Back to Journals » Clinical Ophthalmology » Volume 16
Visual Outcome for Children with Optic Pathway Gliomas Treated with Systemic Chemotherapy
Authors Mohammad M, Alrawashdeh HM, Mehyar M, Amayiri N, Abu Laban D, Alnawaiseh I ![]() , Yousef Y
, Yousef Y
Received 19 May 2022
Accepted for publication 14 July 2022
Published 1 September 2022 Volume 2022:16 Pages 2933—2942
DOI https://doi.org/10.2147/OPTH.S374959
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Mona Mohammad,1,* Hamzeh Mohammad Alrawashdeh,2,* Mustafa Mehyar,1 Nisreen Amayiri,3 Dima Abu Laban,4 Ibrahim Alnawaiseh,1 Yacoub Yousef1
1Department of Surgery, Division of Ophthalmology, King Hussein Cancer Center (KHCC), Amman, Jordan; 2Department of Ophthalmology, Sharif Eye Centers, Irbid, Jordan; 3Department of Pediatric Oncology, King Hussein Cancer Center (KHCC), Amman, Jordan; 4Department of Diagnostic Radiology, King Hussein Cancer Center (KHCC), Amman, Jordan
*These authors contributed equally to this work
Correspondence: Mona Mohammad; Yacoub Yousef, Department of Surgery, Division of Ophthalmology, King Hussein Cancer Center, P.O. Box 1269, Amman, 11941, Jordan, Tel +962 795372321 ; +962 79 100 3333, Fax +962 6 5345 567, Email [email protected]; [email protected]
Purpose: This study aims to report visual acuity outcomes for patients with optic pathway gliomas (OPG) treated with systemic chemotherapy and analyze the associated factors.
Patients and Methods: A retrospective study of 29 children with OPG treated with chemotherapy at King Hussein Cancer Center (KHCC), Amman, Jordan, between May/2005 and August/2020. Details on patient demographics, tumor location, systemic chemotherapy, and progression of disease were extracted from medical records.
Results: Fifty-four eyes of twenty-nine patients were included in this study with a follow-up range from 2 to 17 years. Sixteen patients (55%) had a history of neurofibromatosis-1 (NF1). Most of the eyes (31, 57%) had visual acuity ranges in the moderate or better group. The age group ≥ 5 years at diagnosis, those with hydrocephalus, and patients with non-NF1 presented the worst visual acuity ranges from severe or worse; the p-value was 0.043, 0.0320, and 0.0054, respectively. Following treatment with systemic chemotherapy, visual acuity improved in 5 (17%) patients, remained the same in 23 (79%) patients, and only one patient (3%) had vision deterioration. Of the five patients who showed vision improvement, only one had radiological regression of the tumor. Parallel to this, three (10%) patients showed tumor progression in the final magnetic resonance image (MRI) findings without affecting the final vision.
Conclusion: Children older than 5 years at diagnosis, in sporadic OPG, and those with hydrocephalus had the worst vision at presentation. Treatment with systemic chemotherapy prevented further deterioration of vision, and following treatment with systemic chemotherapy, most of the patients had the same vision; this stability indicates that vision at diagnosis is an important predictor for the final visual outcome.
Keywords: glioma, neurofibromatosis, neuro-oncology, pediatric tumors, vision
Introduction
Among the pediatric age group, low-grade gliomas (LGGs) represent the most common intracranial tumors (accounting for 35–50% of central nervous system tumors). The optic pathway (from the optic nerve to the optic tract) is a common site that is affected by LGGs.1 These tumors are benign neoplasms; therefore, patients have excellent overall survival. However, some survivors may suffer from significant visual impairment as a result of the involvement of vital ocular structures with resultant loss of vision that will badly influence the quality of life for those patients.2 In this regard, preservation of vision is a primary objective to consider when treating children with optic pathway gliomas (OPGs). These tumors principally occur in the first decade of life, and the incidence decreases with increasing age.3
Optic pathway gliomas may occur in association with neurofibromatosis type 1 (NF-1) or as sporadic tumors. It is estimated that 15–20% with NF-1 will develop these tumors, and almost half of those patients will become symptomatic.4 Tumors associated with NF-1 differ from sporadic OPGs; NF1-OPGs were less likely to have associated visual impairment at diagnosis and less likely to exhibit radiographic progression over time.5,6
There is no consensus between different centers as to when to start treatment of OPGs, particularly in patients with NF-1; otherwise, optic gliomas are usually treated in patients with non-NF1 at the time of presentation. This disease has an unpredictable natural history in which tumors may stay dormant for a long time with no growth or may rarely show spontaneous regression. In addition, there is a poor correlation between radiological findings and visual acuity results. Therefore, most physicians will start treatment, mainly with systemic chemotherapy, if there is clinical or radiological progression. In general, treatment is needed if there is a large tumor with major intracranial extension, severe proptosis, loss of vision, visual field loss, optic disc pallor, hydrocephalus, endocrine disturbances, or in the presence of diencephalic syndrome.3,6
In the literature, there is a paucity of data regarding the clinical outcome of children with OPG who receive systemic chemotherapy. The current study aims to report the visual acuity outcomes of a tertiary cancer center in Amman, Jordan, for patients with OPGs, treated with systemic chemotherapy, and to elicit factors related to poor visual prognosis.
