Back to Journals » Biologics: Targets and Therapy » Volume 15
View Point: Disease Modification and Cell Secretome Based Approaches in Parkinson’s Disease: Are We on the Right Track?
Authors Müller T ![]()
Received 2 May 2021
Accepted for publication 19 July 2021
Published 29 July 2021 Volume 2021:15 Pages 307—316
DOI https://doi.org/10.2147/BTT.S267281
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Shein-Chung Chow
Thomas Müller
Department of Neurology, St. Joseph Hospital Berlin-Weissensee, Berlin, 13088, Germany
Correspondence: Thomas Müller
Department of Neurology, St. Joseph Hospital Berlin-Weißensee, Gartenstr. 1, Berlin, 13088, Germany
Tel +49 30 92790223
Fax +49 30 92790703
Email [email protected]; [email protected]
Abstract: The term idiopathic Parkinson’s disease describes an entity of various not well-characterized disorders resembling each other. They are characterized by chronic neuronal dying originating from various disease mechanisms. They result in the onset of motor and related non-motor features, both of which respond to administration of personalized drug combinations and surgical therapies. The unmet need is beneficial disease course modification with repair and neurogenesis. Objectives are to discuss the value of cell secretome based treatments including neuronal graft transplantation and to suggest as an alternative the stimulation of an endogenous available approach for neuronal repair. Chronic neurodegenerative processes result from different heterogeneous, but complementing metabolic, pathological cascade sequences. Accumulated evidence from experimental research suggested neuron transplantation, stem cell application and cell secretome-based therapies as a promising future treatment with cure as an ultimate goal. To date, clinical testing of disease-modifying treatments has focused on substitution or repair of the remaining dopamine synthesizing neurons following diagnosis. At diagnosis, many of the still surviving and functioning, but already affected neurons have lost most of their axons and are primed for cell death. A more promising therapeutic concept may be the stimulation of an existing, endogenous repair system in the peripheral and central nervous systems. The abundant protein repulsive guidance molecule A blocks restoration and neurogenesis, both of which are mediated via the neogenin receptor. Inhibition of the physiological effects of repulsive guidance molecule A is an endogenous available repair pathway in chronic neurodegeneration. Antagonism of this protein with antibodies or stimulation of the neogenin receptor should be considered as an initial repair step. It is an alternative to cell replacement, stem cell or associated cell secretome concepts.
Keywords: transplantation, dopamine, repulsive guidance molecule A, repair, chronic neurodegeneration, secretome, mesenchymal cells
Plain Language Summary
Experimental research provided extensive insights in the mechanisms of chronic neurodegeneration and developed concepts for new disease-modifying therapies.
Translation of these treatments into clinical practice failed in the heterogeneous disease entity Parkinson’s disease with its multiple pathophysiological origins of chronic neuronal dying.
As an alternative, antagonism of the repulsive guidance molecule A pathway is a uniform endogenous available repair and neurogenesis enhancing approach.
Stimulation of this mechanism should be considered for further future experimental and clinical research on disease modification in Parkinson’s disease.
Introduction to Parkinson’s Disease
Disease or Disease Entity?
Several lines of evidence suggest that the term “idiopathic Parkinson’s disease (PD)” does not reflect one disorder, but a disease entity. Its clinically not well-characterized subtypes resemble each other.1,2 More than 20 predisposing so-called “PD genes” with a different extent of penetration are currently known. To a certain extent, they are allocated as responsible components for onset of sporadic PD forms. Genetic alterations and mutations in familial PD, such as SNCA, PARK2, LRRK2, DJ-1, PINK-1, UCHL-1, only account for approximately 10% of idiopathic PD patients. Age of onset and clinical symptoms are variable, e.g. convincingly demonstrated in glucocerebrosidase (GBA) mutation carriers.3–7
Dopamine Decline: The Common Characteristic
All these PD forms have one feature in common. The essential, neurochemical characteristic is an ongoing progression of dopamine decline due to chronic death of dopamine-generating pigmented neurons in the substantia nigra pars compacta.2 This small structure is located deep in the core of the human midbrain. Dopamine is mainly synthesized from L-3,4-dihydroxyphenylalanine (Levodopa, L-dopa) after the hydroxylation of tyrosine.
Many Factors Cause the Rising Incidence of PD
Exposure to endogenous and exogenous toxins, such as pesticides or herbicides, paraquat, rotenone, various metals (e.g. iron, manganese, lead), gaseous compounds (such as carbon monoxide) and even viruses is discussed as a contributing factor for the future augmentation of PD cases.8,9 Generally, frequency of PD goes up with increasing age. Estimates of PD prevalence revealed a 2.4-fold increase in the past 30 years.10,11 The main reasons are the general rise in human life expectancy, a better public awareness and earlier diagnosis due to better instrumental diagnostic tools, i.e. functional imaging techniques, in combination with enhanced treatment possibilities, which prolong life expectancy with PD.12 Generally, the further future elevation of PD incidence will elevate the financial burden for health-care systems worldwide.11
Principles of Symptomatic Treatment in PD Patients
Considerable research activities in the past 60 years led to the development of therapies to alleviate PD symptoms. The success story of PD treatment was the introduction of the dopamine substitution concept for improvement of motor and to a considerable extent related non-motor symptoms in PD since the 1960s.13,14 The initial essential breakthrough was the implementation of L-dopa, the blood–brain barrier trespassing metabolic precursor of dopamine, for the treatment of PD. A debate on the use of L-dopa is still ongoing in the scientific community due to the onset of fluctuations of motor behaviour, acceleration of ageing processes and assumptions on L-dopa neurotoxicity.15,16 Particularly, plasma fluctuations of L-dopa, which are closely associated with dopamine oscillations in the synaptic cleft, counteract the well-accepted concept of “continuous dopaminergic stimulation” for the treatment of PD with its impairment of motor behaviour.17 There is convincing evidence that continuous stimulation of postsynaptic, nigrostriatal dopamine receptors is an essential precondition for nearly normal movement behaviour in PD patients. In contrast, persistent synaptic dopamine oscillations sooner or later cause onset of so-called motor complications.17 Their characteristics are changes between adequate motor behaviour, recurrence of motor impairment and involuntary movements, termed as dyskinesia.17 They are still the focus of current ongoing drug research in PD. Future continuous, subcutaneous L-dopa delivery to the brain by pump devices will improve this still popular issue of motor fluctuations considerably.18
The Resurgence of Research on Non-Motor Symptoms and Non-Dopaminergic Neurodegeneration
Clinical researchers have again occupied themselves with non-motor PD symptoms in more recent years. The many years of persisting focus on the dopamine deficiency in PD is now replaced by a more widespread view of altered, heterogeneous neurotransmission in PD patients again. Since the 1950s, it is known that an individual different, heterogeneous decline of neurotransmitters, such as serotonin (5-HT), norepinephrine etc., occurs in PD.19,20 The current resurgence of research on non-dopaminergic changes in PD also again discusses the importance of microglial activation and neuroinflammation as disease mechanisms.21–24 Both of them may hypothetically initiate the disease process, but may also occur as secondary, concomitant phenomena of chronic neurodegeneration. To date, extensive experimental and neuropathological research have provided distinct and better insights and understanding of chronic neuronal and associated glial cell death in the past fifty years. The predominant responsible, final mechanism cascades are now well identified.25
Disease Modification in PD is Not Available Yet
Based on the extensive knowledge on processes of neuronal dying, antiapoptotic, neuroprotective or oxidative stress reducing compounds, were successfully tested in experimental PD models.26–32 To date, these therapeutic approaches were more or less not successful in clinical trials with PD patients and accordingly they were not approved as a disease-modifying treatment in PD (e.g.33). Even studies on neuron transplantation or application of neuronal growth factors did not provide convincing benefits (important studies:34,35).
