Back to Journals » Drug Design, Development and Therapy » Volume 11
Virtual screening for potential inhibitors of Mcl-1 conformations sampled by normal modes, molecular dynamics, and nuclear magnetic resonance
Authors Glantz-Gashai Y, Meirson T ![]() , Reuveni E, Samson AO
, Reuveni E, Samson AO
Received 24 January 2017
Accepted for publication 6 April 2017
Published 19 June 2017 Volume 2017:11 Pages 1803—1813
DOI https://doi.org/10.2147/DDDT.S133127
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Yitav Glantz-Gashai,* Tomer Meirson,* Eli Reuveni, Abraham O Samson
Faculty of Medicine in the Galilee, Bar Ilan University, Safed, Israel
*These authors contributed equally to this work
Abstract: Myeloid cell leukemia-1 (Mcl-1) is often overexpressed in human cancer and is an important target for developing antineoplastic drugs. In this study, a data set containing 2.3 million lead-like molecules and a data set of all the US Food and Drug Administration (FDA)-approved drugs are virtually screened for potential Mcl-1 ligands using Protein Data Bank (PDB) ID 2MHS. The potential Mcl-1 ligands are evaluated and computationally docked on to three conformation ensembles generated by normal mode analysis (NMA), molecular dynamics (MD), and nuclear magnetic resonance (NMR), respectively. The evaluated potential Mcl-1 ligands are then compared with their clinical use. Remarkably, half of the top 30 potential drugs are used clinically to treat cancer, thus partially validating our virtual screen. The partial validation also favors the idea that the other half of the top 30 potential drugs could be used in the treatment of cancer. The normal mode-, MD-, and NMR-based conformation greatly expand the conformational sampling used herein for in silico identification of potential Mcl-1 inhibitors.
Keywords: virtual screening, Mcl-1, molecular dynamics, NMR, normal modes
Introduction
Apoptosis is a highly conserved and regulated process for eliminating damaged and surplus cells, such as those generated during normal embryonic development and abnormal cancer.1 Important regulators of this process are the B cell lymphoma 2 (Bcl-2) family of proteins, which include pro- and anti-apoptotic members. Anti-apoptotic (ie, pro-survival) members include Bcl-2, Bcl-xL, Bcl-w, and myeloid cell leukemia-1 (Mcl-1), whereas pro-apoptotic members include Bax-like proteins, such as Bax, Bak, and Bok, and BH3-only proteins, such as Bad, Bim, Bmf, Bik, Hrk, Bid, Puma, and Noxa.2 The interaction of pro- and anti-apoptotic proteins with regulators is a key element of cell survival and death.
Anti-apoptotic proteins are commonly overexpressed in a number of human cancers where they foster the survival of tumor cells. To inhibit anti-apoptosis (ie, promote apoptosis) and interfere with tumor cell survival, several small-molecule drugs that mimic pro-apoptotic BH3 proteins were developed.3 The BH3-mimetics include ABT-7374 and its orally available derivative ABT-263.5 These BH3-mimetics bind selectively to Bcl-2, Bcl-xL, and Bcl-w and interfere with cell survival; however, they do not bind to Mcl-1 and some cancers cannot be treated by these compounds alone. To complicate things further, upregulation of Mcl-1 is a key factor in the development of resistance to ABT-737 and ABT-263.2 Thus, there is an unmet need to design ligands, and in particular new small molecules, that inhibit Mcl-1.6
Mcl-1 is a major cancer target, and Mcl-1 overexpression is often encountered in human cancer.7,8 Mcl-1 overexpression has been reported in breast cancer,9 lung cancer,10 prostate cancer,11 pancreatic cancer,12 cervical and ovarian cancers,13 and leukemia.14 Mcl-1 overexpression leads to resistance against Bcl-2-selective inhibitors and other small-molecule drugs used in chemotherapy.15 Remarkably, in vitro inhibition of Mcl-1 overexpression through RNA silencing inhibits tumor growth16 and abolishes chemoresistance.17 As such, Mcl-1 represents a promising cancer target.
Virtual screening is currently a classical tool in drug discovery applied in the search for novel compounds that target a given protein of interest.18 Computational screening approaches have gained general acceptance because, in comparison with high-throughput screening techniques, they are able to decrease both time and cost by limiting the number of compounds that must be experimentally tested.19 There are two main approaches for virtual screening: 1) ligand-based and 2) structure-based virtual screening. The latter approach is often used if the three-dimensional (3D) structure of a drug target is available from experimental studies. For Mcl-1, several experimental structures are available and are listed in Supplementary materials, Table S1.
To assist virtual screening, several studies have used molecular dynamics (MD) simulations.20 MD simulation is a well-established method for understanding protein dynamics. In most cases, MD simulations provide snapshots that improve virtual screening predictive power over known crystal structures, possibly due to sampling more relevant conformations. Furthermore, unrestrained MD simulations can move conformations previously not amenable to docking into the predictive range.21
To assist virtual screening, several studies have also used normal mode analysis (NMA).22 NMA is one of the standard techniques for studying long-time dynamics and, in particular, low-frequency motions.23 In contrast to MD, NMA provides an analytical and fully detailed description of the dynamics around a local energy minimum,24,25 and the conformation ensemble is generated by perturbing the initial structure along a set of relevant low-frequency normal modes.
To assist virtual screening, several studies have utilized structural ensembles obtained using nuclear magnetic resonance (NMR). Using multiple fixed conformation either experimentally determined by crystallography or NMR is a practical shortcut that may improve docking calculations. In several cases, this approach has led to experimentally validated predictions.26,27 Thus, NMR, MD, and NMA have each been used separately to improve virtual screening. Here, we combine the three to assist virtual screening for Mcl-1 inhibitors.
In this study, we use conformations sampled by three separate methods, namely, NMA, MD simulation, and NMR, and virtually screen for novel ligands that can modulate the activity of Mcl-1. Using this technique with two curated data sets, namely, the US Food and Drug Administration (FDA)-approved drugs and lead-like molecules, we identify novel small molecules that could not have been detected using the unperturbed Protein Data Bank (PDB) structure.
Materials and methods
NMA
For conformational sampling, we used model 1 of the NMR structure of PDB ID 2MHS28 as a starting structure. Usually, NMR structures consist of an average structure and a number of ensemble structures. The NMR ensemble structures are numbered model 1, model 2, model 3, etc. As no average structure was available, we decided to use model 1 of PDB ID 2MHS as a starting structure.
