijms17111785
40. Yap SP, Yuen KH, Wong JW. Pharmacokinetics and bioavailability of α−, γ−and δ− tocotrienols under different food status. J Pharm Pharmacol. 2001;53(1):67–71.
41. Joshi YB, Praticò D. Vitamin E in aging, dementia and Alzheimer’s disease. Biofactors. 2012;38(2):90–97. doi:10.1002/biof.195
42. Weinstein SJ, Peters U, Ahn J, et al. Serum a-tocopherol and g-tocopherol concentrations and prostate cancer risk in the PLCO screening trial: a nested case-control study. PLoS One. 2012;7:e40204. doi:10.1371/journal.pone.0040204
43. Wang X, Quinn PJ. Vitamin E and its function in membranes. Prog Lipid Res. 1999;38(4):309–336.
44. Müller L, Theile K, Böhm V. In vitro antioxidant activity of tocopherols and tocotrienols and comparison of vitamin E concentration and lipophilic antioxidant capacity in human plasma. Mol Nutr Food Res. 2010;54(5):731–742. doi:10.1002/mnfr.200900399
45. Raederstorff D, Wyss A, Calder PC, Weber P, Eggersdorfer M. Vitamin E function and requirements in relation to PUFA. Br J Nutr. 2015;114(8):1113–1122. doi:10.1017/S000711451500272X
46. Serbinova EA, Packer L. Antioxidant properties of α-tocopherol and α-tocotrienol. Meth Enzym. 1994;234:354–366.
47. Huang D, Ou B, Hampsch-Woodill M, Flanagan JA, Deemer EK. Development and validation of oxygen radical absorbance capacity assay for lipophilic antioxidants using randomly methylated β-cyclodextrin as the solubility enhancer. J Agric Food Chem. 2002;50(7):1815–1821.
48. Brigelius-Flohé R. Vitamin E: the shrew waiting to be tamed. Free Radic Biol Med. 2009;46(5):543–554. doi:10.1016/j.freeradbiomed.2008.12.007
49. Yoshida Y, Niki E, Noguchi N. Comparative study on the action of tocopherols and tocotrienols as antioxidant: chemical and physical effects. Chem Phys Lipids. 2003;123(1):63–75.
50. Jiang Q. Natural forms of vitamin E: metabolism, antioxidant, and anti-inflammatory activities and their role in disease prevention and therapy. Free Radic Biol Med. 2014;72:76–90. doi:10.1016/j.freeradbiomed.2014.03.035
51. Saito Y, Nishio K, Akazawa YO, et al. Cytoprotective effects of Vitamin E homologues against glutamate-induced cell death in immature primary cortical neuron cultures: tocopherols and tocotrienols exert similar effects by antioxidant function. Free Radic Biol Med. 2010;49:1542–1549. doi:10.1016/j.freeradbiomed.2010.08.016
52. Lee CY, Wan F. Vitamin E supplementation improves cell-mediated immunity and oxidative stress of Asian men and women. J Nutr. 2000;130(12):2932–2937. doi:10.1093/jn/130.12.2932
53. De la Fuente M, Hernanz A, Guayerbas N. Manuel Victor V, Amalich F. Vitamin E ingestion improves several immune functions in elderly men and women. Free Radic Res. 2008;42(3):272–280. doi:10.1080/10715760801898838
54. Rota C, Rimbach G, Minihane AM, Stoecklin E, Barella L. Dietary vitamin E modulates differential gene expression in the rat hippocampus: potential implications for its neuroprotective properties. Nutr Neurosci. 2005;8(1):21–29. doi:10.1080/10284150400027123
55. Giraldo E, Lloret A, Fuchsberger T, Viña J. Aβ and tau toxicities in Alzheimer’s are linked via oxidative stress-induced p38 activation: protective role of vitamin E. Redox Biol. 2014;2:873–877. doi:10.1016/j.redox.2014.03.002