Objectives
Thus to date, the unmet needs are beneficial disease course modification and cure in PD. The objectives of this review are to discuss cell secretome-based therapeutic approaches including neuronal graft transplantation in PD and to describe a more uniform therapeutic approach for repair as an initial treatment step. A systematic literature search was not performed, as this article is a viewpoint.
Cure or Disease Modification by Dopamine Generating Cell Transplants?
Current available dopamine substituting treatment strategies only temporarily and dose-dependently improve the heterogeneous symptom complex characterized by motor and associated non-motor features during the chronic neurodegenerative process in PD patients.36
Transplants of Dopamine Synthesizing Cells
Replacement of dopamine-generating neurons in the striatum with fetal grafts or human embryonic stem cells was believed to provide symptomatic benefits and to help sparing of dopamine substitution dosing.37 Human, fetal ventral mesencephalon tissue was grafted as a source of dopamine-generating cells into PD patients. Clinical benefits were observed to a certain extent. Postmortem investigations demonstrated integration of these cells in the brain network and their subsequent long-term survival.38 The high incidence of graft-induced dyskinesia was attributed to the presence of 5-HT synthesizing neurons within the transplant. It may hypothetically also result from an uncontrolled dopamine synthesis by the transplants, which independently act and are not adapted to existing brain regulation systems of physiologic neurotransmission.39–41 A further drawback was the observed host-to-graft transfer of PD with uptake of host-derived α-synuclein followed by a subsequent aggregation in Lewy bodies.39,42–45 Generally, enrichment of misfolded α-synuclein in Lewy bodies is looked upon as one of the main responsible and important, neuronal, pathological phenomena in PD. It is well known, that failures within physiological activities of protein metabolism initiate protein degradation and -misfolding. These abnormalities are discussed as a PD specific process and are looked upon as responsible for PD onset and progression.46 However this pathological protein accumulation may also be the result of an unspecific side reaction of the metabolic cascade during chronic neurodegenerative processes. It may hypothetically only represent well-wrapped protein waste as a consequence of physiological defence mechanisms.46
Drawbacks of Tissue Grafting
The therapeutic value of grafting with fetal neuronal tissue in PD may additionally face ethical restrictions due to limited tissue supply.39,42–44 Embryonic porcine ventral mesencephalic tissue, autologous carotid body cells, respectively adrenal medullary tissue and even human retinal pigmentary epithelial cells were also considered.34,43,44 Availability, storage and self-renewal of cell sources appeared to be easier. Laboratory findings were successful in the case of immunological compatibility between the donor cells and the host.47 One has to consider, that different major histocompatibility complex antigens, respectively human leukocyte antigens, may counteract successful transplant integration. When not matched and no immunosuppressant drugs are applied, the transplant will be deemed foreign by the host and rejected by the adaptive human system. It initiates an immune response to remove foreign pathogens.48 In the case of CNS transplants, this reaction is more or less fatal. Thus graft rejection is still a major unresolved issue in the field and limits the successful translation into clinical use, i.e. in PD patients.49
Transplantation - A Promising Therapeutic Tool in the Real World?
However, the transplantation sooner or later of dopamine-generating cells as a replacement strategy faces some problems from the view of clinicians. At the time of PD diagnosis, 50–70% of dopamine-generating neurons in the nigrostriatal system are already gone. One hopes that regenerative therapy will stimulate the differentiation of new cells for the replacement of the ones that are already dead. Reactivation of cells, that are still viable but dysfunctional or in a dormant phase, is also promising. Protection with attenuation of the inflammatory processes, or stimulation of repair systems may be suitable therapeutic mechanisms.21–23,50,51 Replacement by transplantation will focus on dopamine-generating neurons only. Amelioration of motor symptoms is the focus. However, PD also affects other neurotransmitter systems and causes onset of a wide array of vegetative and non-motor features.52 PD is heterogeneous in terms of course and expression of symptoms. There are considerable doubts, whether this disease entity follows a fixed pattern of progression.2,52–56 Nowadays, new therapies are mainly experimentally established in uniform disease models in the laboratory. They aim on specific cell types only. Therefore the translational process will face serious problems due to the heterogeneity of PD patients.1,57,58 Accordingly, transplantation of dopamine-synthesizing cells provided benefits. They resemble the effects provided by a continuous L-dopa delivery, i.e. with the future available subcutaneous L-dopa infusion with a pump device.18 This approach or other currently applied dopamine substitution concepts allow a dosing adaptation to the ongoing neurodegenerative process. This is in contrast to transplantation. Sooner or later the grafts even shared α-synuclein enrichment. Moreover, necessary repeat adaptation to the probably further ongoing, rather heterogeneous, neurodegenerative process in the long term is complex.34,39,42,43,45,53 Thus in the end, the market and the acceptance by PD patients will decide, whether a simple future reversible treatment procedure, such as the subcutaneous L-dopa infusion with a to-be-developed simple device technique similar to insulin application in diabetes, will be favored by patients and payers compared with grafting treatment concepts. Accordingly, both therapies do not encounter the existing unmet need of cure or disease modification in PD in contrast to application of mesenchymal cells, respectively their products.
