Back to Journals » Drug Design, Development and Therapy » Volume 19
Vitamin D Alleviates Osteoarthritis Progression by Targeting Cartilage and Subchondral Bone via Myd88-TAK1-ERK Axis Suppression
Authors Gao X, Min Y, Lin R ![]() , Liang D, Zhang M, Xiao Q, Lu Y, Zhang F, Xu B, Liu Y
, Liang D, Zhang M, Xiao Q, Lu Y, Zhang F, Xu B, Liu Y ![]()
Received 2 April 2025
Accepted for publication 26 June 2025
Published 8 July 2025 Volume 2025:19 Pages 5855—5870
DOI https://doi.org/10.2147/DDDT.S526064
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Muzammal Hussain
Xiang Gao,1,2,* Yalin Min,1,3,* Rui Lin,1,* Dahong Liang,1,3 Min Zhang,1,3 Qixian Xiao,1 Yan Lu,2 Fucheng Zhang,1 Bilian Xu,3,4 Yanzhi Liu1,4
1Key Laboratory of Traditional Chinese Medicine for the Prevention and Treatment of Infectious Diseases, Zhanjiang Central Hospital, Guangdong Medical University, Zhanjiang City, Guangdong Province, 524045, People’s Republic of China; 2Stem Cell Research and Cellular Therapy Center, The Affiliated Hospital of Guangdong Medical University, Zhanjiang City, Guangdong Province, 524001, People’s Republic of China; 3Marine Medical Research Institute of Zhanjiang, School of Ocean and Tropical Medicine, Guangdong Medical University, Zhanjiang City, Guangdong Province, 524023, People’s Republic of China; 4School of Pharmacy, Guangdong Medical University, Zhanjiang City, Guangdong Province, 524023, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yanzhi Liu, Key Laboratory of Traditional Chinese Medicine for the Prevention and Treatment of Infectious Diseases, Zhanjiang Central Hospital, Guangdong Medical University, No. 236, Yuanzhu Road, Chikan Disctrict, Zhanjiang, 524045, People’s Republic of China, Email [email protected] Bilian Xu, Marine Medical Research Institute of Zhanjiang, School of Ocean and Tropical Medicine, Guangdong Medical University, No. 2 Wenmingdong Road, Xiashan District, Zhanjiang, 524023, People’s Republic of China, Email [email protected]
Background: Osteoarthritis (OA) causes irreversible joint damage, but current treatments fail to fully address its complex pathology. Emerging evidence suggests subchondral bone metabolic dysfunction may initiate OA. While vitamin D (VitD) is well-established for bone metabolism regulation in osteoporosis, its therapeutic potential in OA remains unclear despite observational studies suggesting protective effects. Our integrated in vivo/in vitro study demonstrates VitD’s dual chondroprotective and osteogenic actions in OA.
Methods: Sprague-Dawley rats (n=24) were divided into three groups: sham operation (Sham), OA model (OA), and OA+VitD treatment, with 8 rats in each group. Oral cholecalciferol (2.34 μg/kg/day) was administered for 6 weeks post-Monosodium iodoacetate (MIA) induction. The therapeutic potential of vitamin D was evaluated through a series of in vivo experiments. Human chondrocyte C28 cells were pretreated with TNFα (1ng/mL) to model inflammatory injury, followed by 1,25(OH)2 D3 (10− 2 μM) exposure for 72 hours to assess the VitD’s effects of chondrogenesis and further investigate its underlying mechanism.
Results: In OA rats, VitD suppressed femoral cartilage degradation (evidenced by 567.76% increased cartilage area, and 39.13% decreased Osteoarthritis Cartilage Histopathology (OACH) score and enhanced subchondral bone mass (61.81% higher BV/TV). At the molecular level, VitD downregulated the expression of cartilage matrix metalloproteinase 13 (MMP13), with a reduction of 74.72% compared to OA group. Additionally, VitD inhibit inflammatory signaling pathways, particularly through the MyD88-TAK1-ERK axis in chondrocytes, and decrease serum IL-6 level. Mechanistic validation of these findings was demonstrated by protein expression reduction of Myd88 (31.22%), phospho-ERK1/2 (66.11%), AP-1 (61.43%) and NFκB (34.36%) compared to OA group. In vitro, VitD also rescued ethanol-induced C28 cell viability loss while significantly upregulating cartilage anabolic markers.
Conclusion: These findings establish VitD as a multimodal OA therapeutic agent targeting both cartilage catabolism and subchondral bone remodeling through Myd88-TAK1-ERK axis.
Keywords: vitamin D, bone, cartilage, osteoarthritis, rat
Graphical Abstract:

Introduction
Osteoarthritis (OA), affecting >240 million globally, manifests as progressive cartilage degradation, aberrant subchondral bone remodeling, and synovial inflammation - collectively driving irreversible joint dysfunction.1 As the leading cause of chronic disability in aging populations, OA pathogenesis arises from the intrinsic systemic factors (including genetic susceptibility, age-related cellular senescence, metabolic dysfunction-associated obesity, sex-specific biological effects, and nutritional homeostasis disruption) and local joint biomechanical factors (comprising aberrant joint loading dynamics, post-traumatic joint remodeling failure, and articular surface incongruity).2 These elements form an integrated and complex regulatory network that orchestrates OA progression. Current palliative therapies (paracetamol, NSAIDs, et.al) have not effectively addressed disease progression and are associated with significant side effects,3 while surgical interventions carry substantial risks.4 For example, NSAIDs can help alleviate pain in OA patients, but their use often comes with gastrointestinal/renal toxicity challenging side effects. Glucosamine and chondroitin sulfate are thought to alleviate OA progression providing a synthetic substrate for cartilage regeneration while their true effectiveness remains limited.
Mechanical stress and wear, aging, and chronic inflammation, these factors are thought to induce sustained cartilage degradation and release substantial production of degrading enzymes. Mechanical overload-induced cartilage breakdown releases proteases that destabilize subchondral bone architecture.5 Subsequent trabecular loss and plate thickening further disrupt load distribution, accelerating cartilage catabolism and degradation,6,7 creating a vicious cycle that ultimately initiate OA progression. Emerging evidence reveals that subchondral bone remodeling precedes cartilage degeneration.7 Emerging evidence indicates that dysregulated subchondral bone remodeling may precede the onset of cartilage degeneration in osteoarthritis pathogenesis.7 Therefore, the concept that subchondral metabolic dysregulation precedes macroscopic cartilage damage, positioning bone modulation as potential a preventive therapeutic window. Breaking the disturbance connection between subchondral bone remodeling imbalance and cartilage catabolism is promising to mitigate OA disease progression.
1,25-Dihydroxyvitamin D3, the biologically active metabolite of vitamin D exhibits pronounced osteoanabolic properties through its pleiotropic regulation of bone mineralization processes.8 Research demonstrates that concomitant calcium and vitamin D3 supplementation significantly reduces hip fracture incidence in postmenopausal osteoporosis by synergistically enhancing bone mineral density and microarchitecture.9 Our previous research also revealed that VitD significantly alleviated osteopenia associated with type 2 diabetes.10 Furthermore, VitD plays a crucial role in controlling human Th17-mediated synovial inflammation11 and exhibits a significant inhibitory effect on the activation of the NLRP1 inflammasome.12 VitD demonstrated promising potential in alleviating symptoms of OA.13,14 These evidences suggest that VitD emerges as a dual-action candidate for OA therapy through osteoanabolic regulation and anti-inflammatory properties. However, there are limited study comprehensively clarify the role of VitD treatment for the protection of cartilage degradation and OA therapy.
Through integrated in vitro and in vivo studies, we investigate vitamin D3’s therapeutic mechanisms in osteoarthritis using an inflammation-stimulated chondrocyte model to examine the potential pathway modulation and a monosodium iodoacetate (MIA)-induced rat OA model to assess structural and functional outcomes. Our goal is to provide mechanistic evidence supporting vitamin D’s potential repurposing from nutritional supplement to disease-modifying OA therapy.
Materials and Methods
Materials and Reagents
Twenty-four female Sprague-Dawley rats (6 months old, SPF-grade) with a mean body weight of 286 ± 19 g were procured from the Guangxi Medical University Laboratory Animal Center (License No. SCXK Gui 2014–0002). All animals were maintained under standardized housing conditions (the SPF Animal facility of Guangdong Medical University, issued ID: SYXK-Yue-2019-0213) with free access to food and water throughout the study period. Following a one-month acclimatization period, their average weight at the start of the experiment was (295 ± 22) g. The rats were weighed weekly, with food withheld the day prior to each weighing session.
All experimental procedures were performed in strict accordance with the Chinese national standards for laboratory animal welfare and approved by the Institutional Animal Care and Use Committee of Guangdong Medical University (License Number GDY2002272).
The human C28 chondrocyte cells (Product number: ZY-C6492H) were commercially acquired from Shanghai zeye biology science and technology co., ltd. Calcitriol capsules (batch number: 14071716) were sourced from Roche Pharmaceutical Factory Hong Kong Limited. For experimental dosing, we adopted a clinically relevant regimen of 1000 IU/day (25 μg/day) as a reference, corresponding to the therapeutic threshold for vitamin D insufficiency in high-risk populations as established by current clinical guidelines.15,16 This human-equivalent dosage was adjusted to 2.34 μg/kg/day in rats to maintain physiological relevance across species. For preparation of the calcitriol working solution, a single pharmaceutical-grade capsule containing 25 μg 1,25-dihydroxycholecalciferol was initially solubilized in 2 mL of Tween 80 under sterile conditions, with subsequent dilution using ultrapure water to achieve a final concentration of 1 μg/mL in a 25 mL volumetric preparation. The suspension was stored in a dark environment to maintain stability.
