Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 14
Vitamin D/VDR Protects Against Diabetic Kidney Disease by Restoring Podocytes Autophagy
Authors Song Z, Xiao C, Jia X, Luo C, Shi L, Xia R, Zhu J, Zhang S
Received 28 January 2021
Accepted for publication 19 March 2021
Published 16 April 2021 Volume 2021:14 Pages 1681—1693
DOI https://doi.org/10.2147/DMSO.S303018
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ming-Hui Zou
Zhixia Song,1,* Chao Xiao,2,* Xiaoli Jia,2 Chunhua Luo,3 Lang Shi,1 Rong Xia,1 Jiefu Zhu,4 Shizhong Zhang2
1Department of Nephrology, The First Clinical Medical College of Three Gorges University, Center People’s Hospital of Yichang, Yichang, 443000, People’s Republic of China; 2Three Gorges University College of Medical Science, Yichang, 443000, People’s Republic of China; 3Department of Clinical Laboratory, The First Clinical Medical College of Three Gorges University, Center People’s Hospital of Yichang, Yichang, 443000, People’s Republic of China; 4Center of Nephrology and Dialysis, Transplantation, Renmin Hospital of Wuhan University, Wuchang, Hubei, 430060, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jiefu Zhu; Shizhong Zhang Email [email protected]; [email protected]
Objective: The present study is to investigate the effect of vitamin D/Vitamin D Receptor (VDR) signaling on podocyte autophagy in diabetic nephropathy.
Methods: Kidney tissue sections from patients with diabetic nephropathy and nontumor kidney were checked under electronic microscope and VDR immunohistochemistry. Diabetic rat models were induced by intraperitoneal injection of streptozotocin (STZ) (60 mg/kg). Calcitriol treatment was achieved by gavage at dose of 0.1μg/kg/d. Blood, urine and kidney tissue specimens were used for serum, urine biochemistry, histopathology and molecular biology testing. Podocyte cell line MPC-5 was cultured under hyperglycaemic conditions in the absence or presence of 100 nmol/L calcitriol to investigate podocyte injury and autophagy.
Results: VDR and autophagosomes in podocytes were significantly decreased in renal biopsy from patients with diabetic nephropathy, compared to healthy kidney tissue. Rats with STZ treatment developed typical diabetic kidney disease with low VDR expression. Calcitriol, the active form of vitamin D, could activate VDR and attenuate diabetic nephropathy including proteinuria and glomerular sclerosis. Calcitriol treatment also alleviated the podocyte foot process fusion, reduced podocyte injury marker desmin and preserved slit diaphragms proteins in diabetic nephropathy. Reduced LC3II/I, Beclin-1 and elevated p62 in renal homogenate and reduced autophagosomes and LC3II in podocytes indicated podocytes autophagy impairment in diabetic nephropathy. Whereas calcitriol treatment restored podocyte autophagy activities. In cultured podocytes, the protective effect of calcitriol against high glucose induced podocyte injury could be abated by autophagy inhibitor chloroquine.
Conclusion: Our study delivered the evidence that calcitriol/VDR signaling attenuated diabetic nephropathy and podocytes injury by restoring podocytes autophagy. This finding may have potential implication for exploring protective mechanisms of calcitriol/VDR in diabetic nephropathy.
Keywords: vitamin D receptor, calcitriol, diabetic nephropathy, podocyte, autophagy
Introduction
Diabetic nephropathy is a serious microvascular complication of diabetes, which has become a global health issues.1–3 The main clinical manifestation of diabetic nephropathy is progressive proteinuria and weakened renal function.2,3 Podocytes are a vital component of glomerular filtration barrier with a network of foot processes, which is a highly differentiated and delicate structure. It is known that podocyte injuries including foot processes fusion play a crucial role in the development and progression of diabetic nephropathy. However, the mechanism of podocyte injury in diabetic nephropathy remains largely unknown. Due to the restricted proliferation capacity of postmitotic podocytes, autophagy governing the intracellular degradation and recycle system may play a critical role in maintaining the homeostasis of the podocytes, especially under diabetic condition.4
Vitamin D/vitamin D receptor (VDR) signaling regulates calcium and phosphorus metabolism and also has been shown to be associated with various kidney diseases.5,6 Recent studies have demonstrated that VDR signaling defects in podocytes contributed to diabetic nephropathy and vitamin D deficiency has been shown to be an independent risk factor for prognosis in patients with type 2 diabetes.7,8 Calcitriol, the biologically active form of vitamin D, is used for treating calcium absorption and bone diseases clinically by activating VDR.9 In our previous studies10,11 and other studies,8,12 calcitriol was effective in reducing podocyte damage and loss in diabetic animals, and reduced proteinuria. However, the underlying protective mechanism of calcitriol-VDR signaling needs further being explored.
Basal autophagy in the kidneys is vital for the renal homeostasis, especially for podocytes. Several studies have implied the role vitamin D/VDR in regulating the autophagy function under physiological and pathophysiological condition,13–15 however to our best acknowledge, no study has directly addressed the renoprotective role of calcitriol on podocytes autophagy in diabetic kidney disease. This study aimed to investigate the vitamin D/VDR deficiency in diabetic nephropathy and whether restoring VDR signaling by calcitriol can preserve podocyte hemostasis and autophagy, thereby reducing proteinuria and delaying the development of diabetic kidney disease.
Materials and Methods
Clinical Data
Diabetic nephropathy was diagnosed based on renal biopsies at the First Clinical Medical College of Three Gorges University. Exclusion criteria include coexistence of other primary and secondary kidney diseases, such as membranous nephropathy or lupus nephropathy. Non-tumor kidney tissue from surgical specimens of patients who had renal carcinoma was used as normal controls. The study protocols were approved by the Human Subjects Committee of the First Clinical Medical College of Three Gorges University. All patients provided written informed consent. Demographic, laboratory, and renal pathological data were recorded. Renal biopsy specimens were collected for measuring the level of autophagosomes under transmission electron microscopy (TEM) and VDR immunohistochemistry under light microscopy.
Establishing Rat Model of Diabetes
Specific pathogen-free Sprague-Dawley (SD) male rats (180–200 g, 6 weeks old) were provided by the Experimental Animal Center of the Three Gorges University School of Medicine. Animals were given 12 hours of light-darkness cycle at a relative ambient temperature (20 ± 2°C) and a relative air humidity (55 ± 10%) and allowed free access to water and standard rodent food. After one week of adaptation to the environment, the rats were randomly divided into 3 groups: vehicle control group (NC group, n=20), diabetic nephropathy group (DN group, n=20), DN rat treated with calcitriol group (DN+VD group, n=20). Streptozotocin (STZ) (Sigma, USA) was dissolved in 0.1M citrate buffer (pH 4.4) at a concentration of 4 mg/mL, and a single rapid injection into the intraperitoneal at a dose of 60 mg/kg. The control group was injected an equal amount of 0.1M citrate buffer. Three days later, the randomly rat’s tail vein blood glucose was measured ≥16.7mmol/L considered as diabetes rats, and then which were monitored every two weeks using a blood glucose monitoring system (Bayer). Calcitriol (Roche Company, USA) was dissolved in edible peanut oil at a concentration of 0.04μg/mL and was delivered by oral gavage at a daily dose 0.1 μg/kg after starting at 3 days after STZ injection until sacrifice. The DN group and the control group received an equal amount of edible peanut oil.
Cell Culture
The MPC5 conditionally immortalized mouse podocytes clonal cells (kindly provided by Peter Mundel, Mount Sinai School of Medicine through Prof. Aihua Zhang at Nanjing Medical University, Nanjing, China) were cultured and induced to differentiate as described16. In brief, the cells were cultured and grown in DMEM-F12 medium containing 10% fetal bovine serum and mouse recombinant IFN-γ (10 U/mL) at 33°C, 5% CO2. The medium was changed every other day. To induce differentiation, the podocytes were then cultured in medium without IFN-γ at 37°C, 5% CO2 within 10–14 days. The fully differentiated cells with 80% confluence were used for subsequent experiments. For euglycaemic or hyperglycaemic conditions, the cells were cultured in medium containing 5.5mM or 30mM glucose for 24 hours.
