Back to Journals » Journal of Inflammation Research » Volume 19
VIM Gene Variant Association to the Co-Occurrence of Livedoid Vasculopathy, Premature Canities, and Early-Onset Cataracts, and Preliminary Efficacy of Tofacitinib: A Case Report
Received 11 January 2026
Accepted for publication 23 April 2026
Published 16 May 2026 Volume 2026:19 589526
DOI https://doi.org/10.2147/JIR.S589526
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Anish R. Maskey
Zhujing Zhu, Huanru Qu
Department of Rheumatology and Immunology, Longhua Hospital Shanghai University of Traditional Chinese Medicine, Shanghai, 200032, People’s Republic of China
Correspondence: Huanru Qu, Email [email protected]
Background: Livedoid vasculopathy (LV) is a rare thrombotic vasculopathy characterized by ischemic lesions and painful skin ulcerations of the lower limbs. This report describes a male patient who presented with a novel co-presentation of LV, premature canities, and early-onset cataracts (EOC), a triad of symptoms not previously described in the context of vimentin (VIM) variants.
Case Presentation: A 37-year-old male with an 8-year history of histopathologically confirmed LV also presented with premature canities and EOC. His mother had similar manifestations, including LV, premature canities, and EOC, suggesting a possible genetic etiology. Whole-exome sequencing revealed a heterozygous variant in the VIM gene (chr10:17277378) in the patient, while his mother was also heterozygous, and the father was wild-type. After 3 months of tofacitinib therapy, the patient’s skin ulcers significantly improved.
Conclusion: This case report highlights the first described co-presentation of a VIM variant with the triad of LV, premature canities, and EOC. The observed clinical improvement with tofacitinib is a preliminary finding that warrants further investigation as a potential therapeutic option for LV in this unique clinical context.
Keywords: livedoid vasculopathy, early-onset cataract, premature canities, tofacitinib, case report
Introduction
Livedoid vasculopathy (LV) is a chronic, ulcerative dermatosis characterized by a relapsing-remitting clinical course. It predominantly affects the distal lower extremities, including the ankles and dorsal feet.1 A rare disorder with an estimated annual incidence of 1 per 100,000, LV demonstrates a marked female predominance (3:1 female-to-male ratio),2 with peak incidence occurring between 15 and 50 years of age and a mean age at presentation of approximately 32 years.3 Histopathology reveals endothelial cell proliferation and segmental hyalinization, fibrin thrombi within dermal vessels, and minimal leukocytoclastic vasculitis, indicative of a hypercoagulable state.4 While secondary LV may associate with autoimmune disorders (eg., lupus anticoagulant) or hypercoagulable conditions, approximately 50% of cases remain idiopathic, suggesting unidentified genetic drivers.
Recent research has identified the Vimentin (VIM) gene as a potential locus for a novel genetic syndrome.5 This study presents a case report of a 37-year-old male patient diagnosed with LV, premature canities, and early-onset cataracts.
Case Presentation
A 37-year-old male patient presented to Longhua Hospital Shanghai University of Traditional Chinese Medicine in December 2024 with a chief complaint of recurrent skin ulcers on both lower limbs persisting for over 8 years. Throughout this 8-year period, the patient experienced recurrent bilateral lower extremity cutaneous ulcers, which were accompanied by significant pain. The ulcers consistently healed with residual ivory-white atrophy and scarring. The patient’s mother reportedly developed the same triad of conditions (recurrent lower limb ulcers, premature canities, and cataracts) in her twenties, suggesting the possibility of an underlying inherited disorder (Figure 1).