Materials and Methods
This is a longitudinal observational cohort retrospective study that was conducted between May/2005 to August/2020, and was approved by the Institution Review Board (IRB) at King Hussein Cancer Center (KHCC), Amman, Jordan (approval Number. 20 KHCC 110). Fifty-four eyes of twenty-nine patients were included. Inclusion criteria were all children with optic pathway gliomas treated by systemic chemotherapy over the study period. We excluded patients who received radiotherapy, patients with insufficient data, those with less than 6-month follow-up following last treatment, and patients with mild disease who were observed and did not receive treatment.
Patients’ demographics including date of birth, gender, age at diagnosis of OPG, NF-1 status, family history of NF-1, presenting symptoms, vision at diagnosis and at last follow-up, details of chemotherapy, including type, number, and dates of cycles, and brain MRI findings including the location of OPGs and the change in size at diagnosis and at last follow-up were extracted from the medical records.
Visual acuity was measured using the Snellen acuity chart in verbal children and LEA cards were used to assess vision for younger children. We considered a two-line decrease in vision compared with the first examination as worsening. Similarly, improvement was defined as a two-line increase in acuity. For each child, if one eye worsened, the outcome was defined as worsening, regardless of the outcome of the other eye. If one eye improved and the other remained stable, the outcome was considered as improved. None of our patients had improvement in one eye and worsening in the other eye. The multiple ranges of vision loss, based on visual acuity, at diagnosis in addition to final vision, were described according to the recommendation by the International Council of Ophthalmology (ICO) (http://www.icoph.org/downloads/visualstandardsreport.pdf).7
For each eye, the vision was classified as normal (0.8 or better), mild (0.3–0.6), moderate (0.125–0.25), severe (0.05–0.1), near blindness (less than 0.05), or blindness (no light perception). We excluded four eyes for four patients with unilateral optic nerve glioma (54 eyes of 29 patients were included in this study). For the sake of comparison, we divided the eyes into two groups: moderate or better group (normal, mild, and moderate loss) versus severe and worse group (severe vision loss, near blindness, and blindness).
All patients had Imaging of the optic pathways with MRI of the brain and orbits, to define the location and extent of the tumor and to compare the size at follow-up scans. Tumor location was described using the Dodge classification: optic nerve alone (stage 1), optic chiasm ±optic nerve (stage 2), and post-chiasm (stage 3).8 According to MRI findings, progression was defined as enlargement of the primary tumor by greater than a 25% or the appearance of new lesions. Regression was defined as a complete response or greater than 25% reduction in tumor size; otherwise, the tumor was labeled as stable.9
All patients were treated according to the standard protocol at KHCC. The usual chemotherapy agents used are first-line monthly vincristine and carboplatin for 15 cycles, second-line weekly vinblastine for 70 weeks, and third-line TPCV protocol (thioguanine, procarbazine, CCNU, vincristine) for 8 cycles (over a year).10–12 None of the study patients was treated with targeted therapy.
We monitor patients with brain MRI every 3 months during therapy and the first year of therapy, then every 6 months for 2 years, then yearly with close monitoring during the pubertal time. We also monitor vision and hormonal profiles at the same frequency.10
Statistical Analysis
The SPSS (version 25; Chicago, IL, USA) was used to conduct the statistical analyses. Descriptive statistics were carried out for all variables, including patients’ demographics, tumor location, visual acuity at diagnosis, final visual acuity, and final MRI finding using frequencies and percentages. Fisher exact test was used to assess the difference between the discrete variables. A p-value of <0.05 was considered statistically significant.
