Back to Journals » International Journal of Nanomedicine » Volume 12
Vaccination with poly(D,L-lactide-co-glycolide) nanoparticles loaded with soluble Leishmania antigens and modified with a TNFα-mimicking peptide or monophosphoryl lipid A confers protection against experimental visceral leishmaniasis
Authors Margaroni M, Agallou M ![]() , Athanasiou E, Kammona O, Kiparissides C, Gaitanaki C, Karagouni E
, Athanasiou E, Kammona O, Kiparissides C, Gaitanaki C, Karagouni E
Received 4 May 2017
Accepted for publication 20 June 2017
Published 23 August 2017 Volume 2017:12 Pages 6169—6184
DOI https://doi.org/10.2147/IJN.S141069
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Maritsa Margaroni,1,2 Maria Agallou,1 Evita Athanasiou,1 Olga Kammona,3 Costas Kiparissides,3,4 Catherine Gaitanaki,2 Evdokia Karagouni1
1Laboratory of Cellular Immunology, Department of Microbiology, Hellenic Pasteur Institute, 2Department of Animal and Human Physiology, School of Biology, National and Kapodistrian University of Athens, Athens, 3Chemical Process & Energy Resources Institute, Centre for Research and Technology Hellas, 4Department of Chemical Engineering, Aristotle University of Thessaloniki, Thessaloniki, Greece
Abstract: Visceral leishmaniasis (VL) persists as a major public health problem, and since the existing chemotherapy is far from satisfactory, development of an effective vaccine emerges as the most appropriate strategy for confronting VL. The development of an effective vaccine relies on the selection of the appropriate antigen and also the right adjuvant and/or delivery vehicle. In the present study, the protective efficacy of poly(D,L-lactide-co-glycolide) (PLGA) nanoparticles (NPs), which were surface-modified with a TNFα-mimicking eight-amino-acid peptide (p8) and further functionalized by encapsulating soluble Leishmania infantum antigens (sLiAg) and monophosphoryl lipid A (MPLA), a TLR4 ligand, was evaluated against challenge with L. infantum parasites in BALB/c mice. Vaccination with these multifunctionalized PLGA nanoformulations conferred significant protection against parasite infection in vaccinated mice. In particular, vaccination with PLGA-sLiAg-MPLA or p8-PLGA-sLiAg NPs resulted in almost complete elimination of the parasite in the spleen for up to 4 months post-challenge. Parasite burden reduction was accompanied by antigen-specific humoral and cellular immune responses. Specifically, injection with PLGA-sLiAg-MPLA raised exclusively anti-sLiAg IgG1 antibodies post-vaccination, while in p8-PLGA-sLiAg-vaccinated mice, no antibody production was detected. However, 4 months post-challenge, in mice vaccinated with all the multifunctionalized NPs, antibody class switching towards IgG2a subtype was observed. The study of cellular immune responses revealed the increased proliferation capacity of spleen cells against sLiAg, consisting of IFNγ-producing CD4+ and CD8+ T cells. Importantly, the activation of CD8+ T cells was exclusively attributed to vaccination with PLGA NPs surface-modified with the p8 peptide. Moreover, characterization of cytokine production in vaccinated–infected mice revealed that protection was accompanied by significant increase of IFNγ and lower levels of IL-4 and IL-10 in protected mice when compared to control infected group. Conclusively, the above nanoformulations hold promise for future vaccination strategies against VL.
Keywords: nanovaccine, soluble Leishmania antigen, visceral leishmaniasis, immune response, T cells, cytokines
Plain language summary
Visceral leishmaniasis (VL) persists as a major public health problem, and since existing drugs have proved to be ineffective, research effort is driven towards the development of an effective vaccine. In the present study, poly(D,L-lactide-co-glycolide) nanoparticles (NPs) surface-modified with a TNFα-mimicking peptide, encapsulating soluble Leishmania antigens and the adjuvant monophosphoryl lipid A, were evaluated as vaccine candidates against experimental VL. Vaccination with the above multifunctionalized NPs resulted in significant reduction of the parasite burden in the liver and spleen, the main target organs of Leishmania. According to our results, protection was related to the existence of antigen-specific cellular immune responses and more specifically to the activation of IFNγ-producing CD4+ and CD8+ T cells, followed by switching of IgG antibody subclasses towards IgG2a. Moreover, significantly lower levels of cytokines related with disease exacerbation were observed in vaccinated and infected mice. Conclusively, the nanoformulations used in the present study hold promise for future vaccination strategies against VL.
Introduction
Leishmaniasis is a vector-borne disease caused by the intracellular parasites of the genus Leishmania and displays a spectrum of clinical manifestations from self-healing cutaneous to life-threatening visceral form of disease.1 The disease is prevalent in larger areas of tropical, subtropical and the Mediterranean countries, and according to the WHO, 310 million people are at risk. The most severe form of the disease is visceral leishmaniasis (VL) with nearly 300,000 new cases and 20,000 deaths per year.2 Available chemotherapy is far from satisfactory, as existing drugs have many side effects and practically show limited efficacy in some endemic areas due to the development of resistant parasites.3,4 Hence, there is an urgent need to develop an effective vaccine against leishmaniasis.
Vaccination against leishmaniasis has been pursued using different strategies, ranging from inoculation of virulent parasites to immunization with killed parasite preparations, live attenuated parasites, recombinant proteins or plasmid DNA coding for defined Leishmania antigens.5,6 Development of an effective vaccine relies not only on the selection of the appropriate antigen but also on selecting the right adjuvant and/or delivery vehicle.