Establishment of OA Model
Preparation of monosodium iodoacetate (MIA) working solution: Weigh 240 mg of MIA into a sterile EP tube. Add 3 mL of normal saline and mix thoroughly until the powder is completely dissolved.17 Ensure the entire procedure is carried out in the dark. Based on the body weight of rats, twenty-four rats were assigned to three groups randomly (n=8/group): Sham control group (Sham), Intra-articular injection of 50 μL sterile saline (0.9% NaCl); OA model group (OA), Monosodium iodoacetate (MIA, 50 μL working solution) via intra-articular injection; Therapeutic intervention group (OA+VitD) group, MIA-induced OA followed by daily oral administration of 1,25-(OH)2D3 (2.34 μg/kg/day).
Pathological Evaluation
Remove the redundant muscles around the femur and tibia, and closely examine the changes in joint shape, cartilage thickness, and color. The gross observation of the articular cartilage was scored according to the criteria outlined in reference standard.18
Serum Biomarkers
Serum concentrations of key biomarkers were quantitatively analyzed using standardized enzyme-linked immunosorbent assays (ELISA, Wuhan Yilai Ruite Biotechnology Co., Ltd), including:
- pro-inflammatory cytokine interleukin-6 (IL-6),
- cartilage-degrading enzyme matrix metalloproteinase-13 (MMP-13),
- angiogenesis marker CD31 (platelet endothelial cell adhesion molecule-1),
- bone formation indicator PINP (N-terminal propeptide of type I procollagen),
- hypertrophic chondrocyte marker collagen X (COL10A1),
- bone resorption marker β-CTX (C-terminal telopeptide of type I collagen).
All assays were performed in technical duplicates following the manufacturer’s standardized protocols.
μ-CT Assessment
We immersed the upper tibia in meglumine diatrizoate solution and heated it to 37°C using a water bath. The sample was maintained at a constant temperature for 20 minutes before being removed for μ-CT three-dimensional scanning. The scanning parameters were as follows: an image matrix of 2048×2048, 200 ms integration time per projection, energy/intensity settings (45 kVp, 177 μA, 8 W). Density calibration: Standardized to 1200 mg hydroxyapatite (HA)/cm³ phantom. Additionally, a three-dimensional μ-CT scan was conducted on the distal femur with the scanning parameters were as follows: energy/intensity settings (70 kVp, 114 μA, 8 W), and other scanning conditions are consistent with upper tibia. Three-dimensional reconstruction and quantitative analysis were performed after CT scan. From these analyses, the following physical parameters were obtained: bone volume/tissue volume (BV/TV), thickness of subchondral bone plate, trabecular separation (Tb.Sp), cartilage thickness, and trabecular thickness (Tb.Th).
Histopathological Analysis
Femoral specimens underwent systematic processing beginning with demineralization in 12% ethylenediaminetetraacetic acid (EDTA) solution (pH 7.4) under controlled ambient conditions for 42 days. Post-demineralization, tissues were subjected to a standardized gradient ethanol dehydration protocol. After the dehydration steps, the tissue samples were rinsed with xylene, embedded in wax, and sectioned into thin slices. To complete the histological evaluation, tissue sections were stained using a safranin O-fast green dual staining system (G1371, Solarbio, China), which selectively highlights proteoglycan-rich cartilage matrix. Quantitative morphometric analysis was performed using Image Pro Plus 6.0 to obtain precise measurements of cartilage thickness and surface area in stained sections. Concurrently, a comprehensive semi-quantitative assessment of cartilage degeneration was conducted through systematic light microscopic examination (Olympus) following established histological grading criteria. The Osteoarthritis Cartilage Histopathology (OACH) score was calculated by multiplying the damage stage by the classification grade, providing a comprehensive assessment of cartilage degeneration.19
Immunohistochemistry
Antigen retrieval was achieved via thermal treatment in citrate buffer (pH 6.0, 95°C ×20 min), with subsequent peroxidase inhibition using 3% hydrogen peroxide (15 min, RT).
Following this, incubate the slice with the recombinant Anti-MMP13 antibody (ab219620, Abcam, American) overnight at 4°C. After the primary antibody incubation, apply the secondary antibody and incubate for 1 hour. To visualize the antibody binding, use the DAB chromogenic kit (DA1010, Solarbio, Beijing, China). After developing the color, perform a slight counterstain with hematoxylin for 20 seconds. Rinse thoroughly, air-dry the slice, and seal it with neutral resin.
Tissue sections were subsequently incubated with recombinant monoclonal anti-MMP-13 antibody (ab219620; Abcam, USA) at 4°C overnight in a humidified chamber. Following primary antibody incubation, sections were treated with species-matched horseradish peroxidase (HRP)-conjugated secondary antibody for 60 minutes at room temperature. Antigen-antibody complexes were visualized using 3,3’-diaminobenzidine (DAB) chromogen (DA1010; Solarbio, China). Nuclei were counterstained with Mayer’s hematoxylin (20 seconds). Rinse thoroughly, air-dry the slice, and seal it with neutral resin. Finally, digital morphometric analysis was conducted with Image-Pro Plus wherein protein-positive areas were segmented, followed by IOD quantification of target epitopes and areal normalization (IOD/Area).
Cell Viability Assay
Human C28 chondrocytes were seeded in 96-well plate and were treated with 0.1% EtOH-dissolved VitD with concentration gradients (D1530, Sigma, American) across three temporal intervals (24, 48, and 72 hours). Cell viability was determined via Cell Counting Kit-8 assay (C0038, Beyotime, China).
TNFα Induction
To model inflammatory chondrocyte injury, C28 chondrocytes were exposed to recombinant human TNF-α (1 ng/mL, 10602-HNAE, Sino Biological, China) in complete DMEM/F-12 medium supplemented with 10% FBS. The experimental design incorporated three treatment groups:
(a) Vehicle control: Complete DMEM/F12 medium (CON)
(b) Inflammatory model: 1 ng/mL TNFα stimulation (TNFα)
(c) Therapeutic intervention: TNFα (1 ng/mL) + 1,25-dihydroxyvitamin D3 (10−2μM; D1530, Sigma) co-treatment (TNFα+VitD)
Quantitative Reverse Transcription-PCR
Total chondrocyte RNA was isolated with TRIzol (Takara, Japan) followed by cDNA synthesis with the PrimeScript RT Master Mix (Perfect Real Time; Takara, Japan). Quantitative gene expression analysis was conducted on an ABI 7500 Fast Real-Time PCR System (Applied Biosystems, USA) using SYBR Green chemistry (TB Green Premix Ex Taq II, Takara, Japan). The following cartilage-specific markers were quantified: collagen type II alpha 1 (Col2a1), SRY-box transcription factor 9 (SOX9), TAK1, and Aggrecan (ACAN). Amplification conditions consisted of an initial denaturation at 95°C for 30 sec, followed by 40 cycles of 95°C for 5 sec and 60°C for 34 sec, with GAPDH serving as the endogenous reference gene.
Western Blotting
Cellular proteins were extracted using ice-cold RIPA lysis buffer (Beyotime, China) supplemented with protease and phosphatase inhibitors. Lysates were clarified by centrifugation at 4°C (10000 × g, 10 min) and protein concentrations were determined via bicinchoninic acid (BCA) assay. Equal amounts of protein (30 μg per lane) were resolved and separated on 10% SDS-polyacrylamide gels and subsequently electro transferred to polyvinylidene difluoride (PVDF) membranes. Samples separated by SDS-PAGE (10% gel) and transferred to PVDF. Membranes were blocked with 5% (w/v) BSA for 1 h at room temperature, followed by incubation with primary antibodies (detailed in Table 1) overnight at 4°C with gentle agitation.
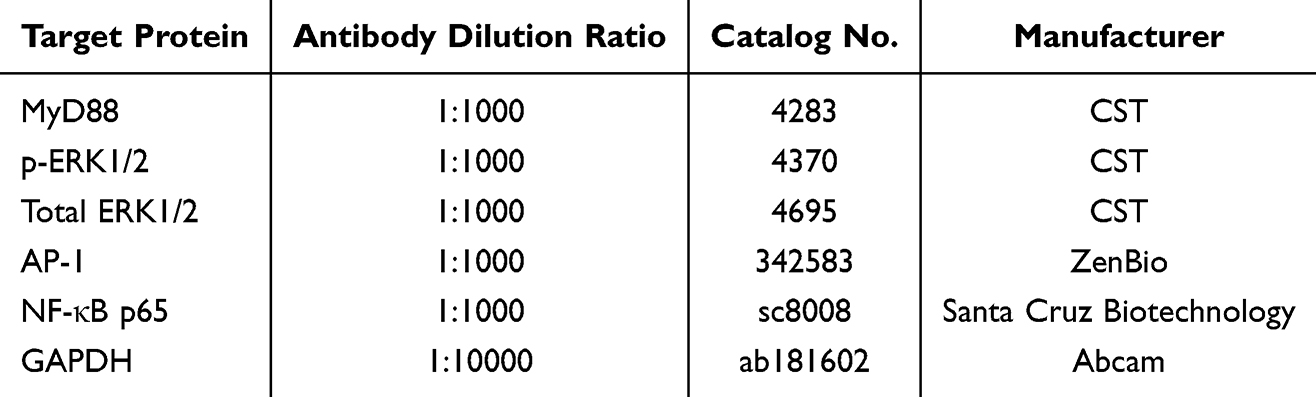 |
Table 1 Primary Antibody Used in Western Blot |
HRP-anti-rabbit IgG (1:5000, Abbkine) used for detection. Imaging via FluorChem® Q (ProteinSimple) with ImageJ analysis.
