Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Value of phagocyte function screening for immunotoxicity of nanoparticles in vivo
Authors Fröhlich E ![]()
Received 18 February 2015
Accepted for publication 18 March 2015
Published 26 May 2015 Volume 2015:10(1) Pages 3761—3778
DOI https://doi.org/10.2147/IJN.S83068
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Eleonore Fröhlich
Center for Medical Research, Medical University of Graz, Graz, Austria
Abstract: Nanoparticles (NPs) present in the environment and in consumer products can cause immunotoxic effects. The immune system is very complex, and in vivo studies are the gold standard for evaluation. Due to the increased amount of NPs that are being developed, cellular screening assays to decrease the amount of NPs that have to be tested in vivo are highly needed. Effects on the unspecific immune system, such as effects on phagocytes, might be suitable for screening for immunotoxicity because these cells mediate unspecific and specific immune responses. They are present at epithelial barriers, in the blood, and in almost all organs. This review summarizes the effects of carbon, metal, and metal oxide NPs used in consumer and medical applications (gold, silver, titanium dioxide, silica dioxide, zinc oxide, and carbon nanotubes) and polystyrene NPs on the immune system. Effects in animal exposures through different routes are compared to the effects on isolated phagocytes. In addition, general problems in the testing of NPs, such as unknown exposure doses, as well as interference with assays are mentioned. NPs appear to induce a specific immunotoxic pattern consisting of the induction of inflammation in normal animals and aggravation of pathologies in disease models. The evaluation of particle action on several phagocyte functions in vitro may provide an indication on the potency of the particles to induce immunotoxicity in vivo. In combination with information on realistic exposure levels, in vitro studies on phagocytes may provide useful information on the health risks of NPs.
Keywords: immunotoxicity, phagocytes, cytokines, respiratory burst, nitric oxide generation, phagocytosis
Introduction
Nanoparticles (NPs) are used in many industrial applications and consumer products, and they are also being developed for targeted drug delivery, imaging, and implants in the medical sector. In addition to cytotoxicity, NPs can act on the immune system. Potential immunotoxic effects of NPs are relevant for human health because the immune system is present at all potential portals of entry of NPs and a variety of immunomodulatory actions of NPs has been proposed.1 The immunmodulatory action of a compound usually describes a desired change in the immune system – for instance, for therapeutic intervention – while “immunotoxicity” is used for adverse immunomodulation indicating nondesired effects on the immune system. Immunotoxicity includes interactions with blood (hemolysis, coagulation, and protein binding), accumulation in the mononuclear phagocyte system (MPS), adjuvant properties, binding of haptens, interference with phagocytosis, and modulation of the Th2/Th1 response to antigens. Epidemiological studies in regions with increased concentrations of ultrafine particles suggested that NPs could influence the immune system. High levels of airborne particles caused worsening of asthma and pneumonia in exposed individuals.2–5 Ultrafine particles in the atmosphere do not meet the size requirements of NPs because their upper size limit is usually 2.5 μm, but the reports stimulated further studies on size-related particle effects and raised the awareness that the large surface area of NPs was the reason for their high biological reactivity and toxicity.6
In contrast to cytotoxicity, the role of in vitro immunotoxicity testing is not well established. This is firstly due to general problems in simulating the complexity of the immunological system in vitro, as well as in the extrapolation of in vitro and animal data to human reactions and, secondly, to NP-specific problems. The immune system is redundant and has the capacity to compensate for minor immunotoxicological effects. High interindividual variations of the immune system further complicate the identification of a link between NP exposure and immunotoxicity in humans. Due to the high proliferation rate and compensation capacity of the immune system, only extreme alterations will result in clinical symptoms. On the other hand, decreased immunosurveillance may have long-term consequences, which cannot be directly linked to immunotoxicity. One example of such effects is the three- to fourfold increase in cancer incidence by immunosuppression with cyclosporine A for 5 years.7
Engineered NPs, to which humans might be exposed, comprise titanium dioxide (TiO2) and zinc oxide (ZnO) NPs in consumer products, silver (Ag) NPs in clothing, and silica (SiO2) NPs in food. For medical products, gold (Au), carbon nanotubes (CNTs), and iron oxide are likely candidates. The main exposure routes are dermal for NPs in consumer products and oral for NPs in food and intravenous for NPs in medical use. The exposure of humans to engineered NPs, due to the different use of these products, is expected to be highly variable. Site-specific composition and reaction of the immune system (lung, skin, blood, etc) affords exposure-specific models because the same NPs might cause no immune effects when applied by the oral and dermal route, but they may induce sensitization after intradermal injection.8 This creates a high number of different testing scenarios and renders the testing of all variations in vivo ethically and financially problematic. In this situation, prescreening by in vitro assays, similar to cytotoxicity screening for systemic toxicity, would be helpful. Of course, in vitro testing has the limitation that only one or a few cell types can be evaluated. Data produced after exposure to high doses for a short period are not representative for the exposure to most NPs.9 Furthermore, the protective mechanisms of the body – for instance, mucociliary clearance in the lung and radical scavenging by glutathione in the blood – will mitigate the toxic effect observed in vitro.
According to the Agence Française de Sécurité Sanitaire des Produits de Santé (AFSSAPS), immunotoxicity testing of NPs should focus on macrophages, granulocytes, and dendritic cells (DCs), and the testing should use cytokines as readout parameters.10 Since phagocytes are involved in the unspecific defense, as well as in the specific immune response, impairment of phagocyte function can indicate a decreased reserve of the immune system in NP-exposed individuals.
Therefore, phagocytes appear to be suitable for discriminating between NPs interfering or not interfering with the immune system. Several studies report interference with phagocyte function by iron oxide particles, but the iron oxide NPs, which have been approved for medical use (such as Ferumoxtran-10 [Sinerem®]), did not influence the different aspects of phagocyte function. The secretion of proinflammatory cytokines, oxidative burst, phagocytosis, and chemotaxis was not affected by the exposure to the particles in vitro.11 The few studies in which the same NPs were assessed by animal exposure and by exposure of cells to Ag and SiO2 NPs show that impairment of phagocytes function in vitro accords with immune inflammation in vivo.12,13 Proinflammatory action was seen in vivo as well as in macrophages isolated from animals exposed to TiO2 and ZnO NPs.14,15
This review is focused on plain (not pegylated or formulated) metal and metal oxide NPs, such as SiO2, iron oxide, Ag, Au, TiO2, and ZnO NPs, and single-walled CNTs (SWCNTs) and multiwalled CNTs (MWCNTs). These NPs are relevant for humans because they are used in a variety of consumer products and as imaging reagents in medicine. Their classification as non- or low biodegradable NPs is often used to differentiate these particles from the enzymatically degradable NPs, such as liposomes, poly(lactic-co-glycolic acid), dendrimers, and so on, which can cause additional effects by their degradation products. However, it should not be forgotten that metal and metal oxide release ions which can interact with proteins and induce inflammation.16 Nevertheless, the NPs mentioned in this review form a more homogeneous group than nanocarriers for drug delivery, which consist of different materials and possess different surface charges and functionalization. Polystyrene (PS) particles are included in this review because they are often used as model particles for nonbiodegradable NPs.17
Role of phagocytes in the immune system
Professional phagocytes are a group of immune cells that share the feature that they can ingest 0.5–10 μm sized particles better than epithelial cells. Since they are key players in the immune defense, they are represented in almost all organs.18,19 Mononuclear phagocytes are derived from myeloid progenitor cells in bone marrow and develop into granulocytes and monocytes. Monocytes circulate in the blood and differentiate into macrophages (Mφ) in the tissue, where they reside as peritoneal Mφ, alveolar Mφ, mesangial phagocytes of the kidney, synovial type A cells, bone marrow stromal Mφ, splenic red pulp and splenic white pulp Mφ, osteoclasts in the bone, histiocytes in the connective tissue, and as microglia in the brain.20 DCs are a specific lineage of monocytic phagocytes and are mainly present as myeloid and plasmacytoid DCs in the blood, as interstitial DCs in many organs, and as interdigitating DCs in the lymphatic organs. Based on the history of their discovery, some of them received specific names, such as the DCs in the epidermis (Langerhans cells) and Mφs in the liver (Kupffer cells). Phagocytes express different surface markers and differ in their optimum size of phagocytosis. Peritoneal macrophages and monocytes in the peripheral blood optimally phagocytose 0.3–1.1 μm particles. The optimal size for phagocytosis by alveolar macrophages is 3–6 μm particles.21–23 Granulocytes are classified into neutrophilic, eosinophilic, and basophilic granulocytes. The phagocytosis of invading pathogens is the main role of neutrophilic granulocytes. After self-destruction, they are the main component of pus. Compared to neutrophilic granulocytes, eosinophilic and basophilic granulocytes have only a low potential for phagocytosis and act mainly against pathogens by the release of enzymes, as well as toxic and proinflammatory substances.
Macrophages possess a variety of receptors for the binding of bacterial constituents (Figure 1). Complement C3b and the Fc fragment of immunoglobulin (Ig)G enable better uptake of opsonized particles. Distinct adhesion molecules, intercellular adhesion molecule (ICAM)-1 (CD54), ICAM-2 (CD102), lymphocyte function-associated antigen (LFA)-1 (CD11a), and LFA-3 (CD58), together with costimulatory molecules B7.1 (CD80), B7.2 (CD86), or CD40, and processed cytosolic proteins presented by major histocompatibility complex (MHC) I or extracellular proteins presented by MHC II, molecules activate T-cells.24 Cytokines such as tumor necrosis factor-alpha (TNF-α) and interferon (IFN)-gamma (IFN-γ), as well as their interaction with lipopolysaccharide (LPS)-binding protein, activate macrophages. Phagocytes ingest a variety of pathogens, such as bacteria, mycobacteria, virus, fungi, and nonpathogenic particles (for instance, dyes and dust) in an unspecific way. On the other hand, they fulfill a definite function as antigen-presenting cells for the correct function of the specific immune system.
 | Figure 1 Receptors linked to main functions of phagocytes. |
In vitro assays to study phagocyte function
A panel of in vitro assays of different complexities can assess phagocyte function. Cytokine secretion, chemotaxis, phagocytosis, and respiratory burst can be measured in all phagocytes. Nitric oxide generation is used only for monocytes and macrophages, whereas the detection of the release of myeloperoxidase and elastase is specific for neutrophilic granulocytes.25 The evaluation of DC function is more complex because it requires interactions with T-cells. Cell isolation, cell exposure, and the detection platform for the performance of the respective assays are described in the core publication and in the supplements of Current Protocols in Immunology.26
Cytokine secretion
A wide spectrum of cytokines is being used in immunotoxicity studies, and phagocytes isolated from exposed animals or cultures of primary cells, and cell lines are equally suitable for these analyses.27 In the presence or absence of the test substance, the release of cytokines/chemokines can be analyzed by enzyme-linked immunosorbant assays, enzyme-linked immunosorbent spot assays, antibody array assays, and bead-based assays. To identify proinflammation responses, interleukin (IL)-1, IL-6, IL-8, and TNF-α are routinely used.28 The classification of allergic responses is based on the type of lymphocyte helper cells that are activated. IL-4 and IL-5 identify TH2 responses, while marker cytokines for TH1 responses are IFN-γ and TNF-β.