Polymeric micro- and nanoparticulate formulations are currently considered as ideal vaccine delivery systems. Among them, biodegradable poly(D,L-lactide-co-glycolide) (PLGA) nanoparticles (NPs) have attracted considerable attention due to their biocompatibility and have been already approved by the US Food and Drug Administration (FDA) and European Medicines Agency (EMA) as drug carriers.7 PLGA-based vaccine delivery systems possess several advantages such as sustained and controlled antigen release, adjuvant co-encapsulation, protection from degradation by enzymes, as well as the ability of specific tissue targeting.8,9 Several studies have demonstrated that these nano-sized delivery systems can induce both humoral and cell-mediated specific immune responses via activation of dendritic cells (DCs) in animals in combination with tumor antigenic peptides,10,11 or parasitic12 or viral antigenic molecules.13,14
Several types of micro- or nanoformulations (ie, liposomes, chitosan, PLGA) and antigens have been tested up till now for vaccine development against leishmaniasis with encouraging results.15,16 In particular, whole heat-inactivated Leishmania major parasites, the causative agent of cutaneous leishmaniasis (CL), or their soluble antigens17,18 as well as specific parasitic antigens (eg, KMP-11)19 in combination with adjuvants have been evaluated in experimental models of CL or VL with promising results.
Immune response against leishmaniasis depends on cell-mediated immunity and is associated with the development of different T cell populations.20 CD4+ Th1 cells are critical for the control of Leishmania infections, due to their ability to produce IFNγ, which activates macrophages and DCs, leading to parasite killing. Moreover, CD8+ T cells that produce IFNγ also hold an important role in protection development against VL.21 On the contrary, certain T cell populations (eg, regulatory T cells) have been proved to be important in disease development through production of suppressive cytokines. Recent studies demonstrate that IL-10 is the major immunosuppressive cytokine in VL.22,23 It has been shown that downregulation of those T cell populations increases the potential of vaccine success. Thus, an exclusive generation of a vaccine-induced Th1 or CD8+ response is insufficient to ensure protection and cannot predict vaccine success in experimental VL.24,25
According to previous results from our group, nanoformulations based on PLGA NPs or PLGA NPs surface-modified with a TNFα-mimicking eight-amino-acid peptide (p8) loaded with a crude mix of proteins from L. infantum promastigotes (soluble Leishmania antigens, sLiAg) and/or monophosphoryl lipid A (MPLA), a known TLR4 ligand serving as adjuvant, were efficiently internalized by DCs, which in turn induced the development of antigen-specific T cell populations in vitro.26 Taking into consideration these data, we designed this study to investigate whether vaccination with the above multifunctionalized nanoformulations would induce similar immune response in the in vivo as those induced in the in vitro system and subsequently confer protection in the murine model of VL. Furthermore, the type of the immune responses elicited in mice vaccinated with the above nanoformulations and infected with L. infantum was also studied.
Materials and methods
Experimental animals, parasite maintenance and sLiAg preparation
Female BALB/c mice (6–8 weeks old) used in the present study were obtained from the breeding unit of the Hellenic Pasteur Institute (HPI; Athens, Greece) and reared in institutional facilities under specific pathogen-free environmental conditions at an ambient temperature of 25°C. Mice were provided with sterile food and water ad libitum, and all animal protocols were conducted in strict accordance with the National Law 56/2013, which adheres to the European Directive 2010/63/EU for animal experiments, and complied with the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines. The study was approved by the Animal Bioethics Committee of the HPI (Approval Number: 4455/10-07-2014).
L. infantum strain GH8 (MHOM/GR/2001/GH8), originally isolated from a Greek patient suffering from VL,27 was kept in a virulent state by continuous passage in BALB/c mice. A homogenized spleen tissue sample from an infected BALB/c mouse was cultured in RPMI-1640 medium (Biochrom AG, Berlin, Germany) supplemented with 24 mM NaHCO3, 2 mM L-glutamine, 100 u/mL penicillin, 100 μg/mL streptomycin, 10 mM HEPES (Thermo Fisher Scientific, Waltham, MA, USA) and 10% (v/v) fetal bovine serum (FBS; Applichem, Darmstadt, Germany). Stationary-phase promastigotes were harvested by centrifugation at 650× g for 10 min at 4°C. The pellet was washed by phosphate-buffered saline (PBS: 4.3 mM Na2HPO4, 1.4 mM KH2PO4, 2.7 mM KCl, 137 mM NaCl) and resuspended at a concentration of 2×108 cells/mL. A volume of 100 μL of this preparation was injected intravenously in the lateral tail vein of each mouse.
Preparation of sLiAg was carried out as previously described.26
Vaccination of mice and challenge infection
Female BALB/c mice (6–8 weeks old; five animals/group/time point) were vaccinated subcutaneously, into the scruff and the base of tail, with the following particle formulations: (i) PLGA-sLiAg, (ii) PLGA-sLiAg-MPLA, (iii) p8-PLGA-sLiAg, and (iv) p8-PLGA-sLiAg-MPLA, which will be referred to as multifunctionalized NPs, as well as with PLGA, p8-PLGA or the soluble form of sLiAg + MPLA as control groups. The above nanoformulations were synthesized and characterized according to Margaroni et al.26 Mice injected with 200 μL of PBS served as negative control. Two booster vaccinations followed at 2-week intervals. Vaccinated mice received 15 μg of encapsulated or free sLiAg and 2 μg of encapsulated or free MPLA (Sigma Aldrich, St Louis, MO, USA) per 200 μL of PBS; those immunized with PLGA or p8-PLGA received an equivalent of PLGA encapsulating sLiAg weight of NPs in PBS. Two weeks after the second booster vaccination, mice were challenged with 2×107 freshly transformed stationary-phase L. infantum promastigotes in 100 μL PBS intravenously via the lateral tail vein.