To calculate normal modes of the Mcl-1 structure PDB ID 2MHS,28 model 1, two programs were utilized, namely, STAND23 and ElNémo.29 For STAND, both real normal modes (REA) and Tirion modes (TIR) were calculated. For speed, the STAND option of coarse graining, 1 point, which accelerates the calculations yet does not flaw the results, was used, and default values of deformation amplitude were used. For ElNémo, default values of DQMIN −100 and DQMAX 100 were utilized. The DQMIN and DQMAX parameters correspond to the deformation amplitude in the direction of a single normal mode. For both STAND and ElNémo, only the three non-trivial lowest frequency modes were calculated. For each of these three modes, six PDB models were generated by STAND and six model structures were generated by ElNémo all fully distorted along the particular mode. These 12 model structures were subsequently used for molecular docking.
MD simulation
We run unrestrained MD of the Mcl-1 structure (PDB ID 2MHS, model 1), with the AMBER simulation package.30 The simulation parameters were obtained from force fields leaprc.ff99SB for the protein and leaprc.gaff for organic molecules, since we used a water and ethanol solvent mixture. Our MD simulation comprises three stages, namely, system heating using constant number, constant volume, and constant temperature (NVT), system equilibration using constant number, constant pressure, and constant temperature (NPT), and production using NVT. System heating uses NVT (ie, constant volume) since at low temperatures, pressure calculation may lead to barostat overcorrection and system instabilities. System equilibration uses NPT (ie, constant pressure) and allows system density to equilibrate before production. Production uses NVT (ie, constant volume). Our MD simulation is based on a protocol similar to the one described by Romero-Durana et al.31 Shortly, each time step was set to 2 fs, heating time was 200 ps, equilibration time was 1 ns, and production time was 200 ns (see Supplementary materials, Figure S1).
MD clustering
To obtain hierarchal MD clusters, the biomolecular simulation trajectory and data analysis program cpptraj,32 which is part of the AMBER package, were used. Clustering is a means of partitioning data so that data points inside a cluster are more similar to each other than they are to points outside a cluster. In the context of molecular simulation, this means grouping similar conformations together. Similarity is determined by a distance metric – the smaller the distance, the more similar the structures. One commonly used distance metric is coordinate root mean square deviation (RMSD). Application of this procedure resulted in four main clusters, from which one centroid conformation PDB structure was chosen for subsequent molecular docking.
NMR conformations
The NMR conformational ensemble of PDB ID 2MHS28 was used. This PDB entry contains 20 experimentally observed structures, which were all used for molecular docking.
Virtual screening
To identify potential inhibitors of Mcl-1, two molecular data sets were virtually screened and docked on to Mcl-1 using the AutoDock Vina program.33 The molecular data sets include a lead-like subset and an FDA-approved subset. The lead-like data set was based on the standard lead-like of the ZINC database (version 12, September 29, 2014),34 which contains 6,053,287 molecules from which a subset of 2,300,000 molecules were randomly selected. The lead-like library has been pre-filtered based on the properties of molecular weight between 250 and 350 g/mol, predicted partition constant (× LogP) ≤3.5, and number of rotatable bonds ≤7. The FDA-approved subset contained 1,790 FDA- and internationally approved drugs and was kindly provided by Dominique Douguet from the Institut de Pharmacologie Moléculaire et Cellulaire.35
To identify potential Mcl-1 inhibitors, we used multiple conformations of the protein. The multiple conformation comprised 20 NMR structures conformation of Mcl-1 (PDB ID 2MHS),28 12 models obtained using NMA, and four models obtained using MD simulations. First, AutoDock calculation was performed on a representative structure of the NMR ensemble (model 1 of PDB ID 2MHS). Then, we picked the 1,000 ligands with the lowest binding energies and docked them on the multiple conformations sampled by NMR, NMA, and MD.
In all cases, AutoDock Vina (1.1.2 for linux) was run on our Ahalama cluster equipped with 960 Intel E5645 processors. In all cases, the default parameters of AutoDock Vina were as follows: the exhaustiveness of the global search was 8, the maximum number of binding modes to generate was 9, and the maximum energy difference between the best and worst binding mode displayed was 3 kcal/mol. For each ligand, only the best pose was retained. In all cases, the binding site on Mcl-1 was defined and limited by a box measuring 16, 18, and 20 Å. The X, Y, and Z coordinates of the center of the docking box were −2.2, −19.8, and 2.9, respectively. The box encompassed the entire binding groove of Mcl-1 and included the P1, P2, P3, and P4 binding sites defined by Belmar and Fesik.2
Results
Mcl-1 conformations
Combined, the NMR structures, NMA distorted models, and MD simulation models span more of the conformational space of Mcl-1 than on their own. The MD models with an RMSD of 1.34 Å cover a larger area of the conformational space than the NMR and NMA models with RMSD values of 0.82 and 0.72 Å, respectively. The NMA and MD models each share less in common than with the NMR structure from which they were derived. In contrast to MD, NMA provides a detailed description of the dynamics around a local energy minimum. Combined, these conformations span a larger area of the conformational space of Mcl-1 than on their own.
Many PDB structures are available for Mcl-1 as listed in Supplementary materials, Table S1. For molecular docking, we chose PDB ID 2MHS, as it was the only structure solved without a peptide or a ligand, and we believe it is closest to the native state of the protein and it does not suffer from induced fit distortions.
Validation of in silico assay
To test the validity of our in silico assay, we virtually docked several ligands known to bind to Mcl-1 and compared their predicted binding energy with their experimental binding constants. These ligands included the small molecules bound to Mcl-1 in PDB IDs 5FDO, 5FDR, 4HW3, 4ZBI, 4ZBF, and 3WIX, and the Bcl-2 ligand, ABT-737, which does not bind to Mcl-1.36 The ligands were virtually docked to the multiple Mcl-1 conformations calculated herein using AutoDock Vina.33 The ligands displayed predicted attractive (negative) binding energies in the following order: −4.97 Kcal/mol (3WIX), −4.86 Kcal/mol (4HW3), −4.82 Kcal/mol (4ZBI), −4.74 Kcal/mol (4ZBF), −2.95 Kcal/mol (5FDO), −0.5 Kcal/mol (5FDR), and repulsive (positive) binding energy +16.6 Kcal/mol (ABT-737). Remarkably, the ligands were ranked in accordance with their experimental binding constants, namely, <1 nM (3WIX), ~10 nM (4HW3), ~10 nM (4ZBI), ~10 nM (4ZBF), ~100 nM (5FDO), ~100 nM (5FDR), and no binding (ABT-737). The small molecule, ABT-737, which does not bind to Mcl-1, provided a positive repulsive energy. Assuming that the positive binding energy was an error, the Pearson’s correlation coefficient of the predicted and observed binding is 0.90. Thus, in a small data set, the predictive capacity of our in silico assay was confirmed, and we set out to test it in large data sets.