Results
Over the study period, a total of twenty-nine patients met inclusion criteria, and the majority of them were females (19, 66%), and aged ≥ 5 years (17, 59%). The median age at diagnosis of OPG was 5.75 years (range: 3–15 years). Patients were followed over a period of 2 to 17 years (mean 3.9 years). The age of children at the last follow-up ranged from 4 to 20 years of age. Sixteen patients (55%) had a history of NF1, and of those 4 patients (14%) were familial. Twelve patients (41%) had optic nerve glioma, and only four of them had unilateral involvement, the others (8 patients) had tumors involving both optic nerves. Therefore, the total number of eyes included was 54 eyes for 29 patients. Only 2 (7%) patients had a history of hydrocephalus. Patients’ demographics and tumor location at diagnosis are shown in Table 1.
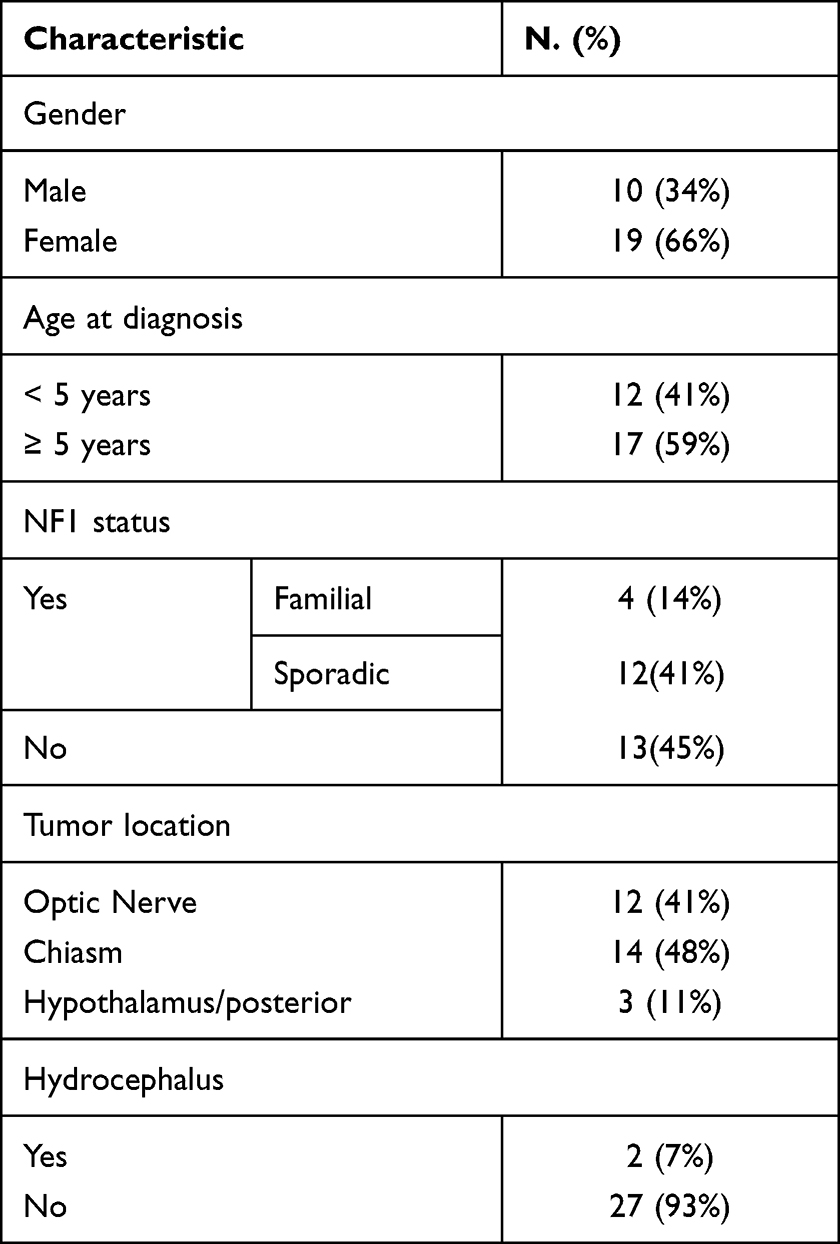 |
Table 1 Patients’ Demographics and Tumor Location at Diagnosis |
The multiple ranges of vision loss, based on visual acuity, at diagnosis in addition to final vision, were described according to the recommendation by the ICO. Table 2 shows visual acuity ranges at diagnosis in relation to patients’ demographics and tumor location. We considered patients with normal, mild, and moderate vision loss as one group (moderate or better group), and compared them to patients with severe, near blindness, and blindness (severe or worse group).
 |
Table 2 Ranges of Visual Acuity at Diagnosis in Relation to Patients’ Demographics and Tumor Location |
The Severity of Visual Loss
Most of the eyes (31/54, 57%) had visual acuity ranges in the moderate or better group, of which females were the majority (17/54, 31.5%). This difference in initial vision between females and males was not statistically significant (P-value = 0.16).