Determination of antibody response
Two weeks after the final booster vaccination, as well as 2 and 4 months post-challenge, serums from vaccinated and infected animals were analyzed by enzyme-linked immunosorbent assay (ELISA) for the presence of sLiAg-specific antibodies.28 In brief, 96-well microtiter plates (Greiner, Kremsmuenster, Austria) were coated with 5 μg/mL sLiAg diluted in PBS overnight at 4°C. The plates were blocked with 2% bovine serum albumin (BSA) (Sigma Aldrich) in PBS at 37°C for 1.5 h to prevent nonspecific binding. After washing with PBS containing 0.05% Tween 20, the plates were incubated with serial dilutions of sera (1:50 to 1:51,200) for 1.5 h at 37°C. Then, the plates were incubated with biotinylated anti-mouse IgG1 and IgG2a antibodies (AbD Serotec, Oxford, UK) according to the manufacturer’s instructions for 1 h at 37°C and after washing were treated with horse radish peroxidase (HRP) streptavidin solution (AbD Serotec) for 1 h at 37°C. The plates were washed and developed with 3,3’,5,5’-Tetramethylbenzidine (TMB) substrate (Pierce, Waltham, MA, USA). Absorbance was read at 450 nm. Results are expressed as titer, that is, the maximum dilution in which the OD value of the sample is higher than the cut-off OD (OD of blank ±2 SD).
Spleen cell proliferation and cytokine assays
Five mice from each group were sacrificed 2 weeks after the last booster vaccination and 2 and 4 months post-challenge, and the spleens were aseptically removed and mechanically teased into single-cell suspension in RPMI-1640 medium. Spleen cells were cultured in triplicates in 96-well U bottom plates (Greiner) at a density of 2×106 cells/mL and stimulated with 12.5 μg/mL of sLiAg. The cells incubated with 3 μg/mL Con A (Sigma Aldrich) or medium alone served as negative or positive control, respectively. The cells were incubated for 96 h in 5% CO2 at 37°C and pulsed for the final 18 h with 0.5 μCi of [3H]thymidine (PerkinElmer, Waltham, MA, USA). The cells were harvested, and lymphocyte proliferation was determined by radioactivity incorporation on a microplate scintillation and luminescence counter (Microbeta Trilux; Wallac, Turku, Finland). For cytokine determination, after 72 h incubation, culture supernatants, from similar cultures as described above, were collected and analyzed using a Milliplex mouse cytokine detection system for IFNγ, IL-4 and IL-10 according to the manufacturer’s instructions (EMD Millipore, Billerica, MA, USA). Each sample was assayed in duplicate, and cytokine standards and quality controls supplied by the manufacturer were run on each plate. Data were acquired on a Luminex 200™ (Luminex Corporation, Austin, TX, USA) and analyzed using xPONENT software (Luminex).
Flow cytometry assay for cell phenotyping and cytokine production in vaccinated and infected mice
Five mice from each group were sacrificed 2 weeks after the second booster vaccination, as well as 2 and 4 months post-challenge, and the spleens were aseptically removed. Single-cell suspension was prepared as described above. Spleen cells were cultured in 24-well flat-bottom plates (Greiner) at a density of 1×106 cells/mL and stimulated with 12.5 μg/mL of sLiAg for 48 h. The cells were incubated with 2.5 μg/mL of Brefeldin A (Sigma Aldrich) for 4 h. The cells were fixed using 2% paraformaldehyde in PBS for 15 min at room temperature and then were triple-stained with allophycocyanin (APC)-conjugated hamster anti-mouse CD3e (clone 145-2C11), fluorescein isothiocyanate (FITC)-conjugated rat anti-mouse CD4 (clone RM4-5) or CD8a (clone 53-6.7) and phycoerythrin (PE)-conjugated rat anti-mouse IFNγ (clone XMG1.2) for 30 min at 4°C in permeabilization buffer (PBS supplemented with 3% [v/v] FBS and 0.1% [v/v] saponin). All antibodies were from BD Pharmingen (San Diego, CA, USA). Each sample was run in triplicate, and 20,000 cells/sample were analyzed on a FACS Calibur (Becton Dickinson, Franklin Lakes, CA, USA), and the data acquired were processed with FlowJo software version 10.0 (Tree Star Inc, Ashland, OR, USA). Results are presented as a percentage (%) of positive cells ± SD from three independent experiments.
Determination of parasite burden with quantitative real-time polymerase chain reaction (qPCR)
DNA was extracted from about 10 mg of spleen or liver tissue from infected mice using QIAamp® DNA Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. The quantity and purity of the DNA were determined with the spectrophotometer NanoDrop® 2000 (Thermo Fischer Scientific).
