Back to Journals » Clinical Pharmacology: Advances and Applications » Volume 11
Vitamin B12 deficiency presenting as pseudo-thrombotic microangiopathy: a case report and literature review
Authors Fahmawi Y ![]() , Campos Y, Khushman M
, Campos Y, Khushman M ![]() , Alkharabsheh O
, Alkharabsheh O ![]() , Manne A, Zubair H, Haleema S, Polski J, Bessette S
, Manne A, Zubair H, Haleema S, Polski J, Bessette S ![]()
Received 1 March 2019
Accepted for publication 20 August 2019
Published 27 August 2019 Volume 2019:11 Pages 127—131
DOI https://doi.org/10.2147/CPAA.S207258
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Arthur E. Frankel
Yazan Fahmawi,1 Yesica Campos,1 Moh’d Khushman,2 Omar Alkharabsheh,2 Ashish Manne,2 Haseeb Zubair,2 Saadia Haleema,3 Jacek Polski,3 Sabrina Bessette1
1Department of Internal Medicine, University of South Alabama, Mobile, AL, USA; 2Department of Hematology-Oncology, Mitchell Cancer Institute, University of South Alabama, Mobile, AL, USA; 3Department of Pathology, University of South Alabama, Mobile, AL, USA
Correspondence: Yazan Fahmawi
Department of Internal Medicine, University of South Alabama, 2451 USA Medical Center Drive, Mobile, AL 36617, USA
Tel +1 251 471 7891
Fax +1 251 470 1652
Email [email protected]
Abstract: Pseudo-thrombotic microangiopathy (pseudo-TMA) is a recognized, yet uncommon, clinical presentation of vitamin B12 deficiency. Patients with pseudo-TMA present with microangiopathic hemolytic anemia (MAHA), thrombocytopenia and schistocytes. They are often misdiagnosed as thrombotic thrombocytopenia purpura (TTP) and receive unnecessary therapy. Here, we report a case of a 60-year-old male who presented with thrombocytopenia and normocytic normochromic anemia. Anemia work-up was remarkable for severe B12 deficiency (<60 pg/mL) and a positive non-immune hemolysis panel. Peripheral smear was reviewed and showed anisocytes, poikilocytes, schistocytes and hypersegmented neutrophils. Vitamin B12 replacement (1000 mcg IM daily) was started, ADAMTS13 activity was sent and daily plasmapheresis was initiated. Over the next 3 days, the patient’s hemoglobin and platelets were stable and the hemolysis panel showed gradual improvement. On day 4, ADAMTS13 activity results came back normal at 61%. Accordingly, plasmapheresis was discontinued, parenteral B12 replacement was continued and that resulted in gradual improvement and eventually cessation of hemolysis and normalization of hemoglobin and platelets. In this patient, parietal cell autoantibodies were positive and so the diagnosis of pernicious anemia was made. Patients with severe vitamin B12 deficiency may present with features mimicking TTP such as MAHA, thrombocytopenia and schistocytosis. An early and accurate diagnosis of pseudo-TMA has a critical clinical impact with respect to administering the correct treatment with vitamin B12 replacement and avoiding, or shortening the duration of, unnecessary therapy with plasmapheresis.