The largest group of visual loss was the mild visual loss, which affected 13/54 eyes (24.1%) followed by near blindness, which affected 11 eyes (20.4%), normal vision (10/54 eyes, 18.5%), moderate (8/54 eyes, 14.8%), then severe and blindness (6 eyes for each group, 11.2%). Those over ≥5 years old, and patients with non-NF1, presented the worst visual acuity ranging from severe or worse (P-value = 0.043, and 0.0054 respectively).
The Impact of Tumor Location
Regarding the association between the location of glioma and vision loss, we observed that the more posterior the glioma is, the higher the chance for severe visual loss; however, this finding was not statistically significant (P-value =0.128). Two patients in our study had hydrocephalus, and for both patients, the initial vision was in the severe or worse group, which was statistically significant compared to patients without hydrocephalus (P-value =0.0320).
Treatment Outcome
Following treatment with chemotherapy, visual acuity was improved in 5/29 patients (17.2%), remained the same in 23/29 patients (79.3%), and only one patient (3.4%) had deterioration of vision. Two out of 12 patients (6.90%) with only optic nerve glioma, one out of 14 patients (3.4%) with chiasmal, and two out of 3 patients (6.9%) with hypothalamic glioma showed improvement in their vision. Hydrocephalus was found in two patients (6.90%), one of whom had hypothalamic glioma, and the other had an optic nerve and tectal glioma. The final visual acuity was improved in the patient with hydrocephalus and hypothalamic glioma, while it was stable in the other patient. Regarding the final MRI findings, the visual acuity was improved in 5 patients (17.2%): four (13.8%) with stable tumor features, and one (3.4%) with tumor regression in the final MRI findings. Three patients (10.3%) had tumor progression in the final MRI without affecting the final visual acuity. Table 3 shows final visual acuity outcomes in relation to patients’ demographics, tumor location, and final MRI findings. Figure 1 shows an example of a brain MRI scan for a patient with optic chiasm glioma, whose tumor showed enlargement of optic pathway glioma causing a mass effect upon the hypothalamic structures and third ventricle with a stable eye exam, and no drop in vision.
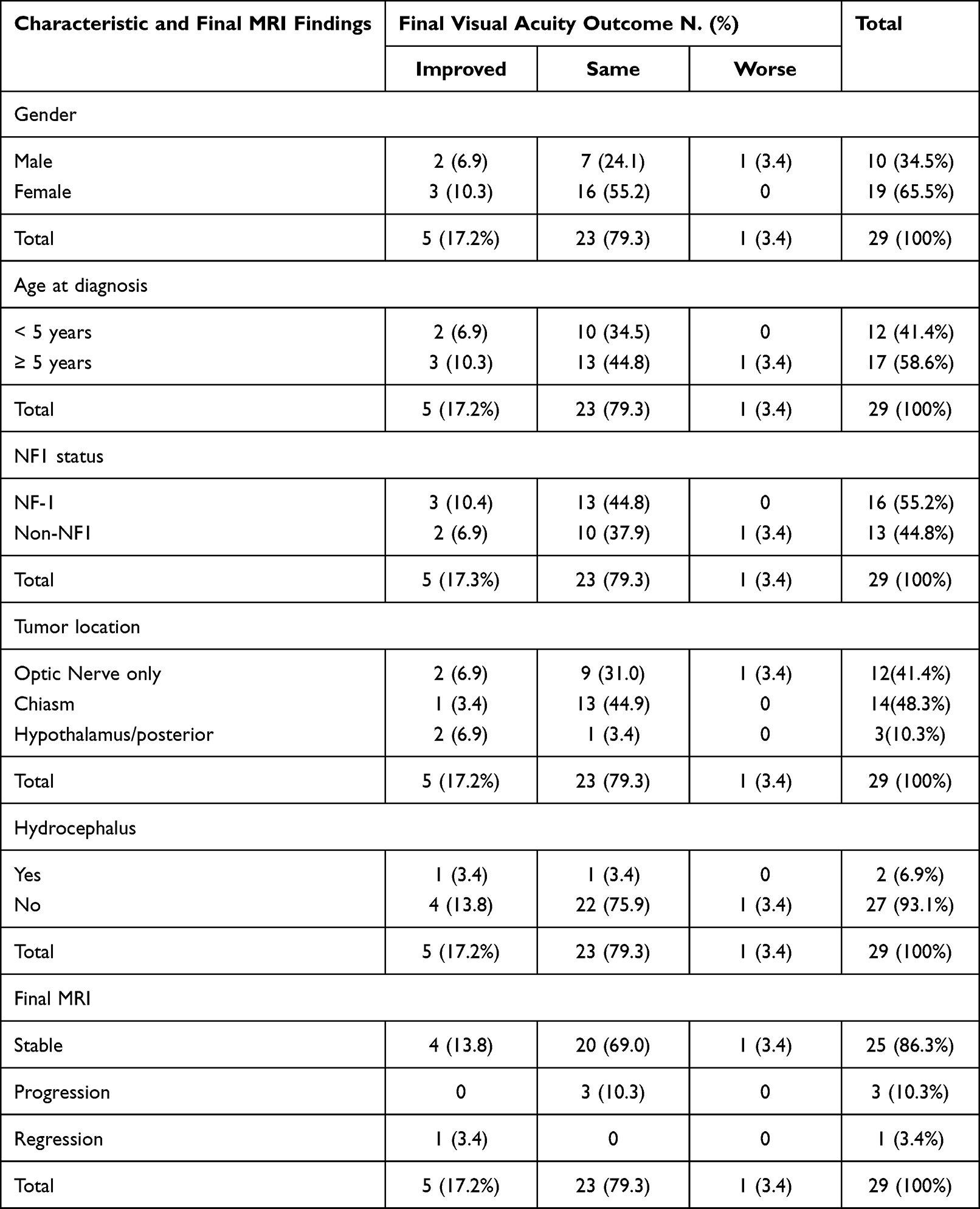 |
Table 3 Final Visual Acuity Outcomes in Relation to Patients’ Demographics, Tumor Location, and Final MRI Findings |
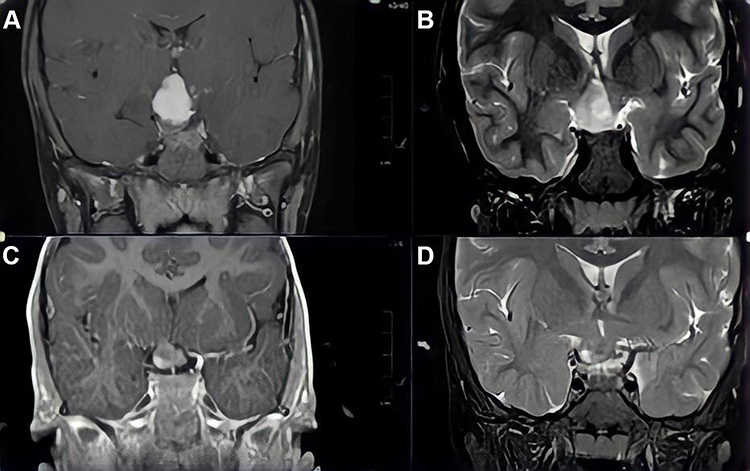 |
Figure 1 Selected coronal brain MRI images from T1-weighted (A and C) and Short Tau Inversion Recovery (STIR) sequences (B and D), showing an optic chiasm glioma. The more recent images (A and B) show enlargement of optic pathway glioma causing a mass effect upon the hypothalamic structures and third ventricle. |
Discussion
OPGs are benign tumors that affect children, particularly those with NF-1. Chemotherapy represents the main modality of treatment; the treatment journey is prolonged (usually over a year for a single protocol) and may need multiple lines of interventions. Different studies showed variable outcomes in relation to factors of severe visual loss and following treatment with systemic chemotherapy. To the best of our knowledge, this is the first study in Jordan that reports visual acuity outcomes for patients with optic pathway gliomas treated with systemic chemotherapy.
