Back to Journals » Neuropsychiatric Disease and Treatment » Volume 15
Vascular cognitive impairment: pathophysiological mechanisms, insights into structural basis, and perspectives in specific treatments
Authors Parfenov VA ![]() , Ostroumova OD, Ostroumova TM
, Ostroumova OD, Ostroumova TM ![]() , Kochetkov AI
, Kochetkov AI ![]() , Fateeva VV, Khacheva KK
, Fateeva VV, Khacheva KK ![]() , Khakimova GR, Epstein OI
, Khakimova GR, Epstein OI ![]()
Received 4 December 2018
Accepted for publication 14 February 2019
Published 21 May 2019 Volume 2019:15 Pages 1381—1402
DOI https://doi.org/10.2147/NDT.S197032
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Roger Pinder
Vladimir A Parfenov,1 Olga D Ostroumova,2,3 Tatiana M Ostroumova,1 Alexey I Kochetkov,2 Victoria V Fateeva,4 Kristina K Khacheva,4 Gulnara R Khakimova,5 Oleg I Epstein6
1Department of Neurology, Federal State Autonomous Educational Institution of Higher Education, I.M. Sechenov First Moscow State Medical University of the Ministry of Health of the Russian Federation (Sechenov University), Moscow, Russian Federation; 2Laboratory of Clinical Pharmacology and therapy, Federal State Budgetary Educational Institution of Higher Education “N.I. Pirogov Russian National Research Medical University” of the Ministry of Health of the Russian Federation, Russian Clinical and Research Center of Gerontology, Moscow, Russia; 3Department of Clinical Pharmacology, Internal Medicine and Propaedeutics I.M. Sechenov First Moscow State Medical University (Sechenov University), Moscow, Russia; 4Medical Information Department, OOO NPF Materia Medica Holding, Moscow, Russian Federation; 5Research and Analytical Division of Scientific Research and Development Department, Moscow, Russian Federation; 6Laboratory of Physiologicaly Active Substances, Department of Molecular and Cellular Pathophysiology, Research Institute of General Pathology and Pathophysiology, Moscow, Russian Federation
Abstract: Vascular cognitive impairment (VCI) and vascular dementia are the most common forms of cognitive disorder associated with cerebrovascular disease and related to increased morbidity and mortality among the older population. Growing evidence suggests the contribution of blood-pressure variability, cardiac arrhythmia, hyperactivation of the renin–angiotensin–aldosterone system, endothelial dysfunction, vascular remodeling and stiffness, different angiopathies, neural tissue homeostasis, and systemic metabolic disorders to the pathophysiology of VCI. In this review, we focus on factors contributing to cerebrovascular disease, neurovascular unit alterations, and novel approaches to cognitive improvement in patients with cognitive decline. One of the important factors associated with the neuronal causes of VCI is the S100B protein, which can affect the expression of cytokines in the brain, support homeostasis, and regulate processes of differentiation, repair, and apoptosis of the nervous tissue. Since the pathological basis of VCI is complex and diverse, treatment affecting the mechanisms of cognitive disorders should be developed. The prospective role of a novel complex drug consisting of released–active antibodies to S100 and to endothelial NO synthase in VCI treatment is highlighted.
Keywords: vascular cognitive impairment, cerebrovascular disease, neurovascular unit, endothelial dysfunction, S100 protein
Cerebrovascular disease (CBVD) is a major cause of morbidity and mortality among the older population.1 There is growing evidence suggesting the contribution of blood-pressure instability, cardiac dysrhythmia (atrial fibrillation), hyperactivation of the renin–angiotensin–aldosterone system, endothelial dysfunction, vascular remodeling and stiffness, angiopathy of different etiology, patient lifestyle (including smoking and drinking), nervous tissue disturbances (eg, amyloid β and τ protein in Alzheimer’s disease, which have a negative impact on neuronal functional processes), and systemic metabolic disorders (particularly diabetes mellitus and dyslipidemia) to CBVD development. Vascular cognitive impairment (VCI) refers to a cognitive disorder form associated with CBVD. Treatment of VCI is currently focused on vascular risk factors. Understanding the pathogenesis of VCI will pave the way for the development of treatments targeting disease-underlying processes. In this review, we focus on factors contributing to CBVD, neurovascular unit alterations, and novel approaches to cognitive improvement in patients with VCI.
Introduction
With increasing life expectancy, cognitive impairment and dementia are becoming an important public health problem. In the Canadian Study of Health and Aging,2 prevalence of mild vascular cognitive impairment (VCI) among respondents aged 65–84 years was higher than that of vascular dementia. Patients with VCI have higher mortality2,3 and institutionalization rates.2
VCI refers to all forms of cognitive deficits of vascular origin, ranging from mild cognitive impairment to dementia. VCI can be classified as vascular mild cognitive impairment (amnesic, amnesic plus other domains, nonamnestic single domain, and nonamnestic multiple domain) and vascular dementia.
In this article we have analyzed VCI concepts focusing on pathophysiological mechanisms and possible treatment options.
Vascular cognitive impairment and cerebral small-vessel disease
The diagnostic criteria for probable VCI according to a statement from the American Heart Association–American Stroke Association include: neuroimaging evidence of сerebrovascular disease (CBVD) and either a temporal relationship between CBVD and cognitive deterioration or a relationship between the severity of cognitive deficits and presence of subcortical vascular lesions, and cognitive impairment should not progress gradualy (to exclude neurodegeneration).4
CBVD is the most common cause of VCI5 and dementia.6 The term CBVD includes a spectrum of disorders causing large-artery and small-vessel disease (SVD). Cerebral SVD is the main cause of cognitive impairment progression in older people,7 and increases the risk of dementia and stroke.8–10
Cerebral SVD includes a variety of conditions with various etiologies that affect the small arteries, arterioles, venules, and capillaries of the brain.11
Hypertensive vasculopathy (HV) and cerebral amyloid angiopathy (CAA) are the two most common forms of cerebral SVD.12
HV includes a great variety of functional and structural changes in small arteries that occur due to hypertension. It involves vascular remodeling, inflammation, endothelial dysfunction, and increased contractility.13 Long-standing hypertension can lead to lipofibrohyalinosis, arteriosclerosis, and arteriolosclerosis of deeply penetrating vessels in the brain, causing diffuse white-matter lesions14 and cerebral microbleeds.15 Some consider that blood–brain barrier (BBB) dysfunction may impact HV-associated cerebral SVD.16
HV can also affect the venous system of the brain. In several studies, it was associated with venous collagenosis.17,18 Thickening of the walls of periventricular veins and venules with collagen subtypes I and III is more frequent in brains with leukoaraiosis and increases with age.19
CAA indicates amyloid β (Aβ) accumulation in vessel walls, and is known to be a major cause of lobar intracerebral hemorrhage.20 CAA prevalence is age-dependent. In the population-based Vantaa 85+ study (253 women, 53 men, mean age at death 92.3 years), 69.6% of participants had CAA, with the highest prevalence in the parietal lobe.21
In pathological studies, CAA has been associated with cortical watershed microinfarcts in both Alzheimer’s disease (AD) and vascular dementia.22,23 These multiple microinfarctions may cause сerebral blood flow (CBF) disturbances, due to capillary occlusion,24 which can lead to progression of white matter hyperintensity (WMH).25
Cortical superficial siderosis can also be found in patients with CAA,26 which is thought to be more indicative of CAA than HV, as in CAA mainly superficial cortical and leptomeningeal vessels are involved.27 Cortical superficial siderosis is more frequently observed in patients with cognitive impairment and АD.28,29
CAA is also one of the major causes of cerebral microbleeds.30 At 7 T magnetic resonance imaging (MRI), cerebral microbleeds were observed in 78% of patients with AD or mild cognitive impairment in one study.31 In the Rotterdam Study, the presence of microbleeds was associated with cognitive decline and increased risk of dementia (HR 2.02, 95% CI 1.25–3.24).32 It is hypothesized that cerebral microbleeds in strategic areas of the brain may damage cortical and subcortical tracts33 or signify microvascular damage that leads to VCI.34 Increasing evidence suggests that CAA can also contribute to VCI, even in the absence of AD.35 In a prospective cohort study, 79% of CAA participants had mild cognitive impairment, and their scores for executive function and processing speed were lower than those of ischemic stroke controls.36 Higher MRI WMH volume was associated with lower processing-speed scores36,37 and executive function37 in CAA. Small-vessel microstructural damage in CAA can make an independent contribution to cognitive impairment, as CAA is also associated with cognitive decline before symptomatic intracerebral hemorrhage.38
Vascular cognitive impairment pathophysiological mechanisms
Сardiovascular risk factors
A large number of publications have been published on the role of cardiovascular risk factors in the occurrence and progression of cognitive impairment. Cardiovascular disease is a well-known risk factor for cognitive impairment and dementia.39 In particular, conditions that increase cardiovascular risk, including diabetes, essential hypertension (EH), hyperuricemia, and smoking, also increase risk of VCI, which is a common cause of cognitive impairment.40 ЕH-induced cеrebral SVD promotes arterial and arteriolar lesions in subcortical white matter surrounding basal ganglia. Recent evidence suggests that subcortical vessels are vulnerable to the damaging effect of increased blood pressure, due to their specific anatomical strucrure: a short straight-flow section after branching out from the brain base arteries.41 WMH detected using MRI is highly predictive of cerebral SVD.8 EH-associated cerebral SVD manifests with arteriosclerosis, characterized by smooth muscle–cell death, deposition of hyaline material in vessel walls, and lipohyalinosis. In more severe cases, fibrinoid necrosis of vascular walls leads to their rupture and hemorrhage.
The exact etiology of cerebral SVD remains unclear; however, it is known that cerebral ischemia promotes its onset. Garry et al showed42 that endothelial release of endogenous nitric oxide (NO) is an obligatory condition for optimal CBF. In the case of cerebral SVD, the concentration of asymmetric dimethylarginine (ADMA) increases. ADMA is NO synthase (NOS) inhibitor that blocks positive vasodilation and endothelioprotective effects of NO.43 Therefore, there is a link between ED and CBF reduction. Furthermore, the increased level of ADMA is considered a risk factor for atherosclerosis and a key factor in cardiovascular disease development.44
Сognitive impairment and serum uric acid
Increased serum uric acid (UA) is an additional cardiovascular risk factor, regardless of EH or diabetes presence.45 It is considered a predictor of cardiovascular events, even when serum UA is high normal.46 In contrast, the pathophysiological relationship between serum UA and cognitive decline in patients with and without history of dementia needs further investigation.46–48 UA's pathophysiological contribution to different types of dementia (eg, AD, Parkinson's disease, and VCI) differs, and also remains unclear.
Interesting results were obtained in the population-based Rotterdam Scan Study,47 which included 814 participants (mean age 62.0 years). This study aimed to evaluate a relationship among UA levels, brain atrophy, and cognitive functioning. Higher UA levels were associated with white-matter atrophy (difference in Z-score of white-matter volume per SD increase in uric acid –0.07 [95% CI –0.12 to –0.01]). This was particularly marked when comparing participants with elevated and normal serum UA (Z-score difference –0.27 [–0.43 to –0.11)]. Persons with elevated UA also had worse cognitive scores (–0.28 [–0.48 to –0.08]).
Nevertheless, recent data allow us to consider that UA after all has a positive impact on cognition, rather than triggers its deterioration. Engel et al48 assessed the relationship between hyperuricemia and dementia in regard to antihyperuricemic treatment. This case–control study included 27,528 patients diagnosed with dementia and 110,112 controls. Among all the participants, 22% had hyperuricemia or gout and 17% received antihyperuricemic treatment. The minimum follow-up was 3 years. Authors reported slightly lower dementia risk in patients with hyperuricemia (OR 0.94, 95% CI 0.89–0.98), and this risk reduction was even more marked among patients receiving antihyperuricemic treatment (OR 0.89, 95% CI 0.85–0.94).
There can be multiple pathophysiological mechanisms for a UA neuprotective effect. Primarily, its antioxidant activity deserves attention.46 A recent meta-analysis49 showed a significant reduction in antioxidant-system activity and serum UA in patients with dementia. Furthermore, essential antioxidant concentrations, including α- and β-carotene, lycopene, lutein, and vitamins A, C, and E, which can facilitate oxidative stress, tend to decrease in cases of low serum UA.50 UA has effects similar to ascorbate in the body, which is another important antioxidant. In addition, UA has the ability to eliminate oxygen and hydroperoxyl radicals, singlet oxygen, and oxoheme oxidants, and can make stable complexes with iron ions.46 Several authors have found a linear association between serum and cerebrospinal fluid (CSF) UA and between impaired BBB and UA concentration in CSF blood, which supports the hypothesis that UA can have an impact on the central nervous system (CNS) and cognition.51
At the same time, prooxidave properties of UA have been described.46 Such dual (pleiotropic/ambiguous) chemical properties of UA are assumed to be due to the influence of the environment in which this substance is included in biochemical processes, including the presence of metal ions.
UA may affect Aβ metabolism, but direct mechanisms are not yet known. Some authors have explained UA neurotoxicity by its ability to potentiate proapoptotic Aβ effects and expression,52,53 while others54 have found that increased UA levels lessen the harmful effects of CSF Aβ1–42 and higher UA reduce the harmful effects of Aβ1–42, a CSF biomarker of cognitive function, and observed an improvement in Mini–Mental State Examination (MMSE) and Alzheimer's Disease Assessment Scale — cognitive subscale scores.