Chemotaxis
The migration of leukocytes from an upper chamber across a membrane to a lower chamber containing a chemoattractant is termed chemotaxis. Human serum-derived complement 5a, human lymphocyte-derived chemotactic factor, monocyte chemoattractant protein 1, or N-formyl-methionyl-leucyl-phenylalanine are commonly used attractants.29 All leukocytes are able for chemotaxis, but monocytes, either as primary cells or as cell lines, are used most frequently. In conventional assays, membrane-containing inserts separating the upper from the lower chamber are used. The amount of cells that passed the membrane and reached the lower chamber is counted or quantified by viability assays. Alternatively, an impedance-based system (eg, xCELLigence and ECIS/Taxis) can be used.30,31
Phagocytosis
The phagocytosis assay evaluates the phagocytic activity of fluorescein-labeled bacteria (Staphylococcus aureus, Escherichia coli) in macrophages, monocytes, and polymorphonuclear neutrophils exposed to the test compound.32
Respiratory burst (reactive oxygen production)
This assay can be performed in macrophages, monocytes, and polymorphonuclear neutrophils by the detection of reactive oxygen species (ROS), which is produced upon phagocytosis. For the assays, mostly unlabeled E. coli is used as the phagocytic stimulus. Either chemiluminescent detection by lucigenin or the oxidation of dyes to fluorescent products (eg, rhodamine 123) can be employed for the quantification of the produced oxygen species.33
Nitric oxide (NO) generation
Murine macrophages are routinely used because, when compared to human monocytes, they possess a much higher production of NO.34 An additional advantage of their use is that, in contrast to human macrophages, they do not need a differentiation step. Differentiation of monocytes with the commonly used phorbol 12-myristate 13-acetate or vitamin D3 cannot reproduce the phenotype of human macrophages in vivo, and it introduces additional variations in the assay.35 The common and very reliable detection method of NO uses the Griess reagent.36
Release of elastase and myeloperoxidase
These enzymes are used as indicators for neutrophilic granulocyte activation.37 Assays are performed in whole blood or in neutrophilic granulocytes isolated from peripheral blood. These cells only rarely show direct effects to conventional chemicals, but they are activated by particles.38,39 The relevance of granulocyte activation for immunotoxicity in vivo, however, is currently unclear.
Function of dendritic cells
DCs for testing cannot be obtained directly from the blood in sufficient amounts, but they require differentiation in vitro. CD14+ mononuclear cells isolated from peripheral blood mononuclear cells (PBMCs) are treated with recombinant (rh) granulocyte macrophage colony-stimulating factor and IL-4 for 7 days. Maturation to DCs induced by LPS in the presence and absence of the test compound is verified by the surface expression of CD80, CD83, CD86, and human leukocyte antigen-DR, and by the secretion of IL-12.
DC function requires a mixed lymphocyte culture, which analyzes the ability of T-cells to recognize allogenic cells as not belonging to the organism (nonself) as a result of the presence and proliferation of different MHC class II antigens on their surface. This assay is used to identify sensitizing agents. A DC to T-cell ratio of 1:100 is sufficient to initiate vigorous and optimal responses.40 Splenocytes or lymph node cells from treated animals (responder cells) with genetically dissimilar cells (stimulator cells) are cocultured. The assay is usually performed in mice, where cells from another strain can be used as stimulators.41 Stimulator cells are inactivated by irradiation or treatment with a DNA intercalating agent such as mitomycin C. After incubation for several days, proliferation of the responder cells is measured using 3H-thymidine uptake.42 The reaction can also be performed using human PBMC-derived DCs mixed with allogenic lymphocytes,43 and the proliferation of the responder T-cells after contact with allogenic lymphocytes is assessed using a viability (formazan bioreduction) assay. Human myeloid leukemia-derived MUTZ-3 cells have the ability to differentiate into DCs,44 and this assay is in the process of validation as an alternative to the in vivo identification of sensitizing agents.45
Specific issues in the assessment of NPs
The specific nature of NPs, mainly linked to their high surface reactivity, complicates their assessment by in vitro assays. The adsorption of molecules (either bacterial proteins or macromolecules from the body to the particle surface) holds importance for the in vivo and in vitro testing of phagocyte function.
In vivo and in vitro – binding of endotoxin
NPs may bind endotoxin, an LPS and pyrogenic compound of the wall of Gram-negative bacteria. Endotoxin is a strong stimulant of the immune response and causes a pyrogenic reaction in the human body.46 Endotoxin contamination of metal and metal oxide NPs and CNTs is less expected because synthesis often includes steps that kill bacteria. However, contamination is often difficult to exclude because endotoxin can be present in distilled water.47 Due to the strong stimulation of endotoxin, its presence in the sample does not allow for the identification of NP effects. The detection of endotoxin is usually achieved by evaluation in the limulus amebocyte lysate assay, one of the accepted alternatives to the in vivo endotoxin detection assays.48 This assay can be performed in different formats, generally as clotting tests and by colorimetric detection.49 Unfortunately, several NPs interfere with this assay. While for some NPs (TiO2, Ag, CaCO3, SiO2 NPs), interference with the gel-clotting assay was more prominent,50 for other particles (Au NPs), interference with the colorimetric limulus amebocyte lysate assay has been reported.51 The release of inflammatory cytokines (IL-6, IL-8, IL-1) from PBMCs produced variable results and it has been suggested that NPs and endotoxin compete against each other in the induction of cytokines.52
In vivo and in vitro – protein corona
High surface activity leads to the binding of macromolecules to the particle surface once they get into contact with physiological solutions. This coating consists mainly of proteins and has been termed “protein corona”.53 It is hypothesized that the composition of the protein corona determines the trafficking and biological effects of NPs. For a description of the composition and variability of the protein corona, the reader is referred to reviews focusing on this topic.54,55 The physicochemical parameters of the NPs and the composition of the biological fluid are the main factors determining the composition of the protein corona. As a general rule, hydrophobic particles bind more proteins than do hydrophilic particles,56 and abundant proteins in the incubation solution are bound faster on the NP surface than the low abundant proteins.57 Dependence on size and shape, as well as surface charge, has been reported in the following way: Au and SiO2 NPs >10 nm bound more proteins than particles <10 nm; more proteins were attached to TiO2 nanospheres than to nanorods and nanotubes; and binding to positively charged Au, PS, and carbon black particles was higher than to particles without charged groups.58–62 While the composition of the inner coating (hard corona) appeared to be more stable, the composition of the outer part (soft corona) was dynamic and changed in its composition when the particle was transferred from one medium to the other.63 The passage through various media left a fingerprint of the protein composition of the previous media on the NP.64 NPs retained the protein corona during endocytosis; the coat was subsequently removed in lysosomes.65
The role of the protein corona composition for biological effects is still not entirely clear. The reduction of toxic effects, such as cytotoxicity and hemolysis, by protein coating of NPs has been observed in several studies of nonphagocytic cells.66–70 This decreased effect was linked to reduced cellular uptake. Bovine serum albumin (BSA) bound to the surface of carboxyl-functionalized PS, quantum dot (Qdot), and Au NPs decreased cell uptake. The opposite was observed for BSA bound to these types of NPs when they were functionalized with amine groups instead of carboxyl groups.71 All BSA-coated NPs displayed the same effective surface charge, but apparently the BSA structure was influenced by the binding in such a way that different groups were visible for the cells. As a result, BSA-coated carboxylated NPs bound to the albumin receptor, while BSA-coated amine-functionalized NPs were ingested after binding to the cellular scavenger receptor.
Protein-coated NPs are expected to produce more pronounced immunological effects because coating with serum increased the uptake by phagocytes.72 The secretion of proinflammatory cytokines by DCs was higher for spherical-than sheet-shaped ZnO NPs, which also bound more proteins on their surface.73 While increased protein binding might have caused the higher secretion of cytokines, the opposite behavior has also been observed: coating of SiO2 NPs with serum decreased cytokine secretion of murine macrophages.74 The presence of complement in the protein corona plays a specific role because the binding of complement C3b and IgG increases uptake by phagocytes by binding to the complement and Fc-receptors. Responses to complement binding were variable; firstly, complement proteins could be activated or inactivated by the binding, and secondly, increased uptake could lead to the activation or inhibition of phagocytes.75,76 Changes in protein conformation appear to be the reason for the different effects; binding of fibrinogen to negatively charged poly(acrylic acid)-conjugated Au NPs induced activation of the Mac-1 receptor on THP-1 monocytes, resulting in a proinflammatory response.77 While these studies support a specific role of the bound proteins, other studies do not support the hypothesis of a protein corona-specific effect because the composition of the protein corona did not correlate with hemocompatibility.78
In vitro – cellular doses
Dose-dependent effects are more difficult to identify for NPs than for conventional compounds because cellular uptake is influenced by the diffusion and sedimentation of the single NPs and agglomerates of the NPs. Several mathematical models have been developed to calculate the deposition of particles suspended in liquids on adherent cells.79,80 Particle-dependent minimal deposition was seen between 50–200 nm, while larger and smaller particles deposited at higher rates.79 Small changes in the dispersion factor caused considerable variations in the deposited dose.80 The differences are due to the formation of agglomerates, but the extent of agglomeration and its effect on deposition are difficult to quantify by mathematical models. The measured deposition of 50–1,000 nm plain PS particles on macrophages increased over time and showed a minimum for 100 nm particles.17 Carboxyl PS particles of 20–1,000 nm showed the cellular uptake of 25%–40% in macrophages with a minimum at 100 nm.81 The cellular dose of the same type of particles with sizes of 20–500 nm in endothelial cells increased from 4.6% to 28.4%, demonstrating higher particle uptake by phagocytic cells, as compared to nonphagocytic cells, in general.82 When adherent cells were cultured upside-down, they ingested much less NPs than the cells cultured in the standard orientation.83 Further complications arise when cells are exposed to aerosolized NPs because cell contact is dependent on the used exposure system, as well as on the variations in the size and concentration of the aerosol; great variations in deposition rates between 0.037% and 30% of the applied dose per well for different particles have been reported.84–88 Furthermore, the influence on flow has to be considered when assessing NP uptake from the systemic blood circulation.89 Endothelial cells best ingested Qdots and SiO2 NPs at a shear stress of 0.05 Pa, which corresponds to postcapillary venules and peripheral arteries.89
In vitro – assay interference
The interference of NPs with several assay systems can strongly influence the results (Table 1). The absorbance of NPs could lead to false-negative results (absence of cytotoxicity is detected, although the NP is cytotoxic) because the metabolic activity (according to absorbance) is estimated to be higher than it actually is.90 Enzyme inhibition by NPs could also cause false-negative results. Lactate dehydrogenase (LDH) is released into the supernatant of cells when the plasma membrane integrity is lost. Its enzymatic activity correlates to the amount of damaged cells. If LDH activity is inhibited by NPs, a lower degree of cell damage will be determined.