The majority of patients included were females (66%) and our results showed no difference between males to females in relation to initial visual loss. Contrary to this, Diggs-Andrews et al conducted a study to determine the impact of patient sex in children with NF-1-associated optic gliomas. They compared the outcome for 205 males and 226 females and found that while boys and girls with NF1 exhibited similar frequency of optic glioma, girls with NF1-associated optic gliomas were three times more likely than boys to require treatment due to visual decline (P value=0.0007). The increase in visual loss secondary to optic glioma in girls was not attributable to differences in patient age or tumor location and did not reflect an increased prevalence of these tumors in girls with NF1.13
More than half of our patients were 5 years old and older (59% of patients), and our results showed that for this age group the chance for severe loss of vision at diagnosis was higher, this may indicate that our patients present with more advanced disease and irreversible loss of vision due to delayed diagnosis. Contrary to this, Fisher et al evaluated risk factors for poor visual outcome following treatment with systemic chemotherapy for NF-1 OPG, and they found that young age at diagnosis (less than 2 years of age) is an indicator of poor outcome.14
In contrast to previous thoughts that children aged 6 years old and younger are at the greatest risk of NF-1 OPGs, Segal et al found that OPGs can present and progress beyond the preschool years.15 Listernick et al suggested extending annual screening for OPGs in NF-1 patients until the age of 7 years and every second year between ages 8 and 18 years.3 Our results showed that more than half of patients were five years of age and older, in four out of 29 patients (14%) OPGs were diagnosed at an age older than 10 years. Thus, we recommend extending the ophthalmological screening until the age of 10 years or later if the diagnosis of OPG was established at an older age.
Many studies showed that sporadic OPGs have an aggressive course compared to NF-1-associated OPGs. Singhal et al compared the natural history of OPG between both groups and found that symptomatic OPGs were more aggressive and presented with more impaired vision in sporadic gliomas than in NF-1 patients.16 Similarly, our results showed a significant difference between both groups; for NF-1 patients (23 eyes, 43%) were in the moderate or better vision group compared to 9 eyes (16%) in the sporadic group with significant P value (P=0.0054).
We did not find a difference in relation to tumor location in the impact of vision loss, however, we observed that for post-chiasmal tumors, 67% of eyes (4 out of 6 eyes) were in the severe group compared to 25% (5 out of 20 eyes) in patients with optic nerve gliomas. Balcer et al reviewed the MRI scans for 43 children with NF-1 OPG, and found a significant chance for severe visual loss in post-chiasmal gliomas, compared to pre-chiasmal tumors (P-value =0.048).17 Also, Fisher et al found a similar aggressive behavior for posterior gliomas.14 In the current study, all children with hydrocephalus presented with severe loss of vision, this is probably due to the fact that with the presence of optic pathway tumor, the optic nerves become more vulnerable to increased damage.18
Over the last years, many studies were conducted to assess and estimate the benefit of systemic chemotherapy in the preservation of vision in patients with optic pathway gliomas (OPGs). Those studies showed variable results. In 2010, Moreno et al carried out a systemic review of the literature published between 1990 and 2008 to find out the visual outcome following chemotherapy in children with OPG. Only studies that include ten or more children with OPGs treated with chemotherapy as the main modality of treatment along with a proper description for visual outcome were included. Of the 85 related studies, only 8 (five retrospective studies and three single-arm trials) were included in the meta-analysis. The analysis includes 174 children of whom 25 (14.4%) showed a visual improvement, 82 (47.1%) showed visual stability and 67 (38.5%) experienced deterioration of vision following chemotherapy. Moreover, the duration of visual response was not documented in any study in this analysis.19
Another retrospective study for 59 patients was designed in 2016 to assess the long-term visual outcome of children with sporadic OPGs in a single institute between 1990 and 2014. Wan et al aimed for the visual acuity at final follow-up and the risk factors associated with a poor visual outcome as well as the rate of tumor progression over a mean follow-up period of 5.2 years. About 40 patients encountered a progression of the tumor during or after treatment, and the majority of children experienced significant long-term visual impairment. Young age at diagnosis, the extent of the tumor, and the presence of optic nerve head pallor were associated with a poor visual outcome in this cohort study.20
Fisher et al conducted a retrospective multicenter analysis of visual outcomes in 88 children treated with chemotherapy for NF1-OPG, vision changes were improved, worsened, and stable in 32%, 28%, and 40% retrospectively. Visual and imaging outcomes were dissociated, which is consistent with previous observations.14
Bennebroek et al conducted a systemic review of the various types of systemic chemotherapeutic agents used to treat OPGs and concluded that the impact of systemic chemotherapy in OPGs on visual function is still unclear. Reports lacked uniform definitions and outcome parameters, which highlights the need for prospective studies that avoid the observed heterogenicity to enable an accurate estimate of the effect of systemic chemotherapy in OPG.21
Consistent with the previous observations, our results showed that the majority of children 23/29 (79.3%) had a stable vision in the last follow-up assessment. Only 5 (17.24%) patients had improvement in vision. For one patient (3.4%), vision dropped during treatment with systemic chemotherapy. Since most patients treated with systemic chemotherapy had stable vision compared to pre-treatment value, we believe that vision at diagnosis is an important indicator for the final visual outcome, and it is very important to diagnose those tumors at a stage where vision preservation is possible since the effect of systemic chemotherapy in vision improvement is still questionable. Chemotherapy may prevent further loss of vision but it may not revert the damage that is already caused by large tumors.
