")
Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 8
Variations in telomere maintenance and the role of telomerase inhibition in gastrointestinal cancer
Authors Heeg S
Received 11 May 2015
Accepted for publication 29 October 2015
Published 4 December 2015 Volume 2015:8 Pages 171—180
DOI https://doi.org/10.2147/PGPM.S52808
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Steffen Heeg
Department of Medicine II, Gastroenterology, Hepatology, Endocrinology, and Infectious Diseases, Medical Center – University of Freiburg, Freiburg, Germany
Abstract: Immortalization is an important step toward the malignant transformation of human cells and is critically dependent upon telomere maintenance. There are two known mechanisms to maintain human telomeres. The process of telomere maintenance is either mediated through activation of the enzyme telomerase or through an alternative mechanism of telomere lengthening called ALT. While 85% of all human tumors show reactivation of telomerase, the remaining 15% are able to maintain telomeres via ALT. The therapeutic potential of telomerase inhibitors is currently investigated in a variety of human cancers. Gastrointestinal tumors are highly dependent on telomerase as a mechanism of telomere maintenance, rendering telomeres as well as telomerase potential targets for cancer therapy. This article focuses on the molecular mechanisms of telomere biology and telomerase activation in gastrointestinal cancers and reviews strategies of telomerase inhibition and their potential therapeutic use in these tumor entities.
Keywords: telomere based therapy, gastrointestinal cancer, telomere maintenance, telomerase inhibition
Introduction
Circumvention of telomere-based senescence via activation of telomere maintenance mechanisms has been defined as a hallmark of cancer. During the last decade, our understanding of telomere biology and mechanisms of telomere maintenance in normal tissue and cancer development has improved significantly and fostered the development of telomere and telomerase-based therapeutic strategies. In this article, the genetic and molecular underpinnings of telomere maintenance in gastrointestinal cancer and their therapeutic implications are reviewed. As epithelial tumors, gastrointestinal cancers predominantly utilize telomerase to maintain telomeres. However, the molecular mechanisms that induce activation of telomerase differ remarkably among cancers of the gastrointestinal tract including transcriptional and posttranscriptional mechanisms as well as receptor tyrosine kinase signaling. Current telomerase-based therapies do either target telomerase directly to inhibit its enzyme function or use telomerase as an antigen to elicit antitumor immunity. Preclinical and early clinical trials have revealed promising results in gastrointestinal cancers and might provide novel therapeutic options in the future.
Telomere biology and mechanisms of telomere maintenance
Telomere biology
The current understanding that chromosome ends are important for the maintenance of chromosomal stability originated almost 80 years ago with two independent publications by McClintock and Muller.1,2 In the 1980s, it was demonstrated that the DNA sequences at the end of chromosomes consisted of DNA repeats comprising TTAGGG in humans.3,4 These terminal DNA sequences were named telomeres, and later experiments in yeast showed that artificial chromosomes could be stabilized by the addition of these telomere repeats, indicating their crucial role in the maintenance of chromosomal stability. To exert this function, one of two higher-order structures named T-loop and G-quadruplex, respectively, is formed by a 150–250 nucleotide long single-stranded G-rich 3′ overhang.5,6 The T-loop structure is stabilized by a complex of six specialized proteins known as shelterins that cap the telomeres and protect the single-stranded overhangs from DNA-damage repair and end-to-end fusions7 (Figure 1A).
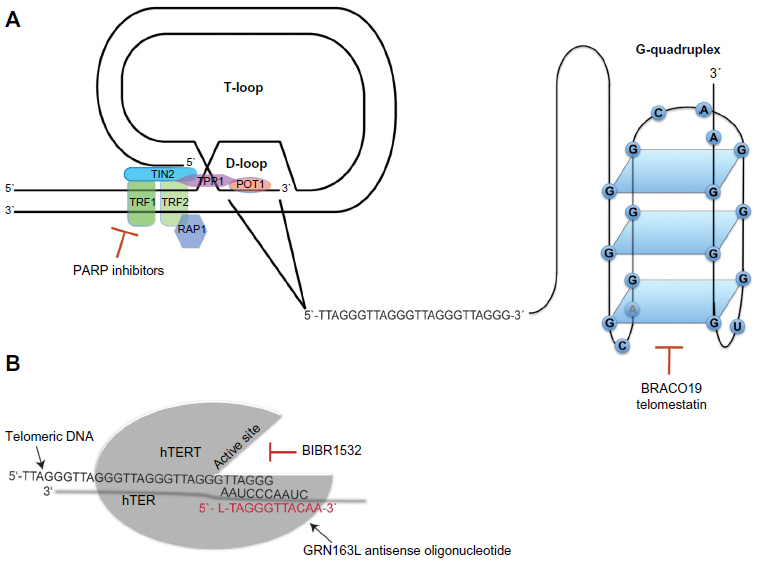 | Figure 1 Telomeres and telomerase as therapeutic targets. |
The G-quadruplex is formed by stacked guanosine quartets incorporating a 16-nucleotide d(GGGTTAGGGTTAGGGT) and a 6-nucleotide d(TAGGGT) sequence of telomeric 3′ overhang, folded via hydrogen bonding (Figure 1A). G-quadruplexes protect the telomeric DNA from being accessed by telomerase, regulating its catalytic activity.8 Moreover, recent data indicate that G-quadruplexes can protect telomeres from DNA-damage signaling.9 In normal cells, telomeres shorten by 50–150 bp with each cell division. After 60–80 cell divisions, telomeres normally reach a critical length that impairs their protective function, leading to the activation of DNA-damage responses very similar to those induced by DNA double-strand breaks.10 The induction of DNA-damage responses results in cellular growth arrest called “replicative senescence”.11 Thus, the stepwise loss of telomeric DNA with aging restricts the life span of each individual cell and serves as an essential mechanism of tissue homeostasis. Moreover, replicative senescence forms an important barrier against tumorigenesis that can only be overcome when cells acquire the ability to maintain their telomeres in the process of malignant transformation.