Parasite burden was determined using TaqMan-based qPCR assay as previously described29 with slight modifications. Briefly, the assay was performed in a final volume of 20 μL containing 4 μL DNA template (150 ng), 10 μL of TaqMan master mix (2×) (Kapa Biosystems, Wilmington, MA, USA), 15 pmol of forward (5′-GGCGGCGGTATTATCTCGAT-3′) and 5 pmol of reverse (5′-ACCACGAGGTAGATGACAGACA-3′) primers (VBC Biotech, Vienna, Austria) (targeting a 74-bp region of the L. major gene encoding the arginine transporter AAP3 gene) and 25 pmol of TET-labeled TaqMan® probe (TET-5′-ATGTCGGGCATCATC-3′-BHQ; VBC Biotech). Each qPCR test was run in triplicate on a SaCycler-96 RUO cycler (Sacace Biotechnologies, Como, Italy). The cycling conditions were 95°C for 10 min, followed by 40 cycles at 95°C, 15 s and 62°C, 60 s. The standard curve method for absolute quantification of parasite number was used. Quantification was performed using standard curves prepared from DNA extracted from tenfold serial dilution of L. infantum parasites (range 1–1×105).
Statistical analysis
Data shown are representative of at least three independent experiments. Differences were assessed by one-way analysis of variance (ANOVA) using Tukey post-test. Analyses were conducted using Prism software (version 5.0; GraphPad Software, Inc, La Jolla, CA, USA). Statistical significance was set at the level of P<0.05.
Results
Vaccination with multifunctionalized PLGA NPs confers protection against experimental VL
We have previously reported that by improving DCs targeting via surface modification with the p8 peptide or by enhancing stimulatory activity via MPLA, PLGA NPs loaded with sLiAg induces a mature and fully functionalized DCs phenotype. These fully mature DCs, in turn, induce the development of different antigen-specific T cell populations in vitro.26 To determine the protective efficacy of these synthesized multifunctionalized PLGA NPs, age-matched BALB/c mice were vaccinated with various nanoformulations, soluble form of sLiAg + MPLA or PBS as indicated in the “Materials and methods” section. Subsequently, 2 weeks after the second booster vaccination, mice were challenged with highly virulent L. infantum promastigotes. Parasite burden was estimated in target visceral organs, the liver and spleen, 2 and 4 months post-challenge by qPCR (Figure 1A). Continuous reduction of parasite burden in both the liver and spleen of PLGA NPs-vaccinated mice was observed at both time points post-challenge. Specifically, mice receiving sLiAg encapsulated in PLGA NPs acquired significantly enhanced resistance to hepatic infection at 2 months (P<0.001), although this did not exceed 50% reduction in peak parasite burden detected in PBS or PLGA control groups (Figure 1B and D), reaching 67% reduction at 4 months post-challenge (Figure 1C and E). Importantly, the co-encapsulation of MPLA or surface modification of PLGA NPs loaded with sLiAg with the p8 peptide resulted in further reduction of parasite burden in the liver 2 months post-infection as compared to mice vaccinated with PLGA-sLiAg (PLGA-sLiAg-MPLA: 2.06±0.44 vs 13.63±0.81×106 parasites/liver, 92% parasite reduction, P<0.001, and p8-PLGA-sLiAg: 1.32±0.12 vs 13.63±0.81×106 parasites/liver, 91% parasite reduction, P<0.001), which was kept stable till 4 months post-challenge. It must be noted that the simultaneous presence of p8 and MPLA in PLGA nanoformulations did not enhance resistance to infection, since a 84% parasite reduction was detected in PBS control mice (P<0.001) (Figure 1B–E).