Statistical Analysis
Data were visualized using GraphPad Prism 8.0. Statistical analyses were performed using SPSS 19.0, with the sample size (N) for each experiment specified in the corresponding figure captions. Each experimental replication was conducted as an independent observation. Normality and homogeneity of variances were first assessed using Levene’s tests, respectively. Significant differences between two groups were analyzed with an independent-samples t-test (assuming equal variances) or Welch’s t-test (for unequal variances). Comparisons among multiple groups were evaluated via one-way ANOVA. Post hoc analyses utilized Fisher’s Least Significant Difference (LSD) test when variances were homogeneous, and Tamhane’s T2 test when variances were heterogeneous. A significance threshold of P<0.05 was considered statistically significant.
Results
TNFα Caused Inflammatory Damage in Chondrocytes, While OA Rats Led to Considerable Bone and Cartilage Damage
The OA animal model was successfully induced in SD rats through intra-articular administration of monosodium iodoacetate (MIA) solution. Macroscopic evaluation revealed distinct pathological changes in the OA group compared to sham-operated controls, the knee cartilage in OA rats exhibited a rough and uneven surface and displayed significant cartilage defects (Figure 1A). Histomorphometry analysis demonstrated a statistically significant 22-fold elevation in osteoarthritis lesion severity scores in MIA-induced rats relative to sham controls (P<0.001) (Figure 1C). Serum biomarker profiling revealed marked upregulation of multiple pathogenic mediators (MMP13, IL-6, CD31, COL10A, CTX-I and PINP) in the OA rats (Figure 2).
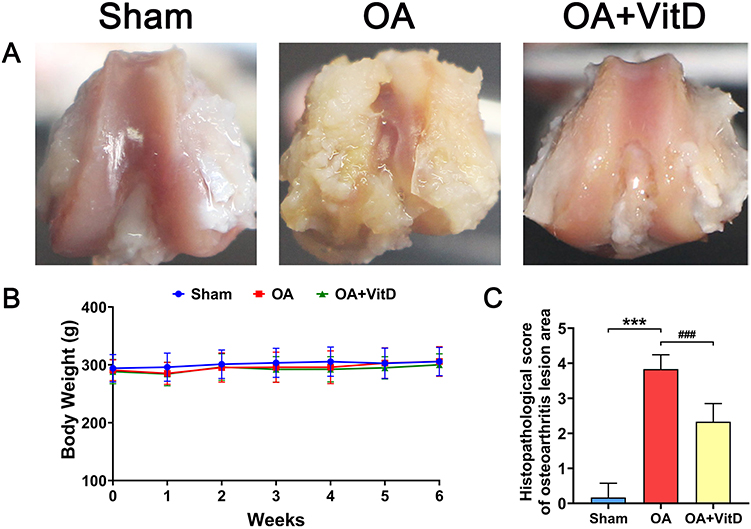 |
Figure 1 Effect of 2.34 μg/kg/day VitD on body weight and knee joint lesion in OA rats. (A) Representative images of the knee joint in OA rats treated with 2.34 μg/kg/day VitD. (B) Body weight measurements of OA rats following treatment with 2.34 μg/kg/day VitD, N=8. (C) Histopathological scores of the osteoarthritis lesion areas in OA rats treated with 2.34 μg/kg/day VitD, N=8. Note: ***P<0.001 vs Sham; ###P<0.001 vs OA. Values are presented as mean ± SD. |
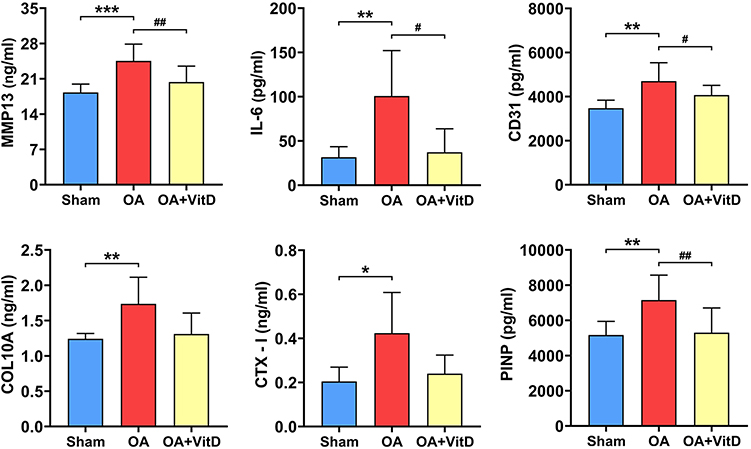 |
Figure 2 Serum biochemical markers in OA rats treated with 2.34 μg/kg/day VitD. Note: N=8, *P<0.05, **P<0.01, ***P<0.001 vs Sham; #P<0.05, ##P<0.01, OA. Values are presented as mean ± SD. |
Micro-computed tomography (μ-CT) quantification demonstrated profound structural alterations in the OA model group, articular cartilage degeneration manifested as a 61.33% reduction in mean thickness relative to sham controls (P<0.001) (Figure 3A), subchondral bone remodeling characterized by a 37.49% increase in plate thickness (P<0.01) (Figure 3B). In addition, we conducted a three-dimensional reconstruction of the femoral cancellous bone, focusing on a 3 mm-thick cancellous bone located 1 mm away from the femoral growth plate, followed by quantitative analysis. Quantitative microarchitectural analysis revealed significant osseous alterations in OA rats compared to sham controls. Femoral trabecular bone volume fraction decreased by 10.54% (Figure 3C), mean trabecular thickness (Tb. Th) was mildly reduced (Figure 3D) and trabecular separation (Tb. Sp) increased by 14.61% (Figure 3E). Histological analysis with safranin O fast green staining revealed a significant increase of 2633.33% in OACH score of OA rats (P<0.001) (Figure 4A). Morphometric analysis further revealed substantial cartilage degeneration in OA rats, characterized by a 48.11% reduction in cartilage mean thickness (P<0.001), and an 87.53% decrease in total articular cartilage surface area (P<0.001) compared to sham controls (Figure 4A). Immunohistochemical quantification showed marked upregulation of matrix metalloproteinase-13 (MMP13) in OA cartilage, with a 277.24% increase in positive staining intensity compared to sham-operated animals (P<0.01, Figure 4B), indicating enhanced extracellular matrix degradation in the disease state.