Impaired cerebrovascular microcirculation can also provide a pathophysiological link between UA and cognitive functioning, especially in patients with VCI. Elevated serum UA levels may trigger inflammatory responses to oxidative stress, endothelium dysfunction, and cerebral microvasculature damage and remodeling, which in turn can explain an increased risk of vascular dementia.46,55,56 Recent data also support the relationship between high levels of UA, inflammation, and vascular dementia. Positive correlations between higher levels of CRP, IL6, and serum UA,57 and between inflammatory markers and WMH, lower gray matter, and hippocampal volume, which are indirect markers of cerebral atrophy, have been observed.58 In animal studies, inhibition of NFκB-signaling pathways, which causes a reduction inUA-related hippocampal inflammation, improved cognitive functioning. Moreover, hippocampal gliosis in both humans and rats was associated with serum UA levels. The authors have also found a significant increase in hippocampal gliosis related to serum-UA levels both in humans and rats.59
Impaired biochemical processes in nervous tissue: the S100 protein family
VCI is also considered an outcome of impaired biochemical processes in neurons, including synaptic transmission failure. Therefore, recent studies have focused on the broad family of Са2+-binding proteins with the EF–hand structural motif called the S100 proteins, and in particular on the S100B brain protein involved in synaptic processes.60 S100 proteins were discovered by Moore in 1965.61 They dissolve completely in 100% saturated ammonium sulfate solution of at pH 7.2, which explains the name of this group.
S100-family proteins are expressed in various tissue types and perform diverse functions. S100 proteins interact with intracellular effectors in tissue, regulate contraction, mobility, growth, and differentiation of cells, play a role in membrane organization, cytoskeleton dynamic composition, and protein phosphorylation and secretion, and protect against oxidation.60
S100 proteins are considered “calcium sensors” like calmodulin and troponin C, without any internal catalytic activity. Upon binding to calcium or (less often) to copper and zinc ions, S100 proteins undergo conformational changes.62 For instance, the recognition of target proteins (ie, τ) is not a calcium-dependent but a zinc-dependent process, and the implementation of neuroprotective properties requires copper ions.60,63 Some of the S100 proteins, including S100B, act like cytokines. S100B may cause both neurotrophic (in physiological nanomolar concentrations) and neurotoxic (in micromolar concentrations) effects.
Astroglia, Schwann cells, neurons, and satellite glial cells, as well as melanocytes, chondrocytes, adipocytes, skeletal muscle fibers, dendritic cells, and some populations of lymphocytes, express S100B.64 This protein stimulates proliferation and migration of cells and inhibits apoptosis and differentiation. Therefore, S100B plays an important role in synaptic process modulation, tissue development and repair, astrocyte activation in neurodegenerative processes, and glioma formation.64 S100B regulates cell proliferation, which may have a positive effect on tissue regeneration and may promote carcinogenesis. An association between chronically elevated S100B levels and Parkinson’s disease has been observed. The mechanisms underlying this association are probably related to a downregulation in the expression of dopamine D2 receptors and G protein–coupled receptor kinase 2, in the acceleration of dopamine metabolism, and in reduction in serotonin concentration.65
In neurotoxic micromolar concentrations, extracellular homo- and heterodimer forms of S100B affect neurons, glial apoptosis, and cell necrosis.66,67 This effect is based on S100B's ability to induce proinflammatory cytokines and oxidative stress–related enzymes, and to amplify other signals directed at neurons and glial cells.66–70
S100B in neurotoxic concentrations enhances the expression of IL1 and interleukin–6 (IL6) in microglia and neurons, changes neuronal metabolism, activates τ-protein hyperphosphorylation, reduces levels of synaptic proteins, and elevates the synthesis and activity of acetylcholinesterase.68 S100B also increases the expression of the Aβ precursor protein in neuronal cell cultures71 and enhances astrocyte activation caused by the Aβ peptide.67 In turn, IL1 induces S100B expression,69 perpetuating the vicious cycle of S100B neurotoxic effects.
Transgenic mice overexpressing S100B have hippocampal dementia-like and behavioral impairment, such as short-term-memory disturbances, partial disability in spatial task solving, spatial and nonspatial memory problems, hyperactivity, ie, exploratory hyperactivity, adaptation disorder, and reduced anxiety.65,72 Recent studies consider S100B as an early and easily measurable marker of cerebral ischemia. S100B can be detected in blood after the release from injured astrocytes into the extracellular space.73 In the study by Gao et al the serum level of S100 protein was measured using enzyme–linked immunosorbent assay in patients with cerebral SVD (n=210) and VCI. Authors provided evidence that plasma level of S100 protein was significantly higher in cerebral SVD patients compared to control group (P<0.05). Significant cognitive impairment was found in cerebral SVD patients, especially in patients with leukoaraiosis (P<0.05; comparing to control group). Significant correlation was found between increased S100 protein level and cognitive decline in patients with leukoaraiosis (P<0.05).7
Concentration of S100B in CSF elevates in acute cerebrovascular events74,75 and correlates with the size of the ischemic area and the clinical outcome.76 It has been shown that S100B concentration reaches a maximum on day 2–3 after ischemic stroke. The concentration of S100B reaches a peak in 2–24 hours after cerebral hypoxia, due to cardiac arrest, and correlates with outcome and coma levels.66 There are data showing S100B concentration increase in CBVD outcomes: subarachnoid hemorrhages and hemorrhagic and ischemic stroke.77,78
Studies have shown possible involvement of S100B in the pathogenesis of AD.69 In AD patients, the level of S100B in the brain is increased, due to activated astrocytes, which are cellular components of amyloid plaques and contain an increased amount of S100B.70 Since S100B stimulates axon growth and neuroprotection,79 its increase in the brain of AD patients is probably initially a compensatory response component. However, overexpression of this protein may have adverse effects. Neurotrophic activity of S100B also promotes aberrant axonal hypertrophy and the formation of large dystrophic neurites, which are found in and near amyloid plaques.80 Chronically elevated levels of S100B in the brain lead to enhanced expression of the Aβ precursor protein,81 which is a source of additional Aβ-peptide accumulation.
An increase in S100B in the brain of AD patients is directly related to τ-positive neuritic pathology.82 There is a parallel overexpression of S100B and the proinflammatory cytokine IL1 in AD and vascular dementia, which plays an important role in the pathogenesis of neuropathological changes.66,68–70 A connection between glial cells overexpressing IL1 and S100B, and an increase in neurofibrillary τ-protein tangles has been found.82
Role of the neurovascular unit in central nervous system diseases
The brain consumes up to 20% of the total amount of oxygen and nutrients (mainly glucose) contained in the blood.83 Neural homeostasis depends on the complex vascular cerebral network. It provides the essential distribution of nutrients and oxygen in the brain in accordance with local metabolic rate.84 Therefore, proper cerebral blood flow is the key factor in neuronal functioning. The brain tissue–blood boundary, referred to as the BBB, plays a decisive role in CNS homeostasis.85 The BBB is formed by endothelial cells with tight junctions between them, constituting an isolating structure that separates circulating blood components from brain tissue. Tight junctions determine the isolating properties of the BBB, as well as contribute to its polarization, leading to different functional features of the internal and external sides, which face the blood flow and brain tissue, respectively.85 The concept of the neurovascular unit (NVU) is closely related to the BBB. Interest in this topic increased significantly in the early 2000s after the publication of the Stroke Progress Review Group report on progression of the increase instroke incidence.86 The NVU consists of neurons, glial cells (astrocytes, microglia, oligodendrocytes), vascular elements (endothelial and smooth-muscle cells, pericytes, basal membrane), and extracellular matrix.87 The NVU integrates neuronal activity with local cerebral perfusion, modulates functional characteristics of the BBB, and interacts with extracellular matrix proteins.88 In addition, the NVU underlies the pathogenesis of several CNS diseases (cerebral stroke, vascular cognitive disorders, dementia, AD, Parkinson’s disease, amyotrophic lateral sclerosis, and multiple sclerosis).89
In the structure of the BBB, highly organized and specialized transport systems (ATP-binding cassette transporters, in particular the A1 subtype, the multidrugresistance protein), perform a detoxifying function and also eliminate the Aβ peptide.90 These transporters also ensure maintenance of CNS homeostasis.88
The other component of the NVU — astrocytes — constitute approximately 50% of brain cells. Studies have shown that astrocytes are involved in all CNS diseases.87 Thousands of processes occur in a single astrocyte, allowing the proper functioning of cerebral microcirculation and synapses and supporting the structure of neuropils. In addition, astrocytes control the ionic balance in the extracellular matrix, as well as development of the vasculature, and synthesize biologically active substances (including neurotrophic factors) that transmit signals to other cells (communicative function). At the same time, astrocytes are able to transform into a reactive state, initiate the production of proinflammatory cytokines and the formation of astroglial scars (gliosis), and suppress axonal regeneration.87
Despite the fact that the concept of the NVU is most applicable for studying processes occurring in gray matter, intercellular interactions are equally important for white matter. As another structural component of the NVU, oligodendrocytes are one of the main subtypes of cells that synthesize myelin, a substance rich in phospholipids that covers axons and is essential for effectively conducting a nerve impulse. Studies have shown that oligodendrocyte–endothelial cell couplings (the so-called oligovascular niche) potentiate angiogenesis and oligodendrogenesis in white matter.87 Once the acute phase of trauma has passed, oligodendrocytes are able to release MMP9, inducing vascular remodeling in white matter.91 The activity of oligodendrocyte-progenitor cells is aimed at the remyelination of damaged white-matter zones in demyelinating diseases, including multiple sclerosis, leukodystrophy, and vascular dementia.92
Another important component of the NVU in terms of VCI pathogenesis are pericytes, which are located around the endothelial layer of capillaries and embrace endothelial cells with their processes. Pericytes perform extremely important functions: integrating, coordinating, and realizing effects on the NVU. Pericytes regulate permeability of the BBB, cerebral perfusion, and eliminate cellular debris. Moreover, pericytes serve as a source of pluripotent stem cells for the CNS.87 As such, pericytes are closely related to endothelial cells and thus support normal functioning of the NVU.
Because the NVU is a vital structure in cerebral homeostasis, the dysfunction of its components may lead to acute conditions, such as traumatic brain injury,94–98 subarachnoid hemorrhage,98 and chronic conditions, such as dementia99,100 and AD.101,102
There are specific morphofunctional changes in all of these pathological processes. The loss of selectivity of the BBB, inflammation, and degradation of the extracellular matrix and basal lamina components are common features observed in NVU dysfunction. A recent study showed that pericyte degeneration due to ischemia contributes to cerebral homeostasis failure.103
Under the concept of the NVU, a focus on neuron pathology as a central link in the pathogenesis of nervous system dysfunction has been transformed into a more integrated view, allowing the creation of a new basis for experimental and clinical research in the field of CNS diseases, including VCI.
Endothelial dysfunction and VCI biomarkers
Several studies have suggest the role of ED in NVU failure and VCI development. In most cases, ED is associated with oxidative stress and results from both ischemia and inflammation. It is known that vascular risk factors enhance ED progression. Under normal conditions, endothelium-derived vasoactive factors take part in the coordination of vasodilatation/vasoconstriction and CBF. Therefore, it is believed that ED leads to diminution of CBF and alteration in BBB stability.104
Under ischemia, the endothelium expresses the adhesion molecules P-selectin, E-selectin, ICAM1, and (VCAM1, which are crucial for leukocyte migration into the perivascular space. Accordingly, ED can be detected using these adhesion molecules, as well as homocysteine, VWF), and MCP1, as biomarkers.105 The detection of these substances has potential utility for early diagnosis and prognosis of CVBD and VCI in particular.106
Under physiological conditions, low concentration of VEGF, which contributes to angiogenesis can be found in the brain. It is known that ischemia promotes VEGF overproduction.107 Tarkowski et al showed increased level of markers of inflammation (TGFβ, VEGF) in VCI, suggesting these substances as potential biomarkers.108,73
VWF is derived mostly from the endothelium. Vasoactive hormones, cytokines, hypoxia, and shear stress induce the production of VWF, and NO indirectly inhibits its secretion in vitro.109 Some studies have reported a correlation between vWF levels and CBVD outcomes (ie, stroke).110,111
Results from a meta-analysis by Quinn et al indicated a relationship between increased levels of ED biomarkers associated with coagulation: thrombin-generation markers (D–dimer and prothrombin fragment 1+2) and VCI.112
MCP1 acts as a key attractant for mononuclear cells and plays a role in collateral vessel formation and blood-flow regulation in response to ischemia in vivo.113,114 There is evidence that MCP1 exerts neuroprotective properties in glial–neuronal cocultures.115 Increased MCP1 levels in CSF and serum have been found in stroke patients, suggesting an association between MCP1 and cerebral ischemia pathogenesis.116,117
Acute-phase CRP is produced in chronic or acute inflammation. Interestingly, studies have shown the role of CRP in MCP1, E–selectin, VCAM1, and ICAM1 expression in cerebral ischemia exaggeration.118,119 Elevated levels of CRP have been found in clinical studies in stroke patients.120,121 Therefore, there is a likelihood that CRP might be also a biomarker of VCI.