Dalla Via et al reported their outcome in the management of NF-1 patients with OPG, and although tumor volume reduction in response to chemotherapy was noticed, this was not accompanied by visual improvement. Therefore, they concluded that the role of systemic chemotherapy in preserving vision is questionable. They assumed that some NF-1 children have progressive visual deterioration for reasons that chemotherapy and radiotherapy cannot control.22 Similarly, our results showed that there was a poor correlation between radiographic and visual outcomes. Five patients in our study showed improvement in the final vision and only one of those had tumor regression in the MRI. Three patients (10.34%) had tumor progression in MRI without an associated drop in vision.
Tow et al found that of 47 patients included in their study, only 3 died of OPG complications. Our study shows a 100% survival rate, none of the patients included was dead at the end of the study period.23
The current study has a few limitations; its retrospective nature and the inclusion of a heterogeneous, small group of patients. In addition, the primary outcome measure used was the impact of treatment on vision, other parameters of visual function like the visual field or color vision were not assessed. This is due to the age of the patients and the difficulty to perform a comprehensive vision assessment for this age group. In spite of those limitations, we believe that our results may be of value. As vision is the most important parameter for the quality of life.
In spite of the small number of patients included in our study, we believe our results may be of value, OPGs are benign tumors with significant morbidity due to damage to vital structures along the visual pathway, and therefore early diagnosis is important before the irreversible loss of vision. It is very important to understand the impact of systemic chemotherapy on final vision, and this should be discussed thoroughly with caregivers to address their expectations following the long journey of treatment. Future large, collaborative, and multi-center studies are recommended.
Conclusion
Children older than 5 years at diagnosis, in sporadic OPG, and those with hydrocephalus had the worst vision at presentation. Treatment with systemic chemotherapy prevented further deterioration of vision, and following treatment with systemic chemotherapy, most of the patients had the same vision; this stability indicates that vision at diagnosis is an important predictor for the final visual outcome.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki and approved by the Institutional Review Board (IRB) of King Hussein Cancer Center (KHCC), Amman, Jordan (approval Number. 20 KHCC 110).
Data Sharing Statement
The data that support the findings of this study are available from the corresponding authors upon reasonable request.
Informed Consent Statement
Informed consent was obtained from all subjects (or their guardians) involved in the study. Written informed consent has been obtained from the patients or their guardians to publish this paper.
Acknowledgments
Special thanks to Ayat Taqash: statistical programmer, Center of Research Shared Resources at King Hussein Cancer Center for her help in data management, and Kawthar Khaleifeh and Raed Ramlawi: clinical nurse coordinators at pediatrics neuro-oncology clinics at King Hussein Cancer Center.
Funding
King Hussein Cancer Center supported this study.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Jahraus CD, Tarbell NJ. Optic pathway gliomas. Pediatr Blood Cancer. 2006;46(5):586–596. doi:10.1002/pbc.20655
2. Opocher E, Kremer LC, Da Dalt L, et al. Prognostic factors for progression of childhood optic pathway glioma: a systematic review. Eur J Cancer. 2006;42(12):1807–1816. doi:10.1016/j.ejca.2006.02.022
3. Listernick R, Ferner RE, Liu GT, Gutmann DH. Optic pathway gliomas in neurofibromatosis-1: controversies and recommendations. Ann Neurol. 2007;61:189–198. doi:10.1002/ana.21107
4. Astrup J. Natural history and clinical management of optic pathway glioma. Br J Neurosurg. 2003;17:327–335. doi:10.1080/02688690310001601216
5. Chateil JF, Soussotte C, Pedespan JM, Brun M, Le Manh C, Diard F. MRI and clinical differences between optic pathway tumours in children with and without neurofibromatosis. Br J Radiol. 2001;74:24–31. doi:10.1259/bjr.74.877.740024
6. Shuper A, Horev G, Kornreich L, et al. Visual pathway glioma: an erratic tumour with therapeutic dilemmas. Arch Dis Child. 1997;76:259–263. doi:10.1136/adc.76.3.259