Transfection and Detection of GFP-LC3
Cells were plated at a density of 4 × 105 at 35-mm dishes and cultured to 60–70% confluence after overnight growth. Then the green fluorescent protein (GFP)-LC3 plasmid was transfected with Lipofectamine 2000 (Invitrogen, Carlsbad, CA) for 36h according to the manufacturer’s recommendation. After treatment cells were briefly fixed 4% paraformaldehyde. Microphotographs of GFP-LC3 labeled cells were obtained with a confocal fluorescence microscopy (Zeiss 780 NY). Image J software was used for GFP-LC3 puncta counting.
Collection of Blood, Urine and Kidney Tissue Specimens
The body weight, urine volume, urinary protein and blood glucose levels were measured every 2 weeks. Blood glucose is monitored by tail vein puncture. Urine was collected and quantified in metabolic cages, and purified by cryo-centrifugation, and stored in ultra-low temperature refrigerator for 24h urine protein detection. The rats were separately sacrificed at 8th, 14th, 18th week (n≥6). Blood samples were collected by abdominal aortic puncture. After kidney removal and weighting, the ratio of kidney weight to body weight (KW/BW) was calculated. The left kidneys were fixed in 4% paraformaldehyde for 48 hours, then dehydrated and embedded in paraffin for morphological analysis. The 1mm3 renal cortex from the right kidney was preserved in 2.5% glutaraldehyde for electronic microscope ultrastructural analysis. The remaining renal cortex was frozen in ultra-low temperature refrigerator for molecular biology detection. The above blood/tissue sampling was performed under pentobarbital anesthesia to reduce animal pain.
Serum, Urine Biochemical Testing
Blood glucose, blood urea nitrogen (BUN), serum creatinine (Scr), serum albumin (ALB), serum calcium (Ca), serum phosphorus (P) were analyzed by the automatic blood biochemistry analyzer (Beckman Instruments, CA, USA) of the Department of Clinical Laboratory of Yichang Central People’s Hospital. Urine protein content was measured by Coomassie Brilliant Blue method, 24h urine protein quantitation (g/24h) = urine protein content (g/L) × 24 hours urine total (L).
Histopathology
Periodic Acid-Schiff (PAS) staining and Masson tricolor staining were done using the kits from Servicebio company (Wuhan, China) according to the standard procedures. Twenty consecutive non-repetitive microscopic pictures were taken for each sample, and the area of mesangial proliferation was analyzed by Image-Pro plus 6.0 software. The final evaluation of each sample was the average of the analysis of 20 glomerulus.
Immunohistochemistry
Histological section of renal (3μm) were used for immunohistochemical staining of podocin (Abeam, UK), nephrin (Abeam, UK), desmin (Abcam, UK), VDR (Santa Cruz, USA), WT1 (Abcam, UK), P62 (Cell signaling, USA) and Beclin-1 (Novus, USA) proteins. After antigen retrieval, sections were incubated with primary antibody, and then the specimen was incubated with a species-specific secondary antibody (Millipore, USA). Subsequently, the sections were visualized with diaminobenzidine (Maixin, China) and the nuclei were counterstained with hematoxylin. The staining intensity was observed under light microscope, and 20 consecutive non-repetitive fields of view were taken from the kidney of each rat. Analyzed with Image-Pro plus 6.0. The final assessment for each sample was the average of the analysis from 20 glomerulus. Pathological evaluation was performed blindly.
Immunofluorescence
Immunofluorescence staining was performed as described previously.10 Briefly, after rehydration and antigen retrieval, kidney tissue sections were incubated with anti-synaptopodin (Santa Cruz, USA) and LC3II (Novus, USA) antibody simultaneously overnight at 4°C. Tissue section was then simultaneously incubated with The TRITC-conjugated and FITC-conjugated secondary antibody (Jackson lab, USA) for 1 hour at room temperature in the dark. After washing with PBS, the nuclei were counterstained with DAPI (Sigma, USA), and then the specimens were observed and evaluated by a laser confocal microscopy (Nikon, Japan).
Transmission Electron Microscope
A 1 mm3 renal cortex was fixed in glutaraldehyde for 24 hours, and then fixed in 1% osmium tetroxide for 1.5 hours (4°C) followed dehydration with gradient ethanol and acetone. The tissue was then permeabilized in a mixture of acetone: epoxy resin 1:1 for 8 hours, sliced (50 nm) with an ultramicrotome (Leica, Germany) and mounted on a copper mesh (200 mesh). The ultrastructural images were observed using a transmission electron microscope (Leica, Germany). The number of the autophagosomes were counted under 10,000/EMP from 8 randomly selected podocytes for each set of tissue samples; each set of samples was derived from five patients or rats. After counting by two certified experienced pathologists, statistics analysis was performed.
Immunoblot
Kidney tissue was homogenized with RIPA lysate and centrifuged, and the supernatant were collected and stored at −70°C. Protein concentration was measured using a BCA kit (Applygen Technologies Inc, China). The protein was denatured (heated at 100°C for 5 minutes), separated by SDS-PAGE electrophoresis and transferred to a PVDF membrane. After blocking with 5% nonfat dry milk TBST for 1 hour, the membrane was incubated with primary antibody overnight at 4°C and secondary antibody at room temperature. The Antigen-antibody complexes were detected with ECL chemiluminescence reagents (Thermofisher, USA).
Statistical Analysis
Statistical analysis was performed using SPSS 18.0 software. All results are expressed as mean ± standard deviation (mean ± SD). The significance of the differences was evaluated by one-way ANOVA followed by Tukey’s post hoc test. P<0.05 was considered statistically significant.
Results
Defect VDR Expression and Podocytes Autophagy in Diabetic Nephropathy
Growing evidences suggest VDR signaling disorders has closely tied to diabatic kidney diseases. In this study, we tested VDR expression difference between healthy kidneys and diabetic kidneys. In healthy kidneys, VDR was mainly expressed in glomerular podocytes with nuclear staining and renal tubular epithelial cells with cytoplasmic staining, while the intensity of VDR signaling was significantly lower in diabetic kidneys (Figure 1A and D). Podocytes exhibited high basal levels of autophagy by clearing cellular debris and recycling substrates for glomerular maintenance.17 To test the clinical relevance of VDR and autophagy in the pathogenesis of diabetic kidney disease, we collected kidney biopsy specimens from 5 patients with diabetic nephropathy and 5 patients with renal cancer. Para-cancer normal kidneys were used as healthy control tissue. Significant reduced numbers of autophagosomes in podocytes were observed in diabetic kidneys compared to para-cancer healthy kidneys (Figure 1B and C). Clinical data of patients are shown in Table 1. These clinical findings indicated both VDR expression and autophagy defects of podocytes in diabetic nephropathy.