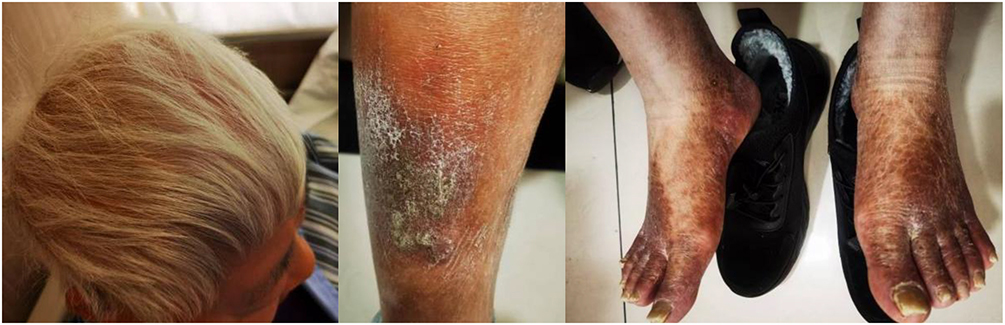 |
Figure 1 A mother with white hair and bilateral leg vasculitis. |
Upon presentation, the patient exhibited multiple painful ulcerations localized to the distal lower legs, ankles, and dorsal aspects of the feet. These lesions were surrounded by petechiae, ecchymoses, and a characteristic reticulated hyperpigmentation, and were notably covered by black eschar. The patient reported severe pain, with a Visual Analog Scale (VAS) score of 8 out of 10, which significantly impaired ambulation. Comprehensive investigations were performed, revealing a negative autoimmune serology panel (including ANA, ENA, RF, CCP, and ANCA). Infectious workup showed no evidence of relevant infections, such as tuberculosis or fungal pathogens, and lower extremity vascular ultrasound identified no significant abnormalities.
A skin biopsy performed in 2016 revealed histopathological features consistent with LV, including perivascular lymphocytic infiltration, fibrinoid necrosis, and occasional thrombus formation. The patient had received long-term treatment with prednisone combined with either hydroxychloroquine or mycophenolate mofetil for the skin ulcers, without significant clinical improvement. He also had a history of premature canities and cataracts, which began in his early 20s (Figure 2). To elucidate the underlying genetic mechanism, trio-based whole-exome sequencing (WES) was performed on the proband and both parents (the patient has no full siblings). The results revealed that the proband harbored a heterozygous variant in the VIM gene, specifically located at chr10:17277378 (Exon 7/10 of VIM), which is annotated as NM_003380.5: c.1219G>A (p.Glu407Lys) according to the standard human gene variation nomenclature. Segregation analysis confirmed: The mother is a heterozygous carrier of the same variant (Figure 3). The father carries the wild-type allele.