Virtual docking with lead-like data set
Virtual docking of the ~2,300,000 lead-like molecules on the representative structure of Mcl-1 (model 1 of the PDB ID 2MHS) took ~7,200 hours. From these, the top 1,000 ligands (Supplementary materials, Table S2) with the lowest binding energy were selected for a more thorough screening using multiple conformations sampled by NMR, NMA, and MD. Virtual docking of the top 1,000 ligands on the 20 NMR ensemble structures of Mcl-1 (PDB ID 2MHS) took ~240 hours. Virtual docking of the top 1,000 ligands on the NMA distorted models of Mcl-1 took ~200 hours. Virtual docking of the top 1,000 ligands on the MD simulated models of Mcl-1 took ~150 hours. In all cases, the binding site on Mcl-1 was defined and limited by a box measuring 16, 18, and 20 Å (Figure 1).
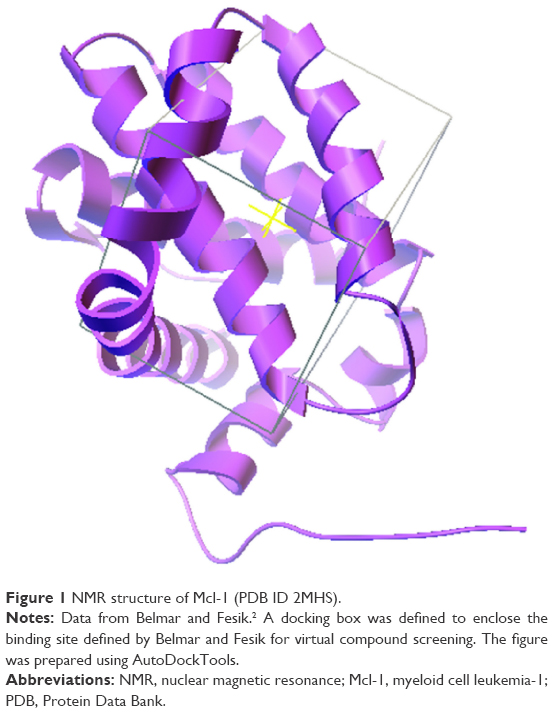 | Figure 1 NMR structure of Mcl-1 (PDB ID 2MHS). |
Table 1 lists the top 10 lead-like molecules (see Supplementary materials, Figure S2) that bind to each Mcl-1 conformation sampled by NMR, NMA, and MD. The top 10 lead-like molecules are ranked according to their average binding energy. Interestingly, the three conformations (NMR, NMA, and MD) give diverse top 10 ranked compounds.
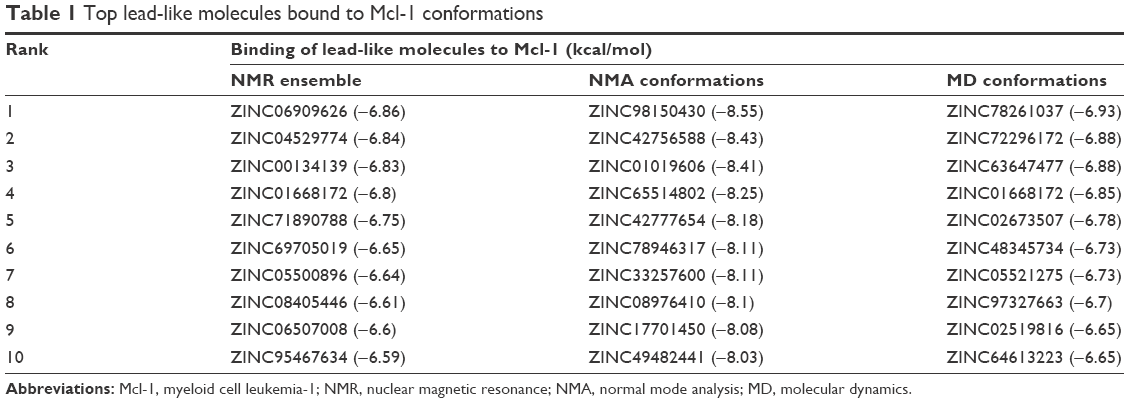 | Table 1 Top lead-like molecules bound to Mcl-1 conformations |
The top 10 ligands with the lowest average binding energies in the NMR conformation ensemble are ZINC06909626, ZINC04529774, ZINC00134139, ZINC01668172, ZINC71890788, ZINC69705019, ZINC05500896, ZINC08405446, ZINC06507008, and ZINC95467634.
ZINC00134139 is structurally related to epinastine. ZINC1019606 is structurally related to S1,37,38 a Bcl-2 family inhibitor that binds to Mcl-1 with nanomolar affinity. ZINC64613223 is structurally related to marinopyrrole A, a natural product isolated from an obligate marine Streptomyces that binds selectively to Mcl-1 (IC50 =10.1 μM) and induces apoptosis in Mcl-1-dependent leukemia39 and melanoma cells.40 ZINC2673507 is structurally related to the indole-2-carboxylic acid 33 (PDB ID 4HW2) that was a potent inhibitor of Mcl-1 (Kd =55 nM).
Virtual screening using lead-like molecules provides little information except the general skeleton of a ligand. In this study, the lead-like molecule mostly has two aromatic rings linked to a hydrocarbon chain. The lead-like molecules often display nitrogen in the hydrocarbon chain and in the polycyclic aromatic rings. In contrast to FDA-approved drugs, the lead-like molecules have little or no bioavailability and pharmacodynamic activity. The lead-like molecules also risk suffering from toxicity and metabolic instability. Thus, the only information provided by the lead-like molecules is the general skeleton that seems to be common to most Mcl-1 inhibitors to date.
Virtual docking with FDA-approved drug data set
Virtual docking of the 1,790 drugs approved by the FDA on the 20 ensemble structures of Mcl-1 (PDB ID 2MHS) took ~300 hours, on the NMA distorted models took ~150 hours, and on the MD simulated models ~80 hours.
Table 2 lists the top 10 FDA-approved drugs that bind to each Mcl-1 conformation ensemble sampled by NMR, NMA, and MD. The top 10 drugs are ranked according to their average binding energy. Remarkably, eight FDA-approved drugs are associated with cancer treatment and could become clinically relevant.