56. Jiang Q, Yin X, Lill MA, Danielson ML, Freiser H, Huang J. Long-chain carboxychromanols, metabolites of vitamin E, are potent inhibitors of cyclooxygenases. Proc Natl Acad Sci USA. 2008;105(51):20464–20469. doi:10.1073/pnas.0810962106
57. Copp RP, Wisniewski T, Hentati F, et al. Localization of alpha-tocopherol transfer protein in the brains of patients with ataxia with vitamin E deficiency and other oxidative stress related neurodegenerative disorders. Brain Res. 1999;822(1–2):80–87. doi:10.1016/s0006-8993(99)01090-2
58. Elkamil A, Johansen KK, Aasly J. Ataxia with vitamin E deficiency in Norway. Mov Disord. 2015;8(1):33. doi:10.14802/jmd.14030
59. Wang H, O’Reilly EJ, Weisskopf MG, et al. Vitamin E intake and risk of amyotrophic lateral sclerosis: a pooled analysis of data from 5 prospective cohort studies. Am J Epidemiol. 2011;173(6):595–602. doi:10.1093/aje/kwq416
60. Etminan M, Gill SS, Samii A. Intake of vitamin E, vitamin C, and carotenoids and the risk of Parkinson’s disease: a meta-analysis. Lancet Neurol. 2005;4(6):362–365. doi:10.1016/S1474-4422(05)70097-1
61. Chin KY, Tay S. A review on the relationship between tocotrienol and Alzheimer Disease. Nutrients. 2018;10:
62. Krycer JR, Phan L, Brown AJ. A key regulator of cholesterol homoeostasis, SREBP-2, can be targeted in prostate cancer cells with natural products. Biochem J. 2012;446(2):191–201. doi:10.1042/BJ20120545
63. Mo H, Yeganehjoo H, Shah A, Mo WK, Soelaiman IN, Shen CL. Mevalonate-suppressive dietary isoprenoids for bone health. J Nutr Biochem. 2012;23(12):1543–1551. doi:10.1016/j.jnutbio.2012.07.007
64. Song B-L, DeBose-Boyd RA. Insig-dependent ubiquitination and degradation of 3-hydroxy-3-methylglutaryl coenzyme a reductase stimulated by delta- and gamma-tocotrienols. J Biol Chem. 2006;281(35):25054–25061. doi:10.1074/jbc.M605575200
65. Khor HT, Ng TT. Effects of administration of alpha-tocopherol and tocotrienols on serum lipids and liver HMGCoA reductase activity. Int J Food Sci Nutr. 2000;51Suppl:S3–S11. doi:10.1080/096374800750049521
66. Zaman Z, Roche S, Fielden P, et al. Plasma concentrations of vitamins and E and carotenoids in Alzheimer’s disease. Age Ageing. 1992;104(6–7):703–710.
67. Jiménez-Jiménez FJ, de Bustos F, Molina JA, et al. Cerebrospinal fluid levels of alpha-tocopherol (vitamin E) in Alzheimer’s disease. J Neural Transm. 1997;21(2):91–94.
68. Sinclair AJ, Bayer AJ, Johnston J, Warner C, Maxwell SR. Alterd plasma antioxidant status in subjects with Alzheimer’s disease and vascular dementia. Int J Geriatr Psychiatry. 1998;13(12):840–845.
69. Foy CJ, Passmore AP, Vahidassr MD, Young IS, Lawson JT. Plasma chain-breaking antioxidants in Alzheimer’s disease, vascular dementia and Parkinson’s disease. Q J Med. 1999;92(1):39–45. doi:10.1093/qjmed/92.1.39
70. Bourdel-Marchasson I, Delmas-Beauvieux MC, Peuchant E, et al. Antioxidant defences and oxidative stress markers in erythrocytes and plasma from normally nourished elderly Alzheimer patients. Age Ageing. 2001;30(3):235–241. doi:10.1093/ageing/30.3.235
71. Polidori MC, Mecocci P. Plasma susceptibility to free radical-induced antioxidant consumption and lipid peroxidation is increased in very old subjects with Alzheimer disease. J Alzheimers Dis. 2002;4(6):517–522.
72. Mecocci P, Polidori M, Cherubini A, et al. Lymphocyte oxidative DNA damage and plasma antioxidants in Alzheimer disease. Arch Neurol. 2002;59(5):794–798.