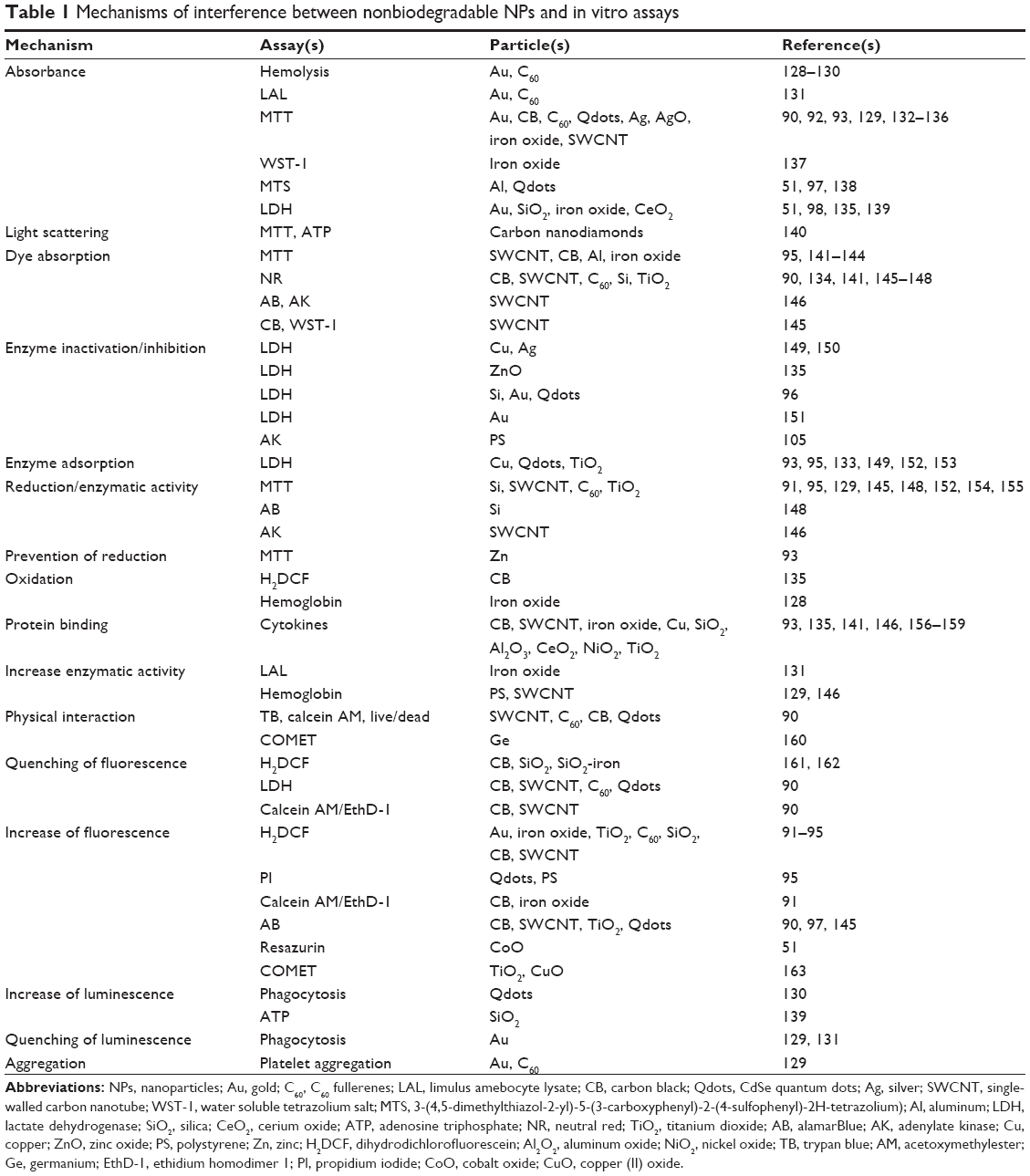 | Table 1 Mechanisms of interference between nonbiodegradable NPs and in vitro assays |
False-positive results (cytotoxicity is detected although the NP is not toxic) are detected when the fluorescent signals of dihydrofluorescein (the detection of oxidative stress) or of propidium iodide (the disruption of membrane integrity) are enhanced by NPs.91–95 Depending on the assays used, the masking of toxic effects and the identification of nonexistent toxicity by NPs can occur simultaneously. Increased absorbance by colored NPs will result in a higher signal of LDH (indicating more dead cells) and in the MTT assay (indicating more viable cells). The use of multiple assays, therefore, helps to reveal assay interference. The addition of protein, mostly BSA, could prevent interference, but it also could increase it. While false-negative results by the inhibition of LDH activity by Si, Au, and CdSe NPs96 was avoided, the addition of BSA caused false-negative effects in protein detection via the Bradford reagent.97 For the identification of potential assay interference, the incubation of NPs with the assay compounds alone (in the absence of cells) and with cells alone (in the absence of assay compounds) can be used. These controls are, however, only useful when the NPs interact with assay compounds and with the readout; assay interactions by Au NPs, which increased the detected amount of dead cells by shuttling the indicator dye, propidium iodide, into the cells, would not have been revealed.98,99 Similarly, the more global effects of NPs on cultured cells, such as the depletion of nutrients by SWCNTs,100 would go unnoticed. Interference can show dose dependency; dye (acridine) fluorescence is increased by low concentrations of Ag NPs and quenched at high concentrations of NPs.101
Some general rules may help identify and prevent the false interpretation of results. The use of low NP concentrations reduces the problem of interference, but the removal of NPs by centrifugation is generally not recommended because analytes adsorbed to the particles might be removed. Assay interference of the colored CNTs, carbon black, C60 fullerenes, and Au NPs, and of the fluorescent Qdots, may occur more frequently than interference with noncolored Si, SiO2, TiO2, and ZnO NPs. Testing of NPs with several assays based on different detection methods can reduce the risk of misinterpretation.90,102 In this regard, immunotoxicity testing poses more problems than cytotoxicity testing because a lower number of assays for a given immunological function are usually available. On the other hand, compared to cytotoxicity testing, NPs are usually studied at much lower NP concentrations, reducing the risk for interference.
Immunotoxicological data from NP exposure
In vivo exposure includes voluntary inhalation and oral application, forced inhalation (intranasal and intratracheal instillation, oropharyngeal administration), forced oral (intragastric/gavage) application as well as noninvasive dermal and invasive (intradermal injection) dermal applications. Parenteral applications include intravenous and intraperitoneal injection. Table 2 shows the general reaction pattern of the immune system after in vivo exposure to NPs.
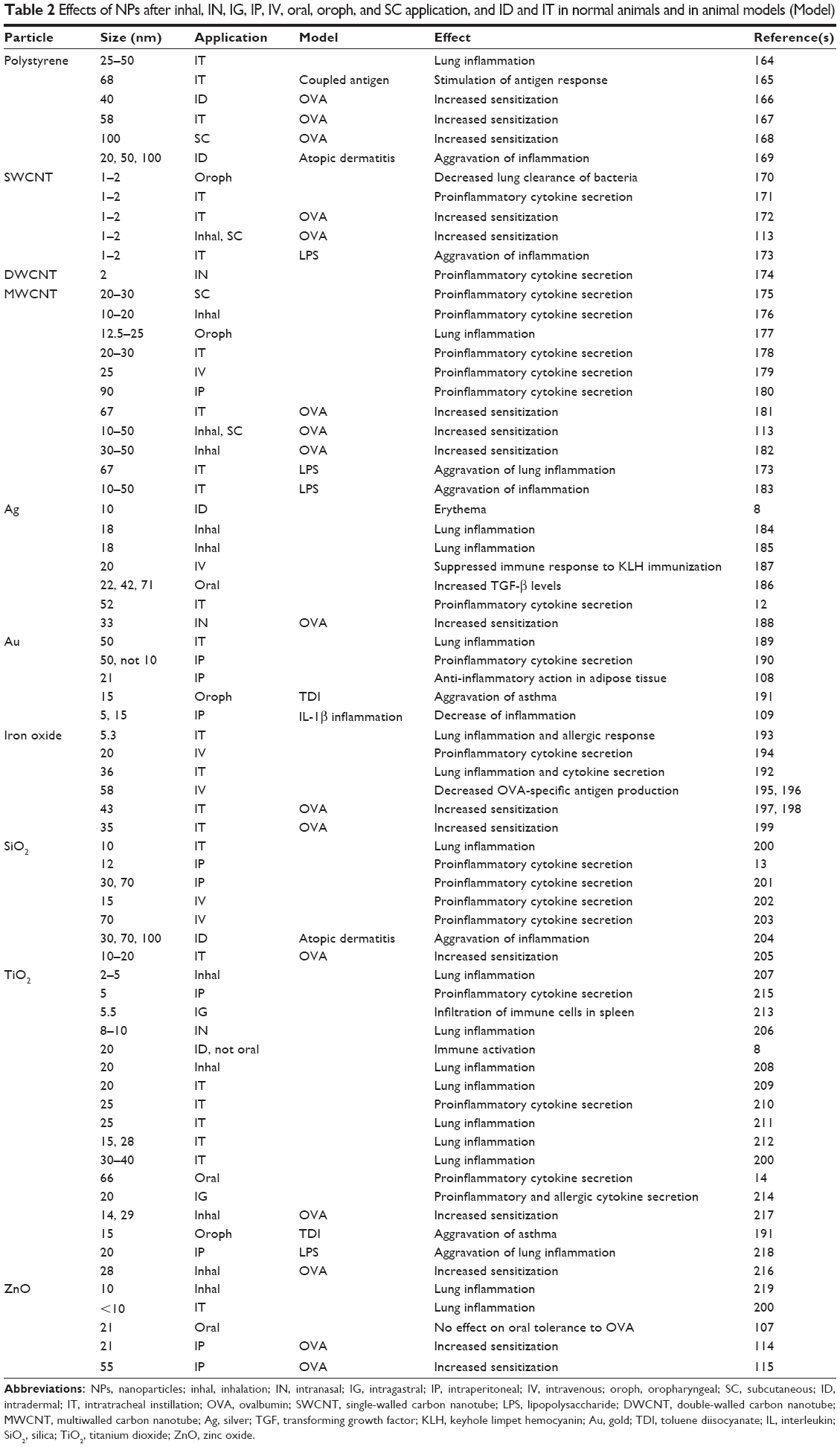 | Table 2 Effects of NPs after inhal, IN, IG, IP, IV, oral, oroph, and SC application, and ID and IT in normal animals and in animal models (Model) |
Systemic immune effects
Effects in the respiratory tract with only a thin epithelium were more pronounced than effects after dermal or oral ingestion exposure, where a horny layer or a thick mucus layer separated NPs from epithelial and immune cells.103
Inflammation in the lung is one of the most frequently reported effects of respiratory exposure to NPs.104 Since cytokines are produced by several cell types, it is not clear whether the reported increases in cytokine secretion and subsequent inflammation were due to specific activation of immune cells, or if they were a consequence of cytotoxic action on alveolar epithelial cells. Heavy metal-containing NPs reacted in a similar manner as PS particles.105,106 Given that heavy metal-containing NPs show ROS generation, and since they are expected to have greater cytotoxicity, the similarity of the reaction does not support the hypothesis of cell death (induced by more cytotoxic heavy metal-containing NPs) as a main inductor of inflammation.
Only a few studies have reported the absence of immunological effects, which could be due to restricted access to immune cells.107 The absence of immune effects after the oral ingestion of and exposure to ZnO and TiO2 NPs could be explained by the hindered assessment of the particles to the cells by mucus.8,107 On the other hand, the low reactivity of intraperitoneally applied Au NPs appears to be due to their high biocompatibility given that few studies have reported on the adverse cellular effects of Au NPs.108,109 This statement is supported by a lack of immunological interference in the cellular assays showing no increased cytokine secretion,110,111 and no effect on DC maturation and activation.94,112
When NPs were applied to diseased animals, the pathology of the disease was aggravated. This aggravation was seen in asthma models, as well as in atopic dermatitis (Table 2). Aggravation of asthma is unlikely to be caused by cytotoxicity of the NPs because exposure by the respiratory tract and by other routes (subcutaneous, intraperitoneal), where no direct contact with the alveolar epithelium occurred, caused the same effects.113–115 The mechanisms for amplifying pre-existing pathologies have been proposed through the following mechanisms:116 pre-existing inflammation in the respiratory tubes could be amplified by enhancing the levels of inflammatory factors or humoral immunity. Second, NPs within the size range of <100 nm were able to stimulate and enhance hypersensitivity, which is primarily mediated by Th2 cells.116
In vitro and ex vivo effects
Phagocyte function after in vitro (cells exposed in wells) and ex vivo (cells harvested from exposed animals) exposure is summarized in Table 3. To evaluate the potential of screening in phagocytes, first, data obtained from ex vivo and in vitro studies have to be compared. Second, the similarity of ex vivo and in vitro exposures to in vivo exposure has to be tested. In vitro data on cytokine secretion and chemotaxis corresponded to the respective ex vivo data (Table 3). NPs showed a similar pattern of interference with phagocyte functions; proinflammatory cytokine secretion (mostly IL-6, IL-1β, and TNF-α) and respiratory burst increased, while phagocytosis and chemotaxis decreased. The degranulation of neutrophilic granulocytes has been shown for a few particles.81,117 The influence on DC maturation and function varied markedly between the particles. MWCNTs inhibited maturation, Au and iron oxide showed no prominent effect, and SiO2 and TiO2 activated DCs.94,112,118,119 The different results could be due to the use of different readouts (maturation and activation).
 | Table 3 Immune effects in isolated phagocytes, either after in vivo treatment with nanoparticles (ex vivo) or by in vitro treatment |
The secretion of proinflammatory cytokines was increased by all NPs when applied by in vitro exposure, and after the ex vivo respiratory exposure, to NPs. The lower sensitivity of phagocytes by the oral route was confirmed in an ex vivo study.120
Uptake of NPs by phagocytes
When NPs are coated with proteins in biological fluids, they are well ingested by phagocytes.121 Phagocytosis of NPs by primary cells, cell lines, macrophages, monocytes, and monocyte-derived macrophages indicated accumulation in the MPS and showed a good correlation to the accumulation of particles in the MPS of the spleen and liver in vivo.122 Due to the crucial function of macrophages and DCs in the specific immune response, the accumulation of NPs in the MPS could result in immunotoxicity. The indication of uptake by the MPS or accumulation in lymphatic organs, however, was not correlated to adverse effects on the immune system in vivo or in vitro.81,123 Accumulation in the spleen was only observed for 30 nm Au particles, while adverse effects on the immune system according to increases in relative spleen weight and immune cell numbers were seen for 5 nm, 10 nm, and 60 nm Au particles.123 Small carboxyl PS particles were ingested in much higher numbers than 1,000 nm particles by macrophages.81 While the 1,000 nm large particles induced oxidative burst and cell damage, particles in the size range between 40 nm and 500 nm were taken up without obvious interference with cell viability and function. Taken together, these data suggest that the uptake of NPs may not result in impaired phagocyte function.