Increased arterial stiffness in the development of cerebral vascular disorders
Increased vascular stiffness is another mechanism in cerebrovascular disorder development. Van Sloten et al122 showed that a decrease in elasticity of the arteries is a predictor of cerebral stroke, regardless of other cardiovascular risk factors or aortic stiffness. Increased stiffness of the carotid arteries may lead to the development of cerebral complications through a variety of mechanisms. Vascular stiffness as a part of CBVD and VCI pathogenesis contributes to pulse pressure that increases the stress on the cerebrovascular system.123–125 The cerebrovascular system is very vulnerable to hemodynamic changes, since it has low resistance potential, allowing high blood pressure to affect the microcirculatory bed. Ultimately, microcirculatory dysfunction manifests with ischemia and hemorrhage. Compensatory remodeling and thickening of the cerebral vascular walls occurs to withstand high pressure in the microcirculatory bed.123,124 Over time, this kind of protective mechanism is transformed into pathology, contributing to vascular reactivity disturbances, hypoperfusion, chronic cerebral ischemia, and VCI. On the other hand, the increased stiffness of elastic arteries (including the carotid arteries) leads to excessive variability in blood pressure,126,127 which increases the sensitivity of organs with high blood flow, including the brain, to pressure fluctuations in the presence of altered reactivity of the microcirculatory system.123 Last but not least, increased stiffness of the carotid arteries mediates the development of CBVD, potentiating the formation of atherosclerotic plaques prone to rupture.125,128
Vascular aging and cognitive dysfunction
Complex and in some cases completely unexplored processes of vascular aging play a role in VCI pathogenesis.129
Oxidative stress and inflammation are pathogenic factors responsible for both CBVD and VCI.127 With aging, the generation of reactive oxygen species (ROS) and hyperactivation of NADPH oxidase lead to oxidative stress and ED.130 Furthermore, the resulting oxidative stress potentiates coronary artery damage, as well as the development of stroke.
ROS generation potentiates the damage of arteries and vasomotor disturbances inhibiting production of NO, the most powerful vasodilator and a crucial factor required for proper endothelial functioning.131 Vasomotor disturbances present as flux-dependent and shear stress–vasodilation impairment that leads to a mismatch between oxygen-supply capacity and tissue demand, causing the development of ischemia. NO exerts vaso- and cardioprotective actions, inhibiting platelet- and inflammatory-cell adhesion to endothelial cells, blocking signaling pathways triggered by proinflammatory cytokines, protecting endothelial progenitor cells by suppressing apoptosis, and regulating tissue metabolism.129 Severe NO deficiency is exacerbated by a lack of tetrahydrobiopterin132 and intracellular L-arginine130 as much as by the age-dependent reduction in endothelial NO synthase (eNOS) expression.131 All these pathological changes promote an intracellular energy deficit, vascular inflammation, atherogenesis, and CBF failure.
A number of experimental studies and clinical data provide evidence that mild chronic inflammatory processes predispose older people to atherosclerosis.133 Studies on experimental models of aging discovered a proinflammatory shift in vascular gene expression resulting in elevations in proinflammatory cytokines, adhesion molecules, and inducible NOS levels.129 In humans, there is a correlation between age and concentration of several inflammatory markers (such as TNFα, VCAM1, E–selectin, IL6, IL18, and MCP1), independently of other cardiovascular risk factors.134–137 Increased concentration of these cytokines creates a proinflammatory microenvironment promoting apoptosis of endothelial cells and vascular dysfunction, and contributes to cognitive decline.138
Activation of RAAS and oxidative stress
Activation of the renin–angiotensin–aldosterone system (RAAS) provokes oxidative stress and mild chronic vascular inflammation and raises vulnerability of cerebral vessels to atherosclerotic lesions. Recent studies have described thickening of the intima–media complex, as well as the remodeling of main arteries under activation of the RAAS in older people.129 Angiotensin II–mediated signal pathways involving Capn1 and MMP2 are associated with migration of vascular smooth-muscle cells139 and artery remodeling in adulthood.
Oxidative stress and inflammation are the key pathogenetic factors responsible for the development of cardiovascular diseases, neurovascular dysfunction, VCI, and dementia.132 Cerebral perfusion autoregulation can be altered in response to changes in systemic blood pressure, particularly in patients with hypertension. This leads to activation of aberrant signaling pathways, by which angiotensin II realizes its adverse vasoactive effects, primarily contributing to the remodeling of blood vessels in the presence of existing blood-pressure dysregulation.140
In addition, angiotensin II might potentiate inflammation by activating leukocytes, cell-adhesion molecules, NADPH oxidase, proinflammatory cytokines, and ROS generation.140,141 A number of experimental studies have shown that generation of ROS activates Toll-like receptors and triggers the inflammatory response. The cascade of inflammatory reactions, for its part, increases oxidative stress by inhibiting antioxidant defense systems.142
It is believed that in this inflammatory–oxidative vicious circle, disruption of the permeability of the BBB is also important, since in this case plasma-complement components and Aβ, which penetrate brain tissue, serve as potential activators of inflammation and production of free radicals.143 As such, regardless of the cause, progressive vascular damage caused by oxidative stress and inflammation probably disrupts the NVU and exacerbates tissue hypoxia, thereby damaging neurons and white matter.
Furthermore, oxidative stress suppresses production of BDNF by the endothelium of vessels,144 which (with the participation of TRKB) provides neuroprotection.145 Сardiovascular risk factors and disruption of the BBB result in inhibition of proliferation, migration, and differentiation of oligodendrocyte-progenitor cells and also interfere with reparative processes in white matter, thus promoting demyelination and local hypoxia.146
Reduced capillary-network density and cerebral perfusion in terms of cognitive dysfunction and dementia
Apoptosis is a possible cause of CBVD-associated cognitive decline.129 The relationship between vascular aging and apoptosis remains unclear. Research has demonstrated age-related increase in the number of endothelial cells undergoing apoptosis.129 NO deficiency, mitochondrial oxidative stress, and elevated TNFα concentration predispose to apoptosis.147 It is thought that apoptosis contributes to age-dependent decrease in capillary-network density in most organs.129 With aging, the decreased microcirculatory network density in certain cerebral areas (eg, the hippocampus) and altered structure of the remaining functioning capillaries are observed. In older patients, these processes either precede or promote cognitive dysfunction in the absence of neurodegeneration.148
Angiogenesis disruption is the mechanism for age-related lesions affecting the capillary network and CBF failure.149 The decline incerebral microcirculatory network density with aging reduces cerebral perfusion. This leads to a decrease in trophic support of neurotransmitter-signaling pathways, especially those with high neuronal activity. Aging mediates the decrease in microvascular plasticity and adequate responsiveness of cerebral capillary blood flow to changes in oxygen and energy-substrate demands. In adults, the development of nervous tissue is coordinated with angiogenesis, and cerebral microvascular plasticity is decreased.148
At any age, EH contributes to cerebral microcirculation disturbances. Patients with EH also have insufficient capillary-network density, vascular stiffness, diminished CBF, and impaired collateral CBF compensation.41
Along with other studies, the results of our open comparative clinical trial confirm the association between EH, cognitive decline, and diminished CBF.150 We enrolled untreated middle-aged patients with stage 1 and 2 EH, and evaluated cerebral perfusion using the arterial spin-labeling technique. We have shown that in contrast to a control group with normal blood pressure (<140/90 mmHg), patients with uncomplicated EH had executive dysfunction and reduced CBF. The observations correlated with vascular age previously estimated in the Framingham study.151 Therefore, early-onset vascular aging plays a role in EH-associated brain damage in middle-aged patients, even at initial stages of the disease.
The cerebral vasculature is the most vulnerable target for elevated blood pressure in the brain. The vast majority of negative EH effects on cerebral vessels ultimately lead to the hypoperfusion, white-matter lesions, and severe CBVD presentations, such as stroke and VCI.41
In experimental animal models and EH patients, vascular wall hypertrophy leads to thickening of walls of arteries and arterioles, internal vascular remodeling, and lumen narrowing.41 Constantly increased hydrostatic blood pressure contributes to collagen and fibronectin deposition, elastin fragmentation, and cerebral artery–wall stiffness. BBB dysfunction results in inflammation, ROS generation, and protease activation.132 Decreased elastic properties of the aorta and stiffness of large cerebral arteries are significant predictors of certain cerebrovascular events and VCI.152–154
In summary, oxidative stress and inflammation resulting from the influence of various pathological vascular factors impaired biochemical processes in nervous tissue and the BBB failure diminish CBF and inhibit the proper functioning of the NVU components, thus promoting severe local hypoxia and VCI progression.146
Treatment of VCI and perspectives of a novel preparation
Due to a wide diversity of vascular factors contributing to VCI, basic antihypertensive, anticoagulant/antiaggregant, and antihyperlipidemic therapies are primal to cognitive impairment prevention. Nevertheless, these types of basic treatment do not significantly affect the already-existing cognitive deficits nor are directly related to restoration of biochemical processes in neurons and glia. The usage of acetylcholinesterase inhibitors in VCI is not a proper choice either, because of the lack of benefit in global functioning seen in patients with VCI and vascular dementia.155 Consequently, attention is drawn to neurotrophic preparations with antioxidant and neuroprotective action.
The combination of released–active antibodies (RAF Abs) to S100 and RAF Abs to eNOS is a novel nootropic preparation for VCI treatment with antioxidant and neuroprotective properties. Released activity is a combination of new properties that forms in an intact solvent during its processing in the presence of the original substance. The technological process is the multiple transfer of a part of the treated solution into an intact solvent accompanied by an external physical action.156 It has been shown that drugs of this class have a fundamentally novel modifying action, since the RAF Abs alter the interaction of the specific antigen (molecule) with its target by a mechanism of conformation modification.156 Compared to other nootropic drugs, the combination of RAF Abs to S100 and RAF Abs to eNOS exerts not only nootropic action but also positively impacts vascular homeostasis and endothelial function, due to RAF Abs to eNOS. The combination of endotheliotropic and neurotropic effects provides new opportunities for VCI treatment.
Treatment perspectives: preclinical trials of novel preparation
Development of the combination preparation RAF Abs to S100 and RAF Abs to eNOS was based on previously discovered pharmacological effects of each separate component (including their different technological versions). A number of studies on in vivo, ex vivo, and in vitro models (Table 1) not only elucidated the pharmacodynamics of RAF Abs to S100, RAF Abs to eNOS and its combination but also provided insight into its mode of action.