73. Rinaldi P, Poliori MC, Metastasio A, et al. Plasma antioxidants are similarly depleted in mild cognitive impairment and in Alzheimer’s disease. Neurobiol Aging. 2003;24(7):915–919.
74. Baldeiras I, Santana I, Proenca MT, et al. Peripheral oxidative damage in mild cognitive impairment and mild Alzheimer’s disease. J Alzheimers Dis. 2008;15:117–128.
75. Mangialasche F, Xu W, Kivipelto M, et al. Tocopherols and tocotrienols plasma levels are associated with cognitive impairment. Nerurobiol Aging. 2012;33(10):2282–2290. doi:10.1016/j.neurobiolaging.2011.11.019
76. Giavarotti L, Simon KA, Azzalis LA, et al. Mild systemic oxidative stress in the subclinical stage of Alzheimer’s disease. Oxid Med Cell Longev. 2013;2013:609019. doi:10.1155/2013/609019
77. Mullan K, Williams MA, Cardwell CR, et al. Serum concentrations of vitamin E and carotenoids are altered in Alzheimer’s disease: a case-control study. Alzheimers Dement Trans Clin Interv. 2017;3:432–439.
78. Schippling S, Kontush A, Arlt S, et al. Increased lipoprotein oxidation in Alzheimer’s disease. Free Radic Biol Med. 2000;28(3):351–360.
79. Ryglewicz D, Rodo M, Kunicki PK, et al. Plasma antioxidant activity and vascular dementia. J Neurol Sci. 2002;203:195–197.
80. Charlton KE, Rabinowitz TL, Geffen LN, Dhansay MA. Lowered plasma vitamin C but not vitamin E concentration in dementia patients. J Nutr Health Aging. 2004;8(2):99–108.
81. Mas E, Dupuy AM, Artero S, et al. Functional vitamin E deficiency in ApoE4 patients with Alzheimer’s disease. Dement Geriatr Cogn Disord. 2006;21(3):198–204. doi:10.1159/000090868
82. Von Arnim CA, Herbolsheimer F, Nikolaus T, et al. Dietary antioxidants and dementia in a population-based case-control study among older people in South Germany. J Alzheimers Dis. 2012;31(4):717–724. doi:10.3233/JAD-2012-120634
83. Jeandel C, Nicolas MB, Dubois F, Nabet-Belleville F. Penin F and Cuny G. Lipid peroxidation and free radical scavengers in Alzheimer’s disease. Gerontology. 1989;35(5–6):275–282. doi:10.1159/000213037
84. Lopes Da Silva S, Vellas B, Elemans S, et al. Plasma nutrient status of patients with Alzheimer’s disease: systematic review and meta-analysis. Alzheimers Dement. 2014;10(4):485–502. doi:10.1016/j.jalz.2013.05.1771
85. Mullan K, Cardwell CR, McGuinness B, Woodside JV, McKay GJ. Plasma antioxidant status in patients with Alzheimer’s disease and cognitively intact elderly: a meta-analysis of case-control studies. J Alzheimers Dis. 2018;62(1):305–317. doi:10.3233/JAD-170758
86. Dong Y, Chen X, Liu Y, et al. Do low-serum vitamin E levels increase the risk of Alzheimer disease in older people? Evidence from a meta-analysis of case-control studies. Int J Geriatr Psychiatry. 2018;33(2):e257–e263. doi:10.1002/gps.4780
87. De Wilde MC, Vellas B, Girault E, Yavuz AC, Sijben JW. Lower brain and blood nutrient status in Alzheimer’s disease: results from meta-analyses. Alzheimers Dement. 2017;3(3):416–431.
88. Liu G, Zhao Y, Jin S, et al. Circulating vitamin E levels and Alzheimer’s disease: a Mendelian randomization study. Neurobiol Aging. 2018;72:189–e1.
89. Mangialasche F, Kivipelto M, Mecocci P, et al. High plasma levels of vitamin E forms and reduced Alzheimer’s disease risk in advanced age. J Alzheimers Dis. 2010;20(4):1029–1037.