Keywords: vitamin B12 deficiency, pseudo-thrombotic microangiopathy, schistocytes
Introduction
Vitamin B12 has a unique structure. It contains a metal ion, cobalt, and thus the name cobalamin. It has essential roles in DNA synthesis, red blood cells, (RBCs) development, and neurologic functions. Cobalamin is a cofactor for the enzymes; methionine synthase and L-methylmalonyl-coenzyme A mutase. Diminished activity of methionine synthase enzyme in patients with cobalamin deficiency impairs the regeneration of tetrahydrofolate (THF), renders folate unusable and that leads to ineffective DNA synthesis in rapidly dividing cells.1 In the bone marrow (BM) that leads to development of megaloblastic anemia (MBA), cytopenia and dysplastic changes. MBA can lead to premature destruction of developing red blood cells in the BM (ineffective erythropoiesis) and peripheral blood (hemolysis).2,3
The clinical presentation of thrombocytopenia and microangiopathic hemolytic anemia (MAHA), which has been reported in cobalamin deficiency, is critical to recognize and evaluate promptly as it may be a manifestation of a life threatening thrombotic microangiopathy (TMA) syndrome. Primary TMA syndromes include thrombotic thrombocytopenia purpura (TTP), hemolytic uremic syndrome (HUS), drug induced TMA, and complement-mediated TMA. These syndromes require urgent treatment that target the primary etiology such as plasmapheresis or monoclonal antibodies that bind complement proteins.4,5 Thrombocytopenia and MAHA are also seen in severe hypertension, HELLP syndrome, systemic infections, malignancies, autoimmune disorders and following solid organ and stem cell transplant. The treatment here should be directed at the underlying disease.6–9
The term pseudo-TMA describes TMA secondary to vitamin B12 deficiency. Patients with pseudo-TMA present with hemolytic anemia, thrombocytopenia, and dysmorphic “fragmented” RBCs. They are often misdiagnosed to have other TMA syndromes and receive unnecessary therapy such as plasmapheresis.10 Here, we report a case of pseudo-TMA in a patient with cobalamin deficiency secondary to pernicious anemia.
Case report
A 60-year‐old African American man presented to the emergency room with a two-week history of dyspnea and profound fatigue. Complete blood count showed severe normocytic normochromic anemia and thrombocytopenia. Hemoglobin (Hb) was 4.2 g/dL (13.5–17.5 g/dL), mean corpuscular volume (MCV) was 90.4FL (80–100FL), and platelets were 67,000/MCL (150,000–450,000/MCL). There was also evidence of leukopenia with the white blood cell count at 2,250/MCL (4,300–10,000/MCL). Anemia work-up was remarkable for severe vitamin B12 deficiency at <60 pg/mL (193–986 pg/mL) and ongoing non-immune hemolysis. Indirect bilirubin was increased at 2.6 mg/dL (0.2–0.7 mg/dL), LDH was increased at 5,901 Unit/L (87–241 Unit/L), haptoglobin was decreased at <10 mg/dL (30–200 mg/dL), and negative direct antiglobulin test. Of note, there was suboptimal BM response as his reticulocyte % was 0.7% (0.5–2.3%). The reticulocyte production index (RPI) was calculated by multiplying the reticulocyte % x Hct/45. The final product was divided by maturation factor 2.5 as Hct was <16. The calculated RPI was 0.1%. His peripheral blood smear was reviewed, and it showed nucleated RBCs, anisopoikilocytes, schistocytes, hypersegmented neutrophils (not captured) and very few platelets (Figure 1). The patient was afebrile and his kidney function was normal. Moreover, there were no neurologic findings. Given the presence of hemolysis, thrombocytopenia and the reported schistcytosis, the diagnosis of TTP was a concern and therefore daily plasmapheresis was initiated. Vitamin B12 (1000 mcg IM daily) was started concomitantly with the plasmapheresis. A Disintegrin and Metalloproteinase with Thrombospondin type-1 motif, member 13 (ADAMTS13) activity, parietal cell antibody and intrinsic factor antibody tests were drawn and sent out to an outside lab. Over the next 3 days, the patient’s hemoglobin and platelets stabilized, and the hemolysis panel showed gradual improvement which was most likely due to vitamin B12 replacement. On day 4, ADAMTS13 activity came back normal at 61% (≥60%). Accordingly, plasmapheresis was discontinued, parenteral B12 replacement was continued which resulted in cessation of hemolysis (Figure 2) and normalization of hemoglobin and platelets (Figure 3). Patient was discharged on weekly parenteral vitamin B12 for one month and with a follow-up with a hematologist. Finally, the parietal cell autoantibodies came back positive consistent with pernicious anemia.