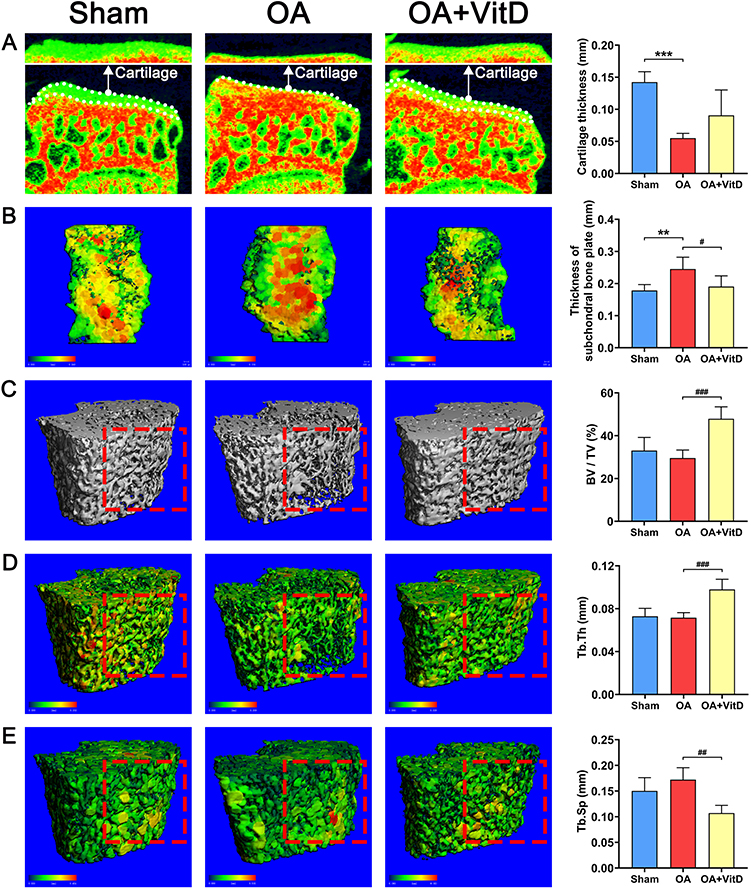 |
Figure 3 Representative μ-CT images in OA rats treated with 2.34 μg/kg/day VitD and the corresponding quantitative analysis. (A) Cartilage from the surface of the medial tibial plateau (B) Subchondral bone plate on the medial tibial plateau (C) Microstructure of the cancellous bone in distal femur (D) Tb. Th of the cancellous bone in the distal femur (E) Tb. Sp of the cancellous bone in the distal femur. Note: The red dotted square shows the areas with significant differences in the representative μ-CT images of each group. N=8, **P<0.01, ***P<0.001 vs Sham; #P<0.05, ##P<0.01, ###P<0.001 vs OA. Values are presented as mean ± SD. |
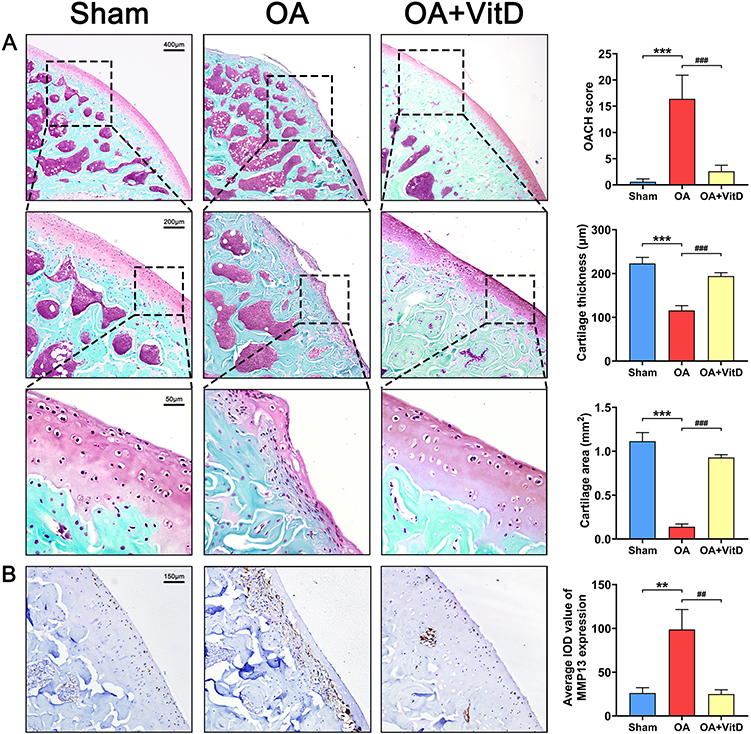 |
Figure 4 (A) Representative images of safranin O fast green staining of knee joint in OA rats treated with 2.34 μg/kg/day VitD and the corresponding quantitative analysis, N=8. (B) Representative images of immunohistochemical staining for MMP13 of knee joint in OA rats treated with 2.34 μg/kg/day VitD and the corresponding quantitative analysis, N=8. Note: **P<0.01, ***P<0.001 vs Sham; ##P<0.01, ###P<0.001 vs OA. Values are presented as mean ± SD. |
VitD Demonstrated Potent Chondroprotective Effects in in vivo Models of Osteoarthritis
In the MIA-induced rat OA model, therapeutic administration of VitD resulted in macroscopic improvement of articular cartilage integrity, evidenced by restoration of smooth articular surfaces, significant mitigation of subchondral bone damage, suggesting disease-modifying potential through dual cartilage-bone protective mechanisms (Figure 1A). During the intragastric administration of VitD, there were no significant differences in the weight of the rats across the various groups (Figure 1B).
Furthermore, VitD treatment elicited significant improvements in OA pathology, as evidenced by a 39.13% reduction in histomorphometry lesion severity scores compared to untreated OA controls (P<0.001) (Figure 1C). Serum biomarker analyses demonstrated significant VitD-mediated downregulation of multiple pathogenic mediators (MMP13, IL-6, CD31, COL10A, CTX-I and PINP) in OA rats (Figure 2). μ-CT quantification demonstrated that VitD treatment exerted significant structural modifications in OA rats, including: (1) 64.50% augmentation of tibial plateau cartilage thickness (Figure 3A); (2) a 22.35% reduction in subchondral bone plate thickness (P<0.05) (Figure 3B); and (3) marked improvements in femoral trabecular microarchitecture, evidenced by a 61.81% increase in bone volume fraction (BV/TV, P<0.001) (Figure 3C), 36.89% elevation in trabecular thickness (Tb.Th, P<0.001) (Figure 3D) and 37.79% decrease in trabecular separation (Tb.Sp, P<0.01, Figure 3E). Histopathological evaluation demonstrated that the OACH score in VitD-treated rats was significantly reduced by 84.15% (P<0.001) (Figure 4A). Cartilage thickness increased by 67.69% (P<0.001), while the cartilage area expanded by 567.76% (Figure 4A) (P<0.001). Immunohistochemical analysis confirmed the therapeutic efficacy of VitD, showing a 74.72% downregulation of MMP13 expression in articular cartilage compared to untreated OA controls (P<0.01) (Figure 4B).
VitD Demonstrated Potent Chondroprotective Effects in Inflammatory Chondrocytes and is Associated with the Inhibition of Myd88-TAK1-ERK Signaling Axis
Following exposure of C28/I2 chondrocytes to graded concentrations of VitD for over 24–72 hours, VitD treatment effectively counteracted the cytotoxic effects of 0.1% ethanol vehicle (Figure 5A). Quantitative cell viability assessment (CCK-8 assay) identified 10−2 μM as the optimal therapeutic concentration for subsequent mechanistic studies. Immunoblotting analysis revealed TNF-α-mediated activation of pro-inflammatory signaling cascades, evidenced by 74.33% upregulation of myeloid differentiation primary response 88 (MyD88, P<0.05); 105.37% increase in phosphorylated ERK1/2 (p-ERK1/2, P<0.01); 125.53% increase in activator protein-1 (AP-1, P<0.01); 109.03% induction of nuclear factor kappa-B (NF-κB p65, P<0.001). VitD intervention potently suppressed these inflammatory mediators, achieving 31.22% reduction in MyD88 expression; 66.11% (P<0.01) decrease in ERK1/2 phosphorylation; 61.43% (P<0.001) decrease in AP-1; 34.36% (P<0.001) downregulation of NF-κB activation (vs TNF-α group, Figure 5 B&C). Transcriptional RT-qPCR profiling demonstrated VitD’s chondroprotective effects through significant upregulation of collagen type II α1 (Col2a1, P<0.01), SRY-box transcription factor 9 (SOX9, P<0.05), and suppression of TGF-β activated kinase 1 (TAK1, P<0.05) (Figure 5D).
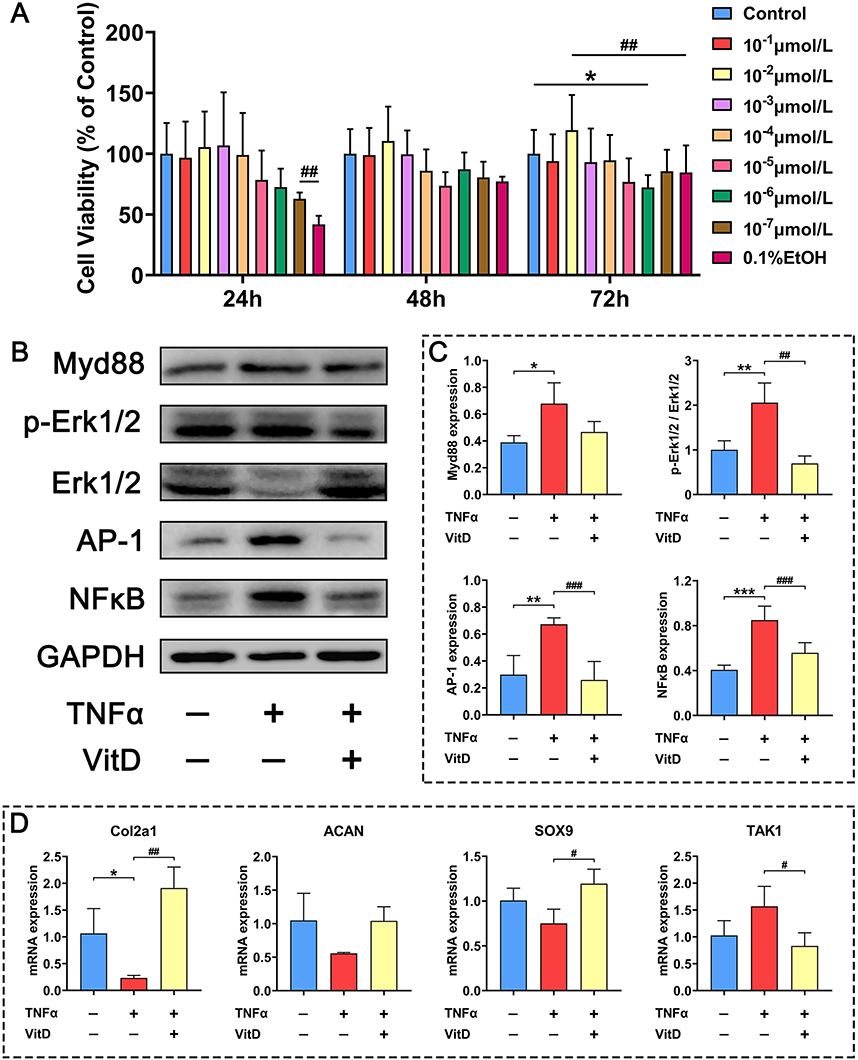 |
Figure 5 Regulation of 10−2μM VitD on Myd88-TAK1-ERK signal axis in C28 chondrocytes induced by 1ng/mL TNFα. (A) Effects of various concentrations of VitD on the viability of C28 chondrocytes, N=6. (B) Western blots show effects of 10−2μM VitD on protein levels of Myd88, p-ERK1/2, AP-1 and NFκB in C28 chondrocytes induced by 1ng/mL TNFα for 72h. (C) Protein levels of Myd88, p-ERK1/2, AP-1 and NFκB, N=5. (D) RT-qPCR show effects of 10−2μM VitD on gene expression of Col2a1, ACAN, SOX9 and TAK1 in C28 chondrocytes induced by 1ng/mL TNFα for 72h, N=6. Note: *P<0.05, **P<0.01, ***P<0.001 vs Control; #P<0.05, ##P<0.01, ###P<0.001 vs TNFα. Values are presented as mean ± SD. |
In summary, these in vitro results establish that VitD enhances chondrocyte viability under inflammatory stress, preserves cartilage-specific gene expression profiles and exerts anti-inflammatory and chondroprotective effects via suppression of the MyD88/TAK1/ERK/NF-κB signaling axis.