Stem Cells and Cell Secretome Based Therapies
Mesenchymal stem cells have a certain potential to serve as a valid therapeutic option with engraftment and subsequent differentiation capacity.59,60 They also deliver bioactive molecules. They are generally referred to as “the secretome”.61 It consists of proteins, such as cytokines, chemokines, and growth factors, lipids.62,63 Their functions are to promote cell survival and differentiation, prevent neuronal cell death, protect other cells from oxidative stress or regulate inflammatory processes.61,62,64 They improved motor impairment in animal models of PD, which mainly reflect the characteristics akinesia, rigidity and tremor and abnormal movement sequences, such as dyskinesia, to a certain extent.59,61,64,65
Cell Secretome
The use of secretome per se is easier compared with conventional stem-cell based techniques and applications.57 Manufacturing, storage and handling is easy. They are ready-to-use biological products. They do not require additional suppression of the immune system by adjuvant therapies.66–68 Therapeutic change to cell-specific effects is possible. Problems such as immune compatibility or induction of tumours, and respectively infection transmission are less.66,69 The secretome aims on disease modification.59,61,62,65 It may solve multiple regulation procedures of key biological processes. Thus it enables neuroprotection, neurodifferentiation or neuroinflammation.22,23 As an example, the secretome may enhance release of brain-derived neurotrophic factor, glial cell line-derived neurotrophic factor or neurotrophin 3.59,61,62,65 These neurotrophins are essential for protection and survival of dopamine-generating neurons. This was shown in experiments in animal models of PD induced by e.g. 6-hydroxydopamine- or rotenone exposure.61,62,65–68,70
Exosomes
Exosomes are bioactive molecules, which are released by mesenchymal stem cells.63 Exosomes are nano-sized extracellular vesicles and can be used as carriers of other therapeutic modalities. They only weakly stimulate defence by immune system activities. They avoid clearance by the reticuloendothelial system. They cross the blood–brain barrier due to their small size.61,62,65,67,68 Exosome exposure was shown as an efficacious mode of action following direct mesenchymal stem cell transplantation. Their beneficial therapeutic effects reduced loss of dopamine neurons with concomitant amelioration of the motor deficits in the 6-hydroxydopamine PD animal model.71–74
Conclusion
Experimental findings repeatedly offer promising and exciting future therapeutic opportunities, which were successfully tested in the laboratory. To date, none of the aforementioned therapies for PD modification or cure survived the translational process into clinical useful treatments for PD patients, and have not yet been approved. The unmet need for cure, disease modification, repair or regeneration still exists in PD. The list of past clinical trial failures is long.
Possible Causes for Failures
One simple reason is that chronic neurodegenerative processes result from different heterogeneous, but complementing metabolic, pathological cascades (fundamental findings:15,75,76; reviews:77,78). They end up in neuronal cell death-inducing events. They are well characterized. A typical example is apoptosis, which is the suicidal cell programme.25,79 However, the processes which initiate onset of PD or are responsible for the progress of chronic neuronal dying, probably vary in PD and have a multifactorial origin. Neuronal death in PD causes an individually pronounced and variable expression of motor and non-motor symptoms. Specific, individual different personality features, socioeconomic factors, such as education, etc., influence the acceptance, expression, adaptation and compensation of clinical deficits in PD patients. All these components complicate the translational process from bench to bedside. The term “neuroplasticity” is used as explaining the phenomenon of the differing capacity to compensate deleterious metabolic processes and to delay symptom onset.80 This heterogeneity in PD, even in more rare genetic PD forms, interferes with the value of assessment tools in clinical trials. High numbers of study participants may not counteract these dilution factors of possible positive effects resulting from a therapeutic disease-modifying intervention. To demonstrate the benefit of disease modification, validated clinical rating scales were used in the past, sometimes even in combination with functional imaging techniques, e.g. visualization of the dopamine neurotransmission in PD (as example:81). However, it is well known that rating scores are biased by examiners and by symptomatic drug effects, e.g. L-dopa or dopamine agonists.
The Importance of Early, Premotor Diagnosis
There is consensus that an essential precondition for PD course modification is an early diagnosis, at a time when the damage resulting from PD is low. Biomarkers or identification of a genetic predisposition for PD may be excellent screening instruments for “prodromal” PD diagnosis or PD-at risk-individuals before the onset of motor symptoms. To date, PD is diagnosed relatively late in the disease process. There are estimates that 60% of dopaminergic axons and 30% of nigral dopaminergic neurons are already gone, when motor symptoms appear.50 Thus in the clinical trials, testing of a disease-modifying therapy takes place with a focus on the remaining 70% dopamine synthesizing neurons in the most affected nigrostriatal area. Many of these still surviving and functioning, but already affected neurons have already lost most of their axons. They are primed for cell death. At this stage, a more promising therapeutic concept may be the stimulation of an existing, endogenous repair system in the peripheral and central nervous system.51,82–86
An Underestimated Concept: Stimulation of Repair?
Antagonism of the repulsive guidance molecule A (RGMa) pathway is worth more widespread consideration. It may serve as initial step for repair both in experimental and clinical research on disease modification in PD.
The RGMa Pathway and PD
Several lines of evidence suggest that RGMa is involved in PD pathophysiology. A RGMa protein increase in the substantia nigra was found following in situ hybridization and immunohistochemistry in neuromelanin positive neurons of post-mortem brain tissue, taken from L-dopa-treated PD patients.83 In view of the ongoing discussion on L-dopa neurotoxicity, one cannot exclude that this outcome may also be associated or at least aggravated by L-dopa administration and the L-dopa exposure associated oxidative stress generation.15,87 Evidence accumulates that RGMa, when located outside of cells, inhibits regeneration of axons and is involved in the acceleration of neuronal demise.88–90 Therefore inhibition of the RGMa pathway with antibodies or antagonism of the neogenin receptor activity, may initiate regenerative repair in the peripheral and central nervous system. In PD, an appropriate time will be after the diagnosis. It shall be an initial step to slow or stop progression of the ongoing disease process and to induce neuronal and glial repair.80,91–94 In view of the multifactorial pathophysiological events, which initiate the disease process in PD, this approach is a more general and thus uniform one.