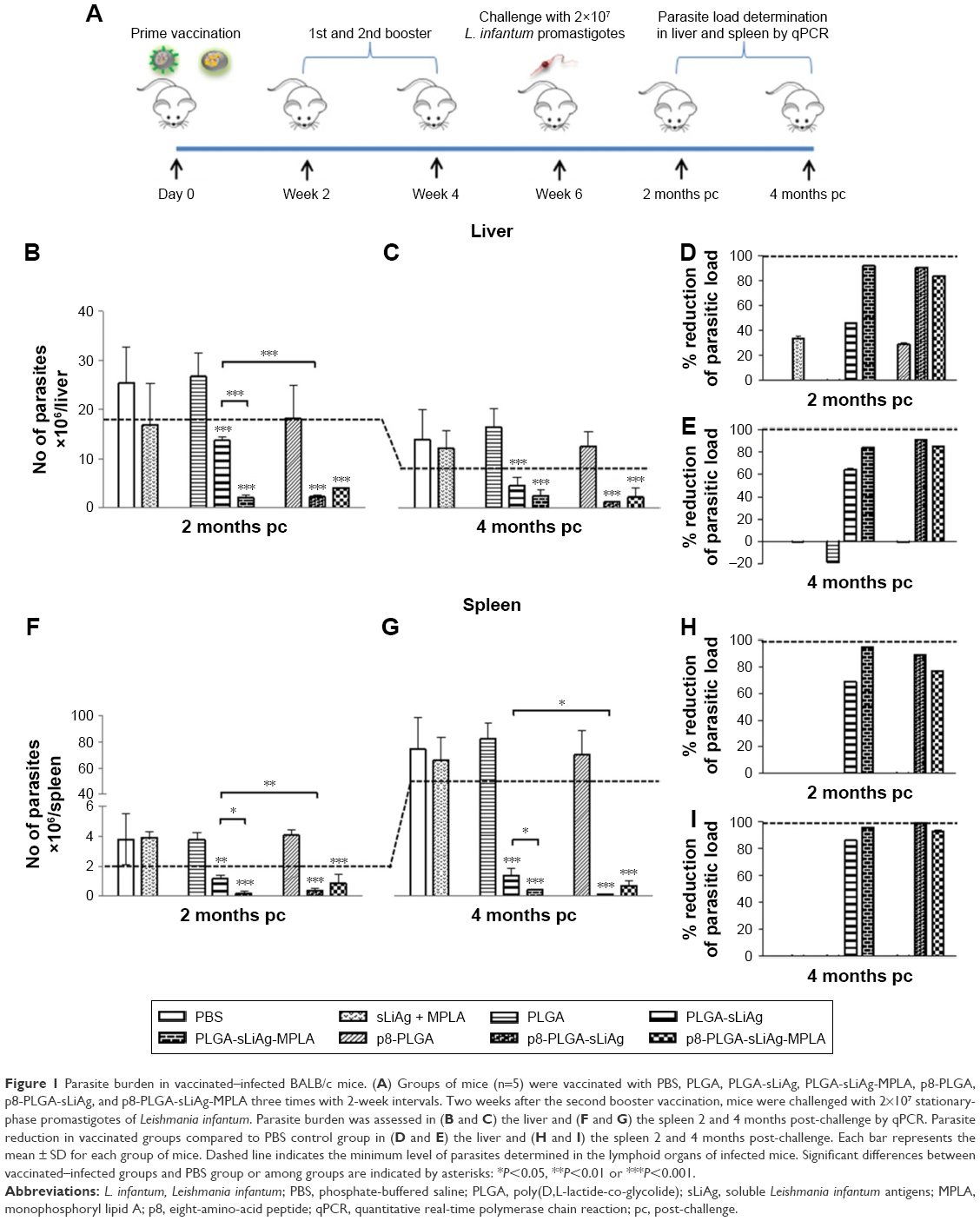 | Figure 1 Parasite burden in vaccinated–infected BALB/c mice. (A) Groups of mice (n=5) were vaccinated with PBS, PLGA, PLGA-sLiAg, PLGA-sLiAg-MPLA, p8-PLGA, p8-PLGA-sLiAg, and p8-PLGA-sLiAg-MPLA three times with 2-week intervals. Two weeks after the second booster vaccination, mice were challenged with 2×107 stationary-phase promastigotes of Leishmania infantum. Parasite burden was assessed in (B and C) the liver and (F and G) the spleen 2 and 4 months post-challenge by qPCR. Parasite reduction in vaccinated groups compared to PBS control group in (D and E) the liver and (H and I) the spleen 2 and 4 months post-challenge. Each bar represents the mean ± SD for each group of mice. Dashed line indicates the minimum level of parasites determined in the lymphoid organs of infected mice. Significant differences between vaccinated–infected groups and PBS group or among groups are indicated by asterisks: *P<0.05, **P<0.01 or ***P<0.001. |
In contrast to naturally acquired resistance to hepatic infection, L. infantum persists in the spleen of BALB/c mice, with the concomitant development of considerable organ-specific pathology similar to that seen in human kala azar. In this aspect, it was important to evaluate the impact of vaccination in this organ. In the spleen, vaccination with PLGA-sLiAg resulted in 69% (P<0.01) reduction of parasite burden at 2 months post-challenge, compared to PBS control group, while encapsulation of MPLA or surface modification of p8 in PLGA NPs (ie, PLGA-sLiAg-MPLA and p8-PLGA-sLiAg) resulted in further reduction of parasitic load as compared to that detected in mice vaccinated with PLGA-sLiAg NPs at 2 months post-challenge, ranging from 66% to 82% (PLGA-sLiAg-MPLA: 0.21±0.15 vs 1.19±0.23×106 parasites/spleen, P<0.05 and p8-PLGA-sLiAg: 0.41±0.13 vs 1.19±0.23×106 parasites/spleen, P<0.01) (Figure 1F and H) (P<0.001). At 4 months post-challenge, when infection is well established, although the parasite load in control infected mice increased, the number of parasites in mice vaccinated with PLGA NPs encapsulating sLiAg remained low reaching 86% of parasite reduction (10.89±3.29 vs 76.06±19.09×106 parasites/spleen, P<0.001). Specifically, it was of interest that vaccination with PLGA-sLiAg-MPLA and p8-PLGA-sLiAg resulted in almost complete elimination of the parasite in the spleen, since protection reached the level of 96% (3.27±0.24 vs 76.06±19.09×106 parasites/spleen, P<0.001) and 99% (0.74±0.45 vs 76.06±19.09×106 parasites/spleen, P<0.001), respectively. Moreover, vaccination with p8-PLGA-sLiAg-MPLA imparted protection from infection, however not at higher levels, that reached 93% (5.25±2.82 vs 76.06±19.09×106 parasites/spleen, P<0.001) (Figure 1G and I). As expected, vaccination with PLGA, p8-PLGA or non-encapsulated sLiAg + MPLA did not affect the levels of parasitic burden which remained high at comparable levels to PBS control mice at both time points.
These data indicated the importance of adjuvant encapsulation or surface modification with the p8 peptide of the selected nanoformulations as reflected in the reduction of parasitic load in both target organs. A confirmatory limiting dilution analysis also verified the presence or absence of live parasites in the liver and spleen of PLGA NPs-vaccinated and infected mice (data not shown).