Discussion
Our study demonstrated that MIA exhibited typical pathological OA changes in rats. These included cartilage damage on the femoral surface, thinning of the cartilage thickness, thickening of the subchondral bone plate, degradation of the bone microstructure at the distal femur, and upregulation of MMP-13 expression in both serum and cartilage on the femoral surface. Collectively, these findings confirm the successful establishment of an OA rat model.
In this study, our findings establish VitD as a potential disease-modifying osteoarthritis drug candidate, exhibiting dual therapeutic efficacy in monosodium iodoacetate-induced osteoarthritis. VitD treatment (2.34 μg/kg/day for 6 weeks) alleviated disease progression by reducing cartilage degradation, evidenced by a 39.13% reduction in OACH scores, a 64.50% increase in tibial cartilage thickness, 74.72% decrease in MMP13, and enhanced subchondral bone remodeling (VitD increased Tb. Th by 36.89% and deceased separation by 37.79%) while normalizing bone plate thickness. Mechanistically, VitD disrupted the Myd88-TAK1-ERK inflammatory cascade, decreasing Myd88, TAK1, and p-ERK1/2 activity, leading to a significant inhibition of NFκB activity by 34.36%. This signaling modulation restored chondrogenic factors, including Col II, Aggrecan, and Sox9. Histological results synergistically match these data, showing improved cartilage repair and reduced MMP-13 expression, along with increased cartilage thickness and area. In vitro, VitD reversed the reduction in C28 cell viability induced by EtOH and mitigated TNFα-induced chondrocyte inflammatory injury by upregulating Col2a1, ACAN, and SOX9 expression, while downregulating TAK1, Myd88, p-ERK1/2, AP-1, and NFκB. These findings establish VitD’s disease-modifying potential through coordinated targeting of cartilage catabolism and aberrant bone remodeling, providing a mechanistic foundation for clinical translation in OA management.
Vitamin D is a lipid-soluble vitamin that primarily includes two forms: vitamin D2 and vitamin D3. Among these, vitamin D3 exhibits higher biological activity and serves as the primary form utilized by the human body. Vitamin D3 undergoes hepatic 25-hydroxylation and subsequent renal 1α-hydroxylation to form its biologically active metabolite, 1,25-dihydroxyvitamin D3, a potent endocrine regulator of calcium-phosphate homeostasis.
As an essential nutritional factor, vitamin D not only supports bone health20 and helps alleviate muscle atrophy,21,22 but also maintains glucose metabolism homeostasis23,24 through increasing secretion of insulin and suppressing inflammation, thereby mitigating type 2 diabetes mellitus (T2DM) risk.25 Our previous study demonstrated that VitD ameliorates osteopenia and enhances musculoskeletal integrity in Goto-Kakizaki rats (T2DM model).10 In addition, previous studies demonstrated that VitD could decrease the risk of OA. Specifically, VitD has been shown to alleviate OA symptoms by regulating autophagy,26 reducing inflammation,27 inhibiting senescence28 and inhibiting the expression of matrix metalloproteinases (MMPs).29
In our study, our experimental data extend these observations by demonstrating that VitD Exerts disease-modifying effects in osteoarthritis through in vitro provide chondroprotective effects in inflammatory chondrocytes and in vivo preservation of cartilage and joint architecture. Further data revealed that VitD mechanistically targets the MyD88/TAK1/ERK signaling axis, representing a novel therapeutic axis for OA intervention.
Myeloid differentiation primary response 88 (MyD88) serves as a pivotal adaptor molecule that orchestrates inflammatory signaling cascades initiated by Toll-like receptors (TLRs) and interleukin-1 receptors (IL-1Rs).30 MyD88’s death domain mediates interactions with interleukin-1 receptor-associated kinases (IRAKs), facilitating sequential recruitment and activation of IRAK4 and IRAK1/IRAK,31,32 forming the myddosome complex, a critical signaling platform.33
Myddosome recruits and activates TRAF634 through phosphorylated IRAK1/IRAK2, forming a cascade of signal transmission and subsequent phosphorylation of TGF-β-activated kinase 1 (TAK1). TAK1 initiated TAK1-TAB1/2/3 complex formation in response to various stimuli, including Pro-inflammatory cytokines (TNF-α, IL-1β) and toll-like receptor ligands.35 This process ultimately triggers the activation of both the NFκB signaling pathway36 and the MAPK signaling pathway.37 Disruption of MyD88 dimerization suppresses synovitis inflammation and attenuates inflammatory arthritis progression.38 Furthermore, as the central signalosome in inflammatory responses, the down-regulation of TAK1 kinase inhibits MAPK cascade (Phosphorylation of JNK, p38, ERK1/2, ATF-2, and c-Jun); Suppress NF-κB pathway: IKK-mediated IκBα degradation to activate p65/p50 nuclear translocation.35,39 Notably, the inhibition of NFκB and ERK maintains cartilage homeostasis, suppresses osteoclast activity in subchondral bone,40 reduces osteochondral pathology, mitigates subchondral bone remodeling.41
Our mechanistic investigations demonstrate that the therapeutic efficacy of VitD on OA is closely associated with its ability to inhibit the Myd88-TAK1-ERK signaling axis. Specifically, VitD primarily exerts its protective role by suppressing inflammatory signal transmission, thereby preventing cartilage degradation. This conclusion is further supported by our results showing a decrease in NFκB protein expression in C28 inflammatory cells and a reduction in MMP13 levels in the cartilage of OA rats following VitD intervention.
Emerging evidence has implicated the MyD88-TAK1 signaling cascade in various inflammatory pathologies (cervical cancer,42 neurons43 and the intestinal barrier44), with therapeutic modulation of this pathway demonstrating efficacy in diverse conditions including cervical cancer (via TLR4-MyD88-TAK1 axis). Additionally, studies have explored its relevance to the treatment of prostatic hyperplasia,45 sepsis46 and pulpitis,47 which are linked to the TAK1-ERK signaling pathway.
Notably, recent work has identified extracellular histones as mediators of trauma-induced inflammation through a MyD88-IRAK1-ERK dependent mechanism, triggering lytic cell death of adipocyte.48 However, despite these advances, the potential role of vitamin D3 in osteoarthritis management through selective inhibition of the MyD88-TAK1-ERK signaling axis remains underexplored, representing a critical gap in our understanding of its chondroprotective mechanisms.
Our study specifically identifies VitD’s pivotal role in suppressing OA progression, establishes VitD as a selective pathway modulator by inhibiting the Myd88-TAK1-ERK axis, providing a solid theoretical foundation to support its clinical application in OA treatment. The clinical translation of VitD in OA management may be further advanced by its therapeutic potential and ease of integration into existing treatment strategies.
a) NSAID-Sparing Effects. Emerging evidence suggests that vitamin D exerts anti-inflammatory effects that provide a synergistic anti-inflammatory effect49 and may reduce dependence on NSAIDs—an important consideration given the gastrointestinal, cardiovascular, and renal risks associated with long-term NSAID use in older adults. VitD suppress COX-2 expression50 and modulates prostaglandin synthesis pathways,51 which reducing reliance on NSAIDs and its associated gastrointestinal and cardiovascular risks while maintaining anti-inflammatory efficacy. In a randomized, double-blind placebo-controlled study, high-dose vitamin D supplementation to analgesic regimens led to faster significant reductions in VAS scores and pro-inflammatory cytokines in patients with musculoskeletal pain, suggesting additive analgesic effects when used with standard therapies.51 Additionally, low serum VitD levels have been correlated with higher NSAID consumption in patients with inflammatory arthritis, further supporting this link.52
b) Augmentation of Physical Therapy. Several studies have demonstrated synergistic benefits when VitD is combined with physical rehabilitation. A clinical trial found that combining vitamin D supplementation with physiotherapy significantly improved pain outcomes in patients with musculoskeletal disorders compared to physiotherapy alone.53 A systematic review and meta-analysis concluded that combined vitamin D and resistance exercise improved lower limb muscle strength, although additional benefits in mobility indices (eg, SPPB, TUG) remain inconclusive.54 These data suggest VitD could potentiate the efficacy of rehabilitative therapies in OA by improving muscle function and reducing inflammation-induced pain.