Current Ongoing Disease Modifying Interventions in PD
Specific trials based on e.g. genetic findings, as for instance the currently tested concept convincingly demonstrated in GBA mutation carriers, are or will be soon be under way in clinical studies.3–7 Similar ones are the current ongoing trials with antibodies against misfolded proteins based on the corresponding neuropathological findings.29,95 Various drug mechanisms are discussed to reduce misfolded α-synuclein and thus disease progression. The focuses are boosting of autophagic/lysosomal clearance, reduction of α-synuclein mRNA by modulating histone deacetylase or RNA interference with decreased expression of α-synuclein.96 Other concepts aim on the impeding of the α-synuclein multimerization with heat shock proteins, dissociation of existing misfolded α-synuclein aggregates with small molecules, blocking of α-synuclein entry through receptor blocking, prevention of α-synuclein transport from cell to cell and immunotherapy with extracellular or intracellular neutralization.97 All these innovative approaches have one, essential, disadvantage. They are based on a more or less singular molecular pathology derived from a neuropathological postmortem finding. Clinical trials, which investigate an α-synuclein antibody, such as BIIB054, were already performed. They reported the pharmacokinetics, safety and tolerability in phase I, prasinezumab did not show relevant clinical benefits according to MDS-UPDRS outcomes.98–101
Inhibition of RGMa: The Future Concept?
In contrast RGMa antagonism considers the existing metabolic similarities both in the peripheral and central nervous system, as it was effective in other nervous system disorders according to experimental findings.51,86,89,102,103 Thus lowering of physiological RGMa effects restores neuronal function in the long term as a more general working concept for repair (Figure 1). It even weakens effects of toxin exposure.80,83,84,91,92,104–106 Currently, two different neutralizing RGMa antibodies (ABT-555 [elezanumab]; MT-3921) are investigated in phase 2 clinical trials in spinal cord injury. In addition ABT-555 is tested in phase 2 clinical trials in progressive and relapse-remitting multiple sclerosis and in ischaemic stroke. Positive results of these clinical studies will support the use of this repair strategy in the chronically damaged human nervous system, like in PD. Transplantation and cell secretome research in PD also aim on neurogenesis. It is worth mentioning, that inhibition of neurogenesis is performed by the RGMa-neogenin pathway, which probably also occurs in PD.51,82,86,107 Neurogenesis also takes place in the adult human brain, in the dentate gyrus or the subventricular zone. RGMa has been shown to block neurogenesis in these areas.89,108 One may postulate that aiming on RGMa function with antibodies may even stimulate neurogenesis in the adult human brain of PD patients. Thus elevation of neurogenesis may also improve motor and non-motor features in PD and beneficially influence the further course of PD. To date it is far from clear, how frequently and long this RGMa pathway modulating therapy has to be repeated in the further course of PD. Further experimental and then clinical research is warranted to test this approach. The future will be a combination of treatment of symptoms and repair following diagnosis (Figure 1). Negative long-term consequences are not known. More detailed pharmacokinetic investigations in humans are needed.
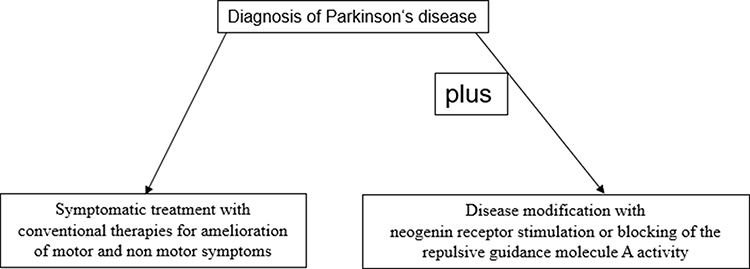 |
Figure 1 Concept for disease modification in Parkinson’s disease with repulsive guidance molecule A pathway modulation. |
Conclusion
RGMa antagonism is an alternative, more simple approach to cell replacement, stem cell and/or associated cell secretome concepts. It does not aim on specific cell types or disease mechanisms, since it represents a more uniform approach for the various disease mechanisms in chronic neurodegeneration, including the disease entity PD.1,81,109–114
Disclosure
The authors report no conflicts of interest in this work.
References
1. Weiner WJ. There is no Parkinson disease. Arch Neurol. 2008;65:705–708.
2. Przuntek H, Müller T, Riederer P. Diagnostic staging of Parkinson’s disease: conceptual aspects. J Neural Transm. 2004;111:201–216. doi:10.1007/s00702-003-0102-y
3. Balestrino R, Tunesi S, Tesei S, Lopiano L, Zecchinelli AL, Goldwurm S. Penetrance of Glucocerebrosidase (GBA) mutations in Parkinson’s disease: a Kin Cohort Study. Mov Disord. 2020;35:2111–2114. doi:10.1002/mds.28200
4. Greuel A, Trezzi JP, Glaab E, et al. GBA variants in Parkinson’s disease: clinical, metabolomic, and multimodal neuroimaging phenotypes. Mov Disord. 2020;35:2201–2210. doi:10.1002/mds.28225
5. Mullin S, Stokholm MG, Hughes D, et al. Brain microglial activation increased in Glucocerebrosidase (GBA) mutation carriers without Parkinson’s disease. Mov Disord. 2021;36:774–779.
6. Straniero L, Asselta R, Bonvegna S, et al. The SPID-GBA study: sex distribution, penetrance, incidence, and dementia in GBA-PD. Neurol Genet. 2020;6:e523. doi:10.1212/NXG.0000000000000523
7. Thaler A, Shenhar-Tsarfaty S, Shaked Y, et al. Metabolic syndrome does not influence the phenotype of LRRK2 and GBA related Parkinson’s disease. Sci Rep. 2020;10:9329. doi:10.1038/s41598-020-66319-9
8. Ahmed H, Abushouk AI, Gabr M, Negida A, Abdel-Daim MM. Parkinson’s disease and pesticides: a meta-analysis of disease connection and genetic alterations. Biomed Pharmacother. 2017;90:638–649. doi:10.1016/j.biopha.2017.03.100
9. Liu X, Ma T, Qu B, Ji Y, Liu Z. Pesticide-induced gene mutations and Parkinson disease risk: a meta-analysis. Genet Test Mol Biomarkers. 2013;17:826–832. doi:10.1089/gtmb.2013.0313
10. de Rijk MC, Launer LJ, Berger K, et al. Prevalence of Parkinson’s disease in Europe: a collaborative study of population-based cohorts. Neurologic Diseases in the Elderly Research Group. Neurology. 2000;54:S21–S23.
11. Deuschl G, Beghi E, Fazekas F, et al. The burden of neurological diseases in Europe: an analysis for the Global Burden of Disease Study 2017. Lancet Public Health. 2020;5:e551–e567. doi:10.1016/S2468-2667(20)30190-0
12. Armstrong MJ, Okun MS. Diagnosis and treatment of Parkinson disease: a review. JAMA. 2020;323:548–560. doi:10.1001/jama.2019.22360
13. Birkmayer W, Hornykiewicz O. [The L-3,4-dioxyphenylalanine (DOPA)-effect in Parkinson-akinesia]. Wien Klin Wochenschr. 1961;73:787–788. [German].