90. Mangialasche F, Westman E, Kivipelto M, et al. Classification and prediction of clinical diagnosis of Alzheimer’s disease based on MRI and plasma measures of α‐/γ‐tocotrienols and γ‐tocopherol. J Intern Med. 2013;273(6):602–621.
91. Mangialasche F, Solomon A, Kàrehold I, et al. Serum levels of vitamin E forms and risk of cognitive impairment in a Finnish cohort of older adults. Exp Gerontol. 2013;48(12):1428–1435.
92. Morris MC, Beckett LA, Scherr PA, et al. Vitamin E and vitamin E supplement use and the risk of incident Alzheimer disease. Alzheimer Dis Assoc Disord. 1998;12:121–126.
93. Zandi PP, Anthony JC, Khachaturian AS, et al. Reduced risk of Alzheimer disease in users of antioxidant vitamin supplements: the Cache County Study. Arch Neurol. 2004;61(1):82–88.
94. Basambombo LL, Carmichael PH, Côté S, Laurin D. Use of vitamin E and C supplements for the prevention of cognitive decline. Ann Pharmacother. 2017;51(2):118–124.
95. Morris MC, Evans DA, Bienias JL, et al. Dietary intake of antioxidant nutrients and the risk of incident Alzheimer disease in a biracial community study. JAMA. 2002;287(24):3230–3237.
96. Engelhart MJ, Geerlings MI, Ruitenberg A, et al. Dietary intake of antioxidants and risk of Alzheimer disease. JAMA. 2002;287(24):3223–3229.
97. Morris MC, Evans DA, Tangney CC, et al. Relation of the tocopherol forms to incident Alzheimer disease and to cognitive change. Am J Clin Nutr. 2005;81(2):508–514.
98. Devore EE, Grodstein F, van Rooij FJ, et al. Dietary antioxidants and long-term risk of dementia. Arch Neurol. 2010;67(7):819–825.
99. Masaki KH, Losonczy KG, Ismirlian G, et al. Association of vitamin E and C supplement use with cognitive function and dementia in elderly men. Neurology. 2000;54(6):1265–1272.
100. Luchsinger JA, Tang MX, Shea S, Maveux R. Antioxidant vitamin intake and risk of Alzheimer disease. Arch Neurol. 2003;60(2):203–208.
101. Gray SL, Anderson ML, Crane PK, et al. Antioxidant vitamin supplement use and risk of dementia or Alzheimer’s disease in older adults. J Am Geriatr Soc. 2008;56(2):291–295. doi:10.1111/j.1532-5415.2007.01531.x
102. Sano M, Ernesto C, Thomas RG, et al. A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer’s disease. New Engl J Med. 1997;336(17):1216–1222. doi:10.1056/NEJM199704243361704
103. Petersen RC, Thomas RG, Grundman M, et al. Vitamin E and donepezil for the treatment of mild cognitive impairment. New Engl J Med. 2005;352(23):2379–2388. doi:10.1056/NEJMoa050151
104. Lloret A, Badía MC, Mora NJ, Pallardó FV, Alonso MD, Viña J. Vitamin E paradox in Alzheimer’s disease: it does not prevent loss of cognition and may even be detrimental. J Alzheimers Dis. 2009;17(1):143–149. doi:10.3233/JAD-2009-1033
105. Dysken MW, Sano M, Asthana S, et al. Effect of vitamin E and memantine on functional decline in Alzheimer disease: the TEAM-AD VA cooperative randomized trial. JAMA. 2014;311(1):33–44. doi:10.1001/jama.2013.282834
106. Kryscio RJ, Abner EL, Caban-Holt A, et al. Association of antioxidant supplement use and dementia in the prevention of Alzheimer’s disease by vitamin E and selenium trial (PREADViSE). JAMA Neurol. 2017;74(5):567–573. doi:10.1001/jamaneurol.2016.5778
107. Miller ER
108. Naeini AA, Elmadfa I, Djazayery A, et al. The effect of antioxidant vitamins E and C on cognitive performance of the elderly with mild cognitive impairment in Isfahan, Iran: a double-blind, randomized, placebo-controlled trial. Eur J Nutr. 2014;53(5):1255–1262. doi:10.1007/s00394-013-0628-1
109. Galasko DR, Peskind E, Clark CM, et al. For Alzheimer disease: a randomized clinical trial with cerebrospinal fluid biomarker measures. Arch Neurol. 2012;69(7):836–841. doi:10.1001/archneurol.2012.85
110. Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11(3):298–300.