 |
Figure 1 The expression of VDR protein and autophagosomes are decreased in podocytes of diabetic patients. Kidney biopsy specimens were from patients with diabetic nephropathy (DN) and non-tumor renal tissue from patients with kidney cancer. (A) Representative pictures of periodic acid-Schiff (PAS) staining, anti-synaptopodin immunofluorescence (IF) and VDR immunohistochemistry (IHC). Bottom panels are enlarged images of the boxed areas in the middle panels. Arrows indicate VDR was mainly expressed in glomerular podocytes with nuclear staining. (B) Representative ultrastructural imagines showing autophagosomes in podocytes. Arrows indicate autophagosomes in podocytes. (C) Semi-quantitative measurement of VDR intensity in the glomeruli in each group. (D) Quantification of autophagosomes in podocytes. All results are presented as mean ± SEM, *P<0.05 considered statistically significant. |
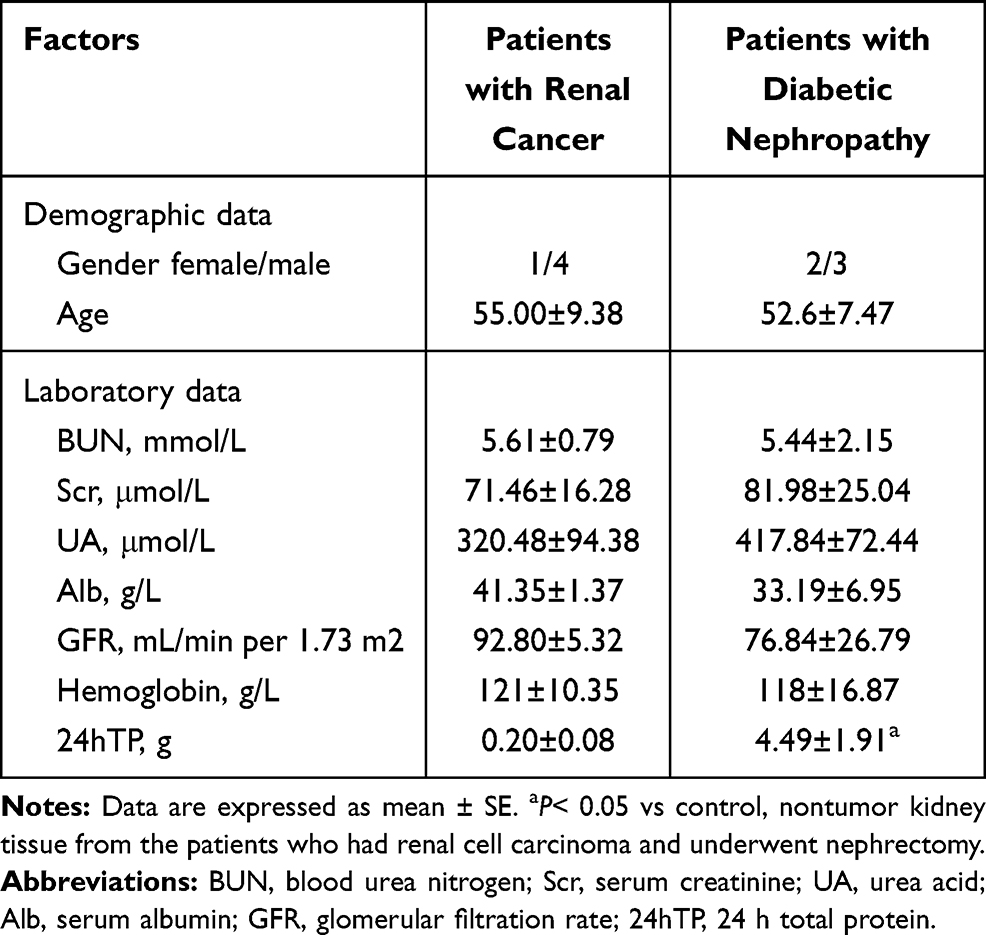 |
Table 1 Demographic and Clinical Laboratory Data of Patients with Renal Cancer and Patients with Diabetic Nephropathy (Each Group n = 5) |
Calcitriol Treatment Upregulates the Expression of VDR Protein in Diabetic Rats
We further investigated the VDR in diabetic nephropathy in experimental animals. Diabetic rats were established by a single rapid injection of STZ into the intraperitoneal at a dose of 60 mg/kg. Expression of VDR in diabetic kidneys was checked by immunohistochemistry and Western blot analysis. In NC group, VDR was mainly expressed in glomerular podocytes with showed nuclear expression. Compared with the NC group, the expression of VDR protein in the glomeruli of the DN group was significantly decreased (Figure 2A–C), consistent to the clinical data in diabetic patients. Calcitriol, a pharmacological activator of VDR, could transcriptionally upregulated the expression of VDR.18 After the daily supplement of calcitriol in diabetic rats, the expression of VDR in diabetic kidney was recovered in diabetic nephropathy in all time points (Figure 2A–C).
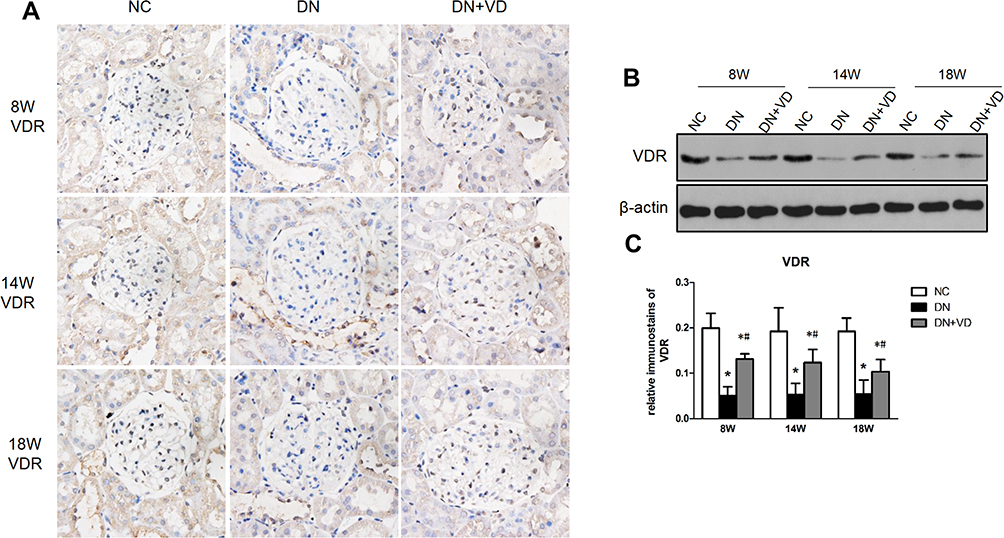 |
Figure 2 Calcitriol normalizes the expression of VDR expression in experimental diabetic nephropathy. SD male rats were intraperitoneally injected with 60 mg/kg streptozotocin. After 3 days, the rats with STZ treatment were garaged with 0.1μg/kg/d calcitriol or vehicle solution daily for consecutive 18 weeks. (A) Representative Immunohistochemical staining of VDR protein (400×) at 8th, 14th, 18th week in three groups. (B) Immunoblot analysis of VDR expression in the renal cortex at different time points in three groups, and β-actin was used as a loading control. (C) Quantification of VDR protein. Values are expressed as mean ± SD (n=6). Significance: *P<0.05 vs NC group, #P<0.05 vs DN group at the same time point, respectively. Abbreviations: NC, normal control group; DN, diabetic nephropathy group; DN+VD, diabetes nephropathy plus calcitriol group. |
Calcitriol Attenuates Proteinuria and Histological Changes in DN Rats
We next test whether reactivation of Vitamin D/VDR could slow down the diabetic disease process. After the induction of STZ, diabetic rats exhibited high blood glucose and body weight loss compared with the controls (Table 2) (Figure 3A and B). As expected, the DN was developed in diabetic rats with proteinuria and diuresis two weeks after STZ injection (Figure 3C and D). During the period of observation, calcitriol treatment effectively attenuated the aberrant albuminuria (Figure 3D). The ratio of KW/BW suggesting renal hypertrophy of the DN group was elevated compared with control group, while was reversed after the intervention of calcitriol (Table 2). Blood ALB, Scr and BUN levels in the DN group were significantly deteriorated, however calcitriol treatment did not alleviate these systemic parameters (Table 2). There were also no significant differences in serum Ca and P levels between the three groups, as illustrated in Table 1. The renal pathological changes were checked by PAS and MASSON staining and the results showed that the expanded mesangial matrix in glomerular area in the DN group (Figure 4A and B). The ultrastructural images demonstrated that the glomerular basement membrane was obviously thickened in diabetic kidneys, approximately 2.5 times than that in non-diabetic kidneys (Figure 4A and C). The above renal histological and ultrastructural lesions were all reversed with calcitriol intervention (Figure 4).