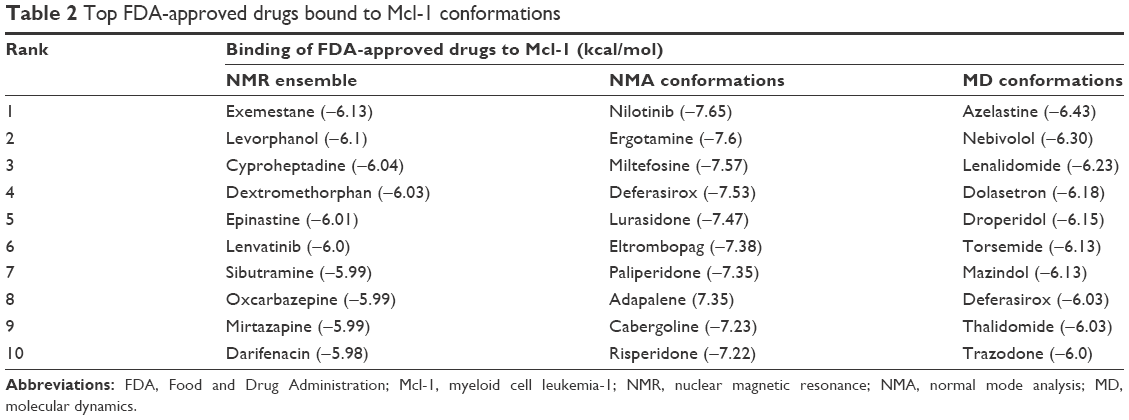 | Table 2 Top FDA-approved drugs bound to Mcl-1 conformations |
The top 10 ligands with the lowest average binding energies in the NMR conformation ensemble are exemestane, levorphanol, cyproheptadine, dextromethorphan, epinastine, lenvatinib, sibutramine, oxcarbazepine, mirtazapine, and darifenacin (Figure 2).
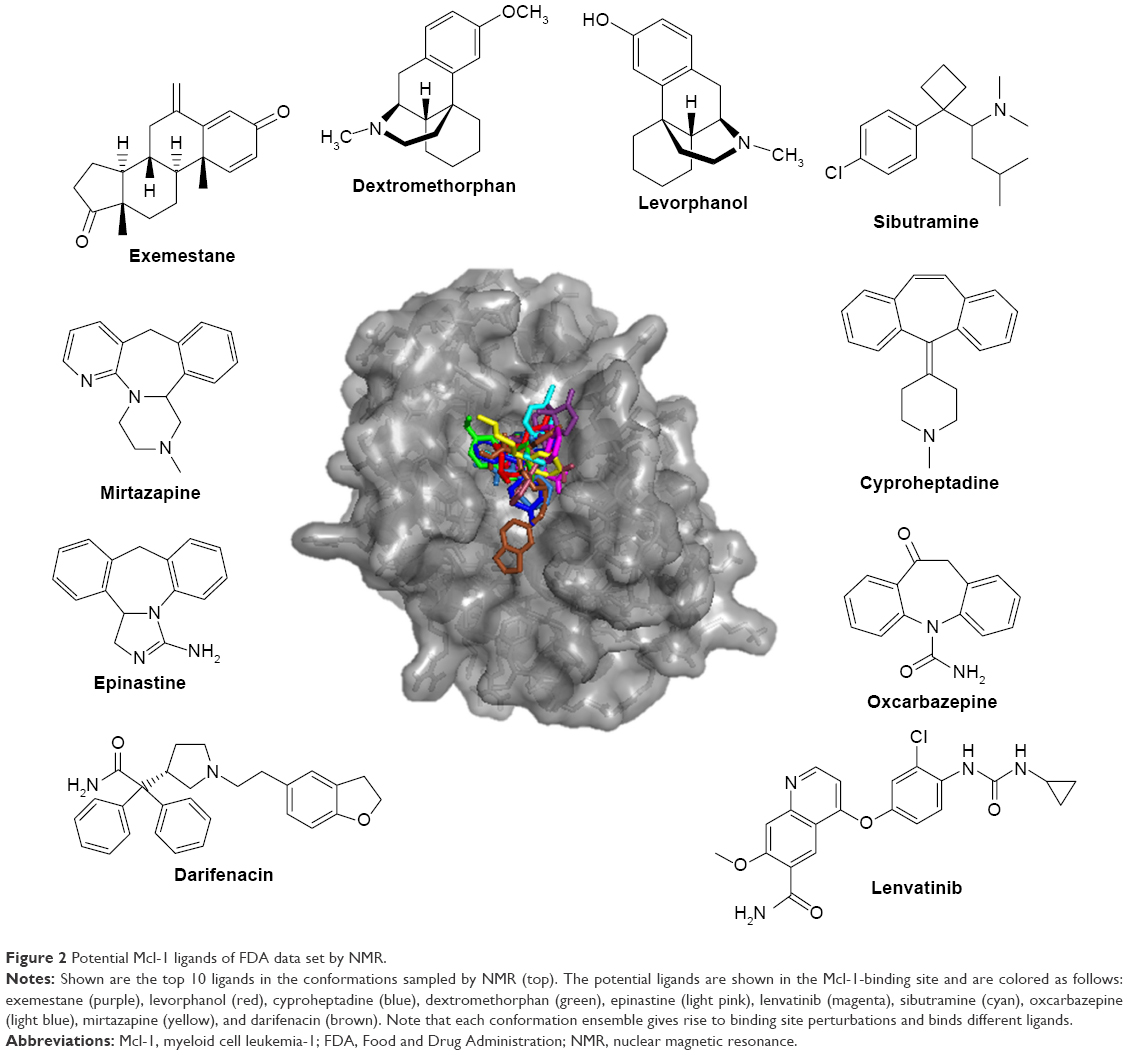 | Figure 2 Potential Mcl-1 ligands of FDA data set by NMR. |
Interestingly, two of these, exemestane and lenvatinib, are clinically used and approved for the treatment of cancer. Exemestane is a third-generation aromatase inhibitor administered in breast cancer,41 and lenvatinib is a vascular endothelial growth factor receptor 1 (VEGFR1), VEGFR2, and VEGFR3 kinase inhibitor administered in thyroid cancer.42 As such, exemestane and lenvatinib are favorable candidates for Mcl-1-targeted cancer therapies. Other drugs among the top 10 ligands include levorphanol, dextromethorphan, and sibutramine, which are morphine-like opioids with some noradrenergic and serotonergic activity. Notably, dextromethorphan is sometimes used to dull pain in cancer, and several clinical trials are currently evaluating its effect in peripheral neuropathy management (ie, ClinicalTrials.gov ID NCT02271893). Dextromethorphan could have an unexpected effect on the outcome of the chemotherapy treatment. Mirtazapine, cyproheptadine, oxcarbazepine, and epinastine are tricyclic compounds with histaminergic and serotonergic activity used to control pain and depression and as antiemetics in cancer. Notably, several clinical trials are currently evaluating them in palliative cancer management (ie, ClinicalTrials.gov IDs NCT01725048 and NCT02336750). If indeed Mcl-1 activity is experimentally shown, then these drugs could serve as adjuvants for cancer therapies. Finally, darifenacin is a muscarinic agonist used for urinary incontinence with little or no known relationship to cancer.
The top 10 ligands with the lowest average binding energies in the NMA conformation ensemble are nilotinib (−7.65), ergotamine (−7.6), miltefosine (−7.57), deferasirox (−7.53), lurasidone (−7.47), eltrombopag (−7.38), paliperidone (−7.35), adapalene (7.35), cabergoline (−7.23), and risperidone (−7.22) (Figure 3).