Mechanisms of telomere maintenance
The observation of immortal tumor cells with an unrestricted capacity of cell divisions implies the ability to stabilize telomere length.12 In 85% of all human tumors, this process is mediated by the enzyme telomerase,13,14 a reverse transcriptase, which contains the catalytic subunit human telomerase reverse transcriptase (hTERT)15 and its own intrinsic RNA moiety human telomerase RNA (hTER), which serves as a template for the synthesis of telomeric DNA16 (Figure 1B). The remaining 15% of telomerase-negative tumors are able to maintain their telomere length by one or more mechanisms referred to as alternative lengthening of telomeres (ALT).17 While the vast majority of carcinomas expresses telomerase, ALT is frequently found in sarcomas and other tumors arising from tissues of mesenchymal origin.14,18 Activity of telomerase and ALT are not mutually exclusive and it has been demonstrated that telomerase and ALT can coexist in human tumors.19,20
Regulation of telomerase activity
Transcriptional regulation of hTERT expression
The major mechanism to regulate telomerase activity in human cells is the transcriptional control of the catalytic subunit hTERT. Analytical studies of the hTERT promoter have identified several transcription factors that directly bind to the hTERT promoter, resulting in the activation of hTERT transcription. Sp1 is a zinc finger transcription factor that binds to five GC-boxes on the hTERT promoter most likely in cooperation with the oncogenic transcription factor c-Myc.21,22 Cellular signaling and regulatory pathways with transcriptional factors as downstream effectors are involved in the transcriptional regulation of hTERT. In this context, phosphatidylinositol-4,5-bisphosphate 3-kinase/AKT (PI3K/AKT) and mitogen-activated protein kinase signaling induces hTERT expression via phosphorylation of the transcriptional repressor Mad1, resulting in its ubiquitinylation and degradation.23,24 Moreover, the receptors of the ErbB family can act as oncoproteins and activate hTERT expression through PI3K/AKT signaling in breast and esophageal cancer as demonstrated by our group and others.25,26 Besides activating factors, a number of negative regulatory factors have been identified. In this context, the downstream E-box located in close proximity to the transcriptional start site was found to mediate the repression of hTERT transcription by Mad1 and USF1.27
CPG-sites as potential targets of methylation can be frequently found within the hTERT promoter, suggesting an epigenetic mechanism in the regulation of hTERT expression. However, a clear correlation between promoter methylation and transcriptional activation of hTERT could not be established. While several groups have demonstrated that promoter methylation is associated with lower hTERT expression reflecting the well-described mechanism of gene silencing through hypermethylation of promoter regions,28–31 others have shown that hypermethylation of the hTERT promoter region leads to increased hTERT expression, whereas demethylation of this region inhibits hTERT transcription.32,33 This discrepancy might at least in part result from the different cell lines and methylation-specific methods that have been used in various studies.31 Moreover, the local distribution of methylation events across the hTERT promoter seems to be critical for the regulation of hTERT expression. In a study by Zinn et al,34 the hTERT promoter was analyzed in telomerase-positive breast, lung, and colon cancer cell lines, which displayed little or no methylation within an area close to the transcriptional start site of hTERT despite abundant methylation within promoter regions further upstream, indicating that demethylation of the core promoter might be required for the transcriptional activation of hTERT in tumor cells. In contrast, observations in normal tumor cells and tissues without hTERT expression revealed un- or hypomethylated promoters indicating that promoter methylation is not required for developmental silencing of hTERT.35 To date, the available data are contradictory, suggesting that changes in the methylation status of the hTERT promoter may not represent a major mechanism regulating hTERT expression.
Posttranslational modifications
Posttranslational regulation of telomerase activity can occur via reversible phosphorylation of hTERT at specific serine/threonine or tyrosine residues.36,37 For example, besides its role in the transcriptional regulation of hTERT expression, AKT can also directly phosphorylate hTERT leading to increased telomerase activity.26,37 Moreover, studies in head and neck as well as in breast cancer cells have demonstrated that telomerase activation can be induced by protein kinase C-dependent phosphorylation of hTERT leading to increased stability of the telomerase holoenzyme.38,39 While phosphorylation of hTERT by AKT and protein kinase C is associated with increased telomerase activity, phosphorylation by the c-ABL kinase at specific tyrosine residues has been reported to decrease telomerase activity and telomere length.40
Nuclear transport, telomerase complex assembly, and telomere binding
According to Tomlinson et al,41 the two essential components of telomerase hTER and hTERT accumulate at distinct intranuclear sites separate from telomeres throughout most of the cell cycle. During S-phase, both components are specifically recruited to subsets of telomeres. The assembly of the telomerase complex involves multiple additional proteins that are crucial for maturation of the holoenzyme and its binding to telomeres. Heat shock protein 90 and p23 are molecular chaperones required for the assembly, maturation, and activation of the telomerase complex. Blocking the adenosine triphosphate-dependent binding of heat shock protein 90 to p23 disrupts the formation of the chaperone complex resulting in telomerase inhibition.42 Moreover, interaction of telomerase with the telomere-binding proteins of the shelterin complex seems to be critical for the regulation of telomerase activity. TRF1 is a telomeric DNA-binding protein that serves as a negative regulator of telomere length by blocking access of telomerase to telomeres. Consequently, removal of TRF1 from telomeres allows telomerase to access and results in telomerase activity and elongation of telomeres.8
ALT – characteristics and regulation
The one common feature of all potential ALT subtypes is the maintenance of telomeres in the absence of telomerase. The initial characteristics described in ALT cells were heterogeneous telomere length within a single cell and the presence of ALT-associated promyelocytic leukemia bodies.43–45 For a subgroup of ALT, more recent data suggest that telomere maintenance is based upon recombination,46,47 which seems to be associated with proteins involved in common reciprocal recombination.48,49 Moreover, ALT cells contain both linear and circular extrachromosomal telomeric repeats representing telomeric templates for recombination.50 Interestingly, the presence of one of the two known telomere maintenance mechanisms varies with specific tumor type. While the majority of epithelial tumors display telomerase activity, cancers of mesenchymal origin such as osteosarcoma or astrocytoma more frequently activate ALT to maintain their telomeres.18,51 The molecular mechanisms involved in the regulation of ALT are not fully understood. However, mutations in genes that physiologically repress DNA recombination such as p53, ATRX, DAXX, and H3F3A seem to facilitate recombination-based telomere maintenance.52 Mutations permissive for ALT include those that are involved in telomerase regulation, and our group and others have recently demonstrated that ALT can be activated by inhibition of telomerase and ALT cells that actively repress telomerase expression.26,53 Moreover, data from human sarcoma indicate that tumors can be mosaic for cells using either telomerase activity or ALT as mechanisms of telomere maintenance.54 These findings suggest that telomerase activity and ALT are not mutually exclusive and can be present in the same tumor and possibly even in the same cell, which might have important implications for telomere-based cancer therapies.