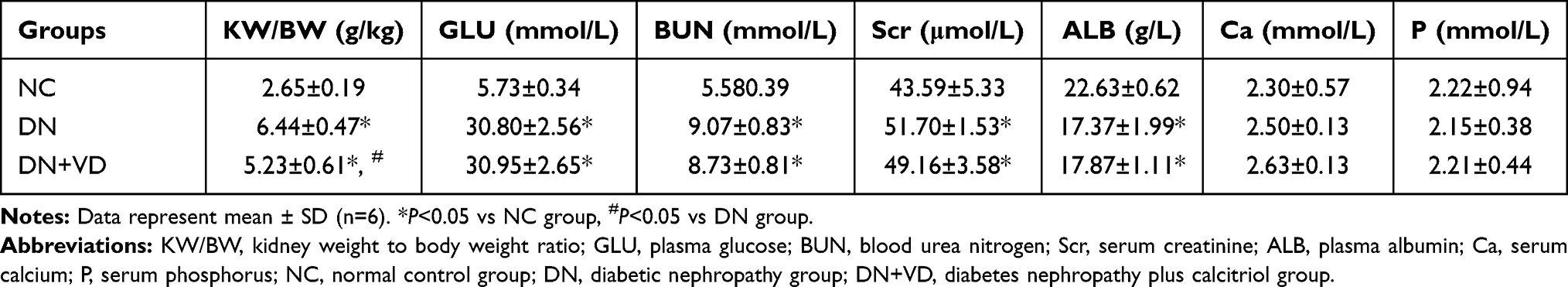 |
Table 2 KW/BW and Blood Biochemical Levels in Diabetic and Control Rats |
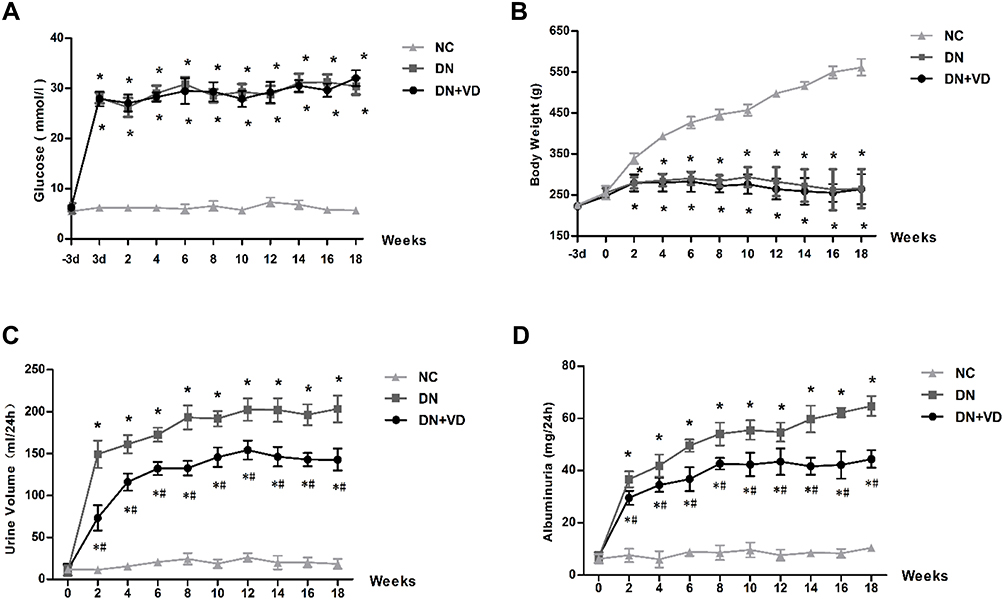 |
Figure 3 Effects of calcitriol on blood glucose, body weight, urine volume and proteinuria in diabetic rats. SD male rats were intraperitoneally injected with 60 mg/kg streptozotocin. After 3 days, the rats with STZ treatment were garaged with 0.1μg/kg/d calcitriol or vehicle solution daily for consecutive 18 weeks. The body weight, 24h urine volume, 24h urinary protein and blood glucose levels were measured every 2 weeks. Time course monitoring of blood glucose (A), body weight (B), 24h urine volume (C) and 24h urinary albumin excretion (D) levels for 18 weeks. Values are mean ± SD (n=6). Significance: *P<0.05 vs NC group, #P<0.05 vs DN group at the same time point, respectively. Abbreviations: NC, normal control group; DN, diabetic nephropathy group; DN+VD, diabetes nephropathy plus calcitriol group. |
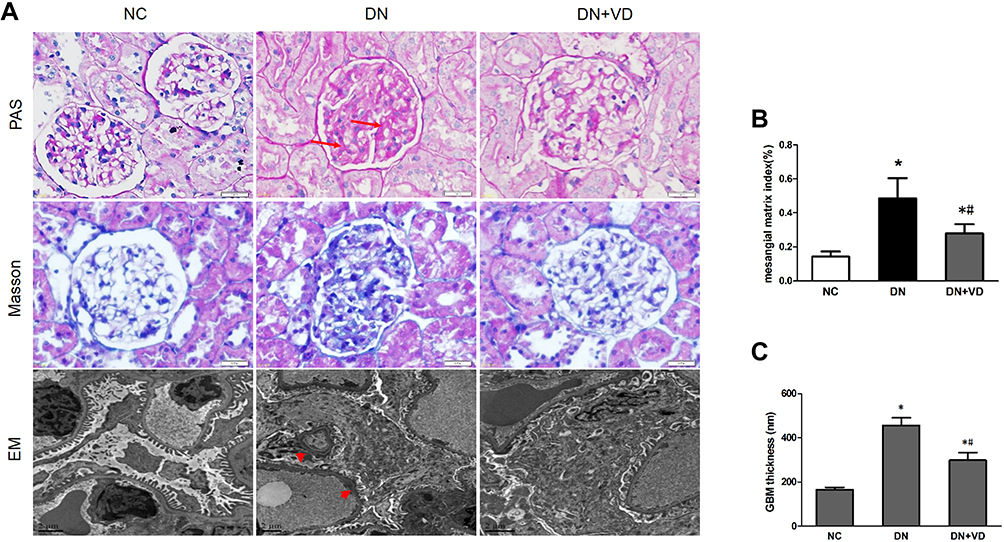 |
Figure 4 Calcitriol attenuates histological abnormalities of diabetic nephropathy. SD male rats were intraperitoneally injected with 60 mg/kg streptozotocin. After 3 days, the rats with STZ treatment were garaged with 0.1μg/kg/d calcitriol or vehicle solution daily for consecutive 18 weeks. Kidney tissues were collected for histology at 18th week. (A) Representative images of Periodic Acid-Schiff (PAS) staining, Masson staining and electron microscopy (EM) of three groups of kidney tissues. Arrows indicate the expanded mesangial matrix in glomerular area and arrowheads indicate the enhanced thickness of the glomerular basement membrane and podocyte effacement. (B) Glomerular mesangial matrix index (%) of three groups. (C) Quantifications of mean GBM thickness. Values are mean ± SD (n=6). Significance: *P<0.05 vs NC group, #P<0.05 vs DN group. Abbreviations: NC, normal control group; DN, diabetic nephropathy group; DN+VD, diabetes nephropathy plus calcitriol group. |
Calcitriol Treatment Attenuated Podocyte Injury
One of the main pathological features of diabetic nephropathy is podocyte injury. Podocyte structural damages play a pivotal role in pathogenesis of diabetic nephropathy. Slit diaphragm proteins like nephrin, podocin and synaptopodin are critical to stabilize glomerular filtration barrier. Desmin is a widely used marker of podocyte injury. As expected, the diabetic nephropathy model showed the increased podocyte injury marker desmin and reduced expression of Slit diaphragm proteins nephrin, podocin and synaptopodin in glomeruli at 8th weeks after STZ initiation (Figures 5A and 6A). Calcitriol treatment could significantly prevent these pathological changes (Figures 5A and 6A). The immunoblot data confirmed the data in immunohistochemistry results (Figure 5B). Podocyte-specific nuclear protein WT1 was often utilized as a molecular marker for evaluating the podocytes number. For quantification, the number of podocytes appeared lower in the diabetic nephropathy compared to non-diabetic kidneys after STZ initiation, however there was no improvement with calcitriol treatment (Figure 5A and C). Morphologically, multiple foot processes underwent fusion or even effacement under electron microscopy in diabetic nephropathy (Figure 4A), which was significantly attenuated by calcitriol treatment. The above phenomenon indicates that calcitriol acts on the slit diaphragm protein to protect DN podocytes from damage (Figure 5).