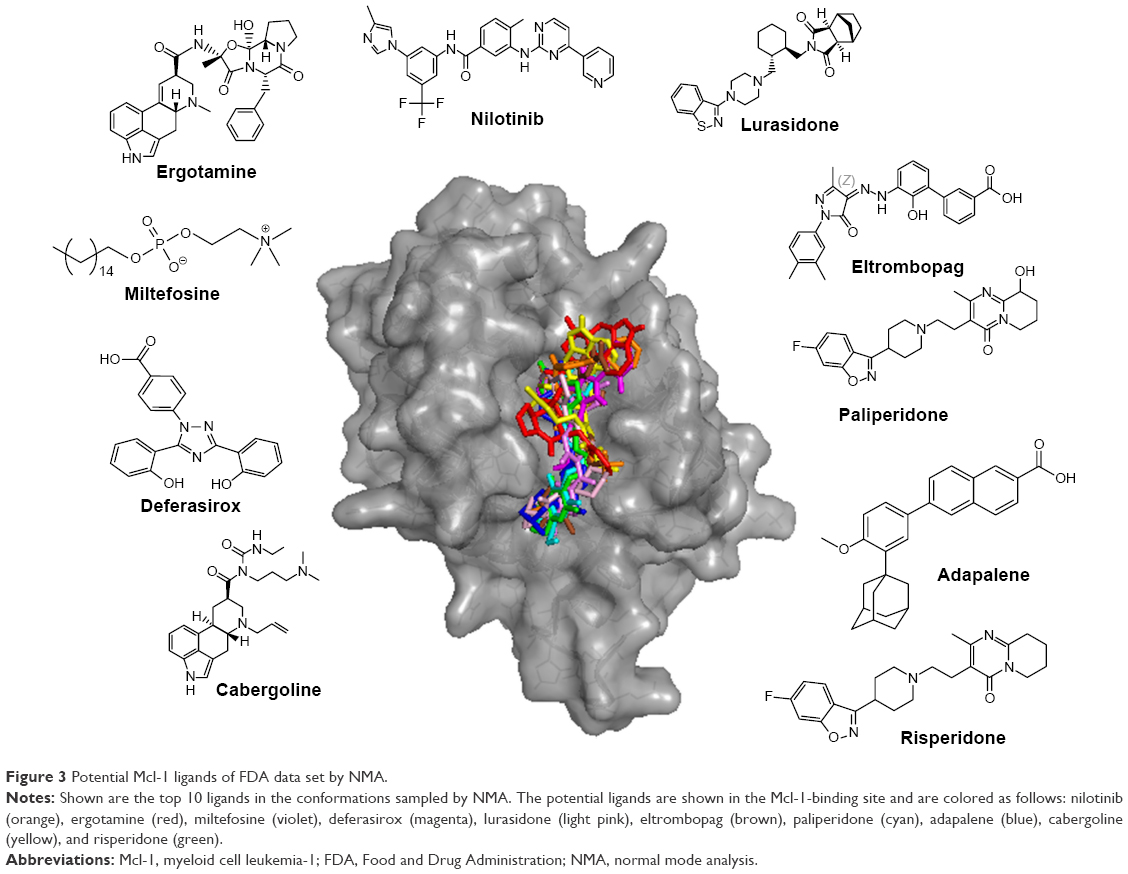 | Figure 3 Potential Mcl-1 ligands of FDA data set by NMA. |
Remarkably, four of these have been associated with the treatment of cancer, namely, nilotinib, cabergoline, miltefosine, and adapalene. Nilotinib is a selective Bcr-Abl tyrosine kinase inhibitor approved for the treatment of chronic myelogenous leukemia. Nilotinib, which shares some structural similarity with ABT-373, is currently undergoing clinical trials in combination with paclitaxel for the treatment of relapsed solid tumors (ClinicalTrials.gov ID NCT02379416). As such, nilotinib is a promising candidate for Mcl-1-targeted cancer therapies. Cabergoline is an ergot derivative and a potent D2 dopamine receptor agonist used in the treatment of prolactinomas and uterine fibroids.43 Cabergoline contains a tricyclic ring that is structurally related to the pan-Bcl-2 family inhibitor, S1,37,38 a Bcl-2 family inhibitor that binds to Mcl-1 and Bcl-2 with nanomolar affinity, disrupts Bax/Bcl-2 and Bak/Mcl-1 complexes, induces Bax/Bak-dependent apoptosis,37 increases oxidative stress,44 and disrupts the interaction of Beclin 1 with Bcl-2.45 Cabergoline is currently undergoing clinical trials in metastatic breast cancer as an adjuvant therapy of tamoxifen (ClinicalTrials.gov ID NCT01730729). Miltefosine is an alkylphosphocholine first studied as a treatment for cancer and approved for leishmaniasis in India. Adapalene is a third-generation topical retinoid that inhibits keratinocyte differentiation and proliferation. Of the top 10 drugs, four are atypical antipsychotics, namely, ergotamine, paliperidone, lurasidone, and risperidone, with serotonergic and dopaminergic activity. They are composed of a tricyclic head and an indole-like tail. Two other drugs do not seem to be associated with cancer and dopamine/serotonin pharmacology. These are deferasirox, an oral iron chelator, used to reduce chronic iron overload, and eltrombopag, a thrombopoietin receptor agonist used in the treatment of thrombocytopenia.
The top 10 ligands with the lowest average binding energies in the MD simulated conformation ensemble are azelastine, florbetapir, nebivolol, lenalidomide, dolasetron, droperidol, torsemide, mazindol, deferasirox, and thalidomide (Figure 4).