Regulation of telomere biology and maintenance in gastrointestinal tumors
Esophageal cancer
Esophageal cancer presents in two distinct histologic subtypes, namely esophageal squamous cell cancer (ESCC) and esophageal adenocarcinoma (EAC). ESCC typically arises from the squamous epithelium in the upper two-thirds of the esophagus with alcohol and tobacco as major risk factors. EAC are frequently located in the lower third of the esophagus and arise from mucus secreting glandular tissues that are reminiscent of an intestinal epithelium and often form after long-term exposure to acid and bile reflux.
Carcinoma in situ of ESCC exhibits telomeres shorter than those in the surrounding normal epithelium, indicating that telomere attrition occurs early in the process of esophageal carcinogenesis55 and is associated with chromosomal instability.56 Telomerase is activated in the majority of ESCC during cancer progression leading to stabilization of telomere lengths. The molecular mechanisms involved in the activation of telomerase in ESCC development are not fully understood. Quante et al57 have demonstrated that genetic alterations frequently found in early esophageal carcinogenesis such as overexpression of cyclin D1 or inactivation of p53 can independently induce transcriptional activation of hTERT through transcriptional activators that are specific for the respective genetic alteration.57 These data indicate that activation of telomerase might occur relatively early in the process of cancer progression and is at least in part regulated on a transcriptional level. Activating mutations within the hTERT promoter have been described in different types of cancer, but their frequency in both types of esophageal cancer is extremely low.58,59 The epidermal growth factor receptor (EGFR)-signaling pathway is frequently activated in ESCC and has been linked to telomerase activation through both transcriptional and posttranslational regulation of hTERT. In this context, EGFR induces hTERT transcription through the transcription factor hypoxia-inducible factor 1α as well as phosphorylation and activation of hTERT via the PI3K/AKT-signaling pathway.26
Chromosomal instability is a hallmark of Barrett’s esophagus, an intestinal metaplasia in the esophagus that forms the precursor lesions of EAC, and is induced by telomere shortening.60 Similar to ESCC, telomerase activity can be frequently detected in EAC and hTERT expression has been found to be gradually increasing during the Barrett’s metaplasia–dysplasia–adenocarcinoma sequence.61,62
HER2, a member of the EGFR family, is overexpressed in approximately 20% of EAC and is successfully used as a therapeutic target by the antibody trastuzumab. HER2 is involved in telomerase activation in breast cancer63 and could play a similar role in the HER2-positive subset of EAC.
Gastric cancer
The risk for the development of gastric cancer is increased in people with shorter telomeres in peripheral blood lymphocytes. Telomere length in peripheral leukocyte DNA reflects cumulative oxidative stress and is associated with Helicobacter pylori positivity, cigarette smoking, and dietary fruit intake.64 A portion of gastric cancers (10%–25%) is characterized by defects in the DNA mismatch repair, resulting in genomic instability characterized by microsatellite instability. These gastric cancers seem to preferentially utilize ALT to maintain telomere length, while tumors with proficient mismatch repair show telomerase activity.65 The mechanisms by which telomerase is activated in gastric cancer remain elusive. Sequencing of the hTERT-promoter in a cohort of almost 800 patients revealed the absence of activating mutations in gastric cancer.66 Similar to esophageal cancer, EGFR-signaling via AKT has been linked to telomerase activation in gastric cancer. AKT activation by epidermal growth factor increased hTERT expression and telomerase activity in gastric cancer cells, while AKT inhibition had the opposite effect. Concurrently, in gastric cancer tissues, significant correlations were found among the levels of phosphorylated AKT, hTERT expression, and telomere length.67
Colorectal cancer
Like in other tumors, telomere shortening in colorectal cancer (CRC) occurs with cell proliferation in preneoplastic lesions and leads to chromosomal instability. Telomerase is activated during the progression of preneoplastic lesions as hTERT levels and telomerase activity increase with the adenoma–carcinoma sequence and are highest in carcinoma.68,69 Telomere lengths may then be stabilized with increasing telomerase activity during tumor progression.70 Both telomere length and telomerase have been extensively studied in CRC.
Several groups have investigated the prognostic value of telomere length correlating telomere length with disease progression in CRC, but the results of these studies are inconsistent. Gertler et al observed a correlation of telomere length with disease progression and more advanced tumor stage in a cohort of 57 patients.71 However, these findings could be confirmed in other cohorts.70,72
While the prognostic value of telomere length in CRC is still debated, it has been recently reported that telomere length might have the potential to serve as a predictive biomarker of sensitivity to anti-EGFR therapy in metastatic CRC. In their study, Bertorelle et al73 demonstrated that among patients with KRAS wild-type tumors treated with anti-EGFR therapy, those with longer telomeres had a superior progression-free survival than those with shorter ones (24.9 vs 11.1 weeks; hazard ratio: 0.31; P=0.048).73 Several studies have revealed that hTERT expression correlates with tumor progression and significantly higher levels of hTERT have been observed in poorly differentiated and higher stage tumors.72 The prognostic value of hTERT expression in CRC has been evaluated in a cohort of 137 patients. Bertorelle et al were able to show that patients with high levels of hTERT messenger RNA (mRNA) in their tumor cells had a significantly higher risk of death during the observation period of 70 months.73 Importantly, high hTERT levels inversely correlated with the disease-free survival as well as overall survival of patients with stage II tumors, indicating that telomerase might be a useful predictive tool to identify CRC patients with a stage II tumor that may benefit from adjuvant chemotherapy.