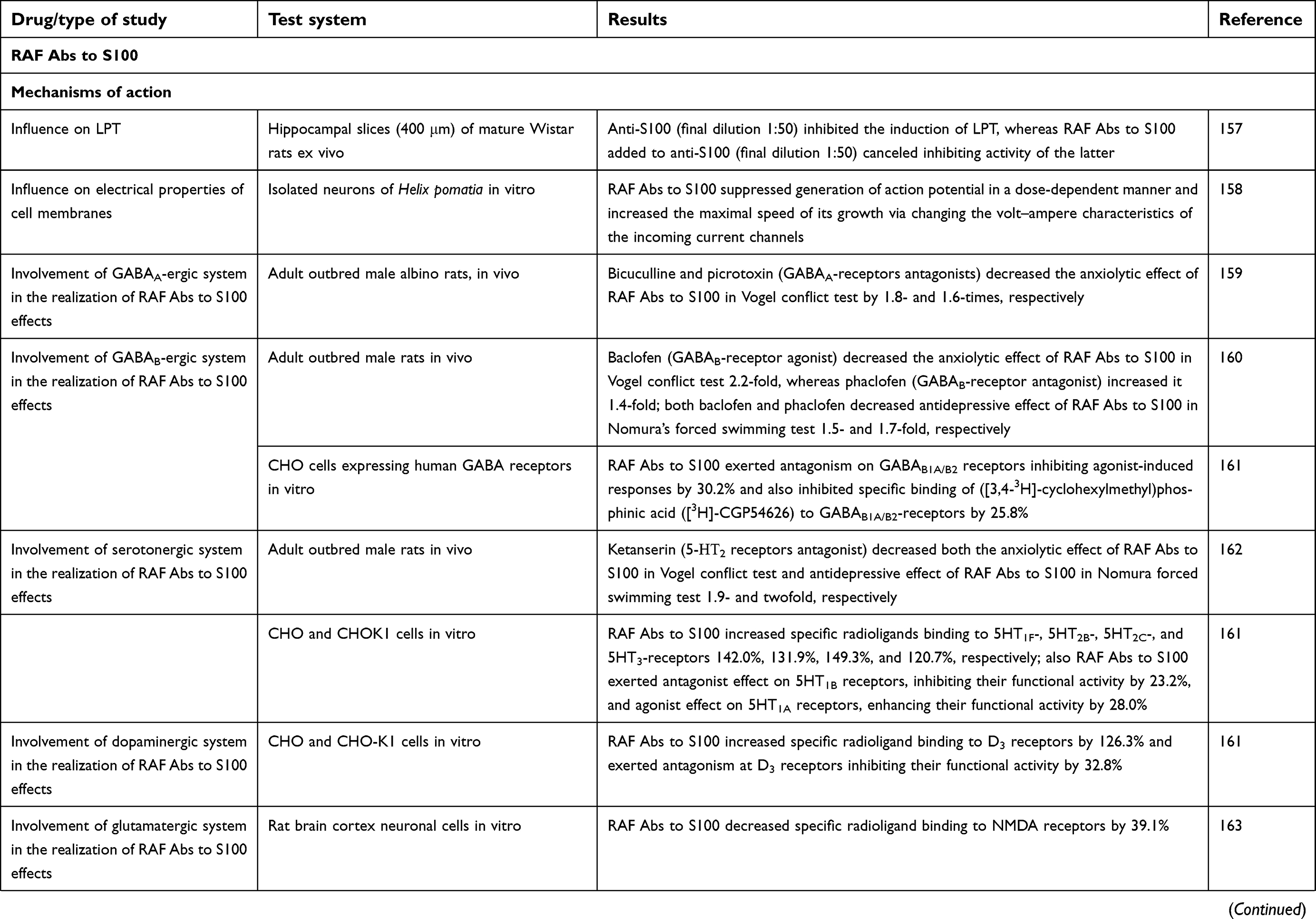 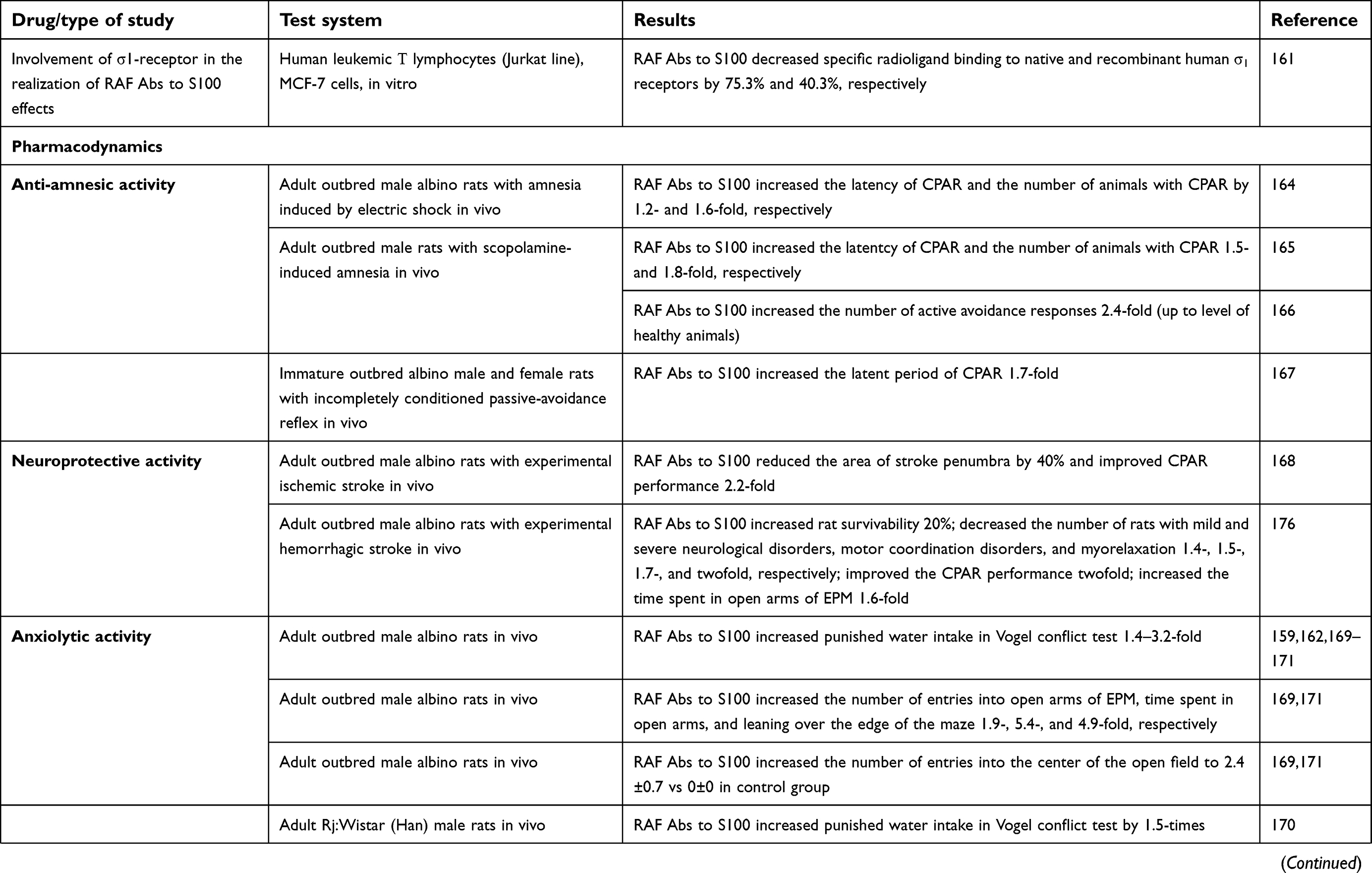 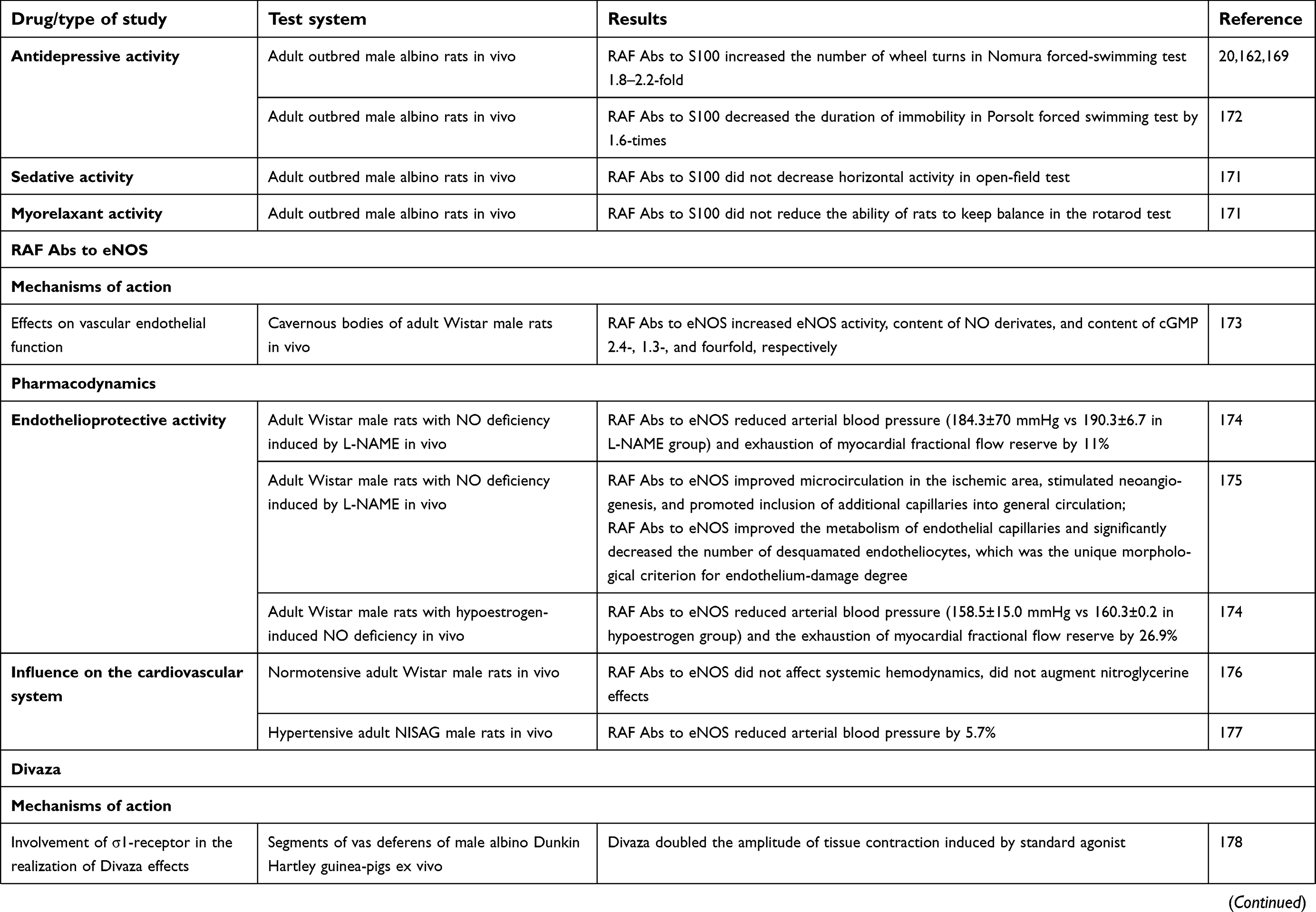 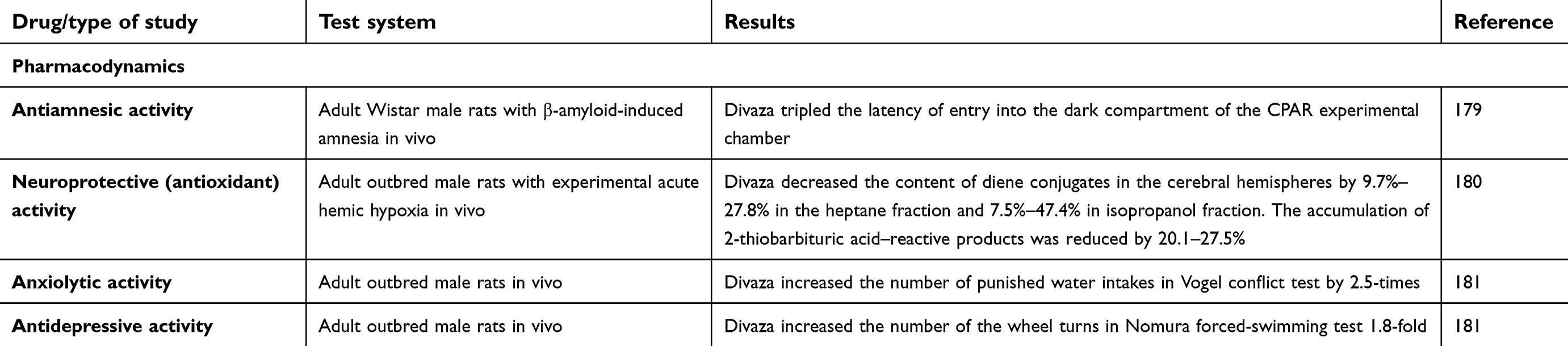 | Table 1 Experimental studies of mechanisms of action and pharmacological activity of RAF Abs to S100, RAF Abs to eNOS and combination drug Divaza |
RAF Abs to S100 modifies the effects of Abs to S100 ex vivo.157 In vitro and in vivo, RAF Abs to S100 has been shown to modify synaptic plasticity and electrical properties of plasma membranes prepared from isolated neurons,158 and exhibited GABA-modulating activity159–161 and effects on the serotonergic,161,162 dopaminergic161 and glutamatergic163 systems.
In addition, RAF Abs to S100 in vitro influence on ligand–receptor interaction of pentazocine (standard) with the σ1 receptor161 might indicate its ability to interfere with other mediator systems that cooperate with these receptors. For example, it is already known that σ1 receptors interact with noradrenergic182 and cholinergic systems.183,184 Also, σ1 receptors exert neuroprotective action,185 and influencing their activity can be considered one of the possible mechanisms of RAF Abs to S100 nootropic effects.
RAF Abs to S100 anti-amnesic activity has been demonstrated in vivo in models of amnesia induced by electric shock164 or scopolamine164,165 and on amodel of incompletely conditioned passive-avoidance reflex.166 RAF Abs to S100 effects were comparable in strength to those of the conventional nootropic drug piracetam.
Neuroprotectivе effects of RAF Abs to S100 have been identified in in vivo models of brain injury: ischemic (photothrombosis-induced)167 and hemorrhagic stroke168 models. The observed effects of RAF Abs to S100 did not differ from those of piracetam, cavinton, or nimodipine.
RAF Abs to S100 psychotropic activity (anxiolytic and antidepressant effects) has been observed in both healthy animals exposed to stress conditions159,162,169–172 and various disease models, eg, cholinergic deficit.165 The anxiolytic and antidepressant activities of RAF Abs to S100 were similar to the effects of diazepam and amitriptyline.169,170,172,186 Noteworthily, RAF Abs to S100 did not cause sedation and/or muscle relaxation.171
Therefore, results of experimental studies of RAF Abs to S100 demonstrate that the drug has neurotropic activity and is able to improve CNS functions under brain injury, as well as in the absence of pathology but under stressful conditions.
Effects of RAF Abs to eNOS
In in vitro biochemical studies, RAF Abs to eNOS stimulated the eNOS–NO–GC–cGMP cascade,173,187 which is responsible for relaxation of vascular smooth muscles and regulation of regional blood flow.
The ability of RAF Abs to eNOS to prevent endothelial damage (endothelioprotective effect) has been observed in in vivo NO deficiency models induced by L-N-nitroarginine methyl ester or hypoestrogen conditions.174,175
A study on RAF Abs to eNOS influence on the cardiovascular system in rats showed that the drug's administration did not affect main hemodynamic parameters in normotensive rats,176 but added to losartan, it decreased arterial pressure in hypertensive rats,177 which makes the use of RAF Abs to eNOS for treatment of cardiovascular diseases promising. Also, RAF Abs to eNOS was shown to cause no additional blood-pressure decrease when combined with nitroglycerin.176
Effects of combination of RAF Abs to eNOS and RAF Abs to S100
The combination of RAF Abs to S100 and RAF Abs to eNOS synthase along with memantine demonstrated antiamnesic effects in a model of Aβ-induced amnesia.179 The drug’s neuroprotective effect, similar to that of RAF Abs to S100 and might be linked to its influence on σ1 receptors,180 was demonstrated in an in vivo model of sodium nitrite–induced acute hypoxia: the combination of RAF Abs to S100 and RAF Abs to eNOS, as well as the reference compound mexidol, prevented or reduced activation of lipid peroxidation in the brains of experimental animals, suggesting an antioxidant-like effect.180
Anxiolytic and antidepressant effects of the combination of RAF Abs to S100 and RAF Abs to eNOS have been demonstrated in healthy animals using the Vogel conflict test and Nomura forced-swim test,181 and were similar to amitriptyline activity.
Safety
Toxicological studies of RAF Abs to S100, RAF Abs to eNOS, and their combination were conducted in accordance with national188,189 and international (ICH M3R2) Guidance on Nonclinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorization for Pharmaceuticals, 2009) guidelines.
The following assessments were performed: single-dose (acute) toxicity, chronic toxicity, reproductive toxicity, genotoxicity, immunotoxicity, local tolerance, and allergenicity. Additionally, local tolerability studies were performed.