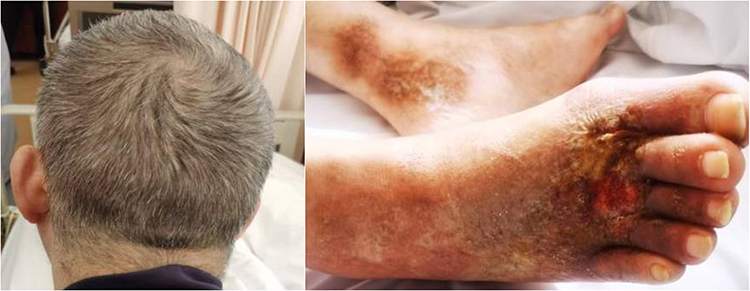 |
Figure 2 The patient with premature canities hair and bilateral leg vasculitis (skin ulceration). |
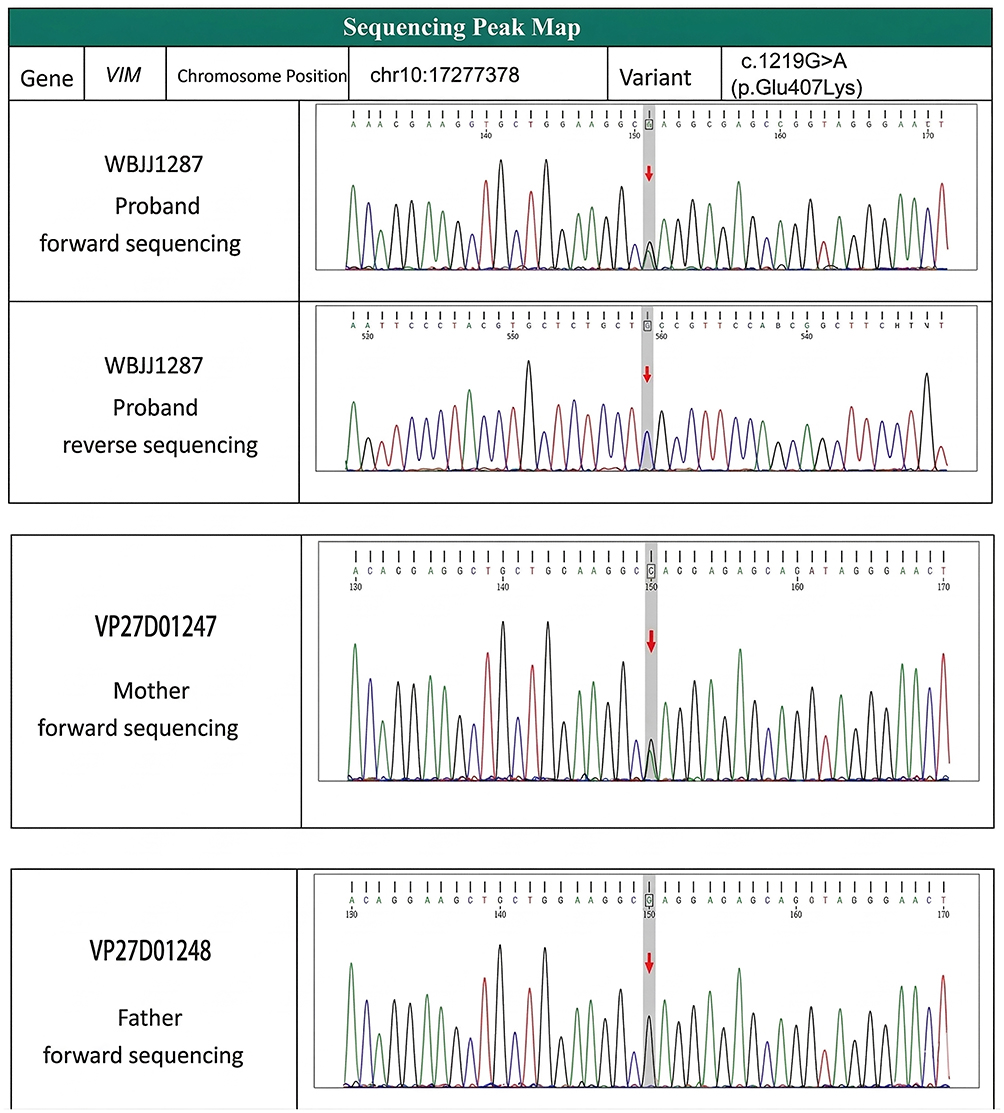 |
Figure 3 Sequencing chromatogram of the VIM gene variant (c.1219G>A) in the proband and his parents. |
Based on these findings, we speculate that the patient’s triad of manifestations may be associated with the VIM gene variant. The patient was initiated on treatment with tofacitinib, a Janus kinase (JAK) inhibitor, at a dose of 5 mg twice daily per os (bid, per os). Tofacitinib was selected for its ability to modulate inflammatory responses and ameliorate microvascular inflammation, with prior reports of efficacy in managing livedo racemosa (a feature associated with livedoid vasculopathy). Following 3 months of therapy, significant clinical improvement in the cutaneous ulcers was observed, characterized by complete re-epithelialization of the ulcerated areas. Pain severity was markedly reduced, with a VAS score decreasing to 2 out of 10 (from a baseline of 8). No new ulcerations developed during the treatment period (Figure 4). Additionally, serial ophthalmic assessments demonstrated no detectable progression of the patient’s cataracts. The patient continued to receive tofacitinib at a dose of 5 mg twice daily (bid) as of January 2026, with a total treatment duration of 14 months (from December 2024 to January 2026); the treatment demonstrated good long-term sustainability, with no recurrence of skin lesions and well-maintained therapeutic response during the follow-up period. However, no hair repigmentation was observed.