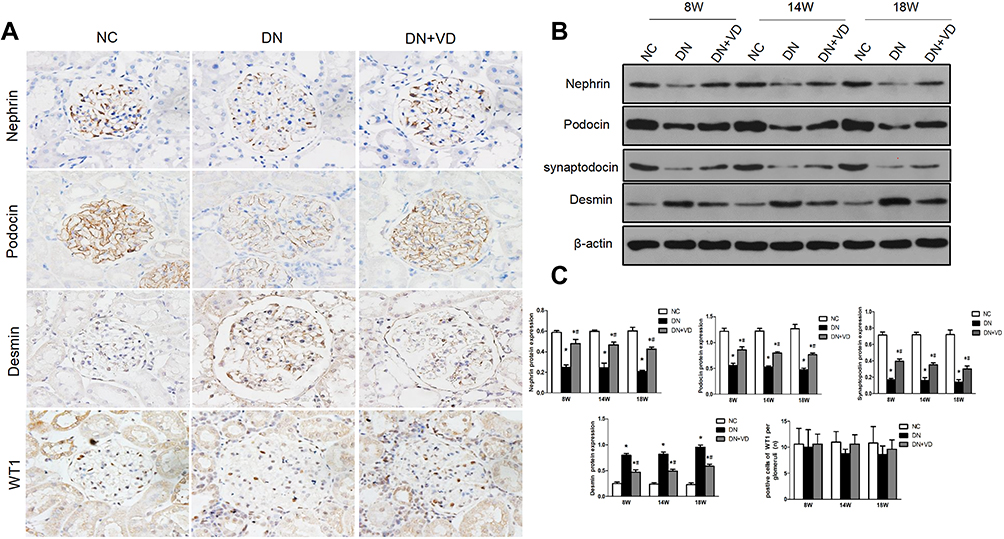 |
Figure 5 Calcitriol treatment preserved slit diaphragm proteins and reduced podocytes injury in diabetic nephropathy. SD male rats were intraperitoneally injected with 60 mg/kg streptozotocin. After 3 days, the rats with STZ treatment were garaged with 0.1μg/kg/d calcitriol or vehicle solution daily for consecutive 18 weeks. Kidney tissues were collected for histology. (A) Representative images of immunohistochemical staining for nephrin, podocin, desmin and WT1 (Wilms’ tumor 1) in renal cortex at 18th week (400×). (B) Immunoblot analysis of nephrin, podocin and desmin at 8th, 14th, 18th week in three groups. β-actin was used as the internal loading control. (C) Densitometry analysis of nephrin, podocin desmin and WT1. Data are expressed as means ± SD (n=6). Significance: *P<0.05 vs NC group, #P<0.05 vs DN group at the same time point, respectively. |
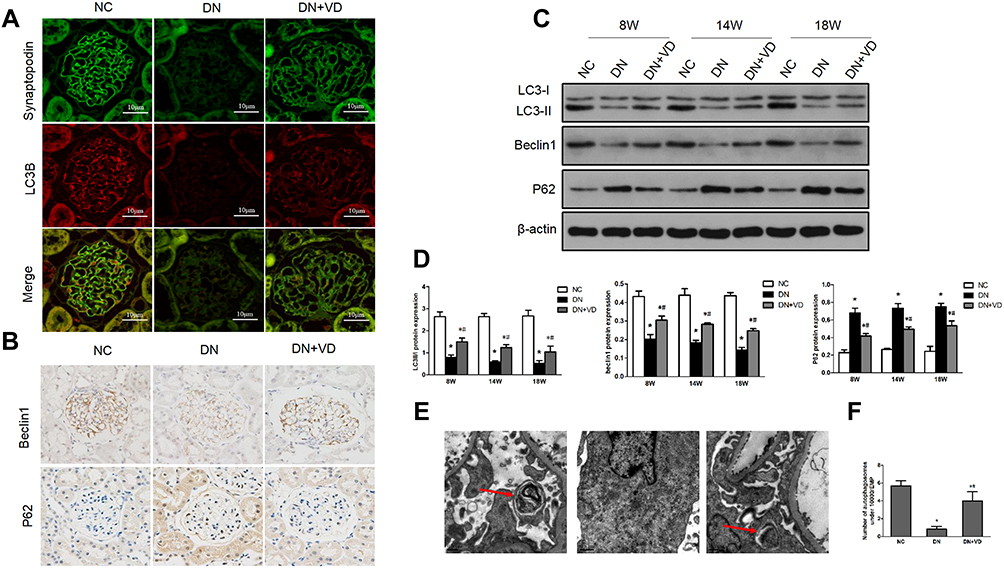 |
Figure 6 Calcitriol treatment restores autophagy activity in diabetic kidney diseases. SD male rats were intraperitoneally injected with 60 mg/kg streptozotocin. After 3 days, the rats with STZ treatment were garaged with 0.1 μg/kg/d calcitriol or vehicle solution daily for consecutive 18 weeks. Kidney tissues were collected for histology or immunoblot analysis at 18th week. (A) Co-staining of synaptopodin (green) and LC3B (red) proteins in glomeruli by double immunofluorescence staining (400×). (B) Immunohistochemical staining of Beclin-1 and P62 in the renal cortex (400×). (C) Immunoblot analysis of autophagy marker proteins LC3 II/I, Beclin-1 and P62 in renal cortex at 8th, 14th, 18th week in three groups, β-actin was used as a loading control. (D) Quantification of LC3 II/I, Beclin-1 and P62 protein. (E) Representative images showing autophagosomes (black arrow) in podocytes by electron microscopy. (F) Quantification of the autophagosomes (under 10,000/EMP from 8 randomly selected podocytes). Values are mean ± SD (n=6). Significance: *P<0.05 vs NC group, #P<0.05 VS DN group at the same time point group, respectively. |
Calcitriol Treatment Preserved Autophagy Activity in DN Rats
The formation of autophagosomes depends on several genes including LC3II, Beclin-1, and other autophagy-related genes.19 Beclin-1 is a core component for autophagy initiation and the autophagy substrate P62 is degraded when autophagy is activated.20 The conversion of LC3-I (cytosolic form) and LC3-II (membrane-bound lipidated form) is an essential event of autophagy. The amount of LC3-II usually correlates well with the number of autophagosomes.20 The immunoblots results showed that higher expression of P62 and lower expression of LC3-II and Beclin-1 in diabetic kidneys compared to non-diabetic kidney suggesting the disruption of autophagy (Figure 6B–D). Calcitriol treatment normalized the expression of these autophagy proteins at all points (Figure 6B–D). We further investigated the activity of autophagy in podocytes, which were identified by the podocyte marker synaptopodin under immunofluorescence. LC3II (red) was diffusely distributed throughout the glomerulus and highly overlapped with synaptopodin signaling (green) in non-diabetic kidney (Figure 6A). The fluorescence intensity of LC3II was diminished in the diabetic kidneys, while calcitriol treatment could recover the overlapping signaling of LC3II and synaptopodin indicating the autophagy repairment of podocytes by calcitriol intervention (Figure 6A). The formation of autophagosomes, the double-membraned vesicles, are the direct evidences of autophagy activity.21 Under transmission electron microscopy, the number of autophagosomes in podocytes were significantly lower in diabetic kidneys than in healthy kidney and autophagosomes formation was corrected after calcitriol intervention (Figure 6E and F). Together, these in vivo data have demonstrated podocytes autophagy disruption in diabetic nephropathy, which could be efficiently preserved by calcitriol treatment.