Safety studies did not reveal any toxic effects (even with doses >100 times recommended human doses; unpublished data). Based on these results, RAF Abs to S100, RAF Abs to eNOS, and their combination can be considered class 4 low-hazard substances (according to the Russian GOST 12.1.007-76).
Treatment perspectives: clinical trials of novel preparation
Clinical studies of the combination of RAF Abs to S100 and RAF Abs to eNOS – were conducted in accordance with the principles of good clinical practice and Declaration of Helsinki requirements. The total number of participants in all studies was 696, and 545 of them received the combination RAF Abs to S100 and RAF Abs to eNOS. The purpose of the clinical trials was to evaluate the efficacy and safety of preparation for CBVD and cognitive impairment treatment.
In the study of the combination of RAF Abs to S100 and RAF Abs to eNOS in patients with CBVD, the 1.9-fold decrease ineNOS (927.5±11.2 to 478.6±13.4 pg/mL, P<0.05 compared to baseline [reference value 450 pg/mL]) and the 1.5–fold decrease in number of circulating (desquamated) endothelial cells (6.98±0.52 to 4.62±0.75 cells/100 μL, P<0.05 compared to baseline [reference value two to four cells/100 μL]) were shown after 12 weeks of therapy. These results indicated that the preparation exerts endothelioprotective action.190
Administration of the combination of RAF Abs to S100 and RAF Abs to eNOS in patients with chronic CBVD led to normalization of ischemia and inflammation biomarkers, such as fibrinogen (–1.6 g/L, P<0.01 compared to baseline) and VWF in plasma (1.5 g/L, P<0.01 compared to baseline) after 3 months of therapy. In addition, statistically significant decreases in CRP (1.7 mg/L; P<0.05 compared to baseline) and the ED biomarker MCP1 (30.1 pg/mL) concentrations were shown, suggesting the endothelioprotective effect of the preparation and the ability of RAF Abs to eNOS + RAF Abs to S100 to reduce the severity of the inflammatory process in vessel walls. No significant fluctuations in VEGF or ET1 concentrations were found, indicating the ability of the combination of RAF Abs to S100 and RAF Abs to eNOS to affect angiogenesis and prevent the progression of CBVD. In addition, the 31.3% decrease in S100 level was found, suggesting the deceleration of neurodegeneration.
Several clinical trials in patients with asthenia and mild cognitive impairment have provided evidence of the nootropic effect of the combination of RAF Abs to S100 and RAF Abs to eNOS. The cognitive improvement manifested in average MMSE-score increase, clock–drawing test, and verbal association test performance. Significant improvement in cognitive functions (according to the MMSE) was shown by the end of the 3-month treatment course with the combination of RAF Abs to S100 and RAF Abs to eNOS.191
Neuroprotective action of the combination of RAF Abs to S100 and RAF Abs to eNOS was shown in a trial, conducted by Parfenov et al: a 10% decrease in asthenia severity in 61% of patients, threefold decrease in 25% of patients, and improvement in sleep and quality of life (according to the SF36 questionnaire) were shown.192
A study on the effect of the combination of RAF Abs to S100 and RAF Abs to eNOS on potentially reversible vascular factors, which play one of the leading roles in the development of cognitive impairment, was performed in the course of the noninterventional observational program Diamant.193 The program was conducted in 30 cities in Russia between 2016 and 2017. Patients with CBVD attending outpatient clinics were treated with the combination of RAF Abs to S100 and RAF Abs to eNOS (two tablets, three times per day). The Montreal Cognitive Assessment (MoCA) scale was used for cognitive ability analysis in patients before and after 3 months of therapy. The study included 2,583 participants with CBVD, and the majority of them (90.7%) experienced symptoms of cognitive impairment (<26 MoCA score). At the end of treatment with the combination RAF Abs to S100 and RAF Abs to eNOS, the mean MoCA score improved from 19.58±5.13 to 23.99±4.21 (P<0.0001). The percentage of patients with normal cognitive function (≥26 MoCA score) increased by 32%. Older and senile patients tolerated the treatment well: <0.6% of adverse events (AEs). The vast majority of doctors (88.4%) noted the effect of the drug as a significant improvement or improvement, and 89.6% of patients evaluated the effect of treatment as excellent or good. The authors concluded that use of the combination of RAF Abs to S100 and RAF Abs to eNOS in patients with CBVD and cognitive impairment was substantiated and promising.
The safety of the combination of RAF Abs to S100 and RAF Abs to eNOS was also evaluated. In total, investigators detected 48 AEs in 43 patients. All of them, according to World Health Organization guidelines, were rare. AEs were not severe, and were related to different organ systems.194 All patients with AEs were monitored until complete resolution (patient recovery). No AEs have been determined to have a certain or probable relationship to the study drug. There were no serious AEs.
A new multicenter, double-blind, placebo-controlled randomized clinical trial of the efficacy and safety of the combination of RAF Abs to S100 and RAF Abs to eNOS in the correction of oxidant disorders in patients with cerebral atherosclerosis (resolution 42 of the Ministry of Health of the Russian Federation, February 5, 2018) has been proposed. The inclusion of at least 124 outpatients (32 in each group) with mild cognitive impairment (MoCA score <26) taking antihypertensive and hypolipidemic therapy at a constant dose and without significant disability (modified Rankin Scale score ≤1) is planned. Within 12 weeks, evaluation of cognitive impairment severity (MoCA scale), oxidative and antioxidant–system laboratory tests, and compensatory endothelial capacity and its ability to regulate vascular tone are going to be performed. Resistance capacity to lipid peroxidation, concentration of lipid peroxidation products (mainly lipid hydroperoxides) and the ability of lipoproteins to be oxidized will be assessed using Fe2+-induced chemiluminescence. Using standard laboratory techniques, the concentration of NO products in serum, platelet aggregation, and thickness of the intima–media complex will be measured. The safety of the combination RAF Abs to S100 and RAF Abs to eNOS will be assessed by the severity of AEs and their relationship to the study drug. Study results will be available at ClinicalTrials.gov (NCT03485495).
Conclusion
In this review, we have considered the wide range of pathophysiological VCI mechanisms. Changes in cerebral vessels in the form of cerebral SVD, ED, a decrease in cerebral capillary-network density, increased stiffness of arterial walls mediated by aging processes, oxidative stress, impact of the RAAS. and systemic blood pressure are the main causes of VCI. We emphasized that VCI, with the advent of the NVU concept, should be considered not only a vascular disorder but also a result of failed interaction between vascular and cellular (primarily neuronal) factors leading to impaired cerebral function. This view is certainly more rational and more correct, since the CNS is an extremely complex structure and its normal functioning is provided by the integrative interaction of vascular and cellular components. It should be mentioned again that one of the important factors associated with the neuronal causes of VCI is S100B, which can affect the expression of cytokines in the brain, support homeostasis, and regulate the processes of differentiation, repair, and apoptosis of nervous tissue.
Since the pathological basis of VCI is complex and diverse and specifically targeted treatment has not yet been found, new methods of treatment affecting all mechanisms of cognitive disorders should be developed.
Highlights
- VCI refers to all forms of cognitive disorder associated with cerebrovascular disease, and its pathogenetic mechanisms are complex and diverse.
- VCI should be considered a result of failed interaction between vascular and cellular (primarily neurotropic) factors leading to impaired cerebral function.
- S100B is an important neurotropic factor associated with VCI.
- The combination of RAF Abs to S100B protein and RAF Abs to eNOS is a safe novel preparation with endotheliotropic and neurotropic effects, providing new opportunities for VCI treatment.
Acknowledgments
The studies of the combination of released–active form of antibodies to S100 protein and released–active form of antibodies to endothelial NO synthase mentioned in this review were funded by a grant from Materia Medica Holding (Moscow, Russia). The statistical analysis was provided by Materia Medica Holding.
Disclosure
VVF, KKK, GRK are employees and OIE is the founder of Materia Medica Holding. Divaza is a preparation manufactured and marketed by Materia Medica Holding. Patents on Divaza belong to OIE. VAP received an investigator grant from Materia Medica Holding to conduct the clinical trials of Divaza mentioned in this review. VVF, KKK and GRK report personal fees from Materia Medica Holding during the conduct of the study and outside the submitted work. OIE reports personal fees from Materia Medica Holding, during the conduct of the study and outside the submitted work. In addition, OIE has patent 1302925.1/GB2496342 (licensed UK patent 2496342). ODO, TMO, and AIK report no conflicts of interest.
References
1. Moran AE. Demographic and epidemiologic drivers of global cardiovascular mortality. N Engl J Med. 2015;14(372):1333–1341. doi:10.1056/NEJMoa1406656
2. Rockwood K, Wentzel C, Hachinski V, Hogan DB, MacKnight C, McDowell I. Prevalence and outcomes of vascular cognitive impairment. Vascular Cognitive Impairment Investigators of the Canadian Study of Health and Aging. Neurology. 2000;54:447–451. doi:10.1212/WNL.54.2.447
3. Brodaty H, Altendorf A, Withall A, Sachdev PS. Mortality and institutionalization in early survivors of stroke: the effects of cognition, vascular mild cognitive impairment, and vascular dementia. J Stroke Cerebrovasc Dis. 2010;19(6):485–493. doi:10.1016/j.jstrokecerebrovasdis.2009.09.006
4. Gorelick PB, Scuteri A, Black SE;
5. Erkinjuntti T. Diagnosis and management of vascular cognitive impairment and dementia. J Neural Transm Suppl. 2002;63:91–109.
6. DeCarli C. The role of cerebrovascular disease in dementia. Neurologist. 2003;9(3):123–136. doi:10.1097/00127893-200305000-00001
7. Gao Q, Fan Y, Mu LY, Ma L, Song ZQ, Zhang YN. S100B and ADMA in cerebral small vessel disease and cognitive dysfunction. J Neurol Sci. 2015;354(1–2):27–32. doi:10.1016/j.jns.2015.04.031
8. Debette S, Markus HS. The clinical importance of white matter hyperintensities on brain magnetic resonance imaging: systematic review and meta–analysis. BMJ. 2010;341(14):36–66. doi:10.1136/bmj.c3666
9. Vermeer SE, Longstreth WT
10. Gorelick PB, Scuteri A, Black SE, et al;
11. Pantoni L. Cerebral small vessel disease: from pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol. 2010;9:689–701. doi:10.1016/S1474-4422(10)70104-6
12. Yael D, van Veluw SJ, Steven MG. Ischemic brain injury in cerebral amyloid angiopathy. J Cereb Blood Flow Metab. 2016;36(1):40–54. doi:10.1038/jcbfm.2015.88
13. Touyz RM, Montezano AC. Hypertensive vasculopathy. PanVascular Med. 1–28. doi: 10.1007/978-3-642-37393-051-1.
14. Englund E. Neuropathology of white matter lesions in vascular cognitive impairment. Cerebrovasc Dis. 2002;13(2):11–15. doi:10.1159/000049144
15. Sergi M-R, Steven M,G, Anand V. Cerebral microbleeds: overview and implications in cognitive impairment. Alzheimers Res Ther. 2014;6:33. doi:10.1186/alzrt263
16. Wardlaw JM. Blood-brain barrier and cerebral small vessel disease. J Neurol Sci. 2010;299:66–71. doi:10.1016/j.jns.2010.08.042
17. Zhou M, Mao L, Wang Y, et al. Morphologic changes of cerebral veins in hypertensive rats: venous collagenosis is associated with hypertension. J Stroke Cerebrovasc Dis. 2015;24(3):530–536. doi:10.1016/j.jstrokecerebrovasdis.2015.05.011
18. Craggs LJ, Hagel C, Kuhlenbaeumer G, et al. Quantitative vascular pathology and phenotyping familial and sporadic cerebral small vessel diseases. Brain Pathol. 2013;23:547–557. doi:10.1111/bpa.12041
19. Brown WR, Moody DM, Challa VR, Thore CR, Anstrom JA. Venous collagenosis and arteriolar tortuosity in leukoaraiosis. J Neurol Sci. 2002;15(203–204):159–163. doi:10.1016/S0022-510X(02)00283-6
20. Finelli PF, Kessimian N, Bernstein PW. Cerebral amyloid angiopathy manifesting as recurrent intracerebral hemorrhage. Arch Neurol. 1984;41(3):330–333. doi:10.1001/archneur.1984.04050150112027
21. Tanskanen M, Mäkelä M, Myllykangas L, et al. Prevalence and severity of cerebral amyloid angiopathy: a population-based study on very elderly Finns (Vantaa 85+). Neuropathol Appl Neurobiol. 2012;38(4):329–336. doi:10.1111/j.1365-2990.2011.01219.x
22. Suter OC, Sunthorn T, Kraftsik R. Cerebral hypoperfusion generates cortical watershed microinfarcts in Alzheimer disease. Stroke. 2002;33:1986–1992.