111. Ghezzi P, Jaquet V, Marcucci F, Schmidt HH. The oxidative stress theory of disease: levels of evidence and epistemological aspects. Br J Pharmacol. 2017;174(12):1784–1796. doi:10.1111/bph.13544
112. Bjelakovic G, Nikolova D, Gluud LL, Simonetti RG, Gluud C. Antioxidant supplements for prevention of mortality in healthy participants and patients with various diseases. Cochrane Database Syst Rev. 2012:(3):CD007176. doi:10.1002/14651858.CD007176.pub2
113. Winterbourn CC. The challenges of using fluorescent probes to detect and quantify specific reactive oxygen species in living cells. Biochim Biophys Acta. 2014;1840(2):730–738. doi:10.1016/j.bbagen.2013.05.004
114. Zielonka J, Hardy M, Michalski R, et al. Recent developments in the probes and assays for measurement of the activity of NADPH oxidases. Cell Biochem Biophys. 2017;75(3–4):335–349. doi:10.1007/s12013-017-0813-6
115. Frijhoff J, Winyard PG, Zarkovic N, Davies SS, Stocker R, Cheng D. Clinical relevance of biomarkers of oxidative stress. Antioxid Redox Signal. 2015;23(14):1144–1170. doi:10.1089/ars.2015.6317
116. Mariani E, Mangialasche F, Feliziani FT, et al. Effects of zinc supplementation on antioxidant enzyme activities in healthy old subjects. Exp Gerontol. 2008;43(5):445–451. doi:10.1016/j.exger.2007.10.012
117. Farina N, Llewellyn D, Isaac MG, Tabet N. Vitamin E for Alzheimer’s dementia and mild cognitive impairment.
118. Corbett A, Ballard C. The value of vitamin E as a treatment for Alzheimer’s disease remains unproven despite functional improvement, due to a lack of established effect on cognition or other outcomes from RCTs. Evid Based Med. 2014;19(4):140. doi:10.1136/eb-2014-101741
119. Ancelin ML, Christen Y, Ritchie K. Is antioxidant therapy a viable alternative for mild cognitive impairment? Examination of the evidence. Dement Geratr Cogn Dis. 2007;24(1):1–9. doi:10.1159/000102567
120. Niki E. Role of vitamin E as a lipid-soluble peroxyl radical scavenger: in vitro and in vivo evidence. Free Radic Biol Med. 2014;66:3–12. doi:10.1016/j.freeradbiomed.2013.03.022
121. Constantinescu A, Han D, Packer L. Vitamin E recycling in human erythrocyte membranes. J Biol Chem. 1993;268(15):10906–10913.
122. Bowry VW, Ingold KU, Stocker R. Vitamin E in human low-density lipoprotein: when and how this antioxidant becomes a pro- oxidant. Biochem J. 1992;288(2):341–344. doi:10.1042/bj2880341
123. Rietjens IM, Boersma MG, de Haan L, et al. The pro-oxidant chemistry of the natural antioxidants vitamin C, vitamin E, carotenoids and flavonoids. Environ Toxicol Pharmacol. 2002;11(3–4):321–333.