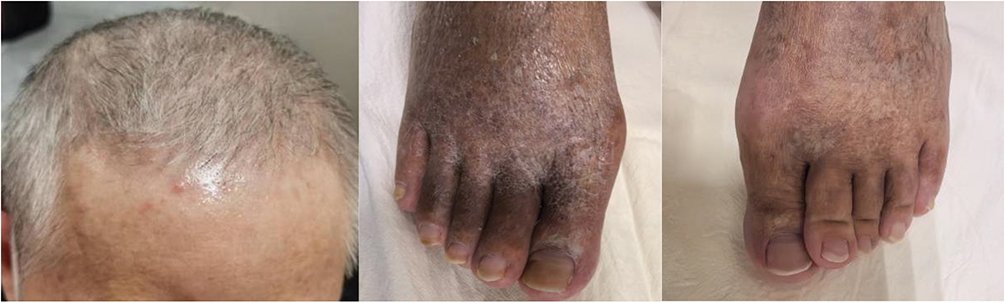 |
Figure 4 The patient’s post-treatment healing of the skin ulcer. |
Discussion
This case report has characterized a novel familial triad of LV, premature canities, and EOC, and reported that VIM gene variants may be the genetic factor for this disease. The successful therapeutic application of the JAK inhibitor tofacitinib, resulting in significant ulcer resolution and pain reduction in the proband. These observations have clinical significance by suggesting a potential targeted therapeutic approach.
LV is a rare, chronic vascular occlusive skin disease characterized by reticular purpura, painful ulcers, and white atrophy. The patient’s ulcers were located on the distal part of the calf, ankle, and dorsum of the foot, which is consistent with the typical clinical features of LV.6 Vimentin mutations have been associated not only with dominant congenital cataracts but also with multisystem progeroid syndromes characterized by premature aging. Missense mutations in the VIM gene have been identified in patients with dominant pulverulent congenital cataracts, caused by the disruption of the vimentin cytoskeleton in lens fiber cells.6,7 Furthermore, a de novo heterozygous missense variant (p.Leu387Pro) has been reported in association with a rare syndrome featuring premature aging, frontonasal dysostosis, abnormal fat distribution, and peripheral nervous system involvement.8 This case reveals a significantly broader phenotypic impact. We report a novel association linking a heterozygous VIM variant (c.1219G>A, p.Glu407Lys) to a unique triad: LV, premature canities, and early-onset cataract. This finding extends beyond the previously recognized associations of VIM variants with cataract and urothelial tumors,9 as no prior reports have connected VIM to LV or premature canities. The co-segregation of this triad in the proband and his mother strongly suggests the identified VIM variant serves as the shared genetic basis for all three manifestations. Mechanistically, vimentin – the intermediate filament protein encoded by VIM – is a critical cytoskeletal component essential for cellular integrity, migration, and signaling in diverse cell types.10,11 The identified VIM variant likely exerts pleiotropic effects across multiple tissues through compromised vimentin function. In endothelial cells, vimentin destabilization predisposes to fibrinoid degeneration, thrombosis, and microvascular inflammation—aligning with the LV pathology observed histopathologically.12 Concurrently, aberrant vimentin disrupts lens cytoskeletal architecture, directly explaining the bilateral cataracts.13 Furthermore, impaired vimentin function may dysregulate melanocyte differentiation or pigment transfer, underlying the familial premature canities. Thus, this single variant concurrently drives pathological changes in vascular, lenticular, and melanocytic tissues, culminating in the distinctive clinical triad.14
Tofacitinib is a selective JAK-STAT pathway inhibitor that primarily inhibits the activity of JAK1 and JAK3, thereby blocking the signaling of various cytokines. Although tofacitinib has been associated with an increased risk of venous thromboembolism, particularly at higher doses and in patients with pre-existing risk factors,15,16 its beneficial effects in this genetically defined LV case likely involve suppression of microvascular inflammation. Impaired vimentin function compromises endothelial stability, potentially sensitizing the microvasculature to inflammatory stimuli. Tofacitinib counters this by potently inhibiting JAK-STAT-dependent T cell activation and proliferation.17 This reduces T-cell recruitment and effector functions within vessel walls, mitigating the localized inflammatory response evident in the patient’s biopsy and alleviating pain. Additionally, vascular destabilization from VIM dysfunction may create a pro-angiogenic milieu. Tofacitinib directly targets this process by suppressing the production of key vascular endothelial growth factors (VEGF, TNC, GLS) and their downstream JAK-STAT signaling.18,19 Furthermore, as demonstrated in arthritis models, tofacitinib exerts direct anti-angiogenic effects,18 likely preventing the aberrant neovascularization associated with chronic ulceration in LV. Therefore, it should be clarified that, despite the known thrombotic risk—a consideration particularly relevant in a condition characterized by livedoid vasculopathy—the therapeutic benefits outweighed the risks in this case, leading to the resolution of dermatologic manifestations without the occurrence of ischemic-thrombotic ulceration.