c) Cartilage Protection and Disease Modification. At the cellular level, vitamin D exhibits cartilage-protective effects through multiple mechanisms. In vitro, 1,25(OH)₂D₃ inhibits TNF-α-induced MMP (MMP-1, −2, −3, −9, and −13) expression and diminish the cytotoxic effect of TNF-α, restores levels of protective mediators like TIMP-1, TIMP-2, and VDR by dose-dependent in human chondrocytes.55 In animal OA models, vitamin D supplementation ameliorated joint inflammation, cartilage degradation, and pain, with additional evidence supporting enhanced autophagic flux and attenuation of inflammatory cell death. Interestingly, the expressions of MMP-13, IL-1β, and MCP-1 in synovial tissues were remarkably attenuated by vitamin D treatment. Vitamin D and celecoxib showed a synergistic effect on antinociceptive and chondroprotective properties in vivo.27 Vitamin D induces chondrocyte autophagy and reduces the loss of proteoglycans in OA, further investigation reveals VitD’s protective actions appear to involve modulation of the NF-κB/AMPK/mTOR axis, offering a mechanistic basis for disease-modifying effects in OA.56
The clinical translation of vitamin D may also warrant more thorough clinical evaluation. Emerging observational data suggest that vitamin D deficiency is associated with the onset and progression of osteoarthritis.57 While promising, vitamin D supplementation should be considered with attention to baseline serum levels.
1,25-Dihydroxyvitamin D deficiency exacerbates aging-related osteoarthritis through Sirtuin 1 (Sirt1) downregulation in murine models. Mechanistically, 1,25(OH)2D3 acts via vitamin D receptor (VDR)-mediated transcriptional regulation to attenuate age-associated knee OA pathogenesis by upregulating Sirt1, an aging-related gene that critical for maintaining articular cartilage homeostasis. This regulatory axis promotes chondrocyte proliferation, enhances extracellular matrix (ECM) protein synthesis, and suppresses cellular senescence and senescence-associated secretory phenotype (SASP).28
Clinically, a prospective cohort analysis of vitamin D-deficient participants (<50 nmol/L serum 25(OH)D) revealed a reduction in NSAID prescription risk among those receiving vitamin D supplementation versus placebo (RR=0.87; p=0.009).58 These findings underscore the necessity of pre-treatment serum 25(OH)D quantification to stratify OA patients, as therapeutic responses to vitamin D supplementation demonstrate significant dose-dependent efficacy in individuals with hypovitaminosis D. Current evidence suggests vitamin D repletion (>40 ng/mL) in deficient populations may potentiate NSAID-sparing effects while improving functional outcomes in early-stage OA.
While VitD and other current OA drug synergies are promising, possible pharmacokinetic interactions, especially for drugs metabolized via CYP3A4, warrant further study to ensure safe co-administration.59
Cost-Effectiveness, Affordability, and Accessibility VitD’s cost-effectiveness is also the advantages when compared to other medicines. From a pharmacoeconomic perspective, vitamin D is a compelling candidate for OA management due to:
a) Affordability. Vitamin D supplements are low-cost and widely available over-the-counter, with costs significantly lower than biologics, intra-articular injections, or long-term NSAID therapy.
b) Global Accessibility. Unlike injectable biologics that require refrigeration and healthcare infrastructure, VitD supplements are shelf-stable at ambient temperatures and accessible in nearly all WHO member states. This confers a distinct implementation advantage, particularly in low-resource settings or rural areas.
c) Ease of Use and Adherence. Vitamin D’s favorable safety profile and simple dosing regimen (typically daily or weekly oral dosing) support higher adherence rates than many OA treatments. Public awareness campaigns around osteoporosis prevention have also indirectly increased compliance with VitD supplementation in aging populations.
In summary, vitamin D represents a biologically active, cost-effective, and implementation-feasible adjunct to existing OA therapies. Its capacity to synergize with NSAIDs, physiotherapy, and exercise—coupled with its global accessibility—positions VitD as a promising component of integrated OA management, particularly for at-risk or resource-limited populations.
While our research primarily focuses on utilizing animal experiments and molecular biology to assess the potential of vitamin D in treating osteoarthritis and to investigate its underlying molecular mechanisms, we noticed that the mixed results observed in clinical trials involving vitamin D. Two randomized clinical trial showed no significant benefits of vitamin D supplementation on cartilage preservation or pain relief in established knee OA when compared with placebo.60,61 However, we also found stratified analyses and post hoc investigations suggesting that maintains sufficient vitamin D levels could be an effective strategy to alleviate OA progression and joint pain. Post-analysis of the results revealed that OA patients who maintained sufficient levels of serum vitamin D exhibited less severe pathological conditions compared to those with persistent deficiency.13 A post hoc study also emphasized that vitamin D supplementation and maintenance of sufficient vitamin D levels improved foot pain in those with knee OA.62 Vitamin D is associated with reduced pain in male patients with KOA.63 The results showed that patients with KOA had lower levels of vitamin D and higher levels of IL-1β, TNF-α, and NF-κB p65 compared with healthy controls. Patients with sufficient vitamin D levels had lower total and physical function WOMAC scores compared with patients with vitamin D insufficiency.64 Some interventional studies also demonstrated that vitamin D supplementation alleviate cancer pain and muscular pain—but only in patients with insufficient levels of vitamin D when starting intervention.65
Consequently, it appears that the clinical therapeutic effect of vitamin D in OA is closely linked to the initial levels of vitamin D in patients. Vitamin D may potential benefits more in early disease and prevention in OA. It is unequivocal that low levels of vitamin D significantly elevate the risk of various diseases, particularly those associated with musculoskeletal health. Furthermore, recent studies have identified the primary risk factors contributing to low vitamin D levels in the general population, which include inadequate environmental ultraviolet radiation, insufficient dietary intake of vitamin D, and genetic polymorphisms.66 Given that the elderly population faces a higher risk of bone tissue diseases and that diminish ability of synthesize vitamin D, taking vitamin D supplements can significantly benefit the improvement of bone tissue metabolic disorders.67 Although individual differences and various environmental factors contribute to the variability in the therapeutic effects of vitamin D, this does not diminish its many benefits. Given the complexity of the pathogenesis of osteoarthritis and its close relationship with the steady state of bone metabolism, it is essential to consider additional influencing factors when evaluating the therapeutic potential of vitamin D for osteoarthritis. To further advance, more in-depth research of the application of vitamin D in osteoarthritis is necessary in the future.
To propel the clinical translation of vitamin D in osteoarthritis management, future research needs to go more in-depth on some critical domains, for example, 1. Precision Dosing Optimization; 2. Combinatorial trials with physical therapy/NSAIDs.
Limitations: First, while our findings demonstrate that vitamin D (VitD) mitigates OA progression through inhibition of the Myd88-TAK1-ERK signaling axis, the precise regulatory mechanisms remain incompletely resolved. Definitive validation of signaling cascade dynamics requires complementary approaches, including in vivo gene knockout models or administration of pathway-specific inhibitors targeting critical nodal components. Second, the VitD dosage employed in this study was extrapolated from clinically recommended human levels,15,16 rather than empirically optimized through preclinical dose-response studies. Consequently, the potential existence of a therapeutic dose-dependent relationship between VitD and OA amelioration remains undefined. Third, although Western blot analyses provided initial evidence supporting VitD-mediated OA alleviation, these findings should be interpreted as preliminary due to limited biological replicates. Future investigations incorporating experiments with expanded samples are required to establish the pathophysiological significance of the observed molecular alterations.
Conclusion
In summary, our findings reveal VitD as a dual-target OA therapeutic with disease-modifying capacity, operating through suppression of the Myd88-TAK1-ERK cascade to concurrently mitigate cartilage catabolism and aberrant subchondral remodeling. While this study mechanistically validates VitD’s multiple pharmacological advantages over conventional OA therapies, future work will delineate dose-response relationships, therapeutic thresholds, and molecular dynamics of VitD-receptor interactions to optimize clinical translation. By repositioning VitD from nutritional adjunct to mechanism-driven OA intervention, this work bridges the gap between observational correlations and actionable therapeutic strategies, offering a cost-effective blueprint for multifactorial joint preservation.
Abbreviations
OA, Osteoarthritis; VitD, Vitamin D; MIA, Monosodium iodoacetate; OACH, Osteoarthritis cartilage histopathology; CD31, platelet endothelial cell adhesion molecule-1; COL10A1, Collagen type X; IL-6, Interleukin 6; PINP, N-terminal propeptide of procollagen type I; CTX-I, C-telopeptide of type I collagen; MMP13, Matrix metalloproteinase 13; BV/TV, Bone volume/Tissue volume; Tb. Th, Trabecular thickness; Tb. Sp, Trabecular separation.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was supported by the National Natural Science Foundation of China (81703584), Guangdong Province Natural Science Foundation of China (No. 2022A1515220166, 2025A1515010196, 2023A1515011091, 2021B1515140054, 2022A1515140138), the Science and Technology Foundation of Zhanjiang (2022A01163, 2022A01099, 2022A01170), Discipline construction project of Guangdong Medical University (CLP2021B012, FYZM001), the Discipline Construction Fund of Central People’s Hospital of Zhanjiang (No. 2022A09), Scientific research project of Guangdong Provincial Bureau of Traditional Chinese Medicine (20245577, 20250047, 20221445).