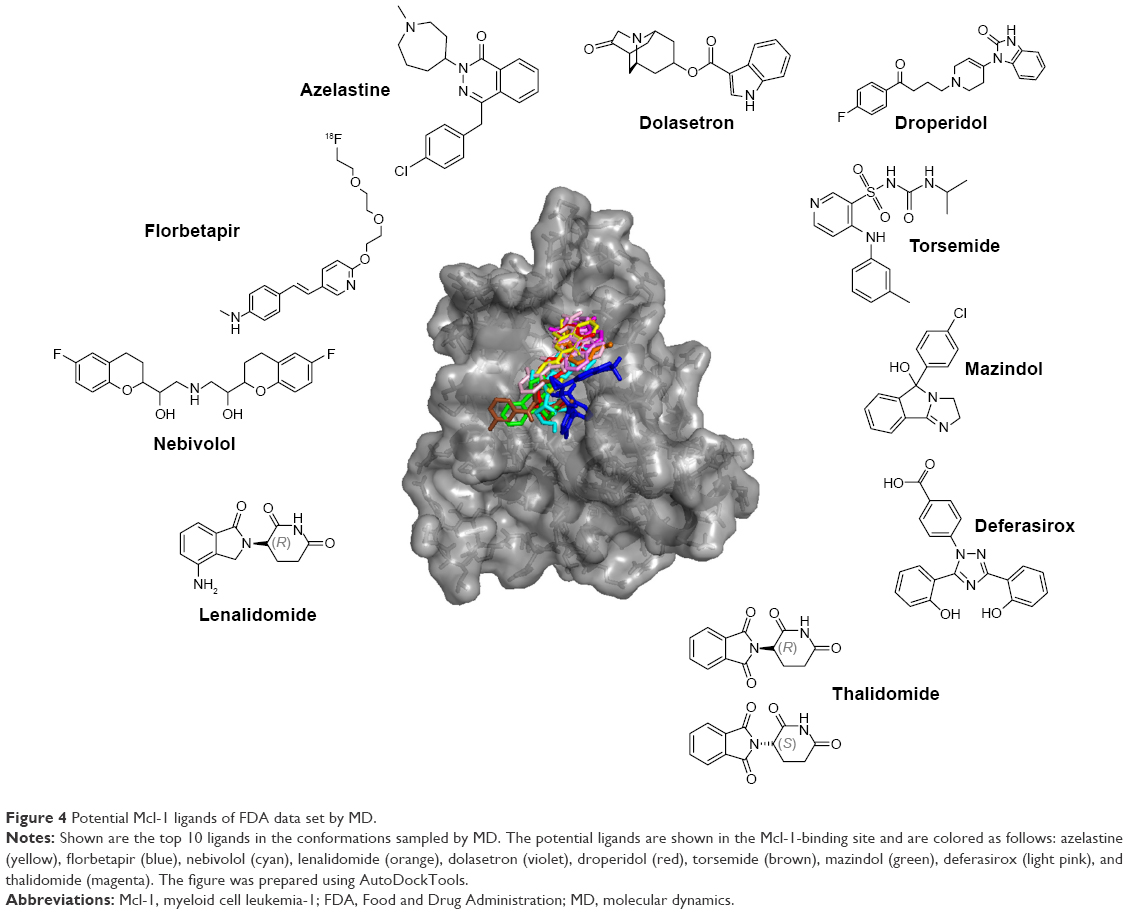 | Figure 4 Potential Mcl-1 ligands of FDA data set by MD. |
Remarkably, two of these are already associated with cancer therapy, namely, lenalidomide and thalidomide, which induce apoptosis and are used in the treatment of multiple myeloma and myelodysplastic syndromes.46,47 Thalidomide and lenalidomide are structural analogs of the rhodanine derivative, BH3I-1, developed by Degterev et al,48 which binds the BH3 binding groove of Mcl-1 with micromolar affinity. Of the top 10 drugs, at least three have dopaminergic and serotonergic activity. Dolasetron is a serotonergic and dopaminergic antagonist used against nausea, droperidol is an antidopaminergic antipsychotic, mazindol is a catecholamine reuptake inhibitor and a central nervous system (CNS) stimulant, trazodone is a serotonergic antagonist and reuptake inhibitor, and nebivolol is a β1-adrenergic receptor blocker with significant dopaminergic pharmacology.49 (Structurally, trazodone is of particular interest as it ranks first on average.) Finally, three other drugs do not seem to be linked with serotonergic and dopaminergic activity. Azelastine is a second-generation H1 histamine receptor antagonist, deferasirox is an oral iron chelator used to reduce chronic iron overload, and torsemide is a pyridine-sulfonylurea-type loop diuretic mainly used in the management of edema associated with congestive heart failure. Interestingly, torsemide shares structural similarity with ABT-737, particularly in the benzyl-sulfonyl-amide moiety, and mandates experimental investigation for its role as an Mcl-1 inhibitor.
Standard deviation of binding energies is low for ligands with few conformations
In Supplementary materials, Table S2, the binding energies of all ligands and their standard deviation are provided. The standard deviation characterizes the variation from the average ligand binding energy to 20 NMR, four MD, and six NMA conformations. AutoDock Vina was applied once for each Mcl-1 protein conformation, but by sampling multiple conformations, the signal-to-noise ratio of true positives is expected to increase. Ligands with low standard deviation of binding energies usually have few rotatable bonds, are rigid, and sometimes symmetric. For example, dihydroartemisinin and artemeter are rigidified by the polycyclic skeleton, whereas carbofenotion contains a C2 axis of pseudosymmetry. Contrarily, ligands with high standard deviations are highly flexible. The standard deviation is an indicator of entropy, and the more conformations a ligand may adopt, the more receptor poses are possible. Often, drug candidates with low standard deviation are preferred as binding is confined, but not always.
Comparison of NMR, NMA, and MD conformations
Remarkably, the rank order of the FDA-approved drug is comparable in all three conformation ensembles. FDA-approved drugs that rank high in the NMR ensemble also rank high in the MD and NMA conformations, and drugs that rank low in the NMR ensemble also have a low rank in the MD and NMA conformations. Figure 5 illustrates the agreement between the drug ranking in the conformations sampled by NMR, NMA, and MD.
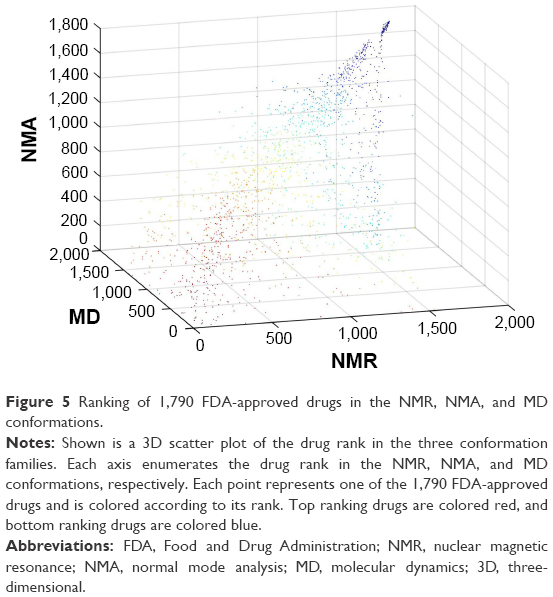 | Figure 5 Ranking of 1,790 FDA-approved drugs in the NMR, NMA, and MD conformations. |
Drug ranking of the 1,790 FDA-approved drugs in the NMR ensemble and in the MD conformations correlates well with a Pearson’s correlation coefficient of 0.87. Drug ranking in the NMR ensemble and in the NMA conformations correlates less well with a Pearson’s correlation coefficient of 0.76. Surprisingly, drug ranking in the NMA conformation and in the MD conformations does not correlate well with a Pearson’s correlation coefficient of 0.54. The latter correlation makes sense in light of the diverging paths of the NMA and MD conformations from the original NMR structure.