Future studies should prioritize expanding pedigrees and screening for VIM variants in unrelated patients to validate genotype-phenotype correlations. Additionally, in vitro/vivo models (eg. CRISPR-edited cells or transgenic animals) should be used to confirm the variant’s functional impact on vascular, lens, and melanocyte biology. Further research should also explore alternative therapeutic agents like other JAK inhibitors through placebo-controlled trials to establish treatment efficacy. These efforts will clarify whether VIM defects constitute a distinct syndrome or part of a broader pathogenic network.
A 37-year-old male with LV, premature canities, and cataracts was found to have a heterozygous VIM gene variant, which was also present in his mother. This variant may be potentially associated with the co-occurrence of these symptoms by affecting vascular structure, lens, and pigment metabolism. Following treatment with tofacitinib, the patient’s skin ulcers showed significant improvement, suggesting that this agent is a possible therapeutic option for LV.
Data Sharing Statement
The anonymized data that support the findings of this study are available from the corresponding author (Huanru Qu) upon reasonable request.
Ethics Approval and Consent to Participate
This case report adheres to the CARE guidelines. I confirm that all methods were performed in accordance with the relevant guidelines. This work has been carried out in accordance with the Declaration of Helsinki of the World Medical Association. This study was approved by the Medical Ethics Committee of Longhua Hospital Shanghai University of Traditional Chinese Medicine. An official ethical approval number was not assigned for this case report.
Consent for Publication
Written informed consent was obtained from the patient for the publication of this case report and any accompanying images.
Author Contributions
Zhujing Zhu: Conceptualization, Investigation, Data curation, Writing - original draft.
Huanru Qu: Conceptualization, Methodology, Supervision, Writing - review and editing.
All authors gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
The authors declare that no funding was received for this study.
Disclosure
The authors declare that they have no competing interests.