Vaccination with multifunctionalized PLGA NPs elicited antigen-specific humoral and cellular immune responses
Antibody isotype profile provides a convenient surrogate marker of Th1 and Th2 CD4+ T cell differentiation. We therefore analyzed the response to sLiAg at the end of vaccination and after challenge infection at predetermined time points in the serum obtained from mice vaccinated with PLGA NPs and PBS control mice groups (Figure 2A). As shown in Figure 2, PLGA-sLiAg and PLGA-sLiAg-MPLA were the only vaccination regimens that induced anti-sLiAg antibody responses, which were exclusively of IgG1 isotype (4,800±2,263 and 2,400±1,131 vs 50±0, P<0.001) (Figure 2B and E), suggesting that vaccination with these nanoformulations stimulated predominantly a Th2 response. Interestingly, mice immunized with p8-surface-modified nanoformulations failed to produce any antibody response to sLiAg before challenge.
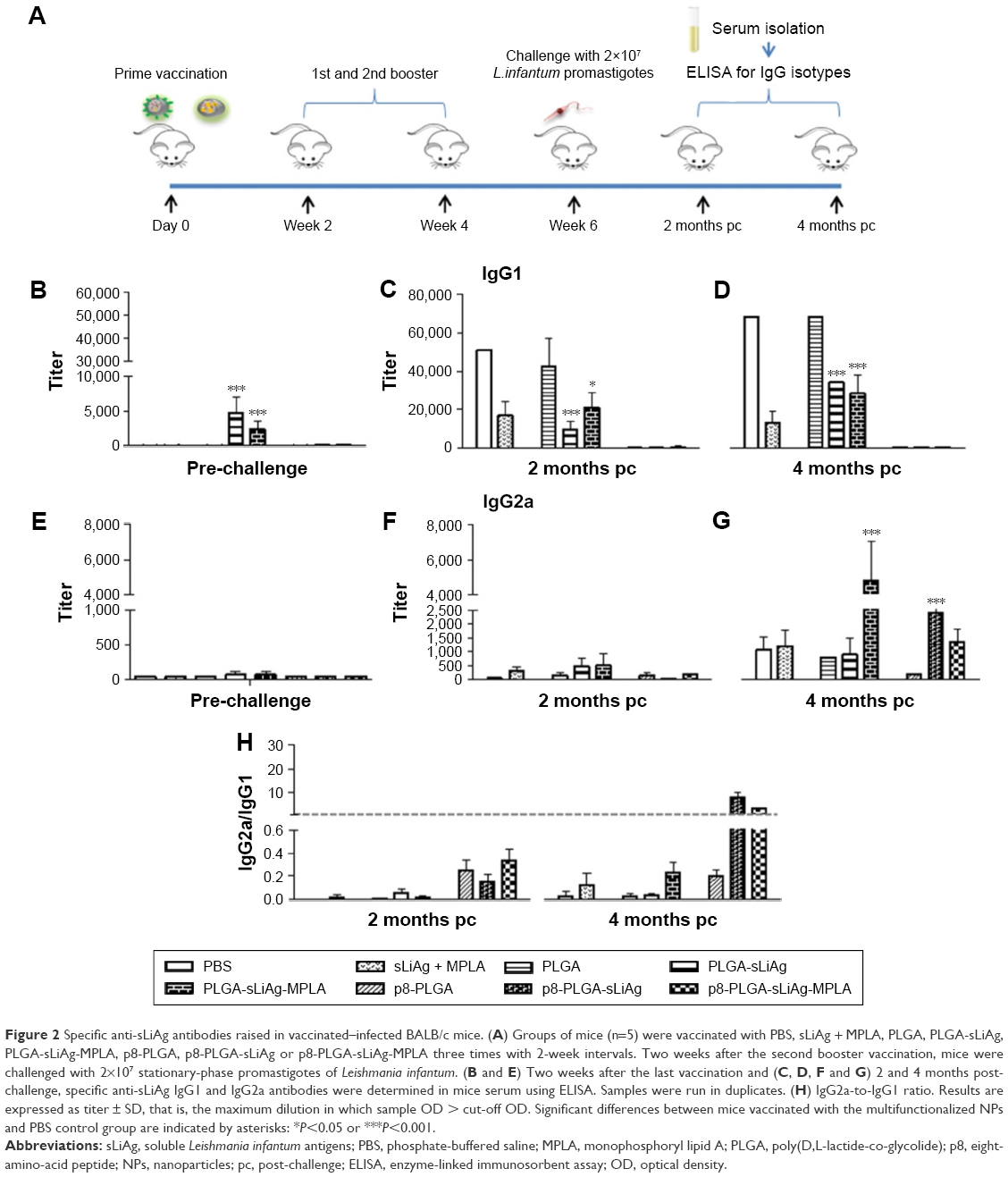 | Figure 2 Specific anti-sLiAg antibodies raised in vaccinated–infected BALB/c mice. (A) Groups of mice (n=5) were vaccinated with PBS, sLiAg + MPLA, PLGA, PLGA-sLiAg, PLGA-sLiAg-MPLA, p8-PLGA, p8-PLGA-sLiAg or p8-PLGA-sLiAg-MPLA three times with 2-week intervals. Two weeks after the second booster vaccination, mice were challenged with 2×107 stationary-phase promastigotes of Leishmania infantum. (B and E) Two weeks after the last vaccination and (C, D, F and G) 2 and 4 months post-challenge, specific anti-sLiAg IgG1 and IgG2a antibodies were determined in mice serum using ELISA. Samples were run in duplicates. (H) IgG2a-to-IgG1 ratio. Results are expressed as titer ± SD, that is, the maximum dilution in which sample OD > cut-off OD. Significant differences between mice vaccinated with the multifunctionalized NPs and PBS control group are indicated by asterisks: *P<0.05 or ***P<0.001. |
Following challenge with L. infantum, there was a small change in the isotype profile in mice vaccinated with PLGA-sLiAg or PLGA-sLiAg-MPLA, with an overall dominance of IgG1 production at 2 months post-challenge, which however was significantly lower to that detected in control mice groups (9,600±4,525 vs 51,200±0, P<0.001 and 25,600±0 vs 51,200±0, P<0.05) (Figure 2C and F). At 4 months, the levels remained stable and still significantly lower than those detected in PBS control group (25,600±0 vs 51,200±0, P<0.001 and 21,333±7,390 vs 51,200±0, P<0.001) (Figure 2D) followed by a substantial increase of sLiAg-specific IgG2a production (PLGA-sLiAg: 900±611 vs 1,067±462 and PLGA-sLiAg-MPLA: 4,800±2,263 vs 1,067±462, P<0.001) (Figure 2G), further suggesting an isotype switching towards Th1 response. The increase of IgG2a antibody production as compared to PBS control mice correlated with the decreasing parasite load in the respective vaccinated mice groups. Moreover, mice vaccinated with the p8-surface-modified NPs continued not to produce sLiAg-specific IgG1 antibodies till the end of the study (4 months post-challenge). Importantly, at this time point, an exclusive, probably Th1-driven IgG2a response towards sLiAg, although at low levels, was detected, which in the case of p8-PLGA-sLiAg-vaccinated mice was significantly enhanced as compared to PBS control mice groups (2,400±1,131 vs 1,067±462, P<0.001) (Figure 2H). Specific anti-sLiAg IgG1 and IgG2a levels induced in control mice groups vaccinated with PLGA or p8-PLGA, pre- or post-challenge, were comparable with those induced in mice vaccinated with PBS.