Hepatocellular carcinoma
Hepatocellular carcinoma (HCC) typically develops in the setting of chronic liver inflammation leading to fibrosis and eventually cirrhosis caused by virus hepatitis, alcohol, nonalcoholic fatty liver disease, and other environmental factors. Patients with chronic hepatitis B infection may even develop HCC in the absence of cirrhosis. Telomere length progressively shortens during carcinogenesis from cirrhotic to low-and high-grade dysplastic nodules and HCC.65 Telomerase activity can be detected in most HCC, and several groups have demonstrated telomerase activation already in precancerous hepatic lesions.74 Oh et al75 have observed shortening of telomeres and activation of telomerase in precancerous dysplastic nodules with a significant induction of hTERT expression in the transition from low-grade dysplastic nodules to high-grade dysplasia. Activating mutations within the hTERT promoter are among the most frequent genetic alterations in HCC and can be found in approximately 60% of these tumors. Consistent with increasing telomerase activity during hepatocellular carcinogenesis, their frequency rapidly increases during the different steps of the transformation from premalignant lesions into HCC.76 Moreover, recent data indicate that somatic hTERT promoter mutations might serve as a biomarker to predict transformation of premalignant lesions into HCC.77 hTERT mRNA can be detected in serum and has been investigated as a diagnostic marker for HCC. In cohort of 300 patients with HCC, serum levels of hTERT mRNA independently correlated with tumor size and tumor differentiation and proved to be superior to α-fetoprotein in the diagnosis of HCC. Moreover, the detection rate of small HCC was superior to α-fetoprotein warranting further evaluation of hTERT as a serum marker for HCC.78
Pancreatic cancer
Pancreatic ductal adenocarcinoma (PDAC) arises from normal pancreatic ductal cells through premalignant lesions called pancreatic intraepithelial neoplasms (PanIN). PanIN are categorized into three grades according to their histopathologic features (PanIN1–3). Telomeres are shorter in PanIN than in normal pancreatic ductal cells, but do not vary significantly between PanIN1–3. The shortest telomeres can be found in PDAC.65 Telomerase activity can be detected in more than 80% of PDAC.79 Similar to other gastrointestinal cancers, promoter mutations are absent in pancreatic cancer and the molecular mechanisms that lead to activation of telomerase in PDAC are largely unknown.80 Activating KRAS mutations are the main driver of tumor progression in more than 90% of PDAC. Downstream effectors of RAS such as MEK and Ets-transcription factors have been identified as activators of hTERT expression in other tumors and might also play a pivotal role in telomerase regulation in PDAC.
Telomerase as therapeutic target in gastrointestinal cancer
Inhibition of telomerase can be mediated either through direct targeting of its catalytic subunit hTERT or its RNA template TER inducing telomere shortening and inhibition of cell proliferation. Indirect targeting aims to block telomerase access to telomeres or binds proteins that are involved in the assembly of the telomerase complex at the telomere. Furthermore, telomerase has been utilized as an antigen for the development of immunotherapies that induces CD8+ cytotoxic T lymphocytes directed against hTERT leading to inhibition of the enzyme function.8,81 The efficacy of therapeutic agents targeting telomerase function in gastrointestinal cancer either by inhibition (GRN163L) or induction of antitumor immunity (GV1001) has been investigated in several preclinical and clinical trials over the last decade.
GRN163L (Imetelstat, Geron Corporation, Menlo Park, CA, USA) is an antisense oligonucleotide directed against the RNA template hTER and has been tested in several preclinical studies on multiple tumor models including breast and lung carcinoma as well as hematologic neoplasias in vitro and in vivo. Inhibition of telomerase by GRN163L resulted in decreased proliferation, increased senescence, and apoptosis. Moreover, reduced tumorigenicity and invasive capacity have been observed in various tumor types including gastrointestinal cancers.8 In EAC cells that were microdissected from human tumor samples, GRN163L successfully inhibited telomerase, induced apoptosis, and resulted in a reduction of tumor size in mouse xenografts.82 In several preclinical studies including esophageal squamous cancer and HCC, GRN163L has shown the potential to sensitize cells with critically short telomeres to radiation and chemotherapy.83,84
Interestingly, in PDAC, GRN163L successfully reduced the number of putative cancer stem cells, a cellular subpopulation that is thought to be involved in both resistance to chemotherapy and in metastatic progression, and significantly impaired the engraftment of PANC1 cells in nude mice.85
GRN163L is currently investigated in several early clinical trials assessing its safety and efficacy in the treatment of various solid tumors and hematologic malignancies. To date, clinical data on the effect of GRN163L on gastrointestinal cancers are lacking. However in late 2014, the manufacturer announced to further develop GRN163L for oncology, including hematologic malignancies, and other human therapeutic uses.
We have recently demonstrated that alternative lengthening of telomeres can be induced by inhibition of telomerase in ESCC, indicating its role as a mechanism of resistance to telomerase inhibition.20 In line with these findings, Shammas et al82 demonstrated that telomerase inhibition prevented telomere elongation, but induced homologous recombination, which contributed to telomere stabilization and reduced therapeutic efficacy. The authors investigated a dual approach targeting both mechanisms in EAC. Combining inhibition of homologous recombination with telomerase inhibition rendered telomeres more vulnerable to degradation and significantly enhanced their attrition, leading to increased apoptosis.86 These data indicate that future telomere-based therapeutic approaches might have to target both telomerase and ALT to inhibit tumor growth.
Despite its expression in normal human tissue, telomerase has been utilized as a tumor antigen for the development of telomere-based anticancer immunotherapies. As outlined, telomerase is present in the majority of cancers and its peptides are capable of producing strong immune responses through the induction of CD8+ cytotoxic T lymphocytes via major histocompatibility complex presentation resulting in cell lysis.8 Several hTERT-based vaccination strategies have been developed over the last decade.
GV1001 is a 16 amino acid major histocompatibility complex class II-restricted hTERT peptide vaccine and is used in combination with an adjuvant to enhance immune responses. GV1001 has completed several Phase I and Phase II clinical trials in patients with advanced stage solid tumors, including HCC and pancreatic cancer. While GV1001 did not induce a significant immune response and antitumor effect in patients with HCC,87 early trials in pancreatic cancer have reported promising results.88,89 For example, Bernhardt et al88 investigated the safety and tolerability of GV1001 in combination with granulocyte-macrophage colony-stimulating factor. GV1001 was well tolerated and immune responses were observed in 63% of the patients, who had a greater median survival (218 days) than those without an immune response (88 days). Subsequently, GV1001 has been tested in patients with locally advanced or metastatic pancreatic cancer in a randomized Phase III trial in combination with chemotherapy. In this trial, the rate of immune responses was significantly lower and no effect of GV1001 on the median survival of patients with pancreatic cancer could be observed.90 For telomerase vaccination strategies to be effective, an active immune response is needed. Pancreatic cancer is a highly proliferative tumor characterized by aggressive local growth and early metastasis with a short median survival time that might be insufficient to establish a stronger antitumor immunity. In addition, pancreatic cancer is generally poorly vascularized and less immunogenic than other tumors. In the future, hTERT vaccination studies will therefore have to focus on adjuvant strategies to enhance immune responses and antitumor effects.
Several other strategies of targeting telomeres and telomerase have been developed and are currently tested in preclinical and early clinical trials with no particular focus on gastrointestinal cancer (Table 1 and Figure 1).