 |
Figure 1 Peripheral blood smear with a nucleated red blood cell, polychromasia, “fragmented” RBCs and thrombocytopenia. The arrow points to a dysmorphic “fragmented” RBC (Wright ‘s stain, original magnification x1000). |
 |
Figure 2 Timeline summary showing gradual improvement of ongoing hemolysis demonstrated by decreasing bilirubin and serum lactate dehydrogenase (LDH) levels. |
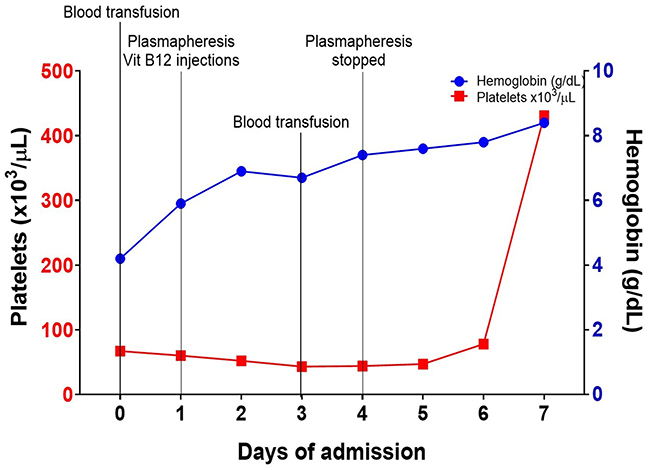 |
Figure 3 Timeline summary showing gradual improvement of anemia and thrombocytopenia demonstrated by increasing hemoglobin (Hb) and platelets. Two packed red blood cells were transfused on day 0 and day 4. |
Discussion
Patients with cobalamin deficiency may present with variable neurologic and neuropsychiatric diseases such as subacute combined degeneration of the spinal cord, mental sluggishness, dementia and psychosis. They may also present with wide spectrum of hematologic manifestations such as macrocytosis, anemia, leukopenia, thrombocytopenia and pancytopenia. Moreover, other less common presentations have been reported such as glossitis and vitiligo.11,12
The variable degree of hematologic manifestations experienced in 201 consecutive patients with cobalamin deficiency was explored. The most common manifestation was anemia (37%). Other manifestations included hypersegmented neutrophils (32%), leukopenia (13.9%) thrombocytopenia (9.9%) and pancytopenia (5%). Pseudo-TMA was experienced in 2.5% of the patients.13
The clinical presentation of primary TMA syndromes can be quite similar to the clinical presentation of pseudo-TMA. TTP is a primary TMA syndrome that has high mortality rate (80–90%) if unrecognized and left untreated.14 When TTP is suspected, while confirmatory test (ADAMTS13 activity) is underway, immediate initiation of plasma exchange should be started. Pseudo-TMA does not respond to plasma exchange. If this diagnosis is not promptly recognized, patients with pseudo-TMA will receive unnecessary, ineffective and potentially harmful therapy.
A systematic review of the existing literature between July 17, 1977 and July 17, 2017 captured 41 patients with pseudo-TMA.10 In this systematic review, the presence of schistocytes was present in 76% of patients. The underlying etiology of schictocytes or dysmorphic “fragmented” RBCs in patients with vitamin B12 deficiency could be multifactorial. Ineffective erythropoiesis secondary to vitamin B12 deficiency could cause severe dysplastic RBCs changes leading to bizarre anisopoikilocytosis. This anisopoikilocytosis can affect the MCV. MCV has been reported to be normal in less than 50% of patients with vitamin B12 deficiency as seen in our case.10 Other postulated cause of the fragmented RBCs is elevated serum homocysteine (was not checked in our patient). Hyerphomocysteinemia has shown to increase the risk of hemolysis in vitamin B12 in vitro as well as it may cause endothelial damage leading to intravascular hemolysis and RBCs fragmentation.15,16 In our case, the serum haptoglobin was low (<10 mg/dL) suggesting the presence of intravascular hemolysis along with intramedullary hemolysis which is seen commonly in vitamin B12 deficiency cases.