As the induction of a strong IgG1 response post-vaccination was unexpected, we directly examined the cellular immune responses raised by PLGA NPs vaccination in vitro (Figure 3A). To understand the nature of immune response generated by PLGA NPs vaccination, we firstly assessed the presence of sLiAg-specific cell-mediated responses. According to conventional recall assays, spleen cells isolated from mice vaccinated with PLGA-sLiAg exhibited significantly higher proliferation levels in response to sLiAg as compared to non-vaccinated mice, 2 weeks after the last vaccination (14.00±0.42 vs 1.00±0.01, P<0.05). Moreover, vaccination with PLGA-sLiAg-MPLA resulted in even higher lymphoproliferative responses, as stimulation index value was about twofold higher (22.25±5.02, P<0.001) over the PLGA-sLiAg-vaccinated mice group. Splenocytes obtained from mice vaccinated with the p8-PLGA-sLiAg (11.26±4.05 vs 1.00±0.01, P<0.05) or p8-PLGA-sLiAg-MPLA (10.18±2.43 vs 1.00±0.01, P<0.05) nanoformulations also exhibited a significant proliferation over the PBS control group, which however was at similar levels to that detected in splenocytes obtained from PLGA-sLiAg-vaccinated group (Figure 3B).
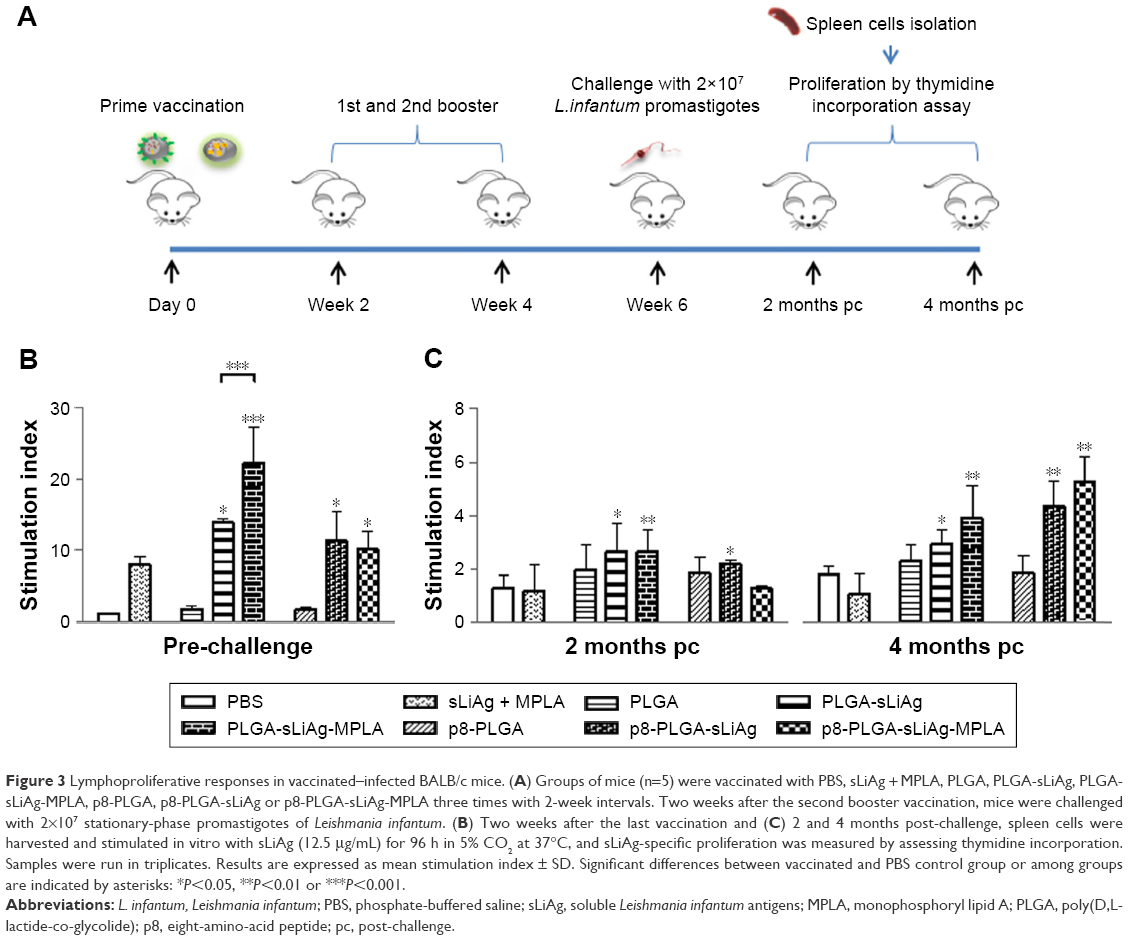 | Figure 3 Lymphoproliferative responses in vaccinated–infected BALB/c mice. (A) Groups of mice (n=5) were vaccinated with PBS, sLiAg + MPLA, PLGA, PLGA-sLiAg, PLGA-sLiAg-MPLA, p8-PLGA, p8-PLGA-sLiAg or p8-PLGA-sLiAg-MPLA three times with 2-week intervals. Two weeks after the second booster vaccination, mice were challenged with 2×107 stationary-phase promastigotes of Leishmania infantum. (B) Two weeks after the last vaccination and (C) 2 and 4 months post-challenge, spleen cells were harvested and stimulated in vitro with sLiAg (12.5 μg/mL) for 96 h in 5% CO2 at 37°C, and sLiAg-specific proliferation was measured by assessing thymidine incorporation. Samples were run in triplicates. Results are expressed as mean stimulation index ± SD. Significant differences between vaccinated and PBS control group or among groups are indicated by asterisks: *P<0.05, **P<0.01 or ***P<0.001. |
Since active VL is characterized by impairment of T cell proliferation, an effective antileishmanial vaccine should be able to restore T cell responses. Accordingly, 2 months post-infection, mice vaccinated with the PLGA-sLiAg, PLGA-sLiAg-MPLA or p8-PLGA-sLiAg were able to maintain the significantly high proliferative capacity of splenocytes, almost twofold higher over PBS control group (PLGA-sLiAg: 2.64±1.27 vs 1.27±0.48, P<0.05, PLGA-sLiAg-MPLA: 2.64±0.81 vs 1.27±0.48, P<0.01 and p8-PLGA-sLiAg: 