Disclosure
The authors declare that there are no conflicts of interest in this work.
References
1. Yue L, Berman J. What is osteoarthritis? JAMA. 2022;327(13):1300. doi:10.1001/jama.2022.1980
2. Palazzo C, Nguyen C, Lefevre-Colau MM, Rannou F, Poiraudeau S. Risk factors and burden of osteoarthritis. Ann Phys Rehabil Med. 2016;59(3):134–138. doi:10.1016/j.rehab.2016.01.006
3. Richette P, Latourte A, Frazier A. Safety and efficacy of paracetamol and NSAIDs in osteoarthritis: which drug to recommend? Expert Opin Drug Saf. 2015;14(8):1259–1268. doi:10.1517/14740338.2015.1056776
4. de l’Escalopier N, Anract P, Biau D. Surgical treatments for osteoarthritis. Ann Phys Rehabil Med. 2016;59(3):227–233. doi:10.1016/j.rehab.2016.04.003
5. Chen L, Zhang Z, Liu X. Role and mechanism of mechanical load in the homeostasis of the subchondral bone in knee osteoarthritis: a comprehensive review. J Inflamm Res. 2024;17:9359–9378. doi:10.2147/jir.S492415
6. Chu L, Liu X, He Z, et al. Articular cartilage degradation and aberrant subchondral bone remodeling in patients with osteoarthritis and osteoporosis. J Bone Mineral Res. 2020;35(3):505–515. doi:10.1002/jbmr.3909
7. Chen Y, Hu Y, Yu YE, et al. Subchondral trabecular rod loss and plate thickening in the development of osteoarthritis. J Bone Mineral Res. 2018;33(2):316–327. doi:10.1002/jbmr.3313
8. Yang R, Chen J, Zhang J, et al. 1,25-dihydroxyvitamin D protects against age-related osteoporosis by a novel VDR-Ezh2-p16 signal axis. Aging Cell. 2020;19(2):e13095. doi:10.1111/acel.13095
9. Liu C, Kuang X, Li K, Guo X, Deng Q, Li D. Effects of combined calcium and vitamin D supplementation on osteoporosis in postmenopausal women: a systematic review and meta-analysis of randomized controlled trials. Food Funct. 2020;11(12):10817–10827. doi:10.1039/d0fo00787k
10. Liang Y, Liu Y, Lai W, et al. 1,25-Dihydroxy vitamin D3 treatment attenuates osteopenia, and improves bone muscle quality in Goto-Kakizaki type 2 diabetes model rats. Endocrine. 2019;64(1):184–195. doi:10.1007/s12020-019-01857-5
11. van Hamburg JP, Asmawidjaja PS, Davelaar N, et al. TNF blockade requires 1,25(OH)2D3 to control human Th17-mediated synovial inflammation. Ann Rheumatic Dis. 2012;71(4):606–612. doi:10.1136/annrheumdis-2011-200424
12. Nakajo T, Katayoshi T, Kitajima N, Tsuji-Naito K. 1,25-Dihydroxyvitamin D(3) attenuates IL-1β secretion by suppressing NLRP1 inflammasome activation by upregulating the NRF2-HO-1 pathway in epidermal keratinocytes. Redox Biol. 2021;48:102203. doi:10.1016/j.redox.2021.102203
13. Zheng S, Jin X, Cicuttini F, et al. Maintaining vitamin D sufficiency is associated with improved structural and symptomatic outcomes in knee osteoarthritis. Am J Med. 2017;130(10):1211–1218. doi:10.1016/j.amjmed.2017.04.038
14. Yang W, Sun C, He SQ, Chen JY, Wang Y, Zhuo Q. The efficacy and safety of disease-modifying osteoarthritis drugs for knee and hip osteoarthritis-a systematic review and network meta-analysis. J Gen Intern Med. 2021;36(7):2085–2093. doi:10.1007/s11606-021-06755-z
15. Uchitomi R, Oyabu M, Kamei Y. Vitamin D and sarcopenia: potential of vitamin D supplementation in sarcopenia prevention and treatment. Nutrients. 2020;12(10):3189. doi:10.3390/nu12103189
16. Chevalley T, Brandi ML, Cashman KD, et al. Role of vitamin D supplementation in the management of musculoskeletal diseases: update from an European Society of Clinical and Economical Aspects of Osteoporosis, Osteoarthritis and Musculoskeletal Diseases (ESCEO) working group. Aging Clin Exp Res. 2022;34(11):2603–2623. doi:10.1007/s40520-022-02279-6
17. Kwon M, Nam D, Kim J. Pathological characteristics of monosodium iodoacetate-induced osteoarthritis in rats. Tissue Eng Regen Med. 2023;20(3):435–446. doi:10.1007/s13770-023-00520-5
18. Wang CJ, Cheng JH, Chou WY, Hsu SL, Chen JH, Huang CY. Changes of articular cartilage and subchondral bone after extracorporeal shockwave therapy in osteoarthritis of the knee. Int J Med Sci. 2017;14(3):213–223. doi:10.7150/ijms.17469
19. Pearson RG, Kurien T, Shu KS, Scammell BE. Histopathology grading systems for characterisation of human knee osteoarthritis--reproducibility, variability, reliability, correlation, and validity. Osteoarthritis Cartilage. 2011;19(3):324–331. doi:10.1016/j.joca.2010.12.005
20. Bouillon R, Marcocci C, Carmeliet G, et al. Skeletal and extraskeletal actions of vitamin D: current evidence and outstanding questions. Endocrine Rev. 2019;40(4):1109–1151. doi:10.1210/er.2018-00126
21. Cheung WW, Hao S, Wang Z, et al. Vitamin D repletion ameliorates adipose tissue browning and muscle wasting in infantile nephropathic cystinosis-associated cachexia. J Cachexia Sarcopenia Muscle. 2020;11(1):120–134. doi:10.1002/jcsm.12497
22. Bollen SE, Bass JJ, Fujita S, Wilkinson D, Hewison M, Atherton PJ. The Vitamin D/Vitamin D receptor (VDR) axis in muscle atrophy and sarcopenia. Cell Signalling. 2022;96:110355. doi:10.1016/j.cellsig.2022.110355
23. Wu J, Atkins A, Downes M, Wei Z. Vitamin D in diabetes: uncovering the sunshine hormone’s role in glucose metabolism and beyond. Nutrients. 2023;15(8). doi:10.3390/nu15081997
24. Szymczak-Pajor I, Drzewoski J, Śliwińska A. The molecular mechanisms by which vitamin d prevents insulin resistance and associated disorders. Int J Mol Sci. 2020;21(18). doi:10.3390/ijms21186644
25. Zhang Y, Tan H, Tang J, et al. Effects of vitamin D supplementation on prevention of type 2 diabetes in patients with prediabetes: a systematic review and meta-analysis. Diabetes Care. 2020;43(7):1650–1658. doi:10.2337/dc19-1708
26. Saengsiwaritt W, Ngamtipakon P, Udomsinprasert W. Vitamin D and autophagy in knee osteoarthritis: a review. Int Immunopharmacol. 2023;123:110712. doi:10.1016/j.intimp.2023.110712
27. Jhun J, Woo JS, Kwon JY, et al. Vitamin D attenuates pain and cartilage destruction in OA animals via enhancing autophagic flux and attenuating inflammatory cell death. Immun Net. 2022;22(4):e34. doi:10.4110/in.2022.22.e34
28. Chen J, Li J, Zhang J, et al. 1,25-dihydroxyvitamin D deficiency accelerates aging-related osteoarthritis via downregulation of sirt1 in mice. Int J Bio Sci. 2023;19(2):610–624. doi:10.7150/ijbs.78785
29. Busa P, Huang N, Kuthati Y, Wong CS. Vitamin D reduces pain and cartilage destruction in knee osteoarthritis animals through inhibiting the matrix metalloprotease (MMPs) expression. Heliyon. 2023;9(4):e15268. doi:10.1016/j.heliyon.2023.e15268
30. Deguine J, Barton GM. MyD88: a central player in innate immune signaling. F1000prime Rep. 2014;6:97. doi:10.12703/p6-97
31. Yadav H, Shirumalla RK. Emerging trends in IRAK-4 kinase research. Mol Biol Rep. 2023;50(9):7825–7837. doi:10.1007/s11033-023-08438-w
32. Pereira M, Gazzinelli RT. Regulation of innate immune signaling by IRAK proteins. Front Immunol. 2023;14:1133354. doi:10.3389/fimmu.2023.1133354
33. Balka KR, De Nardo D. Understanding early TLR signaling through the Myddosome. J Leukocyte Biol. 2019;105(2):339–351. doi:10.1002/jlb.Mr0318-096r
34. Muroi M, Tanamoto K. TRAF6 distinctively mediates MyD88- and IRAK-1-induced activation of NF-kappaB. J Leukocyte Biol. 2008;83(3):702–707. doi:10.1189/jlb.0907629
35. Xu YR, Lei CQ. TAK1-TABs complex: a central signalosome in inflammatory responses. Front Immunol. 2020;11:608976. doi:10.3389/fimmu.2020.608976