124. Pearson P, Lewis SA, Britton J, Young IS, Fogarty A. The pro-oxidant activity of high-dose vitamin E supplements in vivo. BioDrugs. 2006;20(5):271–273. doi:10.2165/00063030-200620050-00002
125. Mecocci P, Polidori MC. Antioxidant clinical trials in mild cognitive impairment and Alzheimer’s disease. Biochim Biophys Acta - Mol Basis Dis. 2012;1822(5):631–638. doi:10.1016/j.bbadis.2011.10.006
126. Bjørneboe A, Bjørneboe GE, Drevon CA. Absorption, transport and distribution of vitamin E. J Nutr. 1990;120(3):233–242. doi:10.1093/jn/120.3.233
127. Richelle M, Enslen M, Hager C, et al. Both free and esterified plant sterols reduce cholesterol absorption and the bioavailability of β-carotene and α-tocopherol in normocholesterolemic humans. Am J Clin Nutr. 2004;80(1):171–177. doi:10.1093/ajcn/80.1.171
128. Goti D, Hammer A, Galla HJ, Malle E, Sattler W. Uptake of lipoprotein‐associated α‐tocopherol by primary porcine brain capillary endothelial cells. J Neurochem. 2000;74(4):1374–1383. doi:10.1046/j.1471-4159.2000.0741374.x
129. Leonard SW, Paterson E, Atkinson JK, Ramakrishnan R, Cross CE, Traber MG. Studies in humans using deuterium-labeled α-and γ-tocopherols demonstrate faster plasma γ-tocopherol disappearance and greater γ-metabolite production. Free Radic Biol Med. 2005;38(7):857–866. doi:10.1016/j.freeradbiomed.2004.12.001
130. Campbell D, Bunker VW, Thomas AJ, Clayton BE. Selenium and vitamin E status of healthy and institutionalized elderly subjects: analysis of plasma, erythrocytes and platelets. Br J Nutr. 1989;62(1):221–227.
131. Shah AA, Khand F, Khand TU. Effect of smoking on serum xanthine oxidase, malondialdehyde, ascorbic acid and α-tocopherol levels in healthy male subjects. Pak J Med Sci. 2015;31(1):146.
132. Gunanti IR, Marks GC, Al-Mamun A, Long KZ. Low serum concentrations of carotenoids and vitamin E are associated with high adiposity in Mexican-American children. J Nutr. 2014;144(4):489–495. doi:10.3945/jn.113.183137
133. Borel P, Desmarchelier C, Nowicki M, Bott R, Tourniaire F. Can genetic variability in α-tocopherol bioavailability explain the heterogeneous response to α-tocopherol supplements? Antioxid Redox Signal. 2015;22:669–678. doi:10.1089/ars.2014.6144
134. Major JM, Yu K, Wheeler W, et al. Genome-wide association study identifies common variants associated with circulating vitamin E levels. Hum Mol Genet. 2011;20(19):3876–3883. doi:10.1093/hmg/ddr296
135. Galmés S, Serra F, Palou A. Vitamin E metabolic effects and genetic variants: a challenge for precision nutrition in obesity and associated disturbances. Nutrients. 2018;10(12):1919. doi:10.3390/nu10121919
136. Galli F, Azzi A, Birringer M, et al. Vitamin E: emerging aspects and new directions. Free Radic Biol Med. 2017;102:16–36. doi:10.1016/j.freeradbiomed.2016.09.017
137. Steiner M. Vitamin E, a modifier of platelet function: rationale and use in cardiovascular and cerebrovascular disease. Nutr Rev. 1999;57(10):306–309. doi:10.1111/j.1753-4887.1999.tb06903.x
138. Schürks M, Glynn RJ, Rist PM, Tzourio C, Kurth T. Effects of vitamin E on stroke subtypes: meta-analysis of randomised controlled trials. BMJ. 2010;341:c5702. doi:10.1136/bmj.c5702
139. Klein EA, Thompson IM, Tangen CM, et al. Vitamin E and the risk of prostate cancer: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA. 2011;306:1549–1556. doi:10.1001/jama.2011.1437
140. Gerss J, Kopcke W. The questionable association of vitamin E supplementation and mortality – inconsistent results of different meta-analytic approaches. Cell Mol Biol. 2009;55:OL1111–Q1120.
141. Jiang S, Pan Z, Li H, Li F, Song Y, Qiu Y. Meta-analysis: low-dose intake of vitamin E combined with other vitamins or minerals may decrease all-cause mortality. J Nutr Sci Vitaminol. 2014;60:194–205.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.