2.19±0.86 vs 1.27±0.48, P<0.05), which was less robust as compared to that detected post-vaccination, since no statistical significant difference was observed among the above groups and PLGA or p8-PLGA control groups, respectively. However, at 4 months post-challenge, a further increase in splenocyte proliferation capacity was detected reaching approximately threefold over PBS control group in mice that were vaccinated with PLGA-sLiAg-MPLA (3.91±1.25 vs 1.81±0.31, P<0.01), p8-PLGA-sLiAg (4.36±0.94 vs 1.81±0.31, P<0.01) or p8-PLGA-sLiAg-MPLA (5.28±0.99 vs 1.81±0.31, P<0.01) and exhibited almost complete elimination of parasite load in the spleen (Figure 3C).
To fully characterize the cellular immune response to sLiAg, we proceeded with intracellular cytokine staining followed by flow cytometry (Figure 4A). According to the results, 2 weeks post-vaccination, the frequency of IFNγ-producing CD4+ T cells was less than 2%, and no differentiation among vaccinated and non-vaccinated mice was observed (Figure 4B and Table 1). The results obtained from analyzing IFNγ-producing CD8+ T cells were quite different. The control, PLGA-sLiAg and PLGA-sLiAg-MPLA group mice had a very low frequency of CD8+ T cells able to produce IFNγ in vitro in response to sLiAg. However, sLiAg stimulation of spleen cells obtained from p8-PLGA-sLiAg and p8-PLGA-sLiAg-MPLA induced significant increase in the number of CD8+ T cells producing IFNγ (Figure 4C and Table 1). Analysis of these populations at 2 months post-challenge showed that PLGA-sLiAg- and PLGA-sLiAg-MPLA-vaccinated mice preserved the same profile regarding IFNγ-producing CD4+ and CD8+ T cells as compared to PBS control mice, a finding that was consistent with the exclusive presence of increased anti-sLiAg IgG1 antibodies during this time period. Importantly, at 4 months post-challenge, a significant increase in the number of IFNγ-producing CD4+ T cells was detected in the spleens of not only PLGA-sLiAg- (3.19%±0.48% vs 1.96%±0.63%, P<0.05) and PLGA-sLiAg-MPLA-vaccinated mice (2.79%±0.39% vs 1.96%±0.63%, P<0.05) but also p8-PLGA-sLiAg-vaccinated mice (2.99%±0.50% vs 1.96%±0.63%, P<0.05), over PBS control mice, consistent with the antibody isotype switching towards IgG2a. It must be noted that the spleens obtained from p8-PLGA-sLiAg − (13.30%±2.54% vs 8.92%±1.05%, P<0.05) and p8-PLGA-sLiAg-MPLA-vaccinated mice (12.22%±2.87% vs 8.92%±1.05%, P<0.05) continued to preserve the significantly enhanced numbers of sLiAg-specific IFNγ-producing CD8+ T cells, which were further increased over those detected at 2 months post-challenge (Figure 5 and Table 1).