23. Okamoto Y, Ihara M, Fujita Y. Cortical microinfarcts in Alzheimer’s disease and subcortical vascular dementia. Neuroreport. 2009;20:990–996. doi:10.1097/WNR.0b013e32832d2e6a
24. Thal DR, Capetillo Zarate E, Larionov S. Capillary cerebral amyloid angiopathy is associated with vessel occlusion and cerebral blood flow disturbances. Neurobiol Aging. 2009;30:1936–1948. doi:10.1016/j.neurobiolaging.2008.01.017
25. Thanprasertsuk S, Martinez-Ramirez S, Pontes-Neto OM, et al. Posterior white matter disease distribution as a predictor of amyloid angiopathy. Neurology. 2014;83(9):794–800. doi:10.1212/WNL.0000000000000732
26. Charidimou A, Jäger RH, Fox Z, et al. Prevalence and mechanisms of cortical superficial siderosis in cerebral amyloid angiopathy. Neurology. 2013;81(7):626−632. doi:10.1212/WNL.0b013e3182a08f2c
27. Masahito Y. Cerebral amyloid angiopathy: emerging concepts. J Stroke. 2015;17(1):17–30. doi:10.5853/jos.2015.17.1.17
28. Wollenweber FA, Buerger K, Mueller C, et al. Prevalence of cortical superficial siderosis in patients with cognitive impairment. J Neurol. 2014;261(2):277−282. doi:10.1007/s00415-013-7161-2
29. Zonneveld HI, Goos JD, Wattjes MP, et al. Prevalence of cortical superficial siderosis in a memory clinic population. Neurology. 2014;82(8):698−704. doi:10.1212/WNL.0000000000000513
30. Yakushiji Y. Cerebral microbleeds: detection, associations and clinical implications. Front Neurol Neurosci. 2015;37:78−92. doi:10.1159/000437115
31. Brundel M, Heringa SM, de Bresser J, et al. High prevalence of cerebral microbleeds at 7Tesla MRI in patients with early Alzheimer’s disease. J Alzheimers Dis. 2012;31(2):259−263. doi:10.3233/JAD-2012-120364
32. Akoudad S, Wolters FJ, Viswanathan A, de Bruijn RF. Cerebral microbleeds are associated with cognitive decline and dementia: the Rotterdam Study. JAMA Neurol. 2016;73(8):934–943. doi:10.1001/jamaneurol.2016.1017
33. Werring DJ, Frazer DW, Coward LJ, et al. Cognitive dysfunction in patients with cerebral microbleeds on T2*-weighted gradient-echo MRI. Brain. 2004;127:2265−2275. doi:10.1093/brain/awh253
34. Werring DJ, Gregoire SM, Cipolotti L. Cerebral microbleeds and vascular cognitive impairment. J Neurol Sci. 2010;299(1−2):131−135. doi:10.1016/j.jns.2010.08.034
35. Steven M, Greenberg MD, Edip G, Jonathan R, Eric E. Amyloid angiopathy–related vascular cognitive impairment. Stroke. 2004;35:2616−2619.
36. Case NF, Charlton A, Zwiers A. Cerebral amyloid angiopathy is associated with executive dysfunction and mild cognitive impairment. Stroke. 2016;47(8):2010−2016. doi:10.1161/STROKEAHA.116.012999
37. Xiong L, Davidsdottir S, Reijmer YD. Cognitive profile and its association with neuroimaging markers of non-demented cerebral amyloid angiopathy patients in a stroke unit. J Alzheimers Dis. 2016;52(1):171−178. doi:10.3233/JAD-150890
38. Banerjee G, Wilson D, Ambler G. Cognitive impairment before intracerebral hemorrhage is associated with cerebral amyloid angiopathy. Stroke. 2018;49(1):40−45. doi:10.1161/STROKEAHA.117.019409
39. Beeri MS, Ravona-Springer R, Silverman JM, Haroutunian V. The effects of cardio-vascular risk factors on cognitive compromise. Dialogues Clin Neurosci. 2009;11:201–212.
40. Alonso A, Jacobs DR
41. Iadecola C, Yaffe K, Biller J, et al.,
42. Garry PS, Ezra M, Rowland MJ, Westbrook J, Pattinson KT. The role of the nitric oxide pathway in brain injury and its treatment—from bench to bedside. Exp Neurol. 2015;263:235–243. doi:10.1016/j.expneurol.2014.10.017
43. Víteček J, Lojek A, Valacchi G, Kubala L. Arginine-based inhibitors of nitric oxide synthase: therapeutic potential and challenges. Mediators Inflamm. 2012;22. doi: 10.1155/2012/318087.
44. Boger RH, Zoccali C. Asymmetric dimethylarginine (ADMA): a novel risk marker in cardiovascular medicine and beyond. Ann Med. 2006;38:126–136. doi:10.1080/07853890500472151
45. Chang CC, Wu CH, Liu LK, et al. Association between serum uric acid and cardiovascular risk in nonhypertensive and nondiabetic individuals: the Taiwan I-Lan Longitudinal Aging Study. Sci Rep. 2018;8:5234. doi:10.1038/s41598-018-22997-0
46. Tana C, Ticinesi A, Prati B, Nouvenne A, Meschi T. Uric acid and cognitive function in older individuals. Nutrients. 2018;10(8):E975. doi:10.3390/nu10080975
47. Verhaaren BF, Vernooij MW, Dehghan A, et al. The relation of uric acid to brain atrophy and cognition: the Rotterdam Scan Study. Neuroepidemiology. 2013;41(1):29−34. doi:10.1159/000346606
48. Engel B, Gomm W, Broich K, Maier W, Weckbecker K, Haenisch B. Hyperuricemia and dementia - a case-control study. BMC Neurol. 2018;18(1):131. doi:10.1186/s12883-018-1136-y
49. Schrag M, Mueller C, Zabel M, et al. Oxidative stress in blood in Alzheimer’s disease and mild cognitive impairment: a meta-analysis. Neurobiol Dis. 2013;59:100–110. doi:10.1016/j.nbd.2013.07.005
50. Mullan K, Cardwell CR, McGuinness B, Woodside JV, McKay GJ. Plasma antioxidant status in patients with Alzheimer’s disease and cognitively intact elderly: a meta-analysis of case-control studies. J Alzheimers Dis. 2018;62:305–317. doi:10.3233/JAD-170758
51. Bowman GL, Shannon J, Frei B, Kaye JA, Quinn JF. Uric acid as a CNS antioxidant. J Alzheimers Dis. 2010;19:1331–1336. doi:10.3233/JAD-2010-1330
52. McFarland NR, Burdett T, Desjardins CA, Frosch MP, Schwarzschild MA. Postmortem brain levels of urate and precursors in Parkinson’s disease and related disorders. Neurodegener Dis. 2013;12:189–198. doi:10.1159/000346370
53. Desideri G, Gentile R, Antonosante A, et al. Uric acid amplifies a amyloid effects involved in the cognitive dysfunction/dementia: evidences from an experimental model in vitro. J Cell Physiol. 2017;232:1069–1078. doi:10.1002/jcp.25509
54. Ye BS, Lee WW, Ham JH, Lee JJ, Lee PH, Sohn YH. Alzheimer’s disease neuroimaging iniziative. Does serum uric acid act as a modulator of cerebrospinal fluid alzheimer’s disease biomarker related cognitive decline? Eur J Neurol. 2016;23:948–957. doi:10.1111/ene.2016.23.issue-5
55. Tanaka A, Kawaguchi A, Tomiyama H, et al. Cross-sectional and longitudinal associations between serum uric acid and endothelial function in subjects with treated hypertension. Int J Cardiol. 2018. doi:10.1016/j.ijcard.2018.06.017
56. Puddu P, Puddu GM, Cravero E, Vizioli L, Muscari A. Relationships among hyperuricemia, endothelial dysfunction and cardiovascular disease: molecular mechanisms and clinical implications. J Cardiol. 2012;59:235–242. doi:10.1016/j.jjcc.2012.01.013
57. Perez-Ruiz F, Becker MA. Inflammation: a possible mechanism for a causative role of hyperuricemia/gout in cardiovascular disease. Curr Med Res Opin. 2015;31(2):9–14. doi:10.1185/03007995.2015.1087980
58. Satizabal CL, Zhu YC, Mazoyer B, Dufouil C, Tzourio C. Circulating IL-6 and CRP are associated with MRI findings in the elderly: the 3C-Dijon Study. Neurology. 2012;78:720–727. doi:10.1212/WNL.0b013e318248e50f
59. Shao X, Lu W, Gao F, et al. Uric acid induces cognitive dysfunction through hippocampal inflammation in rodents and humans. J Neurosci. 2016;36:10990–11005. doi:10.1523/JNEUROSCI.2710-15.2016
60. Sedaghat F, Notopoulos A. S100 protein family and its application in clinical practice. Hippokratia. 2008;12(4):198–204.
61. Moore BW. A soluble protein characteristic of the nervous system. Biochem Biophys Res Commun. 1965;19:739–744.
62. Ikura M. Calcium binding and conformational response in EF-hand proteins. Trends Biochem Sci. 1996;21:14–17.
63. Yu WH, Fraser PE. S100B interaction with tau is promoted by zinc and inhibited by hyperphosphorylation in Alzheimer’s disease. J Neurosci. 2001;21:2240–2246. doi:10.1523/JNEUROSCI.21-07-02240.2001
64. Donato R, Cannon BR, Sorci G, et al. Functions of S100 proteins. Curr Mol Med. 2013;13(1):24–57. doi:10.2174/156652413804486214
65. Liu J, Wang H, Zhang L, et al. S100B transgenic mice develop features of Parkinson’s disease. Arch Med Res. 2011;42:1–7. doi:10.1016/j.arcmed.2011.01.005
66. Adami C, Sorci G, Blasi E. S100B expression in and effects on microglia. Glia. 2001;33:131–142. doi:10.1002/1098-1136(200102)33:2<131::AID-GLIA1012>3.0.CO;2-D
67. Hu J, Ferreira A, Van Eldik LJ. S100B induces neuronal cell death through nitric oxide release from astrocytes. J Neurochem. 1997;69(6):2294–2301. doi:10.1046/j.1471-4159.1997.69062294.x
68. Li Y, Barger SW, Liu L. S100В induction of the pro-inflammatory cytokine interleukin-6 in neurons. J Neurochem. 2000;74:143–150.
69. Griffin WS, Mrak RE. Interleukin-1 in the genesis and progression of and risk for development of neuronal degeneration in Alzheimer’s disease. J Leukoc Biol. 2002;72:233–238.