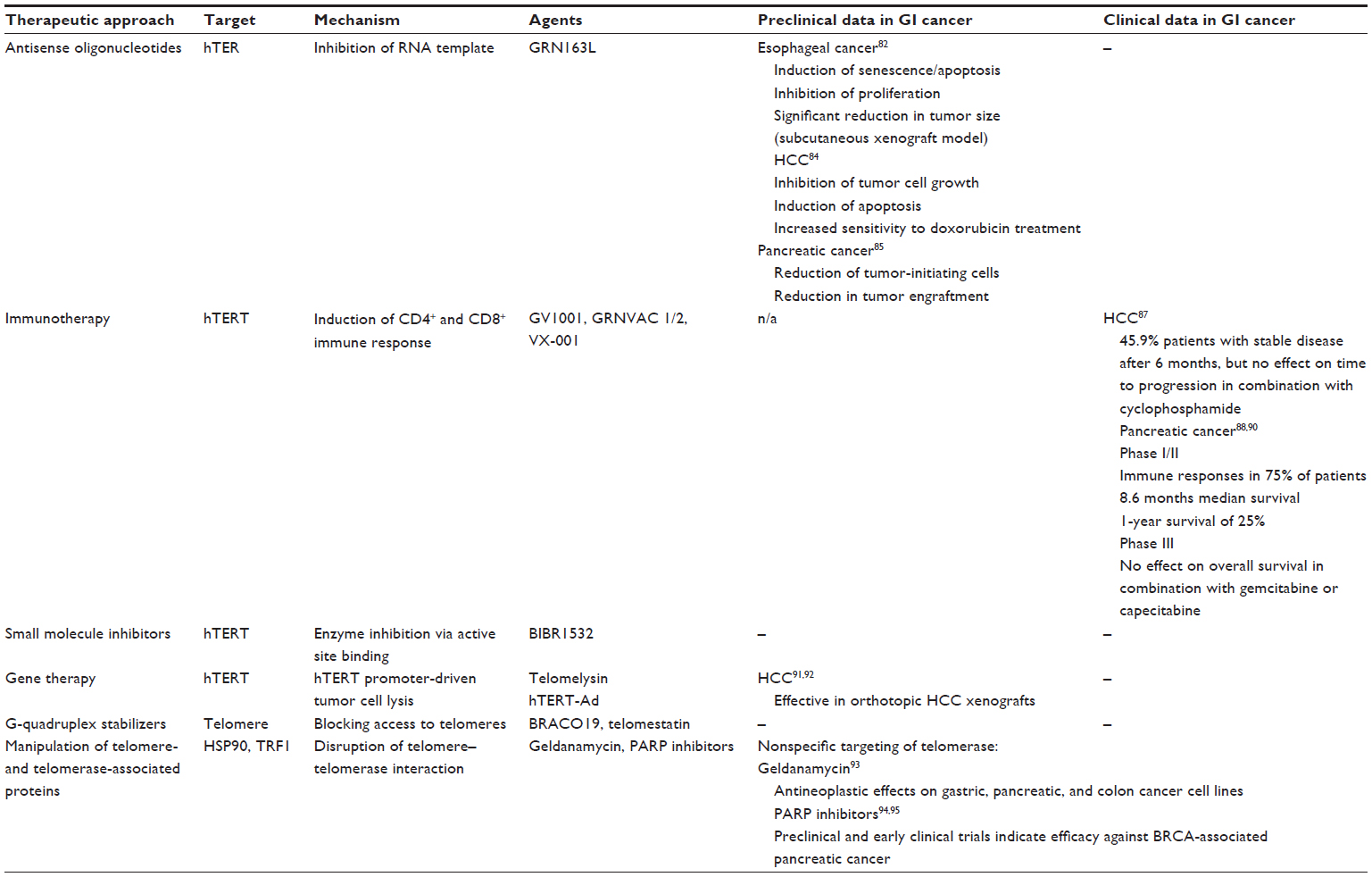 | Table 1 Therapeutic strategies targeting telomeres and telomerase |
Conclusion
Maintenance of telomeres is an extremely complex cellular process that has been extensively studied over the last decades and our understanding of telomerase function and, to a lesser extent, ALT in normal tissue homeostasis and cancer has improved remarkably. Telomerase has been hailed as an ideal target for cancer therapies and several therapeutic strategies targeting telomerase showed potential in preclinical as well as early clinical trials. However, none of these has yet become part of standard therapeutic regimens in gastrointestinal cancer. One possible explanation could be the redundancy of telomerase activity and ALT, with ALT being activated upon inhibition of telomerase. Even though these observations have been made in esophageal cancer, it is very likely that this redundancy is also present in other cancers. In vitro data indicate that this problem might be overcome in the future by dual inhibition of telomerase and ALT.
The molecular mechanisms of telomerase activation in cancers of the gastrointestinal tract vary significantly between tumor types and most likely also between patients. These variations add another layer of complexity to the sophisticated machinery of telomere maintenance in cancer. However, the age of next-generation sequencing, transcriptomics and proteomics might open new perspectives for individualized therapeutic strategies that interfere with the molecular mechanisms of telomerase activation. As outlined in this article, receptor tyrosine kinases and their downstream signaling pathways are involved in the activation of telomerase. Inhibition of these and other receptors and pathways by agents that are already in clinical use for the treatment of cancer might serve as adjuvants or even alternatives to direct targeting of telomerase in the future.
Disclosure
The author reports no conflicts of interest in this work.