In the aforementioned systematic review, LDH was elevated in all patients, haptoglobin was decreased in all patients and bilirubin was increased in 20/23 (87%) patients. The median LDH reported in this systematic review was 3981 Unit/L while the median LDH reported in patients with TTP is 1407–1460 Unit/L.17,18 The LDH in our patient was very high at 5,901 Unit/L. The RPI was the most accurate means for interpreting marrow response. RPI >3.0% was considered an adequate response to anemia. All patients had an RPI <3.0%. The calculated RPI in our patient was 0.1%. Therefore, RPI could help differentiating between hemolytic anemia due to etiologies that affect BM response such as cobalamin deficiency and other etiologies in which BM response is not impaired.17 Together, RPI and LDH represent two laboratory tests that can help to differentiate Pseudo-TMA from TTP.
The underlying etiology of cobalamin deficiency in our patient was pernicious anemia (PA). Apparently, this was the underlying diagnosis in 28/41 patients (68%) as well in the systematic review. Other reported causes of cobalamin deficiency were dietary, exclusive breast feeding in infants, H. pylori gastritis and malabsorption due to ileal resection. ADAMTS13 activity was checked in our patient and the activity was within normal limit. A concern about TTP in the reported patients was encountered and ADAMTS13 activity was checked in 14 patients. In 13/14 (93%) it was detectable. In one patient, the underlying etiology was thought to be a combination of PA and TTP.
Treatment with plasma transfusion and/or exchange was carried out in 14/41 patients (34%). Complications secondary to therapy with plasma exchange were encountered in 2/14 patients (14%). Our patient was treated with plasma exchange until the result of ADAMTS13 activity came back as normal. No complications were encountered in our patient. The mean length of hospital stay in few reports was around 10 days.19 In our case, it was 7 days. Responses within 14 days to parenteral B12 replacement were reported as partial in 13/15 patients (87%) and complete in 2/15 patients (13%). Responses after 14 days to 6 months were reported as partial in 2/15 patients (13%) and complete in 13/15 patients (87%). Our patient achieved complete response within 14 days. Total normal body stores of vitamin B12 are in the range of 2–5 mg. Deficiency takes at least 1–2 years to develop once vitamin B12 intake ceases. The National Institute of health recommends daily intake of vitamin B12 at 2.4 mcg/day in adults. Typical responses to vitamin B12 replacement includes; decrease in hemolysis markers within 1–2 days, and increase in reticulocyte within 3–4 days.20
In conclusion, patients with severe cobalamin deficiency may present with features mimicking TMA syndromes. High LDH level as well as RPI can be useful laboratory tools to differentiate pseudo-TMA from other primary TMA syndromes such as TTP. An early and accurate diagnosis of pseudo-TMA has a critical clinical impact with respect to administering the correct treatment with vitamin B12 replacement and avoiding, or shortening the duration of, unnecessary therapy with plasmapheresis.
Abbreviations
MAHA, Microangiopathic hemolytic anemia; PSEUDO-TMA, Pseudo-thrombotic microangiopathy; TMA, Thrombotic Microangiopathy; TTP, Thrombotic thrombocytopenia purpura; Hb, Hemoglobin; Hct, Hematocrit; RBC, Red blood cell; RPI, Reticulocyte production index; THT, Tetrahydrofolate; BM, Bone marrow; MBA, Megaloblastic anemia; MCV, Mean corpuscular volume; PA, Pernicious anemia.
Previous presentation
Part of the manuscript was accepted for publication at the American College of Physician annual meeting 2019.
Declaration
Written informed consent has been provided by the patient to have the case details published. According to the policy of University of South Alabama IRB approval is not required if the case report includes less than three cases.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Stabler SP. Clinical practice. Vitamin B12 deficiency. N Engl J Med. 2013;368(2):149–160. doi:10.1056/NEJMcp1113996
2. Chhabra N, Lee S, Sakalis EG. Cobalamin deficiency causing severe hemolytic anemia: a pernicious presentation. Am J Med. 2015;128(10):e5–e6. doi:10.1016/j.amjmed.2015.05.048
3. Dimond A, George JN, Hastings C. Severe vitamin B-12 deficiency in a child mimicking thrombotic thrombocytopenic purpura. Pediatr Blood Cancer. 2009;52(3):420–422. doi:10.1002/pbc.21788
4. George JN, Nester CM. Syndromes of thrombotic microangiopathy. N Engl J Med. 2014;371(7):654–666. doi:10.1056/NEJMra1312353
5. Scully M, Cataland S, Coppo P, et al. Consensus on the standardization of terminology in thrombotic thrombocytopenic purpura and related thrombotic microangiopathies. J Thromb Haemost. 2017;15(2):312–322. doi:10.1111/jth.13571
6. McMinn JR, George JN. Evaluation of women with clinically suspected thrombotic thrombocytopenic purpura-hemolytic uremic syndrome during pregnancy. J Clin Apher. 2001;16(4):202–209.