7. Colenbrander A. VISUAL STANDARDS ASPECTS and RANGES of VISION LOSS with Emphasis on Population Surveys.
8. Dodge HW, Love JG, Craig WM, et al. Gliomas of the optic nerves. AMA Arch Neurol Psychiatry. 1958;79:607–621. doi:10.1001/archneurpsyc.1958.02340060003001
9. Fried I, Tabori U, Tihan T, Reginald A, Bouffet E. Optic pathway gliomas: a review. CNS Oncol. 2013;2(2):143–159. doi:10.2217/cns.12.47
10. Gnekow AK, Walker DA, Kandels D, et al. A European randomised controlled trial of the addition of etoposide to standard vincristine and carboplatin induction as part of an 18-month treatment programme for childhood (</ = 16 years) low grade glioma–a final report. Eur J Cancer. 2017;81:206–225. doi:10.1016/j.ejca.2017.04.019
11. Bouffet E, Jakacki R, Goldman S, et al. Phase II study of weekly vinblastine in recurrent or refractory pediatric low-grade glioma. J Clin Oncol. 2012;30(12):1358–1363. doi:10.1200/JCO.2011.34.5843
12. Ater JL, Zhou T, Holmes E, et al. Randomized study of two chemotherapy regimens for treatment of low-grade glioma in young children: a report from the Children’s Oncology Group. J Clin Oncol. 2012;30(21):2641–2647. doi:10.1200/JCO.2011.36.6054
13. Diggs-Andrews KA, Brown JA, Gianino SM, Rubin JB, Wozniak DF, Gutmann DH. Sex Is a major determinant of neuronal dysfunction in neurofibromatosis type 1. Ann Neurol. 2014;75(2):309–316. doi:10.1002/ana.24093
14. Fisher MJ, Loguidice M, Gutmann DH, et al. Visual outcomes in children with neurofibromatosis type 1-associated optic pathway glioma following chemotherapy: a multicenter retrospective analysis. Neuro Oncol. 2012;14(6):790–797. doi:10.1093/neuonc/nos076
15. Segal L, Darvish-Zargar M, Dilenge ME, Ortenberg J, Polomeno RC. Optic pathway gliomas in patients with neurofibromatosis type 1: follow-up of 44 patients. J AAPOS. 2010;14(2):155–158. doi:10.1016/j.jaapos.2009.11.020
16. Singhal S, Birch JM, Kerr B, Lashford L, Evans DG. Neurofibromatosis type 1 and sporadic optic gliomas. Arch Dis Child. 2002;87(1):65–70. doi:10.1136/adc.87.1.65
17. Balcer LJ, Liu GT, Heller G, et al. Visual loss in children with neurofibromatosis type 1 and optic pathway gliomas: relation to tumor location by magnetic resonance imaging. Am J Ophthalmol. 2001;131(4):442–445. doi:10.1016/S0002-9394(00)00852-7
18. Shuper A, Kornreich L, Michowitz S, Schwartz M, Yaniv I, Cohen IJ. Visual pathway tumors and hydrocephalus. Pediatr Hematol Oncol. 2000;17(6):463–468. doi:10.1080/08880010050120818
19. Moreno L, Bautista F, Ashley S, Duncan C, Zacharoulis S. Does chemotherapy affect the visual outcome in children with optic pathway glioma? A systematic review of the evidence. Eur J Cancer. 2010;46(12):2253–2259. doi:10.1016/j.ejca.2010.03.028
20. Wan MJ, Ullrich NJ, Manley PE, Kieran MW, Goumnerova LC, Heidary G. Long-term visual outcomes of optic pathway gliomas in pediatric patients without neurofibromatosis type 1. J Neurooncol. 2016;129(1):173–178. doi:10.1007/s11060-016-2163-4
21. Bennebroek CAM, Wijninga LE, Limpens J, Schouten-van Meeteren AYN, Saeed P. Impact of systemic anticancer therapy in pediatric optic pathway glioma on visual function: a systematic review. PLoS One. 2021;16(10):e0258548. doi:10.1371/journal.pone.0258548
22. Dalla Via P, Opocher E, Pinello ML, et al. Visual outcome of a cohort of children with neurofibromatosis type 1 and optic pathway glioma followed by a pediatric neuro-oncology program. Neuro Oncol. 2007;9(4):430–437. doi:10.1215/15228517-2007-031
23. Tow SL, Chandela S, Miller NR, Avellino AM. Long-term outcome in children with gliomas of the anterior visual pathway. Pediatr Neurol. 2002;28:262–270. doi:10.1016/S0887-8994(02)00628-8
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.