References
McClintock B. The behavior in successive nuclear divisions of a chromosome broken at meiosis. Proc Natl Acad Sci U S A. 1939;25(8):405–416. | |
Muller HJ. The remaking of chromosomes. Collecting Net. 1938;8:182–195. | |
Blackburn EH, Gall JG. A tandemly repeated sequence at the termini of the extrachromosomal ribosomal RNA genes in Tetrahymena. J Mol Biol. 1978;120(1):33–53. | |
Moyzis RK, Buckingham JM, Cram LS, et al. A highly conserved repetitive DNA sequence, (TTAGGG)n, present at the telomeres of human chromosomes. Proc Natl Acad Sci U S A. 1988;85(18):6622–6626. | |
David Wilson W, Paul A. Kinetics and structures on the molecular path to the quadruplex form of the human telomere. J Mol Biol. 2014;426(8):1625–1628. | |
Griffith JD, Comeau L, Rosenfield S, et al. Mammalian telomeres end in a large duplex loop. Cell. 1999;97(4):503–514. | |
Xu L, Li S, Stohr BA. The role of telomere biology in cancer. Annu Rev Pathol. 2013;8:49–78. | |
Ruden M, Puri N. Novel anticancer therapeutics targeting telomerase. Cancer Treat Rev. 2013;39(5):444–456. | |
Ray S, Bandaria JN, Qureshi MH, Yildiz A, Balci H. G-quadruplex formation in telomeres enhances POT1/TPP1 protection against RPA binding. Proc Natl Acad Sci U S A. 2014;111(8):2990–2995. | |
Allsopp RC, Vaziri H, Patterson C, et al. Telomere length predicts replicative capacity of human fibroblasts. Proc Natl Acad Sci U S A. 1992;89(21):10114–10118. | |
Brown JP, Wei W, Sedivy JM. Bypass of senescence after disruption of p21CIP1/WAF1 gene in normal diploid human fibroblasts. Science. 1997;277(5327):831–834. | |
Harley CB, Futcher AB, Greider CW. Telomeres shorten during ageing of human fibroblasts. Nature. 1990;345(6274):458–460. | |
de Lange T. Activation of telomerase in a human tumor. Proc Natl Acad Sci U S A. 1994;91(8):2882–2885. | |
Kim NW, Piatyszek MA, Prowse KR, et al. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994;266(5193):2011–2015. | |
Nakamura TM, Morin GB, Chapman KB, et al. Telomerase catalytic subunit homologs from fission yeast and human. Science. 1997;277(5328):955–959. | |
Shippen-Lentz D, Blackburn EH. Functional evidence for an RNA template in telomerase. Science. 1990;247(4942):546–552. | |
Cesare AJ, Reddel RR. Alternative lengthening of telomeres: models, mechanisms and implications. Nat Rev Genet. 2010;11(5):319–330. | |
Henson JD, Hannay JA, McCarthy SW, et al. A robust assay for alternative lengthening of telomeres in tumors shows the significance of alternative lengthening of telomeres in sarcomas and astrocytomas. Clin Cancer Res. 2005;11(1):217–225. | |
Johnson JE, Varkonyi RJ, Schwalm J, et al. Multiple mechanisms of telomere maintenance exist in liposarcomas. Clin Cancer Res. 2005; 11(15):5347–5355. | |
Queisser A, Heeg S, Thaler M, von Werder A, Opitz OG. Inhibition of telomerase induces alternative lengthening of telomeres during human esophageal carcinogenesis. Cancer Genet. 2013;206(11):374–386. | |
Daniel M, Peek GW, Tollefsbol TO. Regulation of the human catalytic subunit of telomerase (hTERT). Gene. 2012;498(2):135–146. | |
Kyo S, Takakura M, Taira T, et al. Sp1 cooperates with c-Myc to activate transcription of the human telomerase reverse transcriptase gene (hTERT). Nucleic Acids Res. 2000;28(3):669–677. | |
Chou CK, Lee DF, Sun HL, et al. The suppression of MAD1 by AKT-mediated phosphorylation activates MAD1 target genes transcription. Mol Carcinog. 2009;48(11):1048–1058. | |
Zhu J, Blenis J, Yuan J. Activation of PI3K/Akt and MAPK pathways regulates Myc-mediated transcription by phosphorylating and promoting the degradation of Mad1. Proc Natl Acad Sci U S A. 2008;105(18):6584–6589. | |
Goueli BS, Janknecht R. Upregulation of the catalytic telomerase subunit by the transcription factor ER81 and oncogenic HER2/Neu, Ras, or Raf. Mol Cell Biol. 2004;24(1):25–35. | |
Heeg S, Hirt N, Queisser A, et al. EGFR overexpression induces activation of telomerase via PI3K/AKT-mediated phosphorylation and transcriptional regulation through Hif1-alpha in a cellular model of oral-esophageal carcinogenesis. Cancer Sci. 2011;102(2):351–360. | |
Horikawa I, Cable PL, Mazur SJ, Appella E, Afshari CA, Barrett JC. Downstream E-box-mediated regulation of the human telomerase reverse transcriptase (hTERT) gene transcription: evidence for an endogenous mechanism of transcriptional repression. Mol Biol Cell. 2002;13(8):2585–2597. | |
Liu L, Lai S, Andrews LG, Tollefsbol TO. Genetic and epigenetic modulation of telomerase activity in development and disease. Gene. 2004;340(1):1–10. | |
Liu L, Saldanha SN, Pate MS, Andrews LG, Tollefsbol TO. Epigenetic regulation of human telomerase reverse transcriptase promoter activity during cellular differentiation. Genes Chromosomes Cancer. 2004;41(1):26–37. | |
Shin KH, Kang MK, Dicterow E, Park NH. Hypermethylation of the hTERT promoter inhibits the expression of telomerase activity in normal oral fibroblasts and senescent normal oral keratinocytes. Br J Cancer. 2003;89(8):1473–1478. | |
Zhu J, Zhao Y, Wang S. Chromatin and epigenetic regulation of the telomerase reverse transcriptase gene. Protein Cell. 2010;1(1):22–32. | |
Guilleret I, Yan P, Grange F, Braunschweig R, Bosman FT, Benhattar J. Hypermethylation of the human telomerase catalytic subunit (hTERT) gene correlates with telomerase activity. Int J Cancer. 2002;101(4):335–341. | |
Renaud S, Loukinov D, Abdullaev Z, et al. Dual role of DNA methylation inside and outside of CTCF-binding regions in the transcriptional regulation of the telomerase hTERT gene. Nucleic Acids Res. 2007;35(4):1245–1256. | |
Zinn RL, Pruitt K, Eguchi S, Baylin SB, Herman JG. hTERT is expressed in cancer cell lines despite promoter DNA methylation by preservation of unmethylated DNA and active chromatin around the transcription start site. Cancer Res. 2007;67(1):194–201. | |