14. Cotzias GC, Papavasiliou PS, Gellene R. Modification of Parkinsonism–chronic treatment with L-dopa. N Engl J Med. 1969;280:337–345. doi:10.1056/NEJM196902132800701
15. Liedhegner EA, Steller KM, Mieyal JJ. Levodopa activates apoptosis signaling kinase 1 (ASK1) and promotes apoptosis in a neuronal model: implications for the treatment of Parkinson’s disease. Chem Res Toxicol. 2011;24:1644–1652. doi:10.1021/tx200082h
16. Müller T. Detoxification and antioxidative therapy for levodopa-induced neurodegeneration in Parkinson’s disease. Expert Rev Neurother. 2013;13:707–718. doi:10.1586/ern.13.50
17. Olanow CW, Obeso JA, Stocchi F. Continuous dopamine-receptor treatment of Parkinson’s disease: scientific rationale and clinical implications. Lancet Neurol. 2006;5:677–687. doi:10.1016/S1474-4422(06)70521-X
18. Ramot Y, Nyska A, Maronpot RR, et al. Ninety-day local tolerability and toxicity study of ND0612, a novel formulation of levodopa/ carbidopa, administered by subcutaneous continuous infusion in minipigs. Toxicol Pathol. 2017;45:764–773. doi:10.1177/0192623317729891
19. Gannon M, Che P, Chen Y, Jiao K, Roberson ED, Wang Q. Noradrenergic dysfunction in Alzheimer’s disease. Front Neurosci. 2015;9:220. doi:10.3389/fnins.2015.00220
20. Moll G, Gsell W, Wichart I, Jellinger K, Riederer P. Cholinergic and monoaminergic neuromediator systems in DAT. Neuropathological and neurochemical findings. In: Maurer K, Riederer P, Beckmann H, editors. Alzheimer’s Disease. Epidemiology, Neuropathology, Neurochemistry, and Clinics. Vienna: Springer; 1990:235–243.
21. Kuhn W, Müller T, Ilias N, Dieter Ρ. The neuroimmune hypothesis in Parkinson’s disease. Rev Neurosci. 1997;8:29–34. doi:10.1515/REVNEURO.1997.8.1.29
22. Gilhus NE, Deuschl G. Neuroinflammation - a common thread in neurological disorders. Nat Rev Neurol. 2019;15:429–430. doi:10.1038/s41582-019-0227-8
23. Hirsch EC, Standaert DG. Ten unsolved questions about neuroinflammation in Parkinson’s disease. Mov Disord. 2021;36:16–24. doi:10.1002/mds.28075
24. Lane CA, Hardy J, Schott JM. Alzheimer’s disease. Eur J Neurol. 2018;25:59–70. doi:10.1111/ene.13439
25. Toricelli M, Pereira AAR, Souza AG, et al. Mechanisms of neuroplasticity and brain degeneration: strategies for protection during the aging process. Neural Regen Res. 2021;16:58–67. doi:10.4103/1673-5374.286952
26. Eliash S, Shteter N, Eilam R. Neuroprotective effect of rasagiline, a monoamine oxidase-B inhibitor, on spontaneous cell degeneration in a rat model. J Neural Transm. 2005;112:991–1003. doi:10.1007/s00702-004-0254-4
27. Freestone PS, Chung KK, Guatteo E, Mercuri NB, Nicholson LF, Lipski J. Acute action of rotenone on nigral dopaminergic neurons–involvement of reactive oxygen species and disruption of Ca2+ homeostasis. Eur J Neurosci. 2009;30:1849–1859. doi:10.1111/j.1460-9568.2009.06990.x
28. Horger BA, Nishimura MC, Armanini MP, et al. Neurturin exerts potent actions on survival and function of midbrain dopaminergic neurons. J Neurosci. 1998;18:4929–4937. doi:10.1523/JNEUROSCI.18-13-04929.1998
29. Jamal F. Immunotherapies targeting alpha-synuclein in Parkinson disease. Fed Pract. 2020;37:375–379.
30. Kitani K, Minami C, Maruyama W, Kanai S, Ivy GO, Carrillo MC. Common properties for propargylamines of enhancing superoxide dismutase and catalase activities in the dopaminergic system in the rat: implications for the life prolonging effect of (-)deprenyl. In: Advances in Research on Neurodegeneration. Vienna: Springer; 2000:139–156.
31. Maruyama W, Takahashi T, Youdim M, Naoi M. The anti-Parkinson drug, rasagiline, prevents apoptotic DNA damage induced by peroxynitrite in human dopaminergic neuroblastoma SH-SY5Y cells. J Neural Transm. 2002;109:467–481. doi:10.1007/s007020200038
32. Naoi M, Maruyama W, Yi H, Akao Y, Yamaoka Y, Shamoto-Nagai M. Neuroprotection by propargylamines in Parkinson’s disease: intracellular mechanism underlying the anti-apoptotic function and search for clinical markers. In: Neuropsychiatric Disorders An Integrative Approach. Vienna: Springer; 2007:121–131.
33. Beal MF, Oakes D, Shoulson I, et al. A randomized clinical trial of high-dosage coenzyme Q10 in early Parkinson disease: no evidence of benefit. JAMA Neurol. 2014;71:543–552. doi:10.1001/jamaneurol.2014.131
34. Gross RE, Watts RL, Hauser RA, et al. Intrastriatal transplantation of microcarrier-bound human retinal pigment epithelial cells versus sham surgery in patients with advanced Parkinson’s disease: a double-blind, randomised, controlled trial. Lancet Neurol. 2011;10:509–519. doi:10.1016/S1474-4422(11)70097-7
35. Lang AE, Gill S, Patel NK, et al. Randomized controlled trial of intraputamenal glial cell line-derived neurotrophic factor infusion in Parkinson disease. Ann Neurol. 2006;59:459–466. doi:10.1002/ana.20737
36. Müller T, Öhm G, Eilert K, et al. Benefit on motor and non-motor behavior in a specialized unit for Parkinson’s disease. J Neural Transm. 2017;124:715–720. doi:10.1007/s00702-017-1701-3
37. Kopyov OV, Jacques D, Lieberman A, Duma CM, Rogers RL. Clinical study of fetal mesencephalic intracerebral transplants for the treatment of Parkinson’s disease. Cell Transplant. 1996;5:327–337. doi:10.1177/096368979600500221
38. Wenning GK, Odin P, Morrish P, et al. Short- and long-term survival and function of unilateral intrastriatal dopaminergic grafts in Parkinson’s disease. Ann Neurol. 1997;42:95–107. doi:10.1002/ana.410420115
39. Freed CR, Zhou W, Breeze RE. Dopamine cell transplantation for Parkinson’s disease: the importance of controlled clinical trials. Neurotherapeutics. 2011;8:549–561. doi:10.1007/s13311-011-0082-9
40. Politis M, Oertel WH, Wu K, et al. Graft-induced dyskinesias in Parkinson’s disease: high striatal serotonin/dopamine transporter ratio. Mov Disord. 2011;26:1997–2003. doi:10.1002/mds.23743
41. Politis M, Piccini P. In vivo imaging of the integration and function of nigral grafts in clinical trials. Prog Brain Res. 2012;200:199–220.
42. Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat Med. 2008;14:504–506. doi:10.1038/nm1747
43. Kordower JH, Goetz CG, Chu Y, et al. Robust graft survival and normalized dopaminergic innervation do not obligate recovery in a Parkinson disease patient. Ann Neurol. 2017;81:46–57. doi:10.1002/ana.24820
44. Olanow CW, Savolainen M, Chu Y, Halliday GM, Kordower JH. Temporal evolution of microglia and alpha-synuclein accumulation following foetal grafting in Parkinson’s disease. Brain. 2019;142:1690–1700. doi:10.1093/brain/awz104
45. Kordower JH, Dodiya HB, Kordower AM, et al. Transfer of host-derived alpha synuclein to grafted dopaminergic neurons in rat. Neurobiol Dis. 2011;43(3):552–557. doi:10.1016/j.nbd.2011.05.001
46. Sian-Hulsmann J, Monoranu C, Strobel S, Riederer P. Lewy bodies: a spectator or salient killer? CNS Neurol Disord Drug Targets. 2015;14:947–955. doi:10.2174/1871527314666150317225659
47. Tullis GE, Spears K, Kirk MD. Immunological barriers to stem cell therapy in the central nervous system. Stem Cells Int. 2014;2014:507905. doi:10.1155/2014/507905
48. Itakura G, Kawabata S, Ando M, et al. Fail-safe system against potential tumorigenicity after transplantation of iPSC derivatives. Stem Cell Rep. 2017;8:673–684. doi:10.1016/j.stemcr.2017.02.003
49. Simonson OE, Domogatskaya A, Volchkov P, Rodin S. The safety of human pluripotent stem cells in clinical treatment. Ann Med. 2015;47:370–380. doi:10.3109/07853890.2015.1051579
50. Cheng HC, Ulane CM, Burke RE. Clinical progression in Parkinson disease and the neurobiology of axons. Ann Neurol. 2010;67:715–725. doi:10.1002/ana.21995
51. Oda W, Fujita Y, Baba K, Moshizuki H, Niwa H, Yamashita T. Inhibition of repulsive guidance molecule-a protects dopaminergic neurons in a mouse model of Parkinson’s disease. Cell Death Dis. 2021;12:1–15. doi:10.1038/s41419-021-03469-2
52. Carlsson A. Biochemical and pharmacological aspects of Parkinsonism. Acta Neurol Scand Suppl. 1972;51:11–42.
53. Rabey JM, Yarden J, Dotan N, Mechlovich D, Riederer P, Youdim MBH. Creation of a gene expression classifier for predicting Parkinson’s disease rate of progression. J Neural Transm. 2020;127:755–762. doi:10.1007/s00702-020-02194-y
54. Braak H, Rub U, Gai WP, Del TK. Idiopathic Parkinson’s disease: possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J Neural Transm. 2003;110:517–536. doi:10.1007/s00702-002-0808-2
55. Halliday G, McCann H, Shepherd C. Evaluation of the Braak hypothesis: how far can it explain the pathogenesis of Parkinson’s disease? Expert Rev Neurother. 2012;12:673–686. doi:10.1586/ern.12.47
56. Jellinger KA. Is Braak staging valid for all types of Parkinson’s disease? J Neural Transm. 2019;126:423–431. doi:10.1007/s00702-018-1898-9
57. Nickels SL, Modamio J, Mendes-Pinheiro B, Monzel AS, Betsou F, Schwamborn JC. Reproducible generation of human midbrain organoids for in vitro modeling of Parkinson’s disease. Stem Cell Res. 2020;46:101870. doi:10.1016/j.scr.2020.101870
58. Weiner WJ. What do clinical trials tell us about treating patients. Parkinsonism Relat Disord. 2009;15:S34–S37. doi:10.1016/S1353-8020(09)70776-X
59. Mendes-Pinheiro B, Teixeira FG, Anjo SI, Manadas B, Behie LA, Salgado AJ. Secretome of undifferentiated neural progenitor cells induces histological and motor improvements in a rat model of Parkinson’s disease. Stem Cells Transl Med. 2018;7:829–838. doi:10.1002/sctm.18-0009
60. Pires AO, Teixeira FG, Mendes-Pinheiro B, Serra SC, Sousa N, Salgado AJ. Old and new challenges in Parkinson’s disease therapeutics. Prog Neurobiol. 2017;156:69–89.