70. Hu J, Van Eldik LJ. Glial derived proteins activate cultured astrocytes and enhance β–amyloid–induced astrocyte activation. Brain Res. 1999;842(1):46–54. doi:10.1016/S0006-8993(99)01804-1
71. Barger SW, Basile AS. Activation of microglia by secreted amyloid precursor protein evokes release of glutamate by cystine exchange and attenuates synaptic function. J Neurochem. 2001;76:846–854. doi:10.1046/j.1471-4159.2001.00075.x
72. Friend WC, Clapoff S, Landry C, et al. Cell–specific expression of high levels in human S100B in transgenic mouse brain is dependent of gene dosage. J Neurosci. 1992;12:4337–4346. doi:10.1523/JNEUROSCI.12-11-04337.1992
73. Wallin А, Kapaki E, Boban M, et al. Biochemical markers in vascular cognitive impairment associated with subcortical small vessel disease – a consensus report. BMC Neurol. 2017;17:102. doi:10.1186/s12883-017-0877-3
74. Lamers KJB, Van Engelen BGM, Gabreels FJM, Hommes OR, Borm GF, Wevers RA. Cerebrospinal neuron–specific enolase, S–100 and Myelin basic protein in neurological disorders. Acta Neurol Scan. 1995;92:247–251. doi:10.1111/j.1600-0404.1995.tb01696.x
75. Persson L, Hardemark HG, Gustafsson J, et al. S100 protein and neuronspecific enolase in cerebrospinal fluid and serum: markers of cell damage in human central nervous system. Stroke. 1987;18:911–918. doi:10.1161/01.STR.18.5.911
76. Elting JW, De Jager AEJ, Teelken AW. Comparison of serum S-100 protein levels following stroke and traumatic brain injury. J Neurol Sci. 2000;181:104–110. doi:10.1016/S0022-510X(00)00442-1
77. Hardemark HG, Almquist O, Johansson T, Påhlman S, Persson L. S100 protein in cerebrospinal fluid after aneurysmal subarachnoid haemorrhage: relation to functional outcome, late CT and SPECT changes, and signs of higher cortical dysfunction. Acta Neurochir. 1989;99:135–144. doi:10.1007/BF01402322
78. Abraha HD, Butterworth J, Bath PMW, Wassif WS, Garthwaite J, Sherwood RA. Serum S100 protein, relationship to clinical outcome in acute stroke. Ann Clin Biochem. 1997;34:546–550. doi:10.1177/000456329703400405
79. Barger SW, Van Eldik LJ, Mattson MP. S100B protects hippocampal neurons from damage induced by glucose deprivation. Brain Res. 1995;77:167–170. doi:10.1016/0006-8993(95)00160-R
80. Mrak RE, Sheng JG, Griffin WS. Correlation of astrocytic S100B expression with dystrophic neurites in amyloid plaques of Alzheimer’s disease. J Neuropathol Exp Neurol. 1996;55:273–279. doi:10.1097/00005072-199603000-00002
81. Li Y, Wang J, Sheng JG, et al. S100B increases levels of b–amyloid precursor protein and its encoding mRNA in rat neuronal cultures. J Neurochem. 1998;71:1421–1428. doi:10.1046/j.1471-4159.1998.71041421.x
82. Sheng JG, Mrak RE, Griffin WST. Glial–neuronal interactions in Alzheimer disease: progressive association of IL–1α+microglia and S100B+ astrocytes with neurofibrillary tangle stage. J Neuropath Exp Neurol. 1997;56:285–290. doi:10.1097/00005072-199703000-00007
83. Zlokovic BV. The blood–brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. doi:10.1016/j.neuron.2008.01.003
84. Ruitenberg A, Den Heijer T, Bakker SL, et al. Cerebral hypoperfusion and clinical onset of dementia: the Rotterdam Study. Ann Neurol. 2005;57:789–794. doi:10.1002/ana.20493
85. Hermann DM, ElAli A. The abluminal endothelial membrane in neurovascular remodeling in health and disease. Sci Signal. 2012;5(236):re4. doi:10.1126/scisignal.2002886
86. Grotta JC, Moskowitz MA, Jacobs TP, et al. Report of the stroke progress review group. National institute of neurological disorders and stroke; 2002. Availible from:
87. Maki T, Hayakawa K, Pham LD, Xing C, Lo EH, Arai K. Biphasic mechanisms of neurovascular unit injury and protection in CNS diseases. CNS Neurol Disord Drug Targets. 2013;12(3):302–315. doi:10.2174/1871527311312030004
88. ElAli A, Thériault P, Rivest S. The role of pericytes in neurovascular unit remodeling in brain disorders. Int J Mol Sci. 2014;15(4):6453–6474. doi:10.3390/ijms15046453
89. Arai K, Lok J, Guo S, Hayakawa K, Xing C, Lo EH. Cellular mechanisms of neurovascular damage and repair after stroke. J Child Neurol. 2011;26:1193–1198. doi:10.1177/0883073811408610
90. Lam FC, Liu R, Lu P, et al. Beta–Amyloid efflux mediated by p–glycoprotein. J Neurochem. 2001;76:1121–1128. doi:10.1046/j.1471-4159.2001.00113.x
91. Pham LD, Hayakawa K, Seo JH, et al. Crosstalk between oligodendrocytes and cerebral endothelium contributes to vascular remodeling after white matter injury. Glia. 2012;60:875–881. doi:10.1002/glia.22320
92. Franklin RJ, Ffrench–Constant C. Remyelination in the CNS: from biology to therapy. Nat Rev Neurosci. 2008;9:839–855. doi:10.1038/nrn2480
93. Armstead WM, Raghupathi R. Endothelin and the neurovascular unit in pediatric traumatic brain injury. Neurol Res. 2011;33(2):127–132. doi:10.1179/016164111X12881719352138.41
94. Wang YF, Huang SR, Shao SH, Qian L, Ping X. Studies on bioactivities of tea (Camellia sinensis L.) fruit peel extracts: antioxidant activity and inhibitory potential against α-glucosidase and α-amylase in vitro. Ind Crops Prod. 2012;37(1):520–526. doi:10.1016/j.indcrop.2011.07.031
95. Bastide P, Darido C, Pannequin J, et al. Sox9 regulates cell proliferation and is required for Paneth cell differentiation in the intestinal epithelium. J Cell Biol. 2007;178(4):635–648. doi:10.1083/jcb.200704152
96. Fernández-Klett F, Offenhauser N, Dirnagl U, Priller J, Lindauer U. Pericytes in capillaries are contractile in vivo, but arterioles mediate functional hyperemia in the mouse brain. Proc Natl Acad Sci U S A. 2010;107(51):22290–22295. doi:10.1073/pnas.1011321108
97. Ruhrberg C, Bautch VL. Neurovascular development and links to disease. Cell Mol Life Sci. 2013;70(10):1675–1684. doi:10.1007/s00018-013-1277-5
98. Roger T, Koide C, Malcolm FG. Determining place and process: functional traits of ectomycorrhizal fungi that affect both community structure and ecosystem function. New Phytol. 2014;201:433–439. doi:10.1111/nph.12538
99. Carmeliet P. Angiogenesis in health and disease. Nat Med. 2003;9(6):653–660. doi:10.1038/nm0603-653
100. Itoh Y, Ezawa A, Kikuchi K, Tsuruta Y, Niwa T. Protein-bound uremic toxins in hemodialysis patients measured by liquid chromatography/tandem mass spectrometry and their effects on endothelial ROS production. Anal Bioanal Chem. 2012;403(7):1841–1850. doi:10.1007/s00216-012-5929-3
101. Busch KE, Laurent P, Soltesz Z, et al. Tonic signaling from O₂ sensors sets neural circuit activity and behavioral state. Nat Neurosci. 2012;15(4):581–591. doi:10.1038/nn.3061
102. Sagare AP, Bell RD, Zhao Z, et al. Pericyte loss influences Alzheimer-like neurodegeneration in mice. Nature Commun. 2013;4:2932. doi:10.1038/ncomms3932
103. Kisler K, Nelson AR, Rege SV. Pericyte degeneration leads to neurovascular uncoupling and limits oxygen supply to brain. Nat Neurosci. 2017;20(3):406–416. doi:10.1038/nn.4489
104. Deanfield JE, Halcox JP, Rabelink TJ. Endothelial function and dysfunction: testing and clinical relevance. Circulation. 2007;115:1285–1295. doi:10.1161/CIRCULATIONAHA.106.652859
105. Wiseman S, Marlborough F, Doubal F, Webb DJ, Wardlaw J. Blood markers of coagulation, fibrinolysis, endothelial dysfunction and inflammation in lacunar stroke versus non–lacunar stroke and non–stroke: systematic review and meta–analysis. Cerebrovasc Dis. 2014;37:64–75. doi:10.1159/000356789
106. Meissner A. Hypertension and the brain: a risk factor for more than heart disease. Cerebrovasc Dis. 2016;42:255–262. doi:10.1159/000446082
107. Behzadian MA, Wang XL, Shabrawey M, Cadwell RB. Effects of hypoxia on glial cell expression of angiogenesis–regulating factors VEGF and TGF–β. Glia. 1998;24:216–225. doi:10.1002/(SICI)1098-1136(199810)24:2<216::AID-GLIA6>3.0.CO;2-1
108. Tarkowski E, Issa R, Sjogren M, et al. Increased intrathecal levels of the angiogenic factors VEGF and TGF–beta in Alzheimer’s disease and vascular dementia. Neurobiol Aging. 2002;23(2):237–243. doi:10.1016/S0197-4580(01)00285-8
109. Vischer UM. Von Willebrand factor, endothelial dysfunction, andcardiovascular disease. J Thromb Haemost. 2006;4(6):1186–1193. doi:10.1111/j.1538-7836.2006.01949.x
110. Folsom AR, Rosamond WD, Shahar E, et al. Prospective study of markers of hemostatic function with risk of ischemic stroke. The atherosclerosis risk in communities (ARIC) study investigators. Circulation. 1999;100:736–742. doi:10.1161/01.CIR.100.7.736
111. Conway DS, Pearce LA, Chin BS, Hart RG, Lip GY. Prognostic value of plasma von willebrand factor and soluble P‐selectin as indices of endothelial damage and platelet activation in 994 patients with nonvalvular atrial fibrillation. Circulation. 2003;107:3141–3145. doi:10.1161/01.CIR.0000077912.12202.FC
112. Quinn TJ, Gallacher J, Deary IJ, Lowe GDO, Fenton C, Stott DJ. Association between circulating hemostatic measures and dementia or cognitive impairment: systematic review and meta–analyzes. J Thromb Haemost. 2011;9:1475–1482. doi:10.1111/j.1538-7836.2011.04403.x
113. Voskuil M, van Royen N, Hoefer IE, et al. Modulation of collateral artery growth in a porcine hindlimb ligation model using MCP–1. Am J Physiol Heart Circ Physiol. 2003;284(4):
114. Niu J, Wang K, Zhelyabovska O, Saad Y, Kolattukudy PE. MCP–1 induced protein promotes endothelial–like and angiogenic properties in human bone marrow monocytic cells. J Pharmacol Exp Ther. 2013;347(2):288–297. doi:10.1124/jpet.113.207316
115. Bruno V, Copani A, Besong G, Scoto G, Nicoletti F. Neuroprotective activity of chemokines against n–methyl–d–aspartate or beta–amyloid–induced toxicity in culture. Eur J Pharmacol. 2000;399:117–121. doi:10.1016/S0014-2999(00)00367-8
116. Dimitrijevic OB, Stamatovic SM, Keep RF, Andjelkovic AV. Absence of the chemokine receptor CCR2 protects against cerebral ischemia/reperfusion injury in mice. Stroke. 2007;38:1345–1353. doi:10.1161/01.STR.0000259709.16654.8f
117. Losy J, Zaremba J. Monocyte chemoattractant protein–1 is increased in the cerebrospinal fluid of patients with ischemic stroke. Stroke. 2001;32:2695–2696. doi:10.1161/hs1101.097380
118. Pasceri V, Willerson JT, Yeh ET. Direct proinflammatory effect of C-reactive protein on human endothelial cells. Circulation. 2000;102:2165–2168. doi:10.1161/01.CIR.102.18.2165
119. Venugopal SK, Devaraj S, Yuhanna I, Shaul P, Jialal I. Demonstration that C-reactive protein decreases eNOS expression and bioactivity in human aortic endothelial cells. Circulation. 2002;106:1439–1441. doi:10.1161/01.CIR.0000033116.22237.F9
120. Whiteley W, Jackson C, Lewis S, et al. Association of circulating inflammatory markers with recurrent vascular events after stroke: a prospective cohort study. Stroke. 2011;42:10–16. doi:10.1161/STROKEAHA.110.588954
121. Winbeck K, Poppert H, Etgen T, Conrad B, Sander D. Prognostic relevance of early serial С–reactive protein measurements after first ischemic stroke. Stroke. 2002;33:2459–2464.
122. Van Sloten TT, Stehouwer CD. Carotid stiffness: a novel cerebrovascular disease risk factor. Pulse (Basel). 2016;4(1):24–27. doi:10.1159/000445354
123. Mitchell GF. Effects of central arterial aging on the structure and function of the peripheral vasculature: implications for end–organ damage. J Appl Physiol. 2008;105:1652–1660. doi:10.1152/japplphysiol.90549.2008
124. O’Rourke MF, Safar ME. Relationship between aortic stiffening and microvascular disease in brain and kidney: cause and logic of therapy. Hypertension. 2005;46:200–204. doi:10.1161/01.HYP.0000168052.00426.65
125. Tzourio C, Laurent S, Debette S. Is hypertension associated with an accelerated aging of the brain? Hypertension. 2014;63:894–903. doi:10.1161/HYPERTENSIONAHA.113.00147
126. Rothwell PM. Limitations of the usual blood–pressure hypothesis and importance of variability, instability, and episodic hypertension. Lancet. 2010;375:938–948. doi:10.1016/S0140-6736(10)60309-1
127. Schillaci G, Bilo G, Pucci G, et al. Relationship between short–term blood pressure variability and large–artery stiffness in human hypertension: findings from 2 large databases. Hypertension. 2012;60:369–377. doi:10.1161/HYPERTENSIONAHA.112.197491
128. Selwaness M, van Den Bouwhuijsen Q, Mattace–Raso FU, et al. Arterial stiffness is associated with carotid intraplaque hemorrhage in the general population: the Rotterdam study. Arterioscler Thromb Vasc Biol. 2014;34:927–932. doi:10.1161/ATVBAHA.113.302603
129. Ungvari Z. Mechanisms of vascular aging: new perspectives. J Gerontol A Biol Sci Med Sci. 2010;65(10):1028–1041. doi:10.1093/gerona/glq113
130. Donato AJ, Eskurza I, Silver AE, et al. Direct evidence of endothelial oxidative stress with aging in humans. Circ Res. 2007;100(11):1659–1666. doi:10.1161/01.RES.0000269183.13937.e8
131. Ungvari Z, Buffenstein R, Austad SN, Podlutsky A, Kaley G, Csiszar A. Oxidative stress in vascular senescence: lessons from successfully aging species. Front Biosci. 2008;13:5056–5070.
132. Iadecola C. The pathobiology of vascular dementia. Neuron. 2013;80:844–866. doi:10.1016/j.neuron.2013.10.008
133. Sindler AL, Delp MD, Reyes R, Wu G, Muller–Delp JM. Effects of ageing and exercise training on eNOS uncoupling in skeletal muscle resistance arterioles. J Physiol. 2009;587(15):3885–3897. doi:10.1113/jphysiol.2009.172221
134. Berkowitz DE, White R, Li D, et al. Arginase reciprocally regulates nitric oxide synthase activity and contributes to endothelial dysfunction in aging blood vessels. Circulation. 2003;108(16):2000–2006. doi:10.1161/01.CIR.0000092948.04444.C7
135. Matsushita H, Chang E, Glassford AJ, Cooke JP, Chiu CP, Tsao PS. eNOS activity is reduced in senescent human endothelial cells. Circ Res. 2001;89(9):793–798.
136. Franceschi C, Bonafè M, Valensin S, et al. Inflamm–aging: an evolutionary perspective on immunosenescence. Ann N Y Acad Sci. 2000;908(1):244–254.