Calcitriol Attenuates Podocyte Injury Induced by High Glucose in vitro
We next investigated the role of calcitriol on cultured podocytes under high glucose stress. We cultured immortalized MPC5 cell line from mouse podocytes. After differentiation, MPC5 cells were incubated with 30 mM glucose for 24 h to mimic in vivo high glucose micro-environment. High glucose down regulated VDR expression in podocytes, which could be largely rescued by calcitriol supplementation (Figure 7B). We next investigated the expression of the podocyte slit diaphragm proteins and podocyte injury marker in the absence or presence of calcitriol under high glucose conditions. Consistent to in vivo experiment, the expression of slit diaphragm proteins, podocin and nephrin were decreased in podocytes after exposure to high glucose, which could be protected by calcitriol (Figure 7A and C). Furthermore, calcitriol could suppress high glucose induced podocytes injury maker, desmin (Figure 7A and C). Together, these results support in vivo observation that calcitriol attenuated podocyte injury under high glucose stress.
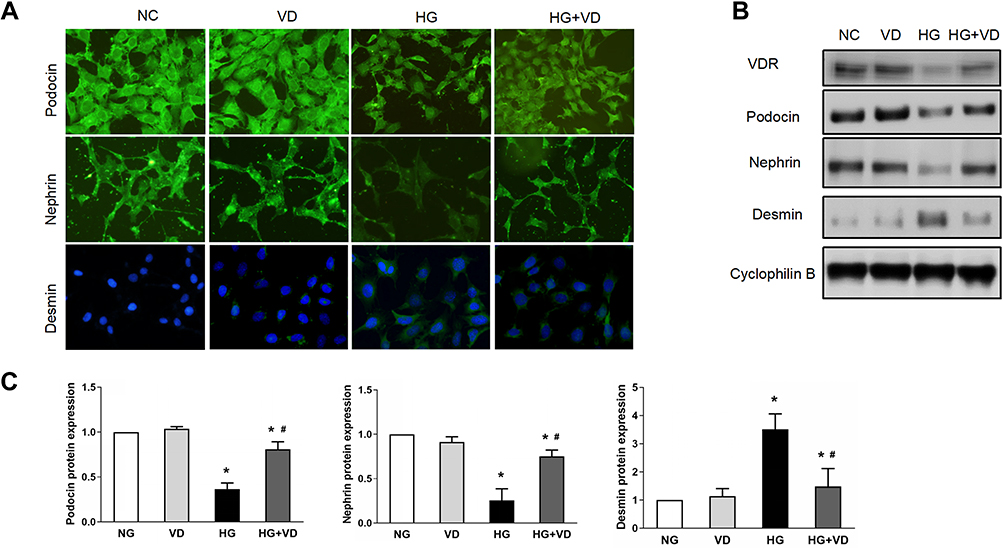 |
Figure 7 Calcitriol attenuates podocyte injury under high glucose. MPC5 were cultured for 24h in medium with 30 mM glucose or 5.5 mM glucose in the absence or presence of 100 nmol/L calcitriol. (A) Immunofluorescence staining of nephrin, podocin and desmin in medium containing NG, VD, HG and HG plus calcitriol for the indicated periods (400×). (B) Western blot was used to detect nephrin, podocin and desmin at different groups, cyclophilin B was used as an internal loading control. (C) The relative densities of the bands in each lane were analyzed and normalized to cyclophilin B in four group. Values are mean ± SD (n=3). Significance: *P<0.05 vs respective control group, #P<0.05 vs DN group at the same time point, respectively. |
The Protection Effect of Calcitriol Was Blocked by Autophagy Inhibition During High-Glucose Treatment MPC5 in vitro
We further investigated the autophagy activity under high glucose in cultured podocyte and the possible regulatory role after calcitriol intervention. High glucose suppressed the expression of LC3-II/I and blocked the degradation of p62. However, calcitriol treatment could normalize the turnover of LC3-II/I and p62 under glucose environment (Figure 8A and B). At the same time, we measured LC3 puncta by GFP-LC3-transfected MPC5. In control or calcitriol treated MPC5, LC3 puncta distributed in the cytosol, while numbers of LC3 was significantly decreased puncta in high glucose conditions, which could be reversed by calcitriol (Figure 8C). Chloroquine (CQ) was used to inhibit endosome-lysosome system acidification and prevent autophagic degradation.22 We demonstrated calcitriol enhanced autophagic influx by increasing LC-II expression and CQ could efficiently block auto lysosomal degradation, being proved by the accumulation of LC3-II (Figure 8D and E). Importantly, immunoblot analysis of podocin, nephrin and desmin indicated that autophagy inhibition by CQ reversed the cytoprotective effect of calcitriol during high glucose treatment of MPC5 cells (Figure 8D and E), indicating the involvement of autophagy in the protective effect of calcitriol during high glucose induced podocyte injury.
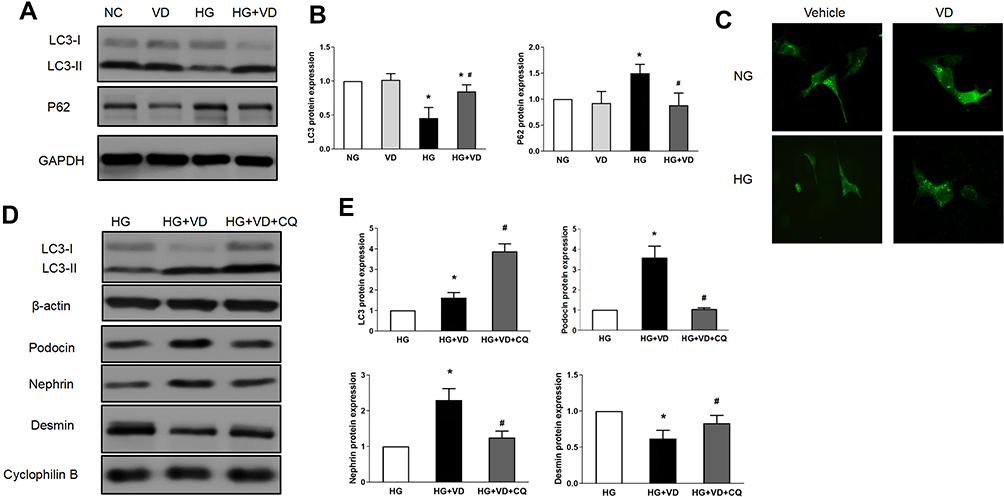 |
Figure 8 The protective effects of calcitriol against high-glucose induced injury are blocked by autophagy inhibitor chloroquine. MPC5 were cultured for 24 h in medium with 30 mM or 5.5 mM glucose in the absence or presence of 100 nmol/L calcitriol. Cells were collected for immunoblot analysis of LC3 and p62 (A and B) or fixed for immunofluorescent microphotographs of GFP-LC3-transfected podocytes (400×) (C). Podocytes were treated with 100 nmol/L calcitriol with and without 20 μM chloroquine (CQ) for 24 hours under high glucose condition. Whole cell lysate were collected for immunoblot analysis of LC3, nephrin, podocin and desmin (D and E). Values are mean ± SD (n=3). Significance: *P<0.05 vs respective control group, #P<0.05 vs DN group at the same time point, respectively. |
Discussion
Diabetic nephropathy is the leading cause for end stage kidney diseases, with huge burden for healthcare systems.2 Podocytes injury plays the central role in the pathogenesis, but the underlying mechanism of podocytes injury is largely unknown. In this study, we have demonstrated that 1. VDR deficiency and autophagy defects in clinical and experimental diabetic nephrology; 2. Restoring vitamin D/VDR signaling by calcitriol attenuated diabetic nephropathy by targeting podocytes; 3. The protective effect of calcitriol was dependent on maintenance of podocytes autophagy under high glucose condition. Our finding expands the role of vitamin D/VDR signaling in diabetic nephropathy and have potential implications for the disease prevention and treatment.