Guidelines for sample preparation and exposure
Physiologically relevant testing is based on sample preparation, as well as on the use of dispersant and intended exposure routes. Most NPs form stable solutions in distilled water, which cannot be used for in vitro studies. The presence of ions and protein in the physiological solution leads to NP agglomerates, which may increase in size, but they may also disintegrate. The surface coating of NPs determines their penetration of barriers, cellular uptake, and immune response.124 The Office of Economic Co-operation and Development (OECD) guidelines for sample preparation and dosimetry had advised that dose should be indicated in terms of mass, surface area, and particle number at a minimum.125 To get information on the stability of the dispersion, repeated measurements are recommended with the documentation of agglomeration and ion release. The dispersants should preferentially contain macromolecules that are present in the target tissue. For exposure with aerosols, and in addition to the NP parameters, the mass median aerodynamic diameter and aerosol concentration should be determined. Guidelines for sample preparation for nanoscale TiO2 are already available,126 and existing guidelines for exposure by spontaneous inhalation, oral gavage, and dermal application are applicable for NP exposure. Moreover, the additional effects of intravenous exposure (for instance, behavior in the syringe) have to be considered.
Freshly prepared solutions from stock solutions prepared in water, diluted in cell culture medium, and treated by sonification should be added to the cells. In the case that no route-specific surfactants, such as 1,2 dipalmitoyl-sn-glycero-3-phosphocholine for pulmonary exposure, are used, BSA appears to be a good choice because this zwitterionic molecule prevents the binding of protein from the solution.
Conclusion
Due to the complexity of the immune system, in vivo testing will remain the gold standard. However, intraindividual variations in the immune system, as well as its compensatory abilities, are major limitations. As has been observed in environmental studies of airborne particles, individuals with impaired immune function were affected by small particle doses, while no effects were observed in the healthy population.2–5
This overview on a variety of carbon, metal, and metal oxide NPs shows that these particles caused relatively similar patterns of immunotoxicity in vivo, which involved inflammation and immunosuppression in healthy animals and aggravation of the pathology in animals with pre-existing diseases. This suggests that the classification of particles as more or less immunotoxic by in vitro screening might be helpful. The extent to which such screening could lead to valid results was studied by comparing data obtained by in vivo exposure, in vitro testing and in vitro data (Table 4). This analysis showed that the results obtained in cells isolated from NP-exposed animals were similar to the data obtained of cells, which were exposed to NPs in vitro. Secondly, NPs that inhibited phagocyte functions in vitro reacted in an immunotoxic manner in vivo (Tables 2 and 4). The data suggest that the in vitro testing of phagocytes might predict the typical immunotoxicity pattern of NPs in vivo. Cellular assays may also be suitable to identify disease-related alterations in the immune reaction to NPs because comparison between reactions of PBMCs from healthy and allergic donors showed that the cells exhibited disease-related differences upon challenge.118
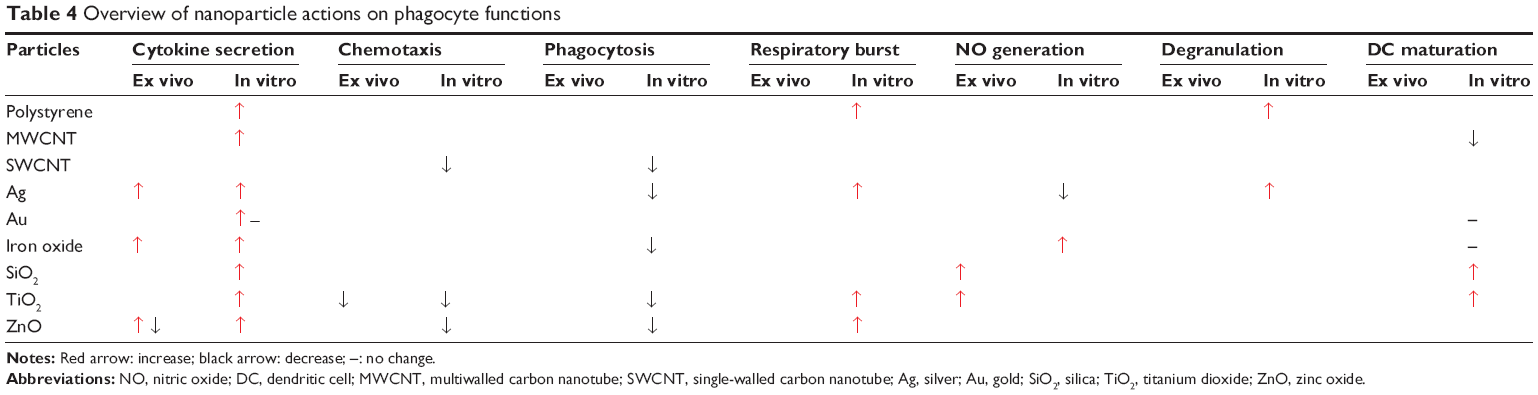 | Table 4 Overview of nanoparticle actions on phagocyte functions |
Due to the specific composition of the immune system at different portals of entry, exposure-specific coculture models including immune cells could serve as a possibility to assess immunotoxicants in vitro. Alveolar epithelial cells and alveolar macrophages in cocultures released inflammatory cytokines at lower concentrations of TiO2 NPs than did the respective monocultures.127 At the expense of greater complexity, these systems could increase the sensitivity of immunotoxicity in vitro screening and enable exposure-specific testing. However, until a correlation of these findings in these systems to data obtained in humans has been shown, their value remains elusive.
Acknowledgments
Support by the European integrated project NMP4-CT-2006-026723 and by the Austrian Science Fund grant P22576-B18 is gratefully acknowledged.
Disclosure
The author reports no conflicts of interest in this work.
References
Dobrovolskaia MA, McNeil SE. Immunological properties of engineered nanomaterials. Nat Nanotechnol. 2007;2(8):469–478. | ||
D’Amato G. Outdoor air pollution in urban areas and allergic respiratory diseases. Monaldi Arch Chest Dis. 1999;54(6):470–474. | ||
Delfino RJ, Zeiger RS, Seltzer JM, Street DH, McLaren CE. Association of asthma symptoms with peak particulate air pollution and effect modification by anti-inflammatory medication use. Environ Health Perspect. 2002;110(10):A607–A617. | ||
Li N, Xia T, Nel AE. The role of oxidative stress in ambient particulate matter-induced lung diseases and its implications in the toxicity of engineered nanoparticles. Free Radic Biol Med. 2008;44(9):1689–1699. | ||
Penttinen P, Timonen KL, Tiittanen P, Mirme A, Ruuskanen J, Pekkanen J. Ultrafine particles in urban air and respiratory health among adult asthmatics. Eur Respir J. 2001;17(3):428–435. | ||
Buzea C, Pacheco II, Robbie K. Nanomaterials and nanoparticles: sources and toxicity. Biointerphases. 2007;2(4):MR17–71. | ||
Sodemann U, Bistrup C, Marckmann P. Cancer rates after kidney transplantation. Dan Med Bull. 2011;58(12):A4342. | ||
Auttachoat W, McLoughlin CE, White KL Jr, Smith MJ. Route-dependent systemic and local immune effects following exposure to solutions prepared from titanium dioxide nanoparticles. J Immunotoxicol. 2014;11(3):273–282. | ||
Warheit DB, Sayes CM, Reed KL, Swain KA. Health effects related to nanoparticle exposures: environmental, health and safety considerations for assessing hazards and risks. Pharmacol Ther. 2008;120(1):35–42. | ||
Claude JR, Domenjoud L, Fattal E, et al. Recommendations for Toxicological Evaluation of Nanoparticle Medicinal Products. Paris, France: Agence Française de Sécurité Sanitaire des Produits de Santé; 2011. Available from: http://ansm.sante.fr/var/ansm_site/storage/original/application/2968a90b774b563b03405379b7d4f4e6.pdf. Accessed April 24, 2014. | ||
Müller K, Skepper JN, Posfai M, et al. Effect of ultrasmall superparamagnetic iron oxide nanoparticles (Ferumoxtran-10) on human monocyte-macrophages in vitro. Biomaterials. 2007;28(9):1629–1642. | ||
Liu H, Yang D, Yang H, et al. Comparative study of respiratory tract immune toxicity induced by three sterilisation nanoparticles: silver, zinc oxide and titanium dioxide. J Hazard Mater. 2013;248–249:478–486. | ||
Park EJ, Park K. Oxidative stress and pro-inflammatory responses induced by silica nanoparticles in vivo and in vitro. Toxicol Lett. 2009; 184(1):18–25. | ||
Nogueira CM, de Azevedo WM, Dagli ML, et al. Titanium dioxide induced inflammation in the small intestine. World J Gastroenterol. 2012;18(34):4729–4735. | ||
Chen JK, Ho CC, Chang H, et al. Particulate nature of inhaled zinc oxide nanoparticles determines systemic effects and mechanisms of pulmonary inflammation in mice. Nanotoxicology. 2015;9:43–53. | ||
Karlsson HL, Cronholm P, Hedberg Y, Tornberg M, De Battice L, Svedhem S, Wallinder IO. Cell membrane damage and protein interaction induced by copper containing nanoparticles–importance of the metal release process. Toxicology. 2013;313(1):59–69. | ||
Ahmad Khanbeigi R, Kumar A, Sadouki F, et al. The delivered dose: Applying particokinetics to in vitro investigations of nanoparticle internalization by macrophages. J Control Release. 2012;162(2):259–266. | ||
Roitt I, Delves P, editors. Roitt’s Essential Immunology. 10th ed. Oxford, UK: Wiley; 2001. | ||
Janeway C Jr, Travers P, Walport M, Shlomchik M, editors. Immunobiology: The Immune System in Health and Disease. 6th ed. New York, NY: Garland Science Pubishing; 2005. | ||
Gordon S, Pluddemann A, Martinez Estrada F. Macrophage heterogeneity in tissues: phenotypic diversity and functions. Immunol Rev. 2014;262(1):36–55. | ||
Hirota K, Hasegawa T, Hinata H, et al. Optimum conditions for efficient phagocytosis of rifampicin-loaded PLGA microspheres by alveolar macrophages. J Control Release. 2007;119(1):69–76. | ||
Kawaguchi H, Koiwai N, Ohtsuka Y, Miyamoto M, Sasakawa S. Phagocytosis of latex particles by leucocytes. I. Dependence of phagocytosis on the size and surface potential of particles. Biomaterials. 1986;7(1):61–66. | ||
Seymour L, Schacht E, Duncan R. The effect of size of polystyrene particles on their retention within the rat peritoneal compartment, and on their interaction with rat peritoneal macrophages in vitro. Cell Biol Int Rep. 1991;15(4):277–286. | ||
Abbas AK, Lichtman H, Pallai S, editors. Cellular and Molecular Immunology. 6th ed. Philadelphia, PA: Saunders; 2007. | ||
Dale DC, Boxer L, Liles WC. The phagocytes: neutrophils and monocytes. Blood. 2008;112(4):935–945. | ||
Coligan JE, Bierer B, Margulies D, Shevach E, Strober W, Kruisbeek A. Current Protocols in Immunology. New York, NY: John Wiley and Sons; 1991. | ||
Krebs FC, Miller SR, Catalone BJ, Fichorova R, Anderson D, Malamud D, Howett MK, Wigdahl B. Comparative in vitro sensitivities of human immune cell lines, vaginal and cervical epithelial cell lines, and primary cells to candidate microbicides nonoxynol 9, C31G, and sodium dodecyl sulfate. Antimicrob Agents Chemother. 2002;46(7):2292–2298. | ||
Borish LC, Steinke JW. 2. Cytokines and chemokines. J Allergy Clin Immunol. 2003;111(2 Suppl):S460–S475. | ||
Falk W, Goodwin RH Jr, Leonard EJ. A 48-well micro chemotaxis assembly for rapid and accurate measurement of leukocyte migration. J Immunol Methods. 1980;33(3):239–247. | ||
Pietrosimone KM, Yin X, Knecht DA, Lynes MA. Measurement of cellular chemotaxis with ECIS/Taxis. J Vis Exp. 2012;(62). pii: 3840. | ||
Iqbal AJ, Regan-Komito D, Christou I, et al. A real time chemotaxis assay unveils unique migratory profiles amongst different primary murine macrophages. PLoS One. 2013;8(3):e58744. | ||