137. Miles EA, Rees D, Banerjee T, et al. Age–related increases in circulating inflammatory markers in men are independent of BMI, blood pressure and blood lipid concentrations. Atherosclerosis. 2008;196(1):298–305. doi:10.1016/j.atherosclerosis.2006.11.002
138. Csiszar A, Labinskyy N, Smith K, Rivera A, Orosz Z, Ungvari Z. Vasculoprotective effects of anti–tumor necrosis factor–α treatment in aging. Am J Pathol. 2007;170(1):388–398. doi:10.2353/ajpath.2007.060708
139. Jiang L, Wang M, Zhang J, et al. Increased aortic calpain–1 activity mediates age–associated angiotensin II signaling of vascular smooth muscle cells. PLoS One. 2008;3(5):e2231. doi:10.1371/journal.pone.0002231
140. Marchesi C, Paradis P, Schiffrin EL. Role of the renin–angiotensin system in vascular inflammation. Trends Pharmacol Sci. 2008;29:367–374. doi:10.1016/j.tips.2008.05.003
141. Faraci FM. Protecting against vascular disease in brain. Am J Physiol Heart Circ Physiol. 2011;300:H1566–H1582. doi:10.1152/ajpheart.01310.2010
142. Gill R, Tsung A, Billiar T. Linking oxidative stress to inflammation: toll–like receptors. Free Radic Biol Med. 2010;48:1121–1132. doi:10.1016/j.freeradbiomed.2010.01.006
143. Carrano A, Hoozemans JJ, van der Vies SM, Rozemuller AJ, van Horssen J, de Vries HE. Amyloid beta induces oxidative stress–mediated blood–brain barrier changes in capillary amyloid angiopath. Antioxid Redox Signal. 2011;15:1167–1178. doi:10.1089/ars.2011.3895
144. Guo S, Kim WJ, Lok J, et al. Neuroprotection via matrix–trophic coupling between cerebral endothelial cells and neurons. Proc Natl Acad Sci USA. 2008;105:7582–7587. doi:10.1073/pnas.0801105105
145. Counts SE, Mufson EJ. Noradrenaline activation of neurotrophic pathways protects against neuronal amyloid toxicity. J Neurochem. 2010;113:649–660. doi:10.1111/j.1471-4159.2010.06622.x
146. Sim FJ, Zhao C, Penderis J, Franklin RJ. The age–related decrease in CNS remyelination efficiency is attributable to an impairment of both oligodendrocyte progenitor recruitment and differentiation. J Neurosci. 2002;22:2451–2459. doi:10.1523/JNEUROSCI.22-07-02451.2002
147. Asai K, Kudej RK, Shen YT, et al. Peripheral vascular endothelial dysfunction and apoptosis in old monkeys. Arterioscler Thromb Vasc Biol. 2000;20(6):1493–1499.
148. Riddle DR, Sonntag WE, Lichtenwalner RJ. Microvascular plasticity in aging. Ageing Res Rev. 2003;2(2):149–168.
149. Rivard A, Fabre JE, Silver M, et al. Age–dependent impairment of angiogenesis. Circulation. 1999;99(1):111–120.
150. Parfenov VA, Ostroumova TM, Perepelova EM, Perepelov VA, Kochetkov AI, Ostroumova OD. Brain perfusion, cognitive function and vascular age in middle-aged patients with essential arterial hypertension. Cardiology. 2018;58(5):23–31. doi: 10.18087/cardio.2018.5.10117. In Russian.
151. D’Agostino RB, Vasan RS, Pencina MJ, et al. General cardiovascular risk profile for use in primary care: the Framingham Heart Study. Circulation. 2008;117:743–753. doi:10.1161/CIRCULATIONAHA.107.699579
152. Henskens LH, Kroon AA, van Oostenbrugge RJ, et al. Increased aortic pulse wave velocity is associated with silent cerebral small–vessel disease in hypertensive patients. Hypertension. 2008;52:1120–1126. doi:10.1161/YPERTENSIONAHA.108.119024
153. Yang EY, Chambless L, Sharrett AR, et al. Carotid arterial wall characteristics are associated with incident ischemic stroke but not coronary heart disease in the Atherosclerosis Risk in Communities (ARIC) study. Stroke. 2012;43:103–108. doi:10.1161/STROKEAHA.111.626200
154. Zeki AL, Hazzouri A, Newman AB, et al;
155. Farooq MU, Min J, Goshgarian C, Gorelick PB. Pharmacotherapy for vascular cognitive impairment. CNS Drugs. 2017;31(9):759–776. doi:10.1007/s40263-017-0459-3
156. Epstein OI. Release–activity: a long way from phenomenon to new drugs. Bull Exp Biol Med. 2012;154(1):54–58. doi:10.1007/s10517-012-1874-6
157. Epshtein OI, Beregovoi NA, Sorokina NS, Starostina MV, Shtark MB. Effect of various dilutions of the potentiated antibodies to the brain-specific protein S-100 on the post-titanic potentiation in surviving hippocampal slices. Bull Exp Biol Med. 1999;3:317–320. In Russian.
158. Epshtein OI, Gainutdinov KL, Shtark MB. Effect of homeopathic doses of antibodies to antigen S–100 on the electric parameters of neuronal membranes. Bull Exp Biol Med. 1999;4:466–467. In Russian.
159. Voronina TA, Molodavkin GM, Sergeeva SA, Epstein OI. GABAergic system in the anxiolytic effect of Proproten: experimental study. Bull Exp Biol Med. 2003;1:125–127.
160. Kheifets IA, Molodavkin GM, Voronina TA, Dugina YL, Sergeeva SA, Epstein OI. Involvement of the GABAB–ergic system in the mechanism of action of ultralow dose antibodies to S–100 protein. Bull Exp Biol Med. 2008;5:552–554. In Russian.
161. Gorbunov EA, Ertuzun IA, Kachaeva EV, Tarasov SA, Epstein OI. In vitro screening of major neurotransmitter systems possibly involved in the mechanism of action of antibodies to S100 protein in released–active form. Neuropsychiatr Dis Treat N Z. 2015;11:2837–2846. doi:10.2147/NDT.S92456
162. Kheifets IA, Dugina YL, Voronina TA, et al. Involvement of the serotoninergic system in the mechanism of action of ultralow dose antibodies to S–100 protein. Bull Exp Biol Med. 2007;5:535–537. In Russian.
163. Ertuzun IA. Mechanisms of Anxiolytic and Antidepressant Action of Tenoten (Experimental Study) [PhD Dissertation Abstract]. Tomsk: Federal State Budgetary Institution “Research Zakusov Institute of Pharmacology” (FSBI “Zakusov Institute of Pharmacology”); 2012:23. In Russian.
164. Voronina TA, Borodavkina MV, Heifets IA, Molodavkin GM, Dugina YL, Sergeeva SA. Experimental study of nootropic and anti–amnestic activity of the tenoten in the test of the conditioned reflex of passive avoidance in rats.
165. Voronina TA, Belopol’skaya MV, Kheyfets IA, Dugina YL, Sergeeva SA, Epshtein OI. Effect of ultralow doses of antibodies to S–100 protein in animals with impaired cognitive function and disturbed emotional and neurological status under conditions of experimental Alzheimer disease. Bull Exp Biol Med. 2009;148(1):533–535. doi:10.1007/s10517-010-0757-y
166. Voronina TA, Molodavkin GM, Borodavkina MV, Kheyfets IA, Dugina YL, Sergeeva SA. Nootropic and antiamnestic effects of tenoten (pediatric formulation) in immature rat pups. Bull Exp Biol Med. 2009;148(1):524–526. doi:10.1007/s10517-010-0754-1
167. Romanova GA, Barskov IV, Ostrovskaia RU, Gudasheva TA, Viktorov IV. Behavioral and morphologic disorders caused by bilateral photoinduced thrombosis of the cerebral vessels of the frontal cortex in rats. Patol Fiziol Eksp Ter. 1998;2:8–10. In Russian.
168. Voronina TA, Kheifets IA, Dugina YL, Sergeeva SA, Epstein OI. Study of the effects of preparation containing ultralow doses of antibodies to S–100 protein in experimental hemorrhagic stroke. Bull Exp Biol Med. 2009;148(1):530–532. doi:10.1007/s10517-010-0756-z
169. Voronina TA, Sergeeva SA, Martyushev–Poklad AV, Dugina JL, Epstein OI. Antibodies to S–100 protein in anxiety–depressive disorders in experimental and clinical conditions. Anim Model Biol Psychiatry Nov Sci Publ. 2006;8:137–152.
170. Kheyfets IA, Voronina TA, Dugina JL, Molodavkin GM, Sergeeva SA. Anxiolytic activity of tenoten and diazepam depends on conditions in vogel conflict test. Bull Exp Biol Med. 2011;151(3):336–339. doi:10.1007/s10517-011-1324-x. In Russian.
171. Voronina TA, Molodavkin GM, Sergeeva SA, Epstein OI. Anxiolytic effect of proproten under conditions of punished and unpunished behavior. Bull Exp Biol Med. 2003;1:120–122.
172. Epstein OI, Molodavkin GM, Voronina TA, Sergeeva SA. Antidepressant properties of proproten and amitriptyline: comparative experimental study. Bull Exp Biol Med. 2003;135(7):123–124. doi:10.1023/A:1024724023144
173. Zhavbert EV Pharmacological Activity of Antibodies to Endothelial NO–Synthase in the Released–Active form on the Model of Erectile Dysfunction [PhD Dissertation Abstract]. Tomsk: Federal State Budgetary Institution “Research Zakusov Institute of Pharmacology” (FSBI “Zakusov Institute of Pharmacology”); 2016:25. In Russian.
174. Belous AU, Arustamova AA, Pokrovsky MV, Korokin MV, Gudyrev OS, Belous VS. L–NAME and hypoestrogen–induced endothelial dysfunction treatment with impaza. Sci bull Belgorod State Univ Ser: Med Pharm. 2011;4(99):116–120. In Russian.
175. Arustamova AA The Anti–Ischemic and Endothelioprotective Effect of Potentiated Antibodies to the Endothelial Growth Factor of Blood Vessels [PhD Dissertation Abstract]. Belgorod; 2011:22. In Russian.
176. Chernyshova GA, Aliev OI, Smoljakova VI, Plotnikov MB, Martyushev AV, Sergeeva SA. Impaza and sildenafil influence on the cardiovascular system and the hypotensive effect of nitroglycerin.
177. Markel’ AL, Zhavbert ES, Tarasov SA, et al. Effect of impaza on cardiovascular system. Bull Exp Biol Med. 2009;148(1):518–519. doi:10.1007/s10517-010-0752-3
178. Kardash EV, Gorbunov EA, Tarasov AV, Yakovleva NN, Tarasov SA. Divaza influence on sigma receptors regulating the basic neurotransmitter systems.
179. Ganinа KK, Duginа YL, Zhavbert ES, et al. Antiamnesic effects divaza and its component model β–amyloid amnesia. Zh Nevrol Psikhiatr Im S S Korsakova. 2016;116(9):69–74. doi: 10.17116/jnevro20161169169-74. In Russian.
180. Zhavbert ES, Guryanova NN, Surkova EI, Dugina YL, Kachaeva EV, Epshtein OI. Divaza influence on the processes of lipid peroxidation.
181. Yakovleva NN, Voronina TA, Suslov NI, et al. Anxiolytic and antidepressant effects of divaza and brizantin. Bull Exp Biol Med. 2015;159(6):753–756. doi:10.1007/s10517–015–3067–6
182. Gonzalez–Alvear GM, Werling LL. Sigma receptor regulation of norepinephrine release from rat hippocampal slices. Brain Res Elsevie. 1995;673(1):61–69. doi:10.1016/0006-8993(94)01394-W
183. Maurice T, Su TP, Privat A. Sigma1-receptor agonists and neurosteroids attenuate B25–35–amyloid peptide–induced amnesia in mice through a common mechanism. Neurosci U S. 1998;83(2):413–428.
184. Hayashi T, Su TP. Sigma–1 receptor ligands: potential in the treatment of neuropsychiatric disorders. CNS Drugs N Z. 2004;18(5):269–284. doi:10.2165/00023210-200418050-00001
185. Hayashi T, Tsai SY, Mori T, Fujimoto M, Su TP. Targeting ligand–operated chaperone sigma–1 receptors in the treatment of neuropsychiatric disorders. Expert Opin Ther Target Engl. 2011;15(5):557–577. doi:10.1517/14728222.2011.560837
186. Castagne V, Lemaire M, Kheyfets I, Dugina JL, Sergeeva SA, Epstein OI. Antibodies to S100 proteins have anxiolytic–like activity at ultra–low doses in the adult rat. J Pharm Pharmacol. 2008;60(3):309–316. doi:10.1211/jpp.60.3.0005
187. Epstein OI. Ultra–Low Doses (History of One Research). Moscow: RAMS Publishing House; 2008:336.
188. Fisenko VP, editor. Guidelines for Experimental (Preclinical) Studies of New Pharmacological Substances. Moscow: «Remedium»; 2000:398. In Russian.
189. Khabriev RU, editor. Guidelines for Experimental (Preclinical) Studies of New Pharmacological Substances. Moscow: «Medicine»; 2005:832. In Russian.
190. Fateeva VV, Shumakher GI, Vorob’eva EN, Khoreva MA, Voskanyan LR. The efficacy and safety of drug therapy Divaza in patients with chronic cerebral ischemia. Zh Nevrol Psikhiatr Im S S Korsakova. 2017;117(2):32–37. doi: 10.17116/jnevro20171172132-37. In Russian.
191. Kamchatnov PR, Vorob’eva OV, Rachin AP. Treatment of emotional and cognitive disorders in patients with chronic cerebral ischemia. Zh Nevrol Psikhiatr Im S S Korsakova. 2014;114(4):52–56. In Russian.
192. Parfenov VА, Kamchatnov PR, Vorobyova ОV, Gustov АV, Glushkov КS, Doronina ОB. Results of multicenter study of efficacy and safety of Divaza in the treatment of the asthenic and mild to moderate cognitive disorders in elderly and senile subjects. Zh Nevrol Psikhiatr Im S S Korsakova. 2017;117(9):43–50. doi: 10.17116/jnevro20171179143-50. In Russian.
193. Parfenov VA, Zhivolupov SA, Nikulina KV, et al. Diagnosis and treatment of cognitive impairment in patients with chronic cerebral ischemia: the results of observational Russian program DIAMANT. Zh Nevrol Psikhiatr Im S S Korsakova. 2018;118(6):15–23. doi: 10.17116/jnevro20181186115. In Russian.
194. Guidelines for preparing core clinical-safety information on drugs, including new proposals for investigators Brochures. Report of CIOMS Working Groups III and V. Geneva (Switzerland): CIOMS. ISBN 92 9036 070 4.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.