7. Booth KK, Terrell DR, Vesely SK, George JN. Systemic infections mimicking thrombotic thrombocytopenic purpura. Am J Hematol. 2011;86(9):743–751. doi:10.1002/ajh.22091
8. George JN. Systemic malignancies as a cause of unexpected microangiopathic hemolytic anemia and thrombocytopenia. Oncology (Williston Park). 2011;25(10):908–914.
9. Laskin BL, Goebel J, Davies SM, Jodele S. Small vessels, big trouble in the kidneys and beyond: hematopoietic stem cell transplantation-associated thrombotic microangiopathy. Blood. 2011;118(6):1452–1462. doi:10.1182/blood-2011-02-321315
10. Tran PN, Tran MH. Cobalamin deficiency presenting with thrombotic microangiopathy (TMA) features: a systematic review. Transfus Apher Sci. 2018;57(1):102–106. doi:10.1016/j.transci.2018.01.003
11. Shipton MJ, Thachil J. Vitamin B12 deficiency - A 21st century perspective. Clin Med (Lond). 2015;15(2):145–150. doi:10.7861/clinmedicine.15-2-145
12. Baker SJ, Ignatius M, Johnson S, Vaish SK. Hyperpigmentation of skin. A sign of vitamin-B12 deficiency. Br Med J. 1963;1(5347):1713–1715. doi:10.1136/bmj.1.5347.1713
13. Andres E, Affenberger S, Zimmer J, et al. Current hematological findings in cobalamin deficiency. A study of 201 consecutive patients with documented cobalamin deficiency. Clin Lab Haematol. 2006;28(1):50–56. doi:10.1111/j.1365-2257.2006.00755.x
14. Joly BS, Coppo P, Veyradier A. Thrombotic thrombocytopenic purpura. Blood. 2017;129(21):2836–2846. doi:10.1182/blood-2016-10-709857
15. Ventura P, Panini R, Tremosini S, Salvioli G. A role for homocysteine increase in haemolysis of megaloblastic anaemias due to vitamin B(12) and folate deficiency: results from an in vitro experience. Biochim Biophys Acta. 2004;1739(1):33–42. doi:10.1016/j.bbadis.2004.08.005
16. Zittan E, Preis M, Asmir I, et al. High frequency of vitamin B12 deficiency in asymptomatic individuals homozygous to MTHFR C677T mutation is associated with endothelial dysfunction and homocysteinemia. Am J Physiol Heart Circ Physiol. 2007;293(1):H860–5. doi:10.1152/ajpheart.01189.2006
17. Noel N, Maigné G, Tertian G, et al. Hemolysis and schistocytosis in the emergency department: consider pseudothrombotic microangiopathy related to vitamin B12 deficiency. QJM. 2013;106(11):1017–1022. doi:10.1093/qjmed/hct142
18. Kremer Hovinga JA, Vesely SK, Terrell DR, Lämmle B, George JN. Survival and relapse in patients with thrombotic thrombocytopenic purpura. Blood. 2010;115(8):
19. Abbott DW, Friedman KD, Karafin MS. Differentiation of pernicious anemia from thrombotic thrombocytopenic purpura: the clinical value of subtle pathologic findings. Transfus Apher Sci. 2016;55(3):318–322. doi:10.1016/j.transci.2016.08.005
20. Stabler SP. Vitamin B12 deficiency. N Engl J Med. 2013;368(21):2041–2042. doi:10.1056/NEJMc1304350
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.