61. Teixeira FG, Panchalingam KM, ssuncao-Silva R, et al. Modulation of the mesenchymal stem cell secretome using computer-controlled bioreactors: impact on neuronal cell proliferation, survival and differentiation. Sci Rep. 2016;6:27791. doi:10.1038/srep27791
62. Marques CR, Marote A, Mendes-Pinheiro B, Teixeira FG, Salgado AJ. Cell secretome based approaches in Parkinson’s disease regenerative medicine. Expert Opin Biol Ther. 2018;18:1235–1245. doi:10.1080/14712598.2018.1546840
63. Marote A, Teixeira FG, Mendes-Pinheiro B, Salgado AJ. MSCs-derived exosomes: cell-secreted nanovesicles with regenerative potential. Front Pharmacol. 2016;7:231. doi:10.3389/fphar.2016.00231
64. Mendes-Pinheiro B, Anjo SI, Manadas B, et al. Bone marrow mesenchymal stem cells’ secretome exerts neuroprotective effects in a Parkinson’s disease rat model. Front Bioeng Biotechnol. 2019;7:294. doi:10.3389/fbioe.2019.00294
65. Teixeira FG, Carvalho MM, Panchalingam KM, et al. Impact of the secretome of human mesenchymal stem cells on brain structure and animal behavior in a rat model of Parkinson’s disease. Stem Cells Transl Med. 2017;6:634–646. doi:10.5966/sctm.2016-0071
66. Assuncao-Silva RC, Mendes-Pinheiro B, Patricio P, et al. Exploiting the impact of the secretome of MSCs isolated from different tissue sources on neuronal differentiation and axonal growth. Biochimie. 2018;155:83–91. doi:10.1016/j.biochi.2018.07.026
67. Vizoso FJ, Eiro N, Cid S, Schneider J, Perez-Fernandez R. Mesenchymal stem cell secretome: toward cell-free therapeutic strategies in regenerative medicine. Int J Mol Sci. 2017;18:1852. doi:10.3390/ijms18091852
68. Vizoso FJ, Eiro N, Costa L, et al. Mesenchymal stem cells in homeostasis and systemic diseases: hypothesis, evidences, and therapeutic opportunities. Int J Mol Sci. 2019;20:3738. doi:10.3390/ijms20153738
69. Pires AO, Mendes-Pinheiro B, Teixeira FG, et al. Unveiling the differences of secretome of human bone marrow mesenchymal stem cells, adipose tissue-derived stem cells, and human umbilical cord perivascular cells: a proteomic analysis. Stem Cells Dev. 2016;25:1073–1083. doi:10.1089/scd.2016.0048
70. Ji C, Xue GF, Lijun C, et al. A novel dual GLP-1 and GIP receptor agonist is neuroprotective in the MPTP mouse model of Parkinson’s disease by increasing expression of BNDF. Brain Res. 2016;1634:1–11. doi:10.1016/j.brainres.2015.09.035
71. Li Q, Wang Z, Xing H, Wang Y, Guo Y. Exosomes derived from miR-188-3p-modified adipose-derived mesenchymal stem cells protect Parkinson’s disease. Mol Ther Nucleic Acids. 2021;23:1334–1344. doi:10.1016/j.omtn.2021.01.022
72. Pinnell JR, Cui M, Tieu K. Exosomes in Parkinson disease. J Neurochem. 2021;157:413–428.
73. Sun T, Ding ZX, Luo X, Liu QS, Cheng Y. Blood exosomes have neuroprotective effects in a mouse model of Parkinson’s disease. Oxid Med Cell Longev. 2020;2020:3807476. doi:10.1155/2020/3807476
74. Yang J, Luo S, Zhang J, et al. Exosome-mediated delivery of antisense oligonucleotides targeting alpha-synuclein ameliorates the pathology in a mouse model of Parkinson’s disease. Neurobiol Dis. 2021;148:105218. doi:10.1016/j.nbd.2020.105218
75. Kumar P, Jha NK, Jha SK, Ramani K, Ambasta RK. Tau phosphorylation, molecular chaperones, and ubiquitin E3 ligase: clinical relevance in Alzheimer’s disease. J Alzheimers Dis. 2015;43:341–361. doi:10.3233/JAD-140933
76. Naoi M, Maruyama W, Yi H, Inaba K, Akao Y, Shamoto-Nagai M. Mitochondria in neurodegenerative disorders: regulation of the redox state and death signaling leading to neuronal death and survival. J Neural Transm. 2009;116:1371–1381. doi:10.1007/s00702-009-0309-7
77. Chaplot K, Jarvela TS, Lindberg I. Secreted chaperones in neurodegeneration. Front Aging Neurosci. 2020;12:268. doi:10.3389/fnagi.2020.00268
78. Gracia P, Camino JD, Volpicelli-Daley L, Cremades N. Multiplicity of alpha-synuclein aggregated species and their possible roles in disease. Int J Mol Sci. 2020;21:8043. doi:10.3390/ijms21218043
79. Boonman Z, Isacson O. Apoptosis in neuronal development and transplantation: role of caspases and trophic factors. Exp Neurol. 1999;156:1–15. doi:10.1006/exnr.1999.7056
80. Mothe AJ, Coelho M, Huang L, et al. Delayed administration of the human anti-RGMa monoclonal antibody elezanumab promotes functional recovery including spontaneous voiding after spinal cord injury in rats. Neurobiol Dis. 2020;143:104995. doi:10.1016/j.nbd.2020.104995
81. Whone AL, Watts RL, Stoessl AJ, et al. Slower progression of Parkinson’s disease with ropinirole versus levodopa: the REAL-PET study. Ann Neurol. 2003;54:93–101. doi:10.1002/ana.10609
82. Isaksen TJ, Yamashita T. Repulsive guidance molecule A regulates adult neurogenesis via the neogenin receptor. Neurosci Insights. 2020;15:2633105520948481. doi:10.1177/2633105520948481
83. Korecka JA, Moloney EB, Eggers R, et al. Repulsive Guidance Molecule a (RGMa) induces neuropathological and behavioral changes that closely resemble Parkinson’s disease. J Neurosci. 2017;37:9361–9379. doi:10.1523/JNEUROSCI.0084-17.2017
84. Muller T, Barghorn S, Lutge S, et al. Decreased levels of repulsive guidance molecule A in association with beneficial effects of repeated intrathecal triamcinolone acetonide application in progressive multiple sclerosis patients. J Neural Transm. 2015;122:841–848. doi:10.1007/s00702-014-1308-x
85. Robinson RA, Griffiths SC, van de Haar LL, et al. Simultaneous binding of guidance cues NET1 and RGM blocks extracellular NEO1 signaling. Cell. 2021;184:2103–2120.e31. doi:10.1016/j.cell.2021.02.045
86. Satoh J, Tabunoki H, Ishida T, Saito Y, Arima K. Accumulation of a repulsive axonal guidance molecule RGMa in amyloid plaques: a possible hallmark of regenerative failure in Alzheimer’s disease brains. Neuropathol Appl Neurobiol. 2013;39:109–120. doi:10.1111/j.1365-2990.2012.01281.x