References
1. Lacerda PN, Garcia LC, IFdS M, Miot HA, Abbade LPF. Livedoid vasculopathy, calciphylaxis, and Martorell’s hypertensive ulcer: update on ischemic ulcers due to impaired microcirculation of the lower limbs. An Bras Dermatol. 2025;100(2):215–6. doi:10.1016/j.abd.2024.09.004
2. Franco Marques G, Criado PR, TC ABM, Cajas García MS. The management of livedoid vasculopathy focused on direct oral anticoagulants (DOACs): four case reports successfully treated with rivaroxaban. Int J Dermatol. 2018;57(6):732–741. doi:10.1111/ijd.13997
3. Micieli R, Alavi A. Treatment for livedoid vasculopathy. JAMA Dermatol. 2018;154(2):237. doi:10.1001/jamadermatol.2017.4374
4. Soulages A, Maisonobe T, Auzou P, et al. Peripheral neuropathy and livedoid vasculopathy. J Neurol. 2022;269(7):3779–3788. doi:10.1007/s00415-022-11007-z
5. Schneider Y, Koller A, Schweigert A, et al. Vimentin network dysregulation mediates neurite deficits in SNCA duplication Parkinson’s patient-derived midbrain neurons. Sci Adv. 2025;11(23):eadq2742. doi:10.1126/sciadv.adq2742
6. Shakshouk H, Hines A, Kody S, Fett N, Alavi A, Ortega-Loayza AG. Inflammatory and vaso-occlusive ulcers: part I – clinical presentation and diagnosis. J Am Acad Dermatol. 2024;91(6):1035–1048. doi:10.1016/j.jaad.2024.01.083
7. Zhai Y, Li J, Yu W, et al. Targeted exome sequencing of congenital cataracts related genes: broadening the mutation spectrum and genotype-phenotype correlations in 27 Chinese Han families. Sci Rep. 2017;7(1):1219. doi:10.1038/s41598-017-01182-9
8. Benjamin C, Cogné J, Bouameur JE, et al. A dominant vimentin variant causes a rare syndrome with premature aging. Eur J Hum Genet. 2020;28:1218–1230. doi:10.1038/s41431-020-0583-2
9. Usman S, Waseem NH, Nguyen TKN, et al. Vimentin is at the heart of epithelial mesenchymal transition (EMT) mediated metastasis. Cancers. 2021;13(19):4985. doi:10.3390/cancers13194985
10. Vakhrusheva A, Endzhievskaya S, Zhuikov V, et al. The role of vimentin in directional migration of rat fibroblasts. Cytoskeleton. 2019;76(9–10):467–476. doi:10.1002/cm.21572
11. Strube F, Infanger M, Wehland M, et al. Alteration of cytoskeleton morphology and gene expression in human breast cancer cells under simulated microgravity. Cell J. 2020;22(1):106–114. doi:10.22074/cellj.2020.6537
12. Langlois B, Belozertseva E, Parlakian A, et al. Correction: vimentin knockout results in increased expression of sub-endothelial basement membrane components and carotid stiffness in mice. Sci Rep. 2018;8(1):10153. doi:10.1038/s41598-018-22187-y
13. Schaedel L, Lorenz C, Schepers AV, Klumpp S, Köster S. Vimentin intermediate filaments stabilize dynamic microtubules by direct interactions. Nat Commun. 2021;12(1):3701. doi:10.1038/s41467-021-23523-z
14. Heindl LM, Platzl C, Wolfmeier H, et al. Choroidal melanocytes: subpopulations of different origin? Ann Anat. 2021;238:151775. doi:10.1016/j.aanat.2021.151775
15. Charles-Schoeman C, Fleischmann R, Genovese MC, et al. Risk of venous thromboembolism with tofacitinib versus tumor necrosis factor inhibitors in cardiovascular risk-enriched rheumatoid arthritis patients. Arthritis Rheumatol. 2024;76:1218–1229. doi:10.1002/art.42846
16. Zhou J, Cai R, Peiguang Y, et al. Venous thromboembolism events associated with tofacitinib in rheumatoid arthritis patients: a real-world study from FAERS database. Naunyn Schmiedebergs Arch Pharmacol. 2025;399:3961–3967. doi:10.1007/s00210-025-04642-6
17. Hasni SA, Gupta S, Davis M, et al. Phase 1 double-blind randomized safety trial of the Janus kinase inhibitor tofacitinib in systemic lupus erythematosus. Nat Commun. 2021;12(1):3391. doi:10.1038/s41467-021-23361-z
18. Di Benedetto P, Ruscitti P, Berardicurti O, et al. Blocking Jak/STAT signalling using tofacitinib inhibits angiogenesis in experimental arthritis. Arthritis Res Ther. 2021;23(1):213. doi:10.1186/s13075-021-02587-8
19. Zisman D, Sabtan H, Rahat MM, et al. Tofacitinib regulates endostatin via effects on CD147 and cathepsin S. Int J Mol Sci. 2024;25(13):7267. doi:10.3390/ijms25137267
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.