Discussion
This study provides a compendium of potential Mcl-1 inhibitors. The potential inhibitors were identified through virtual screening of FDA-approved drugs and lead-like molecules in multiple Mcl-1 conformations. The study is based on in silico observations and does not claim in vitro and in vivo activity. The study notes that many of the potential Mcl-1 inhibitors are currently undergoing clinical trials in the treatment of cancer, supports their use, and proposes a complementary mode of action. The study does not recommend clinical trials with any of the other potential Mcl-1 inhibitors. The study brings to light potential Mcl-1 inhibitors and exposes them to the scientific community for further investigation.
Supplementary mode of action
This study postulates an additional target for some FDA-approved drugs, which may in fact also act on Mcl-1. Some of these drugs are already undergoing clinical trials as noted earlier. As such, this study paves the way for more informed clinical trials, in vivo and in vitro studies, experimental binding assays, and so on. Importantly, if a drug is already approved by the FDA for the treatment of cancer, then the clinical use of the potential Mcl-1 inhibitors could be very promising.
Caveats
In our experience, we cannot know how a docked ligand will modulate the target structure and whether it will act as an agonist or an antagonist. In the past, eg, we inadvertently designed Mcl-1-binding peptides that induced proliferation instead of apoptosis (unpublished results). Here too, it remains unknown if the potential Mcl-1 inhibitors proposed herein act as agonists or antagonists in the Mcl-1-binding site. The ligands could either stabilize or destabilize a conformation required for interactions and apoptosis, and only experimental data in vitro and in vivo and clinical trials can answer this question.
In AutoDock Vina, ligand binding energies (ΔG) are predicted in kcal/mol. Importantly, however, the binding energies should be viewed as qualitative – and not quantitative – binding indicators. The individual value of a ligand’s binding energy is less informative than the binding energy relative to other ligands. This consideration should be kept in mind when analyzing ΔG values, and relative values (ΔΔG) are more useful.
Cell penetration
Since Mcl-1 is located inside the cytosol, the potential drugs must be able to cross the cell membrane. For most FDA-approved drug candidates listed here, some form of membrane crossing has been reported. For the lead-like compounds, however, cellular penetration data are unavailable, and only estimates based on the predicted octanol/water partition coefficient (LogP) are available.50
Serotonergic, dopaminergic, and histaminergic hypothesis
Serotonin, dopamine, and histamine regulate cell proliferation, migration, maturation, and apoptosis in a variety of cell types, including lung, kidney, endothelial cells, mast cells, neurons, and astrocytes.51,52 Some of the potential Mcl-1 ligands presented here are serotonergic, dopaminergic, and histaminergic modulators (ie, ergotamine, paliperidone, lurasidone, and risperidone) and share structural similarity with serotonin, dopamine, and histamine. We hypothesize that serotonin, dopamine, histamine, and Mcl-1 modulation are associated. Such a coincidence is further supported by the vicious cycle of depression, anxiety, and cancer, which are both characterized by abnormally low levels of serotonin and dopamine.53
Acknowledgments
We acknowledge the help of Prof Izhak Haviv for comments and guidance. We thank Gershon Kunin for system administration. We acknowledge Dr Fabian Glaser for his help with MD simulations. This study was made possible through grants of the Leir Foundation, Katz Foundation, and Ginzburg Family Fund to AOS.
Disclosure
The authors report no conflicts of interest in this work.
References
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. | ||
Belmar J, Fesik SW. Small molecule Mcl-1 inhibitors for the treatment of cancer. Pharmacol Ther. 2015;145:76–84. | ||
Beekman AM, O’Connell MA, Howell LA. Identification of small-molecule inhibitors of the antiapoptotic protein myeloid cell leukaemia-1 (Mcl-1). ChemMedChem. 2016;11(8):840–844. | ||
Oltersdorf T, Elmore SW, Shoemaker AR, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435(7042):677–681. | ||
Tse C, Shoemaker AR, Adickes J, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68(9):3421–3428. | ||
Du H, Wolf J, Schafer B, Moldoveanu T, Chipuk JE, Kuwana T. BH3 domains other than Bim and Bid can directly activate Bax/Bak. J Biol Chem. 2011;286(1):491–501. | ||
Beroukhim R, Mermel CH, Porter D, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463(7283):899–905. | ||
Wei G, Margolin AA, Haery L, et al. Chemical genomics identifies small-molecule MCL1 repressors and BCL-xL as a predictor of MCL1 dependency. Cancer Cell. 2012;21(4):547–562. | ||
Ding Q, He X, Xia W, et al. Myeloid cell leukemia-1 inversely correlates with glycogen synthase kinase-3beta activity and associates with poor prognosis in human breast cancer. Cancer Res. 2007;67(10):4564–4571. | ||
Song L, Coppola D, Livingston S, Cress D, Haura EB. Mcl-1 regulates survival and sensitivity to diverse apoptotic stimuli in human non-small cell lung cancer cells. Cancer Biol Ther. 2005;4(3):267–276. | ||
Krajewska M, Krajewski S, Epstein JI, et al. Immunohistochemical analysis of bcl-2, bax, bcl-X, and mcl-1 expression in prostate cancers. Am J Pathol. 1996;148(5):1567–1576. | ||
Miyamoto Y, Hosotani R, Wada M, et al. Immunohistochemical analysis of Bcl-2, Bax, Bcl-X, and Mcl-1 expression in pancreatic cancers. Oncology. 1999;56(1):73–82. | ||
Brotin E, Meryet-Figuiere M, Simonin K, et al. Bcl-XL and MCL-1 constitute pertinent targets in ovarian carcinoma and their concomitant inhibition is sufficient to induce apoptosis. Int J Cancer. 2010;126(4):885–895. | ||
Derenne S, Monia B, Dean NM, et al. Antisense strategy shows that Mcl-1 rather than Bcl-2 or Bcl-x(L) is an essential survival protein of human myeloma cells. Blood. 2002;100(1):194–199. | ||
Wei SH, Dong K, Lin F, et al. Inducing apoptosis and enhancing chemosensitivity to gemcitabine via RNA interference targeting Mcl-1 gene in pancreatic carcinoma cell. Cancer Chemother Pharmacol. 2008;62(6):1055–1064. | ||