Gille C, Spring B, Tewes L, Poets CF, Orlikowsky T. A new method to quantify phagocytosis and intracellular degradation using green fluorescent protein-labeled Escherichia coli: comparison of cord blood macrophages and peripheral blood macrophages of healthy adults. Cytometry A. 2006;69(3):152–154. | ||
Elsner J, Kapp A. Reactive Oxygen Release. In: Proudfoot A, Wells T, Power C, editors. Methods in Molecular Biology: Chemokine Protocols. Human Press: Totowa; 2000:153–157. | ||
Schneemann M, Schoeden G. Macrophage biology and immunology: man is not a mouse. J Leukoc Biol. 2007;81(3):579; discussion 580. | ||
Daigneault M, Preston JA, Marriott HM, Whyte MK, Dockrell DH. The identification of markers of macrophage differentiation in PMA-stimulated THP-1 cells and monocyte-derived macrophages. PLoS One. 2010;5(1):e8668. | ||
Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8(12):958–969. | ||
Mann BS, Chung KF. Blood neutrophil activation markers in severe asthma: lack of inhibition by prednisolone therapy. Respir Res. 2006;7:59. | ||
Jovanović B, Anastasova L, Rowe EW, Palić D. Hydroxylated fullerenes inhibit neutrophil function in fathead minnow (Pimephales promelas Rafinesque, 1820). Aquat Toxicol. 2011;101(2):474–482. | ||
Vesnina LÉ, Mamontova TV, Mikitiuk MV, et al. [Effect of fullerene C60 on functional activity of phagocytic cells]. Eksp Klin Farmakol. 2011;74(6):26–29. Russian. | ||
Steinman RM, Witmer MD. Lymphoid dendritic cells are potent stimulators of the primary mixed leukocyte reaction in mice. Proc Natl Acad Sci U S A. 1978;75(10):5132–5136. | ||
Kang HG, Lee JE, Yang SH, et al. Donor-strain-derived immature dendritic cell pre-treatment induced hyporesponsiveness against allogeneic antigens. Immunology. 2010;129(4):567–577. | ||
House RV, Thomas PT, Bhargava HN. In vitro evaluation of fentanyl and meperidine for immunomodulatory activity. Immunol Lett. 1995;46(1–2):117–124. | ||
Li Y, Li X, Li Z, Gao H. Surface-structure-regulated penetration of nanoparticles across a cell membrane. Nanoscale. 2012;4(12):3768–3775. | ||
Masterson AJ, Sombroek CC, De Gruijl TD, et al. MUTZ-3, a human cell line model for the cytokine-induced differentiation of dendritic cells from CD34+ precursors. Blood. 2002;100(2):701–703. | ||
Nelissen I, Selderslaghs I, Heuvel RV, Witters H, Verheyen GR, Schoeters G. MUTZ-3-derived dendritic cells as an in vitro alternative model to CD34+ progenitor-derived dendritic cells for testing of chemical sensitizers. Toxicol In Vitro. 2009;23(8):1477–1481. | ||
Magalhães PO, Lopes AM, Mazzola PG, Rangel-Yagui C, Penna TC, Pessoa A Jr. Methods of endotoxin removal from biological preparations: a review. J Pharm Pharm Sci. 2007;10(3):388–404. | ||
Anderson WB, Huck PM, Dixon DG, Mayfield CI. Endotoxin inactivation in water by using medium-pressure UV lamps. Appl Environ Microbiol. 2003;69(5):3002–3004. | ||
Tsuji K, Steindler KA, Harrison SJ. Limulus amoebocyte lysate assay for detection and quantitation of endotoxin in a small-volume parenteral product. Appl Environ Microbiol. 1980;40(3):533–538. | ||
Hofman J. Bacterial endotoxins. In: Shier W, Mebs D, editors. Handbook of Toxicology. New York, NY: Marcel Dekker Inc.; 1990:655–682. | ||
Smulders S, Kaiser JP, Zuin S, et al. Contamination of nanoparticles by endotoxin: evaluation of different test methods. Part Fibre Toxicol. 2012;9:41. | ||
Oostingh GJ, Casals E, Italiani P, et al. Problems and challenges in the development and validation of human cell-based assays to determine nanoparticle-induced immunomodulatory effects. Part Fibre Toxicol. 2011;8(1):8. | ||
Dobrovolskaia MA, McNeil SE. Endotoxin and Engineered Nanomaterials. In: Dobrovolskaia MA, McNeil SE, editors. Handbook of Immunological Properties of Engineered Nanomaterials. Republic of Singapore: World Scientific Pub Co Inc; 2012:77–116. | ||
Cedervall T, Lynch I, Foy M, et al. Detailed identification of plasma proteins adsorbed on copolymer nanoparticles. Angew Chem Int Ed Engl. 2007;46(30):5754–5756. | ||
Monopoli MP, Aberg C, Salvati A, Dawson KA. Biomolecular coronas provide the biological identity of nanosized materials. Nat Nanotechnol. 2012;7(12):779–786. | ||
Rahman M, Laurent S, Tawil N, Yahia LH, Mahmoudi M. Nanoparticle and protein corona. In: Rahman M, Laurent S, Tawil N, Yahia LH, Mahmoudi M, editors. Protein-Nanoparticle Interactions: The Bio-Nano Interface Springer Series in Biophysics, Vol. 15: Berlin, Germany: Springer; 2013:21–44. | ||
Ding HM, Ma YQ. Computer simulation of the role of protein corona in cellular delivery of nanoparticles. Biomaterials. 2014; 35(30):8703–8710. | ||
Izak-Nau E, Voetz M, Eiden S, Duschl A, Puntes VF. Altered characteristics of silica nanoparticles in bovine and human serum: the importance of nanomaterial characterization prior to its toxicological evaluation. Part Fibre Toxicol. 2013;10(1):56. | ||
Deng ZJ, Liang M, Toth I, Monteiro MJ, Minchin RF. Molecular interaction of poly(acrylic acid) gold nanoparticles with human fibrinogen. ACS Nano. 2012;6(10):8962–8969. | ||
Deng ZJ, Mortimer G, Schiller T, Musumeci A, Martin D, Minchin RF. Differential plasma protein binding to metal oxide nanoparticles. Nanotechnology. 2009;20(45):455101. | ||
Fertsch-Gapp S, Semmler-Behnke M, Wenk A, Kreyling WG. Binding of polystyrene and carbon black nanoparticles to blood serum proteins. Inhal Toxicol. 2011;23(8):468–475. | ||
Lundqvist M, Sethson I, Jonsson BH. Protein adsorption onto silica nanoparticles: conformational changes depend on the particles’ curvature and the protein stability. Langmuir. 2004;20(24):10639–10647. | ||
Deng ZJ, Liang M, Toth I, Monteiro M, Minchin RF. Plasma protein binding of positively and negatively charged polymer-coated gold nanoparticles elicits different biological responses. Nanotoxicology. 2013;7(3):314–322. | ||
Monopoli MP, Walczyk D, Campbell A, et al. Physical-chemical aspects of protein corona: relevance to in vitro and in vivo biological impacts of nanoparticles. J Am Chem Soc. 2011;133(8):2525–2534. | ||
Lundqvist M, Stigler J, Cedervall T, et al. The evolution of the protein corona around nanoparticles: a test study. ACS Nano. 2011;5(9):7503–7509. | ||
Wang F, Yu L, Monopoli MP, et al. The biomolecular corona is retained during nanoparticle uptake and protects the cells from the damage induced by cationic nanoparticles until degraded in the lysosomes. Nanomedicine. 2013;9(8):1159–1168. | ||
Baier G, Costa C, Zeller A, et al. BSA adsorption on differently charged polystyrene nanoparticles using isothermal titration calorimetry and the influence on cellular uptake. Macromol Biosci. 2011;11(5):628–638. | ||
De Angelis I, Barone F, Zijno A, et al. Comparative study of ZnO and TiO2 nanoparticles: physicochemical characterisation and toxicological effects on human colon carcinoma cells. Nanotoxicology. 2013;7(8): 1361–1372. | ||
Hsiao IL, Huang YJ. Effects of serum on cytotoxicity of nano- and micro-sized ZnO particles. J Nanopart Res. 2013;15:1829. | ||
Vidic J, Haque F, Guigner JM, Vidy A, Chevalier C, Stankic S. Effects of water and cell culture media on the physicochemical properties of ZnMgO nanoparticles and their toxicity toward mammalian cells. Langmuir. 2014;30(38):11366–11374. | ||
Saha K, Moyano DF, Rotello VM. Protein coronas suppress the hemolytic activity of hydrophilic and hydrophobic nanoparticles. Mater Horiz. 2014;2014(1):102–105. | ||
Fleischer CC, Payne CK. Nanoparticle-cell interactions: molecular structure of the protein corona and cellular outcomes. Acc Chem Res. 2014;47(8):2651–2659. | ||
Ruge CA, Kirch J, Cañadas O, et al. Uptake of nanoparticles by alveolar macrophages is triggered by surfactant protein A. Nanomedicine. 2011;7(6):690–693. | ||
Heng BC, Zhao X, Tan EC, et al. Evaluation of the cytotoxic and inflammatory potential of differentially shaped zinc oxide nanoparticles. Arch Toxicol. 2011;85(12):1517–1528. | ||
Panas A, Marquardt C, Nalcaci O, et al. Screening of different metal oxide nanoparticles reveals selective toxicity and inflammatory potential of silica nanoparticles in lung epithelial cells and macrophages. Nanotoxicology. 2013;7(3):259–273. | ||
Pondman KM, Sobik M, Nayak A, et al. Complement activation by carbon nanotubes and its influence on the phagocytosis and cytokine response by macrophages. Nanomedicine. 2014;10(6):1287–1299. | ||
Ling WL, Biro A, Bally I, et al. Proteins of the innate immune system crystallize on carbon nanotubes but are not activated. ACS Nano. 2011;5(2):730–737. | ||
Deng ZJ, Liang M, Monteiro M, Toth I, Minchin RF. Nanoparticle-induced unfolding of fibrinogen promotes Mac-1 receptor activation and inflammation. Nat Nanotechnol. 2011;6(1):39–44. | ||
Dobrovolskaia MA, Neun BW, Man S, et al. Protein corona composition does not accurately predict hematocompatibility of colloidal gold nanoparticles. Nanomedicine. 2014;10(7):1453–1463. | ||
Hinderliter PM, Minard KR, Orr G, et al. ISDD: A computational model of particle sedimentation, diffusion and target cell dosimetry for in vitro toxicity studies. Part Fibre Toxicol. 2010;7(1):36. | ||
Cárdenas WH, Mamani JB, Sibov TT, Caous CA, Amaro E Jr, Gamarra LF. Particokinetics: computational analysis of the superparamagnetic iron oxide nanoparticles deposition process. Int J Nanomedicine. 2012;7:2699–2712. | ||
Prietl B, Meindl C, Roblegg E, Pieber TR, Lanzer G, Fröhlich E. Nano-sized and micro-sized polystyrene particles affect phagocyte function. Cell Biol Toxicol. 2014;30(1):1–16. | ||
Fröhlich E, Meindl C, Roblegg E, Ebner B, Absenger M, Pieber TR. Action of polystyrene nanoparticles of different sizes on lysosomal function and integrity. Part Fibre Toxicol. 2012;9:26. | ||
Cho EC, Zhang Q, Xia Y. The effect of sedimentation and diffusion on cellular uptake of gold nanoparticles. Nat Nanotechnol. 2011;6(6): 385–391. | ||
Fröhlich E, Bonstingl G, Höfler A, et al. Comparison of two in vitro systems to assess cellular effects of nanoparticles-containing aerosols. Toxicol In Vitro. 2013;27(1):409–417. | ||
Lenz AG, Karg E, Lentner B, et al. A dose-controlled system for air-liquid interface cell exposure and application to zinc oxide nanoparticles. Part Fibre Toxicol. 2009;6:32. | ||