Vitamin D deficiency has been shown as a key feature of chronic kidney disease and a major risk factor for type 2 diabetes disease progress [2]. Calcitriol, a 1,25(OH)2 Vitamin D3, is an active form of vitamin D and could transcriptionally induce VDR expression.18 Several randomized, double-blind, placebo-controlled clinical trials involving diabetic nephropathy patients have shown that supplementation with active vitamin D3 benefited proteinuria control,23,24 although vitamin D3 are recently reported to be ineffective for preventing loss of kidney function in patients with diabetic kidney diseases.25 Consistent to these findings, our study has demonstrated that glomerular VDR differences between diabetic nephropathy and normal control, and calcitriol treatment could restore VDR expression in experimental diabetic nephropathy. Furthermore, calcitriol slowed down the development of diabetic kidney disease by attenuating proteinuria, glomerular hypertrophy and mesangial matrix hyperplasia.
Diabetic nephropathy is characterized by the expansion of the extracellular matrix of the glomerulus, thickening of glomerular and tubular basement membrane, disappearance of podocytes, accompanied by progressive proteinuria and impaired renal function. Notably, the podocyte slit diaphragm proteins including nephrin, podocin and synaptopodin are essential for the maintenance of the skeletal structure and function of podocytes.26 Dysfunction of slit diaphragm proteins could lead to filter dysfunction, thus promoting the development of proteinuria.27 Our previous studies and other studies have demonstrated that therapeutic intervention to preserve of slit diaphragm proteins could reduce urinary protein excretion10,11. The slit diaphragm protein was damaged and intermediate filaments such as desmin (podocyte injury marker) was up regulated in diabetic rats. All above pathological changes can be prevented, as least partially, by calcitriol treatment. However, there was no statistically significant difference in podocyte-specific nuclear protein marker WT1 between the three groups, suggesting that the main effect of calcitriol on podocytes is likely to be on the slit membrane protein rather than preserving the podocyte number. To be notedly, calcitriol did not improve renal function and blood glucose control in diabetic rats. Possible explanations are that the treatment of calcitriol has not yet reached a level that allows renal function levels to be identifiable. The lack of effect of calcitriol on glucose level most likely resulted from the consequence of STZ-induced irreversible beta cell damage.
Podocytes have high levels of basal autophagy activity to maintain their normal function.17 In respond to high glucose, starvation, hypoxia and oxidative stress, autophagy is required for protein hemostasis and autophagy dysfunction could exacerbate cellular damage. For example, specific knockdown of autophagy-related gene such as Atg5 in podocytes can aggravate self-injury and accompanied by a large amount of proteinuria.28 In contrast, therapies targeting the autophagy pathway such as low-dose rapamycin treatment can stabilize podocyte structure and function and delay the progression of diabetic nephropathy.29,30 These evidences support that autophagy-mediated cellular homeostasis may be the key to podocytes in respond to high glucose-induced damage. In this study, we found that the autophagy was inhibited in the kidney of diabetic rats by decreased autophagy related protein, weakened overlapping fluorescence of LC3II (red) and synaptopodin and the reduced number of autophagosomes in podocytes. Strikingly, calcitriol protected against diabetic nephropathy and restored the autophagy activity in podocytes by recovering LC3, Beclin-1 and autophagosome formation. Furthermore, the protective effect of calcitriol against podocytes injury under high glucose was blocked by autophagy inhibitor chloroquine. These results illustrated that calcitriol protected podocyte structure and function depending on maintaining podocytes autophagy in diabetic kidneys.
It is currently clear that how vitamin D/VDR signaling regulates podocyte autophagy. Specific deletion of the VDR gene and Vitamin D deficiency could lead to insufficient autophagy and affect organ function.13,31 Many studies supported that vitamin D/VDR may act as an important transcriptional factor regulating autophagy. Tavera-Mendoza et al demonstrated that VDR could activate an autophagic transcriptional signature in breast cancer cells.32 More specifically, ATG16L1 (autophagy related 16 like 1), as a VDR target gene, could be transcriptionally activated by vitamin D/VDR to initiate autophagy in Paneth cells and intestinal epithelial cells.13–15 Studies has demonstrated that vitamin D can downregulate the expression of mTOR protein, thus inducing autophagy by inhibiting the mTORC1 complex.33 While another study indicated vitamin D3 treatment did not transcriptionally regulate autophagy-related gene in gastric epithelium but store lysosomal acidification and degradation function by enhancing Ca2+ release from lysosomes.34 In this study, we found vitamin D/VDR restored autophagy in diabetic nephropathy and further studies are required for understanding signaling pathway behind it.
Despite the findings, there are several limitations in this work. In this study, the specific mechanisms between autophagy and Vitamin D/VDR signaling in podocytes in diabetic nephropathy were not investigated in depth. As a transcriptional factor, VDR may regulate autophagy by controlling the transcription of autophagy associated gene like Bcl-2, beclin-1, mammalian target of rapamycin. Further studies are needed to explore the precise molecular pathways of autophagy and VDR. Secondly, the effect of calcitriol on autophagy was only investigated in vitro by using CQ as an autophagy inhibitor. However, the association between Vitamin D/VDR signaling and autophagy needs to be further studied with a podocyte-specific autophagy-related genes knockout mice in vivo.
In conclusion, our study delivered the evidence that calcitriol/VDR signaling attenuated diabetic nephropathy and podocytes injury by restoring podocytes autophagy. This finding may have new insight into exploring the detailed protective mechanisms of calcitriol/VDR in diabetic nephropathy.
Abbreviations
DN, diabetic nephropathy; STZ, streptozotocin; VDR, vitamin D receptor; SPF, specific pathogen free; SD, Sprague-Dawley; NC group, vehicle control group; DN group, diabetic nephropathy group; DN+VD group, diabetic nephropathy rat treated with calcitriol group; KW/BW, kidney weight to body weight ratio; BUN, blood urea nitrogen; Scr, serum creatinine; ALB, serum albumin, Ca, serum calcium, P, serum phosphorus; PAS, Periodic Acid-Schiff.
Ethical Statement
The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. The trial was conducted in accordance with the Declaration of Helsinki. The study was approved by Human Subjects Committee of the First Clinical Medical College of Three Gorges University (HEC-KYJJ2019-053-01) and informed consent was taken from all the patients. All animal care and experimental programs are in the compliance with animal management regulations of the Ministry of Health of the People’s Republic of China and approved by the Ethics Committee of the Three Gorges University (2017-0012). The use of the MPC5 cell lines was approved by the Ethics Committee of the Three Gorges University (2017-0012).
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Funding
This work was supported by Grants from the National Youth Science Foundation of China (grant number 81600567), Science and Technology Bureau Project of Yichang (grant number A16-301-02). Zhixia Song and Chao Xiao are co-first authors in this publication.