87. Muller T, Trommer I, Muhlack S, Mueller BK. Levodopa increases oxidative stress and repulsive guidance molecule A levels: a pilot study in patients with Parkinson’s disease. J Neural Transm. 2016;123:401–406. doi:10.1007/s00702-016-1519-4
88. Babitt JL, Zhang Y, Samad TA, et al. Repulsive guidance molecule (RGMa), a DRAGON homologue, is a bone morphogenetic protein co-receptor. J Biol Chem. 2005;280:29820–29827. doi:10.1074/jbc.M503511200
89. Key B, Lah GJ. Repulsive guidance molecule A (RGMa): a molecule for all seasons. Cell Adh Migr. 2012;6:85–90. doi:10.4161/cam.20167
90. Malinauskas T, Peer TV, Bishop B, Mueller TD, Siebold C. Repulsive guidance molecules lock growth differentiation factor 5 in an inhibitory complex. Proc Natl Acad Sci USA. 2020;117:15620–15631. doi:10.1073/pnas.2000561117
91. Kubo T, Tokita S, Yamashita T. Repulsive guidance molecule-a and demyelination: implications for multiple sclerosis. J Neuroimmune Pharmacol. 2012;7:524–528. doi:10.1007/s11481-011-9334-z
92. Mothe AJ, Tassew NG, Shabanzadeh AP, et al. RGMa inhibition with human monoclonal antibodies promotes regeneration, plasticity and repair, and attenuates neuropathic pain after spinal cord injury. Sci Rep. 2017;7:10529. doi:10.1038/s41598-017-10987-7
93. Charish J, Shabanzadeh AP, Chen D, et al. Neogenin neutralization prevents photoreceptor loss in inherited retinal degeneration. J Clin Invest. 2020;130:2054–2068. doi:10.1172/JCI125898
94. Shabanzadeh AP, Tassew NG, Szydlowska K, et al. Uncoupling Neogenin association with lipid rafts promotes neuronal survival and functional recovery after stroke. Cell Death Dis. 2015;6:e1744. doi:10.1038/cddis.2015.109
95. Castonguay AM, Gravel C, Lévesque M. Treating Parkinson's Disease with Antibodies: Previous Studies and Future Directions. J Parkinsons Dis. 2021;11(1):71–92. doi:10.3233/JPD-202221
96. Emamzadeh FN, Surguchov A. Parkinson’s disease: biomarkers, treatment, and risk factors. Front Neurosci. 2018;12:612. doi:10.3389/fnins.2018.00612
97. Fields CR, Bengoa-Vergniory N, Wade-Martins R. Targeting alpha-synuclein as a therapy for Parkinson’s disease. Front Mol Neurosci. 2019;12:299. doi:10.3389/fnmol.2019.00299
98. Teng JS, Ooi YY, Chye SM, Ling APK, Koh RY. Immunotherapies for Parkinson’s disease: progression of clinical development. CNS Neurol Disord Drug Targets. 2021;20. Epub ahead of print. doi:10.2174/1871527320666210526160926
99. Brys M, Fanning L, Hung S, et al. Randomized phase I clinical trial of anti-alpha-synuclein antibody BIIB054. Mov Disord. 2019;34:1154–1163. doi:10.1002/mds.27738
100. Meissner WG, Traon AP, Foubert-Samier A, et al. A phase 1 randomized trial of specific active alpha-synuclein immunotherapies PD01A and PD03A in multiple system atrophy. Mov Disord. 2020;35:1957–1965. doi:10.1002/mds.28218
101. Volc D, Poewe W, Kutzelnigg A, et al. Safety and immunogenicity of the alpha-synuclein active immunotherapeutic PD01A in patients with Parkinson’s disease: a randomised, single-blinded, phase 1 trial. Lancet Neurol. 2020;19:591–600. doi:10.1016/S1474-4422(20)30136-8
102. Song M, Tian F, Xia H, Xie Y. Repulsive guidance molecule a suppresses seizures and mossy fiber sprouting via the FAKp120RasGAPRas signaling pathway. Mol Med Rep. 2019;19:3255–3262.
103. Tanabe S, Yamashita T. Repulsive guidance molecule-a is involved in Th17-cell-induced neurodegeneration in autoimmune encephalomyelitis. Cell Rep. 2014;9:1459–1470. doi:10.1016/j.celrep.2014.10.038
104. Chen J, Shifman MI. Inhibition of neogenin promotes neuronal survival and improved behavior recovery after spinal cord injury. Neuroscience. 2019;408:430–447. doi:10.1016/j.neuroscience.2019.03.055
105. Nakagawa H, Ninomiya T, Yamashita T, Takada M. Treatment with the neutralizing antibody against repulsive guidance molecule-a promotes recovery from impaired manual dexterity in a primate model of spinal cord injury. Cereb Cortex. 2019;29:561–572. doi:10.1093/cercor/bhx338
106. Yang W, Sun P. Promoting functions of microRNA-29a/199B in neurological recovery in rats with spinal cord injury through inhibition of the RGMA/STAT3 axis. J Orthop Surg Res. 2020;15:427. doi:10.1186/s13018-020-01956-4
107. Isaksen TJ, Fujita Y, Yamashita T. Repulsive guidance molecule A suppresses adult neurogenesis. Stem Cell Rep. 2020;14:677–691. doi:10.1016/j.stemcr.2020.03.003
108. Tian C, Shi H, Xiong S, Hu F, Xiong WC, Liu J. The neogenin/DCC homolog UNC-40 promotes BMP signaling via the RGM protein DRAG-1 in C. elegans. Development. 2013;140:4070–4080. doi:10.1242/dev.099838
109. Guarnieri G, Sarchielli E, Vannelli GB, Morelli A. Cell-based therapy in Alzheimer’s disease: can human fetal cholinergic neurons “untangle the skein”? Neural Regen Res. 2018;13:2105–2107. doi:10.4103/1673-5374.241459
110. Liu Z, Cheung HH. Stem cell-based therapies for Parkinson disease. Int J Mol Sci. 2020;21:8060. doi:10.3390/ijms21218060
111. Schweyer K, Ruschoff-Steiner C, Arias-Carrion O, Oertel WH, Rosler TW, Hoglinger GU. Neuronal precursor cells with dopaminergic commitment in the rostral migratory stream of the mouse. Sci Rep. 2019;9:13359. doi:10.1038/s41598-019-49920-5
112. Desplats P, Spencer B, Crews L, et al. Alpha-synuclein induces alterations in adult neurogenesis in Parkinson disease models via p53-mediated repression of Notch1. J Biol Chem. 2012;287:31691–31702. doi:10.1074/jbc.M112.354522
113. Winner B, Regensburger M, Schreglmann S, et al. Role of alpha-synuclein in adult neurogenesis and neuronal maturation in the dentate gyrus. J Neurosci. 2012;32:16906–16916. doi:10.1523/JNEUROSCI.2723-12.2012
114. Winner B, Marchetto MC, Winkler J, Gage FH. Human-induced pluripotent stem cells pave the road for a better understanding of motor neuron disease. Hum Mol Genet. 2014;23:R27–R34. doi:10.1093/hmg/ddu205
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.