Mertes P, Praplan AP, Künzi L, et al. A compact and portable deposition chamber to study nanoparticles in air-exposed tissue. J Aerosol Med Pulm Drug Deliv. 2013;26(4):228–235. | ||
Savi M, Kalberer M, Lang D, et al. A novel exposure system for the efficient and controlled deposition of aerosol particles onto cell cultures. Environ Sci Technol. 2008;42(15):5667–5674. | ||
Tippe A, Heinzmann U, Roth C. Deposition of fine and ultrafine aerosol particles during exposure at the air/cell interface. J Aerosol Sci. 2002;33(2):207–218. | ||
Samuel SP, Jain N, O’Dowd F, et al. Multifactorial determinants that govern nanoparticle uptake by human endothelial cells under flow. Int J Nanomedicine. 2012;7:2943–2956. | ||
Monteiro-Riviere NA, Inman AO, Zhang LW. Limitations and relative utility of screening assays to assess engineered nanoparticle toxicity in a human cell line. Toxicol Appl Pharmacol. 2009;234(2):222–235. | ||
Berg JM, Ho S, Hwang W, et al. Internalization of carbon black and maghemite iron oxide nanoparticle mixtures leads to oxidant production. Chem Res Toxicol. 2010;23(12):1874–1882. | ||
Doak SH, Griffiths SM, Manshian B, et al. Confounding experimental considerations in nanogenotoxicology. Mutagenesis. 2009;24(4): 285–293. | ||
Kroll A, Pillukat MH, Hahn D, Schnekenburger J. Current in vitro methods in nanoparticle risk assessment: limitations and challenges. Eur J Pharm Biopharm. 2009;72(2):370–377. | ||
Pfaller T, Colognato R, Nelissen I, et al. The suitability of different cellular in vitro immunotoxicity and genotoxicity methods for the analysis of nanoparticle-induced events. Nanotoxicology. 2010;4(1):52–72. | ||
Stone V, Johnston H, Schins RP. Development of in vitro systems for nanotoxicology: methodological considerations. Crit Rev Toxicol. 2009;39(7):613–626. | ||
Maccormack TJ, Clark RJ, Dang MK, et al. Inhibition of enzyme activity by nanomaterials: potential mechanisms and implications for nanotoxicity testing. Nanotoxicology. 2012;6(5):514–525. | ||
Ong KJ, MacCormack TJ, Clark RJ, et al. Widespread nanoparticle-assay interference: implications for nanotoxicity testing. PLoS One. 2014;9(3):e90650. | ||
Keene AM, Tyner KM. Analytical characterization of gold nanoparticle primary particles, aggregates, agglomerates, and agglomerated aggregates. Journal of Nanoparticle Research. 2011;13(8): 3465–3481. | ||
Shukla S, Priscilla A, Banerjee M, et al. Porous gold nanospheres by controlled transmetalation reaction: a novel material for application in cell imaging. Chem Mater. 2005;17(20):5000–5005. | ||
Casey A, Herzog E, Lyng FM, Byrne HJ, Chambers G, Davoren M. Single walled carbon nanotubes induce indirect cytotoxicity by medium depletion in A549 lung cells. Toxicol Lett. 2008;179(2):78–84. | ||
Sabatini CA, Pereira RV, Gehlen MH. Fluorescence modulation of acridine and coumarin dyes by silver nanoparticles. J Fluoresc. 2007;17(4): 377–382. | ||
Fröhlich E, Meindl C, Pieber TR. Important issues in the cytotoxicity screening of nano-sized materials. EURO-NanoTox Letters. 2010;1: 1–6. | ||
SCENIHR working group [webpage on the Internet]. The appropriateness of existing methodologies to assess the potential risks associated with engineered and adventitious products of nanotechnologies; 2006. Available from: http://ec.europa.eu/health/ph_risk/committees/04_scenihr/docs/scenihr_o_003b.pdf. Accessed January 1, 2015. | ||
Brashers V. Alterations of pulmonary function. In: Huether S, McCance K, editors. Understanding Pathophysiology. 5th ed. St Louis, MO: Elsevier; 2012:678–699. | ||
Fröhlich E, Samberger C, Kueznik T, et al. Cytotoxicity of nanoparticles independent from oxidative stress. J Toxicol Sci. 2009;34(4): 363–375. | ||
Xia T, Kovochich M, Liong M, Zink JI, Nel AE. Cationic polystyrene nanosphere toxicity depends on cell-specific endocytic and mitochondrial injury pathways. ACS Nano. 2008;2(1):85–96. | ||
Matsumura M, Takasu N, Nagata M, Nakamura K, Kawai M, Yoshino S. Effect of ultrafine zinc oxide (ZnO) nanoparticles on induction of oral tolerance in mice. J Immunotoxicol. 2010;7(3):232–237. | ||
Chen H, Dorrigan A, Saad S, Hare DJ, Cortie MB, Valenzuela SM. In vivo study of spherical gold nanoparticles: inflammatory effects and distribution in mice. PLoS One. 2013;8(2):e58208. | ||
Sumbayev VV, Yasinska IM, Garcia CP, et al. Gold nanoparticles downregulate interleukin-1β-induced pro-inflammatory responses. Small. 2013;9(3):472–477. | ||
Zhang Q, Hitchins VM, Schrand AM, Hussain SM, Goering PL. Uptake of gold nanoparticles in murine macrophage cells without cytotoxicity or production of pro-inflammatory mediators. Nanotoxicology. 2011;5(3):284–295. | ||
Shukla R, Bansal V, Chaudhary M, Basu A, Bhonde RR, Sastry M. Biocompatibility of gold nanoparticles and their endocytotic fate inside the cellular compartment: a microscopic overview. Langmuir. 2005;21(23):10644–10654. | ||
Villiers C, Freitas H, Couderc R, Villiers MB, Marche P. Analysis of the toxicity of gold nano particles on the immune system: effect on dendritic cell functions. J Nanopart Res. 2010;12(1):55–60. | ||
Nygaard UC, Hansen JS, Samuelsen M, Alberg T, Marioara CD, Løvik M. Single-walled and multi-walled carbon nanotubes promote allergic immune responses in mice. Toxicol Sci. 2009;109(1): 113–123. | ||
Matsumura M, Nagata M, Nakamura K, et al. Adjuvant effect of zinc oxide on Th2 but not Th1 immune responses in mice. Immunopharmacol Immunotoxicol. 2010;32(1):56–62. | ||
Roy R, Kumar S, Verma AK, et al. Zinc oxide nanoparticles provide an adjuvant effect to ovalbumin via a Th2 response in Balb/c mice. Int Immunol. 2014;26(3):159–172. | ||
Li Y, Zhang Y, Yan B. Nanotoxicity overview: nano-threat to susceptible populations. Int J Mol Sci. 2014;15(3):3671–3697. | ||
Haase H, Fahmi A, Mahltig B. Impact of silver nanoparticles and silver ions on innate immune cells. J Biomed Nanotechnol. 2014;10(6): 1146–1156. | ||
Laverny G, Casset A, Purohit A, et al. Immunomodulatory properties of multi-walled carbon nanotubes in peripheral blood mononuclear cells from healthy subjects and allergic patients. Toxicol Lett. 2013; 217(2):91–101. | ||
Winter M, Beer HD, Hornung V, Krämer U, Schins RP, Förster I. Activation of the inflammasome by amorphous silica and TiO2 nanoparticles in murine dendritic cells. Nanotoxicology. 2011;5(3):326–340. | ||
Kim CS, Nguyen HD, Ignacio RM, et al. Immunotoxicity of zinc oxide nanoparticles with different size and electrostatic charge. Int J Nanomedicine. 2014;9 Suppl 2:195–205. | ||
Fadeel B. Clear and present danger? Engineered nanoparticles and the immune system. Swiss Med Wkly. 2012;142:w13609. | ||
Caron WP, Lay JC, Fong AM, et al. Translational studies of phenotypic probes for the mononuclear phagocyte system and liposomal pharmacology. J Pharmacol Exp Ther. 2013;347(3):599–606. | ||
Zhang XD, Wu D, Shen X, et al. Size-dependent in vivo toxicity of PEG-coated gold nanoparticles. Int J Nanomedicine. 2011;6: 2071–2081. | ||
Remedios Serrano Lopez, D, Lalatsa A. Active Targeting. In: Uchegbu I, Schätzlein A, Cheng W, Lalatsa A, editors. Fundamentals of Pharmaceutical Nanoscience. Springer: New York; 2013:337–374. | ||
OECD Environment, Health and Safety Publications. [webpage on the Internet]. Guidance on sample preparation and dosimetry for the safety testing of manufactured nanomaterials. Paris, France: Paris Environment Directorate organisation for economic co-operation and development; 2012. Available from: http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=ENV/JM/MONO%282012%2940&docLanguage=En. Acessed January 1, 2015. | ||
Taurozzi J, Hackley V, Wiesner M. Preparation of a nanoscale TiO2 aqueous dispersion for toxicological or environmental testing. Version 1.2. NanoEHS Protocols(NIST Special Publication 1200-3). 2012. Available from: http://nvlpubs.nist.gov/nistpubs/SpecialPublications/NIST.SP.1200-3.pdf. Accessed November 1st, 2014. | ||
Tao F, Kobzik L. Lung macrophage-epithelial cell interactions amplify particle-mediated cytokine release. Am J Respir Cell Mol Biol. 2002; 26(4):499–505. | ||
Dobrovolskaia MA, Aggarwal P, Hall JB, McNeil SE. Preclinical studies to understand nanoparticle interaction with the immune system and its potential effects on nanoparticle biodistribution. Mol Pharm. 2008;5(4):487–495. | ||
Hall JB, Dobrovolskaia MA, Patri AK, McNeil SE. Characterization of nanoparticles for therapeutics. Nanomedicine (Lond). 2007;2(6): 789–803. | ||
Murthy R, Pathak Y. Drug delivery nanoparticles: formulation and characterization. In: Pathak Y, Thassu D, editors. In Vitro Blood Interaction and Pharmacological and Toxicological Characterization of Nanosystems. New York, NY: Informa Health Care; 2009:190–218. | ||
Dobrovolskaia MA, Germolec DR, Weaver JL. Evaluation of nanoparticle immunotoxicity. Nat Nanotechnol. 2009;4(7):411–414. | ||
Malugin A, Ghandehari H; Arnida. Cellular uptake and toxicity of gold nanoparticles in prostate cancer cells: a comparative study of rods and spheres. J Appl Toxicol. 2010;30(3):212–217. | ||
Clift MJ, Rothen-Rutishauser B, Brown DM, et al. The impact of different nanoparticle surface chemistry and size on uptake and toxicity in a murine macrophage cell line. Toxicol Appl Pharmacol. 2008;232(3):418–427. | ||
Pujalté I, Passagne I, Brouillaud B, et al. Cytotoxicity and oxidative stress induced by different metallic nanoparticles on human kidney cells. Part Fibre Toxicol. 2011;8:10. | ||
Kroll A, Pillukat MH, Hahn D, Schnekenburger J. Interference of engineered nanoparticles with in vitro toxicity assays. Arch Toxicol. 2012;86(7):1123–1136. | ||
Zhang LW, Zeng L, Barron AR, Monteiro-Riviere NA. Biological interactions of functionalized single-wall carbon nanotubes in human epidermal keratinocytes. Int J Toxicol. 2007;26(2):103–113. | ||
Ying E, Hwang HM. In vitro evaluation of the cytotoxicity of iron oxide nanoparticles with different coatings and different sizes in A3 human T lymphocytes. Sci Total Environ. 2010;408(20):4475–4481. | ||
Braydich-Stolle L, Hussain S, Schlager JJ, Hofmann MC. In vitro cytotoxicity of nanoparticles in mammalian germline stem cells. Toxicol Sci. 2005;88(2):412–419. | ||
Wahl B, Wahl B, Daum N, Ohrem HL, Lehr CM. Novel luminescence assay offers new possibilities for the risk assessment of silica nanoparticles. Nanotoxicology. 2008;2(4):243–251. | ||