Devereux TR, Horikawa I, Anna CH, Annab LA, Afshari CA, Barrett JC. DNA methylation analysis of the promoter region of the human telomerase reverse transcriptase (hTERT) gene. Cancer Res. 1999;59(24):6087–6090. | |
Cong YS, Wright WE, Shay JW. Human telomerase and its regulation. Microbiol Mol Biol Rev. 2002;66(3):407–425, table of contents. | |
Kang SS, Kwon T, Kwon DY, Do SI. Akt protein kinase enhances human telomerase activity through phosphorylation of telomerase reverse transcriptase subunit. J Biol Chem. 1999;274(19):13085–13090. | |
Chang JT, Lu YC, Chen YJ, et al. hTERT phosphorylation by PKC is essential for telomerase holoprotein integrity and enzyme activity in head neck cancer cells. Br J Cancer. 2006;94(6):870–878. | |
Li H, Zhao L, Yang Z, Funder JW, Liu JP. Telomerase is controlled by protein kinase Calpha in human breast cancer cells. J Biol Chem. 1998; 273(50):33436–33442. | |
Kharbanda S, Kumar V, Dhar S, et al. Regulation of the hTERT telomerase catalytic subunit by the c-Abl tyrosine kinase. Curr Biol. 2000; 10(10):568–575. | |
Tomlinson RL, Ziegler TD, Supakorndej T, Terns RM, Terns MP. Cell cycle-regulated trafficking of human telomerase to telomeres. Mol Biol Cell. 2006;17(2):955–965. | |
Holt SE, Aisner DL, Baur J, et al. Functional requirement of p23 and Hsp90 in telomerase complexes. Genes Dev. 1999;13(7):817–826. | |
Bryan TM, Englezou A, Gupta J, Bacchetti S, Reddel RR. Telomere elongation in immortal human cells without detectable telomerase activity. EMBO J. 1995;14(17):4240–4248. | |
Chung I, Osterwald S, Deeg KI, Rippe K. PML body meets telomere: the beginning of an ALTernate ending? Nucleus. 2012;3(3):263–275. | |
Yeager TR, Neumann AA, Englezou A, Huschtscha LI, Noble JR, Reddel RR. Telomerase-negative immortalized human cells contain a novel type of promyelocytic leukemia (PML) body. Cancer Res. 1999;59(17):4175–4179. | |
Dunham MA, Neumann AA, Fasching CL, Reddel RR. Telomere maintenance by recombination in human cells. Nat Genet. 2000;26(4):447–450. | |
Murnane JP, Sabatier L, Marder BA, Morgan WF. Telomere dynamics in an immortal human cell line. EMBO J. 1994;13(20):4953–4962. | |
Le S, Moore JK, Haber JE, Greider CW. RAD50 and RAD51 define two pathways that collaborate to maintain telomeres in the absence of telomerase. Genetics. 1999;152(1):143–152. | |
Tarsounas M, Muñoz P, Claas A, et al. Telomere maintenance requires the RAD51D recombination/repair protein. Cell. 2004;117(3):337–347. | |
Cesare AJ, Griffith JD. Telomeric DNA in ALT cells is characterized by free telomeric circles and heterogeneous t-loops. Mol Cell Biol. 2004; 24(22):9948–9957. | |
Henson JD, Reddel RR. Assaying and investigating alternative lengthening of telomeres activity in human cells and cancers. FEBS Lett. 2010;584(17):3800–3811. | |
Gocha AR, Harris J, Groden J. Alternative mechanisms of telomere lengthening: permissive mutations, DNA repair proteins and tumorigenic progression. Mutat Res. 2013;743–744:142–150. | |
Hu J, Hwang SS, Liesa M, et al. Antitelomerase therapy provokes ALT and mitochondrial adaptive mechanisms in cancer. Cell. 2012;148(4):651–663. | |
Gocha AR, Nuovo G, Iwenofu OH, Groden J. Human sarcomas are mosaic for telomerase-dependent and telomerase-independent telomere maintenance mechanisms: implications for telomere-based therapies. Am J Pathol. 2013;182(1):41–48. | |
Yu HP, Xu SQ, Lu WH, et al. Telomerase activity and expression of telomerase genes in squamous dysplasia and squamous cell carcinoma of the esophagus. J Surg Oncol. 2004;86(2):99–104. | |
Zheng YL, Hu N, Sun Q, Wang C, Taylor PR. Telomere attrition in cancer cells and telomere length in tumor stroma cells predict chromosome instability in esophageal squamous cell carcinoma: a genome-wide analysis. Cancer Res. 2009;69(4):1604–1614. | |
Quante M, Heeg S, von Werder A, et al. Differential transcriptional regulation of human telomerase in a cellular model representing important genetic alterations in esophageal squamous carcinogenesis. Carcinogenesis. 2005;26(11):1879–1889. | |
Zhao Y, Gao Y, Chen Z, Hu X, Zhou F, He J. Low frequency of TERT promoter somatic mutation in 313 sporadic esophageal squamous cell carcinomas. Int J Cancer. 2014;134(2):493–494. | |
van Nistelrooij AM, Zwarthoff EC, Post E, et al. Absence of TERT promoter mutations in esophageal adenocarcinoma. Int J Cancer. 2014;134(8):2014–2015. | |
Finley JC, Reid BJ, Odze RD, et al. Chromosomal instability in Barrett’s esophagus is related to telomere shortening. Cancer Epidemiol Biomarkers Prev. 2006;15(8):1451–1457. | |
Lord RV, Salonga D, Danenberg KD, et al. Telomerase reverse transcriptase expression is increased early in the Barrett’s metaplasia, dysplasia, adenocarcinoma sequence. J Gastrointest Surg. 2000;4(2):135–142. | |
Morales CP, Lee EL, Shay JW. In situ hybridization for the detection of telomerase RNA in the progression from Barrett’s esophagus to esophageal adenocarcinoma. Cancer. 1998;83(4):652–659. | |
Papanikolaou V, Iliopoulos D, Dimou I, et al. The involvement of HER2 and p53 status in the regulation of telomerase in irradiated breast cancer cells. Int J Oncol. 2009;35(5):1141–1149. | |
Hou L, Savage SA, Blaser MJ, et al. Telomere length in peripheral leukocyte DNA and gastric cancer risk. Cancer Epidemiol Biomarkers Prev. 2009;18(11):3103–3109. | |
Basu N, Skinner HG, Litzelman K, Vanderboom R, Baichoo E, Boardman LA. Telomeres and telomere dynamics: relevance to cancers of the GI tract. Expert Rev Gastroenterol Hepatol. 2013;7(8):733–748. | |
Huang DS, Wang Z, He XJ, et al. Recurrent TERT promoter mutations identified in a large-scale study of multiple tumour types are associated with increased TERT expression and telomerase activation. Eur J Cancer. 2015;51(8):969–976. | |