Disclosure
The authors have no conflicts of interest to declare.
References
1. Thomas MC, Brownlee M, Susztak K, et al. Diabetic kidney disease. Nat Rev Dis Primers. 2015;1(1):15018. doi:10.1038/nrdp.2015.18
2. Levin A, Tonelli M, Bonventre J, et al. Global kidney health 2017 and beyond: a roadmap for closing gaps in care, research, and policy. Lancet (London). 2017;390(10105):1888–1917. doi:10.1016/S0140-6736(17)30788-2
3. Alicic RZ, Rooney MT, Tuttle KR. Diabetic kidney disease: challenges, progress, and possibilities. Clin J Am Soc Nephrol. 2017;12(12):2032–2045. doi:10.2215/CJN.11491116
4. Liapis H, Romagnani P, Anders HJ. New insights into the pathology of podocyte loss: mitotic catastrophe. Am J Pathol. 2013;183(5):1364–1374. doi:10.1016/j.ajpath.2013.06.033
5. Herrmann M, Farrell CL, Pusceddu I, Fabregat-Cabello N, Cavalier E. Assessment of vitamin D status - a changing landscape. Clin Chem Lab Med. 2017;55(1):3–26.
6. Moukayed M, Grant WB. Linking the metabolic syndrome and obesity with vitamin D status: risks and opportunities for improving cardiometabolic health and well-being. Diabetes Metab Syndr Obes. 2019;12:1437–1447. doi:10.2147/DMSO.S176933
7. Fernandez-Juarez G, Luno J, Barrio V, et al. 25 (OH) vitamin D levels and renal disease progression in patients with type 2 diabetic nephropathy and blockade of the renin-angiotensin system. Clin J Am Soc Nephrol. 2013;8(11):1870–1876. doi:10.2215/CJN.00910113
8. Wang Y, Deb DK, Zhang Z, et al. Vitamin D receptor signaling in podocytes protects against diabetic nephropathy. J Am Soc Nephrol. 2012;23(12):1977–1986. doi:10.1681/ASN.2012040383
9. Goltzman D. Functions of vitamin D in bone. Histochem Cell Biol. 2018;149(4):305–312. doi:10.1007/s00418-018-1648-y
10. Song Z, Guo Y, Zhou M, Zhang X. The PI3K/p-Akt signaling pathway participates in calcitriol ameliorating podocyte injury in DN rats. Metabolism. 2014;63(10):1324–1333. doi:10.1016/j.metabol.2014.06.013
11. Zhang X, Song Z, Guo Y, Zhou M. The novel role of TRPC6 in vitamin D ameliorating podocyte injury in STZ-induced diabetic rats. Mol Cell Biochem. 2015;399(1–2):155–165. doi:10.1007/s11010-014-2242-9
12. Garsen M, Sonneveld R, Rops AL, et al. Vitamin D attenuates proteinuria by inhibition of heparanase expression in the podocyte. J Pathol. 2015;237(4):472–481. doi:10.1002/path.4593
13. Wu S, Zhang YG, Lu R, et al. Intestinal epithelial vitamin D receptor deletion leads to defective autophagy in colitis. Gut. 2015;64(7):1082–1094. doi:10.1136/gutjnl-2014-307436
14. Sun J. VDR/vitamin D receptor regulates autophagic activity through ATG16L1. Autophagy. 2016;12(6):1057–1058. doi:10.1080/15548627.2015.1072670
15. Lu R, Zhang YG, Xia Y, Sun J. Imbalance of autophagy and apoptosis in intestinal epithelium lacking the vitamin D receptor. FASEB J. 2019;33(11):11845–11856. doi:10.1096/fj.201900727R
16. Mundel P, Reiser J, Kriz W. Induction of differentiation in cultured rat and human podocytes. J Am Soc Nephrol. 1997;8(5):697–705.
17. Bork T, Liang W, Yamahara K, et al. Podocytes maintain high basal levels of autophagy independent of mtor signaling. Autophagy. 2019;1–17.
18. Zhang Z, Yuan W, Sun L, et al. 1,25-dihydroxyvitamin D3 targeting of NF-kappaB suppresses high glucose-induced MCP-1 expression in mesangial cells. Kidney Int. 2007;72(2):193–201. doi:10.1038/sj.ki.5002296
19. Boya P, Reggiori F, Codogno P. Emerging regulation and functions of autophagy. Nat Cell Biol. 2013;15(7):713–720. doi:10.1038/ncb2788
20. Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140(3):313–326. doi:10.1016/j.cell.2010.01.028
21. Ganley IG, Lane JD. Autophagosome maturation and lysosomal fusion. Essays Biochem. 2013;55:65–78. doi:10.1042/bse0550065
22. Mauthe M, Orhon I, Rocchi C, et al. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy. 2018;14(8):1435–1455. doi:10.1080/15548627.2018.1474314
23. de Borst MH, Hajhosseiny R, Tamez H, Wenger J, Thadhani R, Goldsmith DJ. Active vitamin D treatment for reduction of residual proteinuria: a systematic review. J Am Soc Nephrol. 2013;24(11):1863–1871. doi:10.1681/ASN.2013030203
24. Joergensen C, Tarnow L, Goetze JP, Rossing P. Vitamin D analogue therapy, cardiovascular risk and kidney function in people with Type 1 diabetes mellitus and diabetic nephropathy: a randomized trial. Diabet Med. 2015;32(3):374–381. doi:10.1111/dme.12606
25. de Boer IH, Zelnick LR, Ruzinski J, et al. Effect of vitamin D and omega-3 fatty acid supplementation on kidney function in patients with Type 2 diabetes: a Randomized Clinical Trial. JAMA. 2019;322(19):1899. doi:10.1001/jama.2019.17380
26. Kawachi H, Fukusumi Y. New insight into podocyte slit diaphragm, a therapeutic target of proteinuria. Clin Exp Nephrol. 2020;24(3):193–204. doi:10.1007/s10157-020-01854-3
27. Trimarchi H. Mechanisms of podocyte detachment, podocyturia, and risk of progression of glomerulopathies. Kidney Dis. 2020;6(5):324–329. doi:10.1159/000507997
28. Tagawa A, Yasuda M, Kume S, et al. Impaired podocyte autophagy exacerbates proteinuria in diabetic nephropathy. Diabetes. 2016;65(3):755–767. doi:10.2337/db15-0473
29. Xiao T, Guan X, Nie L, et al. Rapamycin promotes podocyte autophagy and ameliorates renal injury in diabetic mice. Mol Cell Biochem. 2014;394(1–2):145–154. doi:10.1007/s11010-014-2090-7
30. Kaushal GP, Chandrashekar K, Juncos LA, Shah SV. Autophagy function and regulation in kidney disease. Biomolecules. 2020;10(1):100. doi:10.3390/biom10010100
31. Wu S, Sun J. Vitamin D, vitamin D receptor, and macroautophagy in inflammation and infection. Discov Med. 2011;11(59):325–335.
32. Tavera-Mendoza LE, Westerling T, Libby E, et al. Vitamin D receptor regulates autophagy in the normal mammary gland and in luminal breast cancer cells. Proc Natl Acad Sci U S A. 2017;114(11):E2186–E94. doi:10.1073/pnas.1615015114
33. Lisse TS, Hewison M. Vitamin D: a new player in the world of mTOR signaling. Cell Cycle. 2011;10(12):1888–1889. doi:10.4161/cc.10.12.15620
34. Hu W, Zhang L, Li MX, et al. Vitamin D3 activates the autolysosomal degradation function against Helicobacter pylori through the PDIA3 receptor in gastric epithelial cells. Autophagy. 2019;15(4):707–725. doi:10.1080/15548627.2018.1557835
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.