Murdock RC, Braydich-Stolle L, Schrand AM, Schlager JJ, Hussain SM. Characterization of nanomaterial dispersion in solution prior to in vitro exposure using dynamic light scattering technique. Toxicol Sci. 2008;101(2):239–253. | ||
Monteiro-Riviere NA, Inman AO. Challenges for assessing carbon nanomaterial toxicity to the skin. Carbon. 2006;44(6):1070–1078. | ||
Monteiro-Riviere NA, Oldenburg SJ, Inman AO. Interactions of aluminum nanoparticles with human epidermal keratinocytes. J Appl Toxicol. 2010;30(3):276–285. | ||
Wörle-Knirsch JM, Pulskamp K, Krug HF. Oops they did it again! Carbon nanotubes hoax scientists in viability assays. Nano Lett. 2006;6(6):1261–1268. | ||
Gonzales M, Mitsumori LM, Kushleika JV, Rosenfeld ME, Krishnan KM. Cytotoxicity of iron oxide nanoparticles made from the thermal decomposition of organometallics and aqueous phase transfer with Pluronic F127. Contrast Media Mol Imaging. 2010;5(5):286–293. | ||
Casey A, Herzog E, Davoren M, Lyng FM, Byrne HJ, Chambers G. Spectroscopic analysis confirms the interactions between single walled carbon nanotubes and various dyes commonly used to assess cytotoxicity. Carbon. 2007;45(7):1425–1432. | ||
Davoren M, Herzog E, Casey A, et al. In vitro toxicity evaluation of single walled carbon nanotubes on human A549 lung cells. Toxicol In Vitro. 2007;21(3):438–448. | ||
L’azou B, Jorly J, On D, et al. In vitro effects of nanoparticles on renal cells. Part Fibre Toxicol. 2008;5:22. | ||
Low SP, Williams KA, Canham LT, Voelcker NH. Evaluation of mammalian cell adhesion on surface-modified porous silicon. Biomaterials. 2006;27(26):4538–4546. | ||
Han X, Gelein R, Corson N, et al. Validation of an LDH assay for assessing nanoparticle toxicity. Toxicology. 2011;287(1–3):99–104. | ||
Oh SJ, Kim H, Liu Y, et al. Incompatibility of silver nanoparticles with lactate dehydrogenase leakage assay for cellular viability test is attributed to protein binding and reactive oxygen species generation. Toxicol Lett. 2014;225(3):422–432. | ||
Stueker O, Ortega VA, Goss GG, Stepanova M. Understanding interactions of functionalized nanoparticles with proteins: a case study on lactate dehydrogenase. Small. 2014;10(10):2006–2021. | ||
Holder AL, Goth-Goldstein R, Lucas D, Koshland CP. Particle-induced artifacts in the MTT and LDH viability assays. Chem Res Toxicol. 2012;25(9):1885–1892. | ||
Suska F, Gretzer C, Esposito M, et al. In vivo cytokine secretion and NF-kappaB activation around titanium and copper implants. Biomaterials. 2005;26(5):519–527. | ||
Belyanskaya L, Manser P, Spohn P, Bruinink A, Wick P. The reliability and limits of the MTT reduction assay for carbon nanotubes–cell interaction. Carbon. 2007;45(13):2643–2648. | ||
Laaksonen T, Santos H, Vihola H, et al. Failure of MTT as a toxicity testing agent for mesoporous silicon microparticles. Chem Res Toxicol. 2007;20(12):1913–1918. | ||
Brown DM, Dickson C, Duncan P, Al-Attili F, Stone V. Interaction between nanoparticles and cytokine proteins: impact on protein and particle functionality. Nanotechnology. 2010;21(21):215104. | ||
Kocbach A, Totlandsdal AI, Låg M, Refsnes M, Schwarze PE. Differential binding of cytokines to environmentally relevant particles: a possible source for misinterpretation of in vitro results? Toxicol Lett. 2008;176(2):131–137. | ||
Val S, Hussain S, Boland S, Hamel R, Baeza-Squiban A, Marano F. Carbon black and titanium dioxide nanoparticles induce pro-inflammatory responses in bronchial epithelial cells: need for multiparametric evaluation due to adsorption artifacts. Inhal Toxicol. 2009;21 Suppl 1:115–122. | ||
Veranth JM, Kaser EG, Veranth MM, Koch M, Yost GS. Cytokine responses of human lung cells (BEAS-2B) treated with micron-sized and nanoparticles of metal oxides compared to soil dusts. Part Fibre Toxicol. 2007;4:2. | ||
Lin MH, Hsu TS, Yang PM, Tsai MY, Perng TP, Lin LY. Comparison of organic and inorganic germanium compounds in cellular radiosensitivity and preparation of germanium nanoparticles as a radiosensitizer. Int J Radiat Biol. 2009;85(3):214–226. | ||
Aam BB, Fonnum F. Carbon black particles increase reactive oxygen species formation in rat alveolar macrophages in vitro. Arch Toxicol. 2007;81(6):441–446. | ||
Hoet PH, Nemery B, Napierska D. Intracellular oxidative stress caused by nanoparticles: what do we measure with the dichlorofluorescein assay? Nano Today. 2013;8(3):223–227. | ||
Karlsson HL. The comet assay in nanotoxicology research. Anal Bioanal Chem. 2010;398(2):651–666. | ||
Inoue K, Takano H, Yanagisawa R, Koike E, Shimada A. Size effects of latex nanomaterials on lung inflammation in mice. Toxicol Appl Pharmacol. 2009;234(1):68–76. | ||
van Zijverden M, Granum B. Adjuvant activity of particulate pollutants in different mouse models. Toxicology. 2000;152(1–3):69–77. | ||
Fifis T, Gamvrellis A, Crimeen-Irwin B, et al. Size-dependent immunogenicity: therapeutic and protective properties of nano-vaccines against tumors. J Immunol. 2004;173(5):3148–3154. | ||
Nygaard UC, Samuelsen M, Aase A, Løvik M. The capacity of particles to increase allergic sensitization is predicted by particle number and surface area, not by particle mass. Toxicol Sci. 2004;82(2):515–524. | ||
Nygaard UC, Ormstad H, Aase A, Løvik M. The IgE adjuvant effect of particles: characterisation of the primary cellular response in the draining lymph node. Toxicology. 2005;206(2):181–193. | ||
Yanagisawa R, Takano H, Inoue KI, Koike E, Sadakane K, Ichinose T. Size effects of polystyrene nanoparticles on atopic dermatitislike skin lesions in NC/NGA mice. Int J Immunopathol Pharmacol. 2010;23(1):131–141. | ||
Shvedova AA, Fabisiak JP, Kisin ER, et al. Sequential exposure to carbon nanotubes and bacteria enhances pulmonary inflammation and infectivity. Am J Respir Cell Mol Biol. 2008;38(5):579–590. | ||
Park EJ, Roh J, Kim SN, et al. A single intratracheal instillation of single-walled carbon nanotubes induced early lung fibrosis and subchronic tissue damage in mice. Arch Toxicol. 2011;85(9):1121–1131. | ||
Inoue K, Yanagisawa R, Koike E, Nishikawa M, Takano H. Repeated pulmonary exposure to single-walled carbon nanotubes exacerbates allergic inflammation of the airway: Possible role of oxidative stress. Free Radic Biol Med. 2010;48(7):924–934. | ||
Inoue K, Takano H, Koike E, et al. Effects of pulmonary exposure to carbon nanotubes on lung and systemic inflammation with coagulatory disturbance induced by lipopolysaccharide in mice. Exp Biol Med (Maywood). 2008;233(12):1583–1590. | ||
Crouzier D, Follot S, Gentilhomme E, et al. Carbon nanotubes induce inflammation but decrease the production of reactive oxygen species in lung. Toxicology. 2010;272(1–3):39–45. | ||
Meng J, Yang M, Jia F, Xu Z, Kong H, Xu H. Immune responses of BALB/c mice to subcutaneously injected multi-walled carbon nanotubes. Nanotoxicology. 2011;5(4):583–591. | ||
Mitchell LA, Gao J, Wal RV, Gigliotti A, Burchiel SW, McDonald JD. Pulmonary and systemic immune response to inhaled multiwalled carbon nanotubes. Toxicol Sci. 2007;100(1):203–214. | ||
Tkach AV, Shurin GV, Shurin MR, et al. Direct effects of carbon nanotubes on dendritic cells induce immune suppression upon pulmonary exposure. ACS Nano. 2011;5(7):5755–5762. | ||
Park EJ, Cho WS, Jeong J, Yi J, Choi K, Park K. Pro-inflammatory and potential allergic responses resulting from B cell activation in mice treated with multi-walled carbon nanotubes by intratracheal instillation. Toxicology. 2009;259(3):113–121. | ||
Wang X, Podila R, Shannahan JH, Rao AM, Brown JM. Intravenously delivered graphene nanosheets and multiwalled carbon nanotubes induce site-specific Th2 inflammatory responses via the IL-33/ST2 axis. Int J Nanomedicine. 2013;8:1733–1748. | ||
Yamaguchi A, Fujitani T, Ohyama K, et al. Effects of sustained stimulation with multi-wall carbon nanotubes on immune and inflammatory responses in mice. J Toxicol Sci. 2012;37(1):177–189. | ||
Inoue K, Koike E, Yanagisawa R, Hirano S, Nishikawa M, Takano H. Effects of multi-walled carbon nanotubes on a murine allergic airway inflammation model. Toxicol Appl Pharmacol. 2009;237(3): 306–316. | ||
Ryman-Rasmussen JP, Tewksbury EW, Moss OR, Cesta MF, Wong BA, Bonner JC. Inhaled multiwalled carbon nanotubes potentiate airway fibrosis in murine allergic asthma. Am J Respir Cell Mol Biol. 2009;40(3):349–358. | ||
Cesta MF, Ryman-Rasmussen JP, Wallace DG, et al. Bacterial lipopolysaccharide enhances PDGF signaling and pulmonary fibrosis in rats exposed to carbon nanotubes. Am J Respir Cell Mol Biol. 2010; 43(2):142–151. | ||
Sung JH, Ji JH, Park JD, et al. Subchronic inhalation toxicity of silver nanoparticles. Toxicol Sci. 2009;108(2):452–461. | ||
Han YH, Kim SH, Kim SZ, Park WH. Intracellular GSH levels rather than ROS levels are tightly related to AMA-induced HeLa cell death. Chem Biol Interact. 2008;171(1):67–78. | ||
Park EJ, Bae E, Yi J, et al. Repeated-dose toxicity and inflammatory responses in mice by oral administration of silver nanoparticles. Environ Toxicol Pharmacol. 2010;30(2):162–168. | ||
Vandebriel RJ, Tonk EC, de la Fonteyne-Blankestijn LJ, et al. Immunotoxicity of silver nanoparticles in an intravenous 28-day repeated-dose toxicity study in rats. Part Fibre Toxicol. 2014;11:21. | ||
Su CL, Chen TT, Chang CC, et al; Taiwan CardioPulmonary Research Group (T-CPR). Comparative proteomics of inhaled silver nanoparticles in healthy and allergen provoked mice. Int J Nanomedicine. 2013;8:2783–2799. | ||
Gosens I, Post JA, de la Fonteyne LJ, et al. Impact of agglomeration state of nano- and submicron sized gold particles on pulmonary inflammation. Part Fibre Toxicol. 2010;7(1):37. | ||
Khan HA, Abdelhalim MA, Alhomida AS, Al-Ayed MS. Effects of naked gold nanoparticles on proinflammatory cytokines mRNA expression in rat liver and kidney. Biomed Res Int. 2013;2013:590730. | ||
Hussain S, Vanoirbeek JA, Luyts K, et al. Lung exposure to nanoparticles modulates an asthmatic response in a mouse model. Eur Respir J. 2011;37(2):299–309. | ||
Cho WS, Cho M, Kim SR, et al. Pulmonary toxicity and kinetic study of Cy5.5-conjugated superparamagnetic iron oxide nanoparticles by optical imaging. Toxicol Appl Pharmacol. 2009;239(1):106–115. | ||