Sasaki T, Kuniyasu H, Luo Y, et al. AKT activation and telomerase reverse transcriptase expression are concurrently associated with prognosis of gastric cancer. Pathobiology. 2014;81(1):36–41. | |
Naito Y, Takagi T, Handa O, et al. Telomerase activity and expression of telomerase RNA component and catalytic subunits in precancerous and cancerous colorectal lesions. Tumour Biol. 2001;22(6):374–382. | |
Engelhardt M, Drullinsky P, Guillem J, Moore MA. Telomerase and telomere length in the development and progression of premalignant lesions to colorectal cancer. Clin Cancer Res. 1997;3(11):1931–1941. | |
Rampazzo E, Bertorelle R, Serra L, et al. Relationship between telomere shortening, genetic instability, and site of tumour origin in colorectal cancers. Br J Cancer. 2010;102(8):1300–1305. | |
Gertler R, Rosenberg R, Stricker D, et al. Telomere length and human telomerase reverse transcriptase expression as markers for progression and prognosis of colorectal carcinoma. J Clin Oncol. 2004;22(10):1807–1814. | |
Bertorelle R, Rampazzo E, Pucciarelli S, Nitti D, De Rossi A. Telomeres, telomerase and colorectal cancer. World J Gastroenterol. 2014;20(8):1940–1950. | |
Bertorelle R, Briarava M, Rampazzo E, et al. Telomerase is an independent prognostic marker of overall survival in patients with colorectal cancer. Br J Cancer. 2013;108(2):278–284. | |
Wege H, Brummendorf TH. Telomerase activation in liver regeneration and hepatocarcinogenesis: Dr Jekyll or Mr Hyde? Curr Stem Cell Res Ther. 2007;2(1):31–38. | |
Oh BK, Kim YJ, Park YN, Choi J, Kim KS, Park C. Quantitative assessment of hTERT mRNA expression in dysplastic nodules of HBV-related hepatocarcinogenesis. Am J Gastroenterol. 2006;101(4):831–838. | |
Nault JC, Mallet M, Pilati C, et al. High frequency of telomerase reverse-transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat Commun. 2013;4:2218. | |
Nault JC, Calderaro J, Di Tommaso L, et al. Telomerase reverse transcriptase promoter mutation is an early somatic genetic alteration in the transformation of premalignant nodules in hepatocellular carcinoma on cirrhosis. Hepatology. 2014;60(6):1983–1992. | |
Miura N, Osaki Y, Nagashima M, et al. A novel biomarker TERTmRNA is applicable for early detection of hepatoma. BMC Gastroenterol. 2010;10:46. | |
Mizumoto K, Tanaka M. Detection of telomerase activity in patients with pancreatic cancer. Methods Mol Med. 2005;103:199–205. | |
Vinagre J, Pinto V, Celestino R, et al. Telomerase promoter mutations in cancer: an emerging molecular biomarker? Virchows Arch. 2014;465(2):119–133. | |
Beatty GL, Vonderheide RH. Telomerase as a universal tumor antigen for cancer vaccines. Expert Rev Vaccines. 2008;7(7):881–887. | |
Shammas MA, Qazi A, Batchu RB, et al. Telomere maintenance in laser capture microdissection-purified Barrett’s adenocarcinoma cells and effect of telomerase inhibition in vivo. Clin Cancer Res. 2008;14(15):4971–4980. | |
Wu X, Smavadati S, Nordfjäll K, et al. Telomerase antagonist imetelstat inhibits esophageal cancer cell growth and increases radiation-induced DNA breaks. Biochim Biophys Acta. 2012;1823(12):2130–2135. | |
Djojosubroto MW, Chin AC, Go N, et al. Telomerase antagonists GRN163 and GRN163L inhibit tumor growth and increase chemosensitivity of human hepatoma. Hepatology. 2005;42(5):1127–1136. | |
Joseph I, Tressler R, Bassett E, et al. The telomerase inhibitor imetelstat depletes cancer stem cells in breast and pancreatic cancer cell lines. Cancer Res. 2010;70(22):9494–9504. | |
Lu R, Pal J, Buon L, et al. Targeting homologous recombination and telomerase in Barrett’s adenocarcinoma: impact on telomere maintenance, genomic instability and tumor growth. Oncogene. 2014;33(12):1495–1505. | |
Greten TF, Forner A, Korangy F, et al. A phase II open label trial evaluating safety and efficacy of a telomerase peptide vaccination in patients with advanced hepatocellular carcinoma. BMC Cancer. 2010;10:209. | |
Bernhardt SL, Gjertsen MK, Trachsel S, et al. Telomerase peptide vaccination of patients with non-resectable pancreatic cancer: a dose escalating phase I/II study. Br J Cancer. 2006;95(11):1474–1482. | |
Choudhury A, Mosolits S, Kokhaei P, Hansson L, Palma M, Mellstedt H. Clinical results of vaccine therapy for cancer: learning from history for improving the future. Adv Cancer Res. 2006;95:147–202. | |
Middleton G, Silcocks P, Cox T, et al. Gemcitabine and capecitabine with or without telomerase peptide vaccine GV1001 in patients with locally advanced or metastatic pancreatic cancer (TeloVac): an open-label, randomised, phase 3 trial. Lancet Oncol. 2014;15(8):829–840. | |
Lin WH, Yeh SH, Yang WJ, et al. Telomerase-specific oncolytic adenoviral therapy for orthotopic hepatocellular carcinoma in HBx transgenic mice. Int J Cancer. 2013;132(6):1451–1462. | |
Wirth T, Kühnel F, Fleischmann-Mundt B, et al. Telomerase-dependent virotherapy overcomes resistance of hepatocellular carcinomas against chemotherapy and tumor necrosis factor-related apoptosis-inducing ligand by elimination of Mcl-1. Cancer Res. 2005;65(16):7393–7402. | |
Moser C, Lang SA, Stoeltzing O. Heat-shock protein 90 (Hsp90) as a molecular target for therapy of gastrointestinal cancer. Anticancer Res. 2009;29(6):2031–2042. | |
Javle M, Curtin NJ. The role of PARP in DNA repair and its therapeutic exploitation. Br J Cancer. 2011;105(8):1114–1122. | |
Waaler J, Machon O, Tumova L, et al. A novel tankyrase inhibitor decreases canonical Wnt signaling in colon carcinoma cells and reduces tumor growth in conditional APC mutant mice. Cancer Res. 2012; 72(11):2822–2832. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.