Moulding DA, Giles RV, Spiller DG, White MR, Tidd DM, Edwards SW. Apoptosis is rapidly triggered by antisense depletion of MCL-1 in differentiating U937 cells. Blood. 2000;96(5):1756–1763. | ||
Taniai M, Grambihler A, Higuchi H, et al. Mcl-1 mediates tumor necrosis factor-related apoptosis-inducing ligand resistance in human cholangiocarcinoma cells. Cancer Res. 2004;64(10):3517–3524. | ||
Lill M. Virtual screening in drug design. Methods Mol Biol. 2013;993:1–12. | ||
Senderowitz H, Marantz Y. G protein-coupled receptors: target-based in silico screening. Curr Pharm Des. 2009;15(35):4049–4068. | ||
Shoichet BK. Virtual screening of chemical libraries. Nature. 2004;432(7019):862–865. | ||
Nichols SE, Baron R, Ivetac A, McCammon JA. Predictive power of molecular dynamics receptor structures in virtual screening. J Chem Inf Model. 2011;51(6):1439–1446. | ||
Moroy G, Sperandio O, Rielland S, et al. Sampling of conformational ensemble for virtual screening using molecular dynamics simulations and normal mode analysis. Future Med Chem. 2015;7(17):2317–2331. | ||
Levitt M, Sander C, Stern PS. Protein normal-mode dynamics: trypsin inhibitor, crambin, ribonuclease and lysozyme. J Mol Biol. 1985;181(3):423–447. | ||
Kolan D, Fonar G, Samson AO. Elastic network normal mode dynamics reveal the GPCR activation mechanism. Proteins. 2014;82(4):579–586. | ||
Greenberger MM, Samson AO. Normal mode dynamics of voltage-gated K channels: gating principle, opening mechanism, and inhibition. J Comput Neurosci. 2015;38(1):83–88. | ||
Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov. 2004;3(11):935–949. | ||
Anderson AC. The process of structure-based drug design. Chem Biol. 2003;10:787–797. | ||
Liu G, Poppe L, Aoki K, Yamane H, Lewis J, Szyperski T. High-quality NMR structure of human anti-apoptotic protein domain Mcl-1 (171–327) for cancer drug design. PLoS One. 2014;9(5):e96521. | ||
Suhre K, Sanejouand YH. ElNemo: a normal mode web server for protein movement analysis and the generation of templates for molecular replacement. Nucleic Acids Res. 2004;32(Web Server issue):W610–W614. | ||
Case DA, Cheatham TE 3rd, Darden T, et al. The Amber biomolecular simulation programs. J Comput Chem. 2005;26(16):1668–1688. | ||
Romero-Durana M, Pallara C, Glaser F, Fernandez-Recio J. Modeling binding affinity of pathological mutations for computational protein design. Methods Mol Biol. 2017;1529:139–159. | ||
Roe DR, Cheatham TE 3rd. PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J Chem Theory Comput. 2013;9(7):3084–3095. | ||
Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461. | ||
Irwin JJ, Shoichet BK. ZINC – a free database of commercially available compounds for virtual screening. J Chem Inf Model. 2005;45(1):177–182. | ||
Pihan E, Colliandre L, Guichou JF, Douguet D. e-Drug3D: 3D structure collections dedicated to drug repurposing and fragment-based drug design. Bioinformatics. 2012;28(11):1540–1541. | ||
Pelz NF, Bian Z, Zhao B, et al. Discovery of 2-Indole-acylsulfonamide myeloid cell leukemia 1 (Mcl-1) inhibitors using fragment-based methods. J Med Chem. 2016;59(5):2054–2066. | ||
Zhang BZ, Ren HY. [STI571 induces apoptosis of K562 cells through down-regulation of anti-apoptotic protein Mcl-1 and Bcl-xl expression]. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2007;15:1182–1185. | ||
Song T, Li X, Chang X, et al. 3-Thiomorpholin-8-oxo-8H-acenaphtho [1,2-b] pyrrole-9-carbonitrile (S1) derivatives as pan-Bcl-2-inhibitors of Bcl-2, Bcl-xL and Mcl-1. Bioorg Med Chem. 2013;21(1):11–20. | ||
Doi K, Li R, Sung SS, et al. Discovery of marinopyrrole A (maritoclax) as a selective Mcl-1 antagonist that overcomes ABT-737 resistance by binding to and targeting Mcl-1 for proteasomal degradation. J Biol Chem. 2012;287(13):10224–10235. | ||
Pandey MK, Gowda K, Doi K, Sharma AK, Wang HG, Amin S. Proteasomal degradation of Mcl-1 by maritoclax induces apoptosis and enhances the efficacy of ABT-737 in melanoma cells. PLoS One. 2013;8(11):e78570. | ||
Fabian CJ. The what, why and how of aromatase inhibitors: hormonal agents for treatment and prevention of breast cancer. Int J Clin Pract. 2007;61(12):2051–2063. | ||
Schlumberger M, Tahara M, Wirth LJ, et al. Lenvatinib versus placebo in radioiodine-refractory thyroid cancer. N Engl J Med. 2015;372(7):621–630. | ||
Odaka H, Numakawa T, Adachi N, et al. Cabergoline, dopamine D2 receptor agonist, prevents neuronal cell death under oxidative stress via reducing excitotoxicity. PLoS One. 2014;9(6):e99271. | ||
Soderquist R, Bates DJ, Danilov AV, Eastman A. Gossypol overcomes stroma-mediated resistance to the BCL2 inhibitor ABT-737 in chronic lymphocytic leukemia cells ex vivo. Leukemia. 2013;27(11):2262–2264. | ||
Zhong Y, Liao Y, Fang S, Tam JP, Liu DX. Up-regulation of Mcl-1 and Bak by coronavirus infection of human, avian and animal cells modulates apoptosis and viral replication. PLoS One. 2012;7(1):e30191. | ||
Maier SK, Hammond JM. Role of lenalidomide in the treatment of multiple myeloma and myelodysplastic syndrome. Ann Pharmacother. 2006;40(2):286–289. | ||
Mark TM, Bowman IA, Rossi AC, et al. Thalidomide, clarithromycin, lenalidomide and dexamethasone therapy in newly diagnosed, symptomatic multiple myeloma. Leuk Lymphoma. 2014;55(12):2842–2849. | ||
Degterev A, Lugovskoy A, Cardone M, et al. Identification of small-molecule inhibitors of interaction between the BH3 domain and Bcl-xL. Nat Cell Biol. 2001;3(2):173–182. | ||
Nade VS, Shendye NV, Kawale LA, Patil NR, Khatri ML. Protective effect of nebivolol on reserpine-induced neurobehavioral and biochemical alterations in rats. Neurochem Int. 2013;63(4):316–321. | ||
Irwin JJ, Sterling T, Mysinger MM, Bolstad ES, Coleman RG. ZINC: a free tool to discover chemistry for biology. J Chem Inf Model. 2012;52(7):1757–1768. | ||
Azmitia EC. Modern views on an ancient chemical: serotonin effects on cell proliferation, maturation, and apoptosis. Brain Res Bull. 2001;56(5):413–424. | ||
Deckx N, Lee WP, Berneman ZN, Cools N. Neuroendocrine immunoregulation in multiple sclerosis. Clin Dev Immunol. 2013;2013:705232. | ||
Hernandez-Reif M, Field T, Ironson G, et al. Natural killer cells and lymphocytes increase in women with breast cancer following massage therapy. Int J Neurosci. 2005;115(4):495–510. | ||
Morris GM, Huey R, Lindstrom W, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem. 2009;30(16):2785–2791. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.