Park EJ, Kim H, Kim Y, Yi J, Choi K, Park K. Inflammatory responses may be induced by a single intratracheal instillation of iron nanoparticles in mice. Toxicology. 2010;275(1–3):65–71. | ||
Chen BA, Jin N, Wang J, et al. The effect of magnetic nanoparticles of Fe(3)O(4) on immune function in normal ICR mice. Int J Nanomedicine. 2010;5:593–599. | ||
Shen CC, Wang CC, Liao MH, Jan TR. A single exposure to iron oxide nanoparticles attenuates antigen-specific antibody production and T-cell reactivity in ovalbumin-sensitized BALB/c mice. Int J Nanomedicine. 2011;6:1229–1235. | ||
Shen CC, Liang HJ, Wang CC, Liao MH, Jan TR. Iron oxide nanoparticles suppressed T helper 1 cell-mediated immunity in a murine model of delayed-type hypersensitivity. Int J Nanomedicine. 2012;7:2729–2737. | ||
Zhu M, Li Y, Shi J, Feng W, Nie G, Zhao Y. Exosomes as extrapulmonary signaling conveyors for nanoparticle-induced systemic immune activation. Small. 2012;8(3):404–412. | ||
Zhu M, Tian X, Song X, et al. Nanoparticle-induced exosomes target antigen-presenting cells to initiate Th1-type immune activation. Small. 2012;8(18):2841–2848. | ||
Ban M, Langonné I, Huguet N, Goutet M. Effect of submicron and nano-iron oxide particles on pulmonary immunity in mice. Toxicol Lett. 2012;210(3):267–275. | ||
Cho WS, Duffin R, Poland CA, et al. Metal oxide nanoparticles induce unique inflammatory footprints in the lung: important implications for nanoparticle testing. Environ Health Perspect. 2010;118(12):1699–1706. | ||
Morishige T, Yoshioka Y, Inakura H, et al. Cytotoxicity of amorphous silica particles against macrophage-like THP-1 cells depends on particle-size and surface properties. Pharmazie. 2010;65(8):596–599. | ||
Chen Q, Xue Y, Sun J. Kupffer cell-mediated hepatic injury induced by silica nanoparticles in vitro and in vivo. Int J Nanomedicine. 2013;8: 1129–1140. | ||
Nishimori H, Kondoh M, Isoda K, Tsunoda S, Tsutsumi Y, Yagi K. Silica nanoparticles as hepatotoxicants. Eur J Pharm Biopharm. 2009; 72(3):496–501. | ||
Hirai T, Yoshikawa T, Nabeshi H, et al. Amorphous silica nanoparticles size-dependently aggravate atopic dermatitis-like skin lesions following an intradermal injection. Part Fibre Toxicol. 2012;9:3. | ||
Han B, Guo J, Abrahaley T, et al. Adverse effect of nano-silicon dioxide on lung function of rats with or without ovalbumin immunization. PLoS One. 2011;6(2):e17236. | ||
Ambalavanan N, Stanishevsky A, Bulger A, et al. Titanium oxide nanoparticle instillation induces inflammation and inhibits lung development in mice. Am J Physiol Lung Cell Mol Physiol. 2013;304(3): L152–L161. | ||
Grassian VH, O’shaughnessy PT, Adamcakova-Dodd A, Pettibone JM, Thorne PS. Inhalation exposure study of titanium dioxide nanoparticles with a primary particle size of 2 to 5 nm. Environ Health Perspect. 2007;115(3):397–402. | ||
Oberdörster G, Ferin J, Lehnert BE. Correlation between particle size, in vivo particle persistence, and lung injury. Environ Health Perspect. 1994;102 Suppl 5:173–179. | ||
Oberdörster G, Finkelstein JN, Johnston C, et al. Acute pulmonary effects of ultrafine particles in rats and mice. Res Rep Health Eff Inst. 2000;(96):5–74; disc. 75–86. | ||
Roursgaard M, Jensen KA, Poulsen SS, et al. Acute and subchronic airway inflammation after intratracheal instillation of quartz and titanium dioxide agglomerates in mice. Scientific World Journal. 2011;11: 801–825. | ||
Gustafsson Å, Lindstedt E, Elfsmark LS, Bucht A. Lung exposure of titanium dioxide nanoparticles induces innate immune activation and long-lasting lymphocyte response in the Dark Agouti rat. J Immunotoxicol. 2011;8(2):111–121. | ||
Kobayashi N, Naya M, Endoh S, Maru J, Yamamoto K, Nakanishi J. Comparative pulmonary toxicity study of nano-TiO(2) particles of different sizes and agglomerations in rats: different short- and long-term post-instillation results. Toxicology. 2009;264(1–2):110–118. | ||
Sheng L, Wang L, Sang X, et al. Nano-sized titanium dioxide-induced splenic toxicity: a biological pathway explored using microarray technology. J Hazard Mater. 2014;278:180–188. | ||
Sang X, Fei M, Sheng L, et al. Immunomodulatory effects in the spleen-injured mice following exposure to titanium dioxide nanoparticles. J Biomed Mater Res A. 2014;102(10):3562–3572. | ||
Ma L, Zhao J, Wang J, et al. The acute liver injury in mice Caused by nano-anatase TiO2. Nanoscale Res Lett. 2009;4(11):1275–1285. | ||
Larsen ST, Roursgaard M, Jensen KA, Nielsen GD. Nano titanium dioxide particles promote allergic sensitization and lung inflammation in mice. Basic Clin Pharmacol Toxicol. 2010;106(2):114–117. | ||
de Haar C, Hassing I, Bol M, Bleumink R, Pieters R. Ultrafine but not fine particulate matter causes airway inflammation and allergic airway sensitization to co-administered antigen in mice. Clin Exp Allergy. 2006;36(11):1469–1479. | ||
Moon C, Park HJ, Choi YH, Park EM, Castranova V, Kang JL. Pulmonary inflammation after intraperitoneal administration of ultrafine titanium dioxide (TiO2) at rest or in lungs primed with lipopolysaccharide. J Toxicol Environ Health A. 2010;73(5):396–409. | ||
Adamcakova-Dodd A, Stebounova LV, Kim JS, et al. Toxicity assessment of zinc oxide nanoparticles using sub-acute and sub-chronic murine inhalation models. Part Fibre Toxicol. 2014;11:15. | ||
Xia T, Hamilton RF, Bonner JC, et al. Interlaboratory evaluation of in vitro cytotoxicity and inflammatory responses to engineered nanomaterials: the NIEHS Nano GO Consortium. Environ Health Perspect. 2013;121(6):683–690. | ||
Witasp E, Shvedova AA, Kagan VE, Fadeel B. Single-walled carbon nanotubes impair human macrophage engulfment of apoptotic cell corpses. Inhal Toxicol. 2009;21 Suppl 1:131–136. | ||
Greulich C, Kittler S, Epple M, Muhr G, Köller M. Studies on the biocompatibility and the interaction of silver nanoparticles with human mesenchymal stem cells (hMSCs). Langenbecks Arch Surg. 2009;394(3):495–502. | ||
Shin SH, Ye MK, Kim HS, Kang HS. The effects of nano-silver on the proliferation and cytokine expression by peripheral blood mononuclear cells. Int Immunopharmacol. 2007;7(13):1813–1818. | ||
Carlson C, Hussain SM, Schrand AM, et al. Unique cellular interaction of silver nanoparticles: size-dependent generation of reactive oxygen species. J Phys Chem B. 2008;112(43):13608–13619. | ||
Stebounova LV, Adamcakova-Dodd A, Kim JS, et al. Nanosilver induces minimal lung toxicity or inflammation in a subacute murine inhalation model. Part Fibre Toxicol. 2011;8(1):5. | ||
Smulders S, Luyts K, Brabants G, et al. Toxicity of nanoparticles embedded in paints compared with pristine nanoparticles in mice. Toxicol Sci. 2014;141(1):132–140. | ||
Yang EJ, Kim S, Kim JS, Choi IH. Inflammasome formation and IL-1β release by human blood monocytes in response to silver nanoparticles. Biomaterials. 2012;33(28):6858–6867. | ||
Shavandi Z, Ghazanfari T, Moghaddam KN. In vitro toxicity of silver nanoparticles on murine peritoneal macrophages. Immunopharmacol Immunotoxicol. 2011;33(1):135–140. | ||
Hutter E, Boridy S, Labrecque S, et al. Microglial response to gold nanoparticles. ACS Nano. 2010;4(5):2595–2606. | ||
Yen HJ, Hsu SH, Tsai CL. Cytotoxicity and immunological response of gold and silver nanoparticles of different sizes. Small. 2009;5(13): 1553–1561. | ||
Srinivas A, Rao PJ, Selvam G, Goparaju A, Murthy PB, Reddy PN. Oxidative stress and inflammatory responses of rat following acute inhalation exposure to iron oxide nanoparticles. Hum Exp Toxicol. 2012; 31(11):1113–1131. | ||
Settles M, Etzrodt M, Kosanke K, et al. Different capacity of monocyte subsets to phagocytose iron-oxide nanoparticles. PLoS One. 2011; 6(10):e25197. | ||
Hsiao JK, Weng TI, Tai MF, et al. Cellular behavior change of macrophage after exposure to nanoparticles. J Nanosci Nanotechnol. 2009;9(2):1388–1393. | ||
Hsiao JK, Chu HH, Wang YH, et al. Macrophage physiological function after superparamagnetic iron oxide labeling. NMR Biomed. 2008;21(8):820–829. | ||
Liu R, Zhang X, Pu Y, et al. Small-sized titanium dioxide nanoparticles mediate immune toxicity in rat pulmonary alveolar macrophages in vivo. J Nanosci Nanotechnol. 2010;10(8):5161–5169. | ||
Yang EJ, Choi IH. Immunostimulatory effects of silica nanoparticles in human monocytes. Immune Netw. 2013;13(3):94–101. | ||
Waters KM, Masiello LM, Zangar RC, et al. Macrophage responses to silica nanoparticles are highly conserved across particle sizes. Toxicol Sci. 2009;107(2):553–569. | ||
Sun Q, Tan D, Ze Y, et al. Pulmotoxicological effects caused by long-term titanium dioxide nanoparticles exposure in mice. J Hazard Mater. 2012;235–236:47–53. | ||
Liu R, Yin LH, Pu YP, et al. The immune toxicity of titanium dioxide on primary pulmonary alveolar macrophages relies on their surface area and crystal structure. J Nanosci Nanotechnol. 2010; 10(12):8491–8499. | ||
Scherbart AM, Langer J, Bushmelev A, et al. Contrasting macrophage activation by fine and ultrafine titanium dioxide particles is associated with different uptake mechanisms. Part Fibre Toxicol. 2011;8:31. | ||
Lozano-Fernández T, Ballester-Antxordoki L, Pérez-Temprano N, et al. Potential impact of metal oxide nanoparticles on the immune system: The role of integrins, L-selectin and the chemokine receptor CXCR4. Nanomedicine. 2014;10(6):1301–1310. | ||
Hedenborg M. Titanium dioxide induced chemiluminescence of human polymorphonuclear leukocytes. Int Arch Occup Environ Health. 1988;61(1–2):1–6. | ||
Sahu D, Kannan GM, Vijayaraghavan R. Size-dependent effect of zinc oxide on toxicity and inflammatory potential of human monocytes. J Toxicol Environ Health A. 2014;77(4):177–191. | ||
Roy R, Tripathi A, Das M, Dwivedi PD. Cytotoxicity and uptake of zinc oxide nanoparticles leading to enhanced inflammatory cytokines levels in murine macrophages: comparison with bulk zinc oxide. J Biomed Nanotechnol. 2011;7(1):110–111. | ||
Feltis BN, OKeefe SJ, Harford AJ, Piva TJ, Turney TW, Wright PF. Independent cytotoxic and inflammatory responses to zinc oxide nanoparticles in human monocytes and macrophages. Nanotoxicology. 2012;6(7):757–765. | ||
Wilhelmi V, Fischer U, Weighardt H, et al. Zinc oxide nanoparticles induce necrosis and apoptosis in macrophages in a p47phox- and Nrf2-independent manner. PLoS One. 2013;8(6):e65704. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.