36. Cao F, Deliz-Aguirre R, Gerpott FH, Ziska E, Taylor MJ. Myddosome clustering in IL-1 receptor signaling regulates the formation of an NF-kB activating signalosome. EMBO Rep. 2023;24(10):e57233. doi:10.15252/embr.202357233
37. Jia A, James E, Lu HY, et al. Clinical IRAK4 deficiency caused by homozygosity for the novel IRAK4 (c.1049delG, p.Gly350Glufs*15) variant. Cold Spring Harbor Mol Case Stud. 2020;6(3). doi:10.1101/mcs.a005298
38. Ramirez-Perez S, Vekariya R, Gautam S, Reyes-Perez IV, Drissi H, Bhattaram P. MyD88 dimerization inhibitor ST2825 targets the aggressiveness of synovial fibroblasts in rheumatoid arthritis patients. Arthritis Res Ther. 2023;25(1):180. doi:10.1186/s13075-023-03145-0
39. Endale M, Kim TH, Kwak YS, et al. Torilin inhibits inflammation by limiting TAK1-mediated MAP kinase and NF-κB ACTIVATION. Mediators Inflammation. 2017;2017:7250968. doi:10.1155/2017/7250968
40. Zhu M, Xu Q, Yang X, et al. Vindoline attenuates osteoarthritis progression through suppressing the NF-κB and ERK pathways in both chondrocytes and subchondral osteoclasts. Front Pharmacol. 2021;12:764598. doi:10.3389/fphar.2021.764598
41. Jiang T, Gong Y, Zhang W, et al. PD0325901, an ERK inhibitor, attenuates RANKL-induced osteoclast formation and mitigates cartilage inflammation by inhibiting the NF-κB and MAPK pathways. Bioorg Chem. 2023;132:106321. doi:10.1016/j.bioorg.2022.106321
42. He A, Ji R, Shao J, He C, Jin M, Xu Y. TLR4-MyD88-TRAF6-TAK1 complex-mediated NF-κB activation contribute to the anti-inflammatory effect of V8 in LPS-induced human cervical cancer SiHa cells. Inflammation. 2016;39(1):172–181. doi:10.1007/s10753-015-0236-8
43. Chen S, Xiong J, Zhan Y, Liu W, Wang X. Wogonin inhibits LPS-induced inflammatory responses in rat dorsal root ganglion neurons via inhibiting TLR4-MyD88-TAK1-mediated NF-κB and MAPK signaling pathway. Cell Mol Neurobiol. 2015;35(4):523–531. doi:10.1007/s10571-014-0148-4
44. Wang W, Xia T, Yu X. Wogonin suppresses inflammatory response and maintains intestinal barrier function via TLR4-MyD88-TAK1-mediated NF-κB pathway in vitro. Inflamm Res. 2015;64(6):423–431. doi:10.1007/s00011-015-0822-0
45. Jin BR, Ju JY, Nugroho A, Lee M, An HJ. Carica papaya leaf extract inhibits prostatitis-associated prostatic hyperplasia via the TRAF6/TAK1/MEK/NF-κB pathway. Biomed Pharmacothe. 2021;135:111197. doi:10.1016/j.biopha.2020.111197
46. Xie W, Li H, Yu T, et al. Design and synthesis of hederagenin derivatives for the treatment of sepsis by targeting TAK1 and regulating the TAK1-NF-κB/MAPK signaling. J Med Chem. 2025;68(3):2694–2719. doi:10.1021/acs.jmedchem.4c02032
47. Lin SI, Lin LD, Chang HH, et al. IL-1β induced IL-8 and uPA expression/production of dental pulp cells: role of TAK1 and MEK/ERK signaling. J Formosan Med Assoc. 2018;117(8):697–704. doi:10.1016/j.jfma.2018.04.003
48. Roos J, Zinngrebe J, Huber-Lang M, et al. Trauma-associated extracellular histones mediate inflammation via a MYD88-IRAK1-ERK signaling axis and induce lytic cell death in human adipocytes. Cell Death Dis. 2024;15(4):285. doi:10.1038/s41419-024-06676-9
49. Thill M, Reichert K, Woeste A, et al. Combined treatment of breast cancer cell lines with vitamin D and COX-2 inhibitors. Anticancer Res. 2015;35(2):1189–1195.
50. Wang Q, He Y, Shen Y, et al. Vitamin D inhibits COX-2 expression and inflammatory response by targeting thioesterase superfamily member 4. J Biol Chem. 2014;289(17):11681–11694. doi:10.1074/jbc.M113.517581
51. Gendelman O, Itzhaki D, Makarov S, Bennun M, Amital H. A randomized double-blind placebo-controlled study adding high dose vitamin D to analgesic regimens in patients with musculoskeletal pain. Lupus. 2015;24(4–5):483–489. doi:10.1177/0961203314558676
52. Furuya T, Hosoi T, Tanaka E, et al. Prevalence of and factors associated with vitamin D deficiency in 4793 Japanese patients with rheumatoid arthritis. Clin Rheumatol. 2013;32(7):1081–1087. doi:10.1007/s10067-013-2216-4
53. Ali M, Uddin Z, Hossain A. Combined effect of vitamin D supplementation and physiotherapy on reducing pain among adult patients with musculoskeletal disorders: a quasi-experimental clinical trial. Front Nutr. 2021;8:717473. doi:10.3389/fnut.2021.717473
54. Antoniak AE, Greig CA. The effect of combined resistance exercise training and vitamin D(3) supplementation on musculoskeletal health and function in older adults: a systematic review and meta-analysis. BMJ Open. 2017;7(7):e014619. doi:10.1136/bmjopen-2016-014619
55. Avcioglu G, Özbek Ipteç B, Akcan G, et al. Effects of 1,25-Dihydroxy vitamin D(3) on TNF-α induced inflammation in human chondrocytes and SW1353 cells: a possible role for toll-like receptors. Mol Cell Biochem. 2020;464(1–2):131–142. doi:10.1007/s11010-019-03655-z
56. Liu P, Zhou J, Cui H, et al. 1,25(OH)(2)D(3) induces chondrocyte autophagy and reduces the loss of proteoglycans in osteoarthritis through inhibiting the NF-κB pathway. Clin Rheumatol. 2025;44(2):811–822. doi:10.1007/s10067-024-07281-z
57. Shen M, Luo Y, Niu Y, et al. 1,25(OH)2D deficiency induces temporomandibular joint osteoarthritis via secretion of senescence-associated inflammatory cytokines. Bone. 2013;55(2):400–409. doi:10.1016/j.bone.2013.04.015
58. Wu Z, Camargo CA Jr, Malihi Z, et al. Monthly vitamin D supplementation, pain, and pattern of analgesic prescription: secondary analysis from the randomized, double-blind, placebo-controlled Vitamin D Assessment study. Pain. 2018;159(6):1074–1082. doi:10.1097/j.pain.0000000000001189
59. Robien K, Oppeneer SJ, Kelly JA, Hamilton-Reeves JM. Drug-vitamin D interactions: a systematic review of the literature. Nutr Clin Prac. 2013;28(2):194–208. doi:10.1177/0884533612467824
60. Jin X, Jones G, Cicuttini F, et al. Effect of vitamin D supplementation on tibial cartilage volume and knee pain among patients with symptomatic knee osteoarthritis: a randomized clinical trial. JAMA. 2016;315(10):1005–1013. doi:10.1001/jama.2016.1961
61. McAlindon T, LaValley M, Schneider E, et al. Effect of vitamin D supplementation on progression of knee pain and cartilage volume loss in patients with symptomatic osteoarthritis: a randomized controlled trial. JAMA. 2013;309(2):155–162. doi:10.1001/jama.2012.164487
62. Tu L, Zheng S, Cicuttini F, et al. Effects of vitamin D supplementation on disabling foot pain in patients with symptomatic knee osteoarthritis. Arthritis Care Res. 2021;73(6):781–787. doi:10.1002/acr.24371
63. Zuo A, Jia Q, Zhang M, Zhou X, Li T, Wang L. The association of vitamin D with knee osteoarthritis pain: an analysis from the osteoarthritis initiative database. Sci Rep. 2024;14(1):30176. doi:10.1038/s41598-024-81845-6
64. Amirkhizi F, Asoudeh F, Hamedi-Shahraki S, Asghari S. Vitamin D status is associated with inflammatory biomarkers and clinical symptoms in patients with knee osteoarthritis. Knee. 2022;36:44–52. doi:10.1016/j.knee.2021.12.006
65. Helde-Frankling M, Björkhem-Bergman L. Vitamin D in pain management. Int J Mol Sci. 2017;18(10). doi:10.3390/ijms18102170
66. Giustina A, Bilezikian JP, Adler RA, et al. Consensus statement on vitamin D status assessment and supplementation: whys, whens, and hows. Endocrine Rev. 2024;45(5):625–654. doi:10.1210/endrev/bnae009
67. Giustina A, Bouillon R, Dawson-Hughes B, et al. Vitamin D in the older population: a consensus statement. Endocrine. 2023;79(1):31–44. doi:10.1007/s12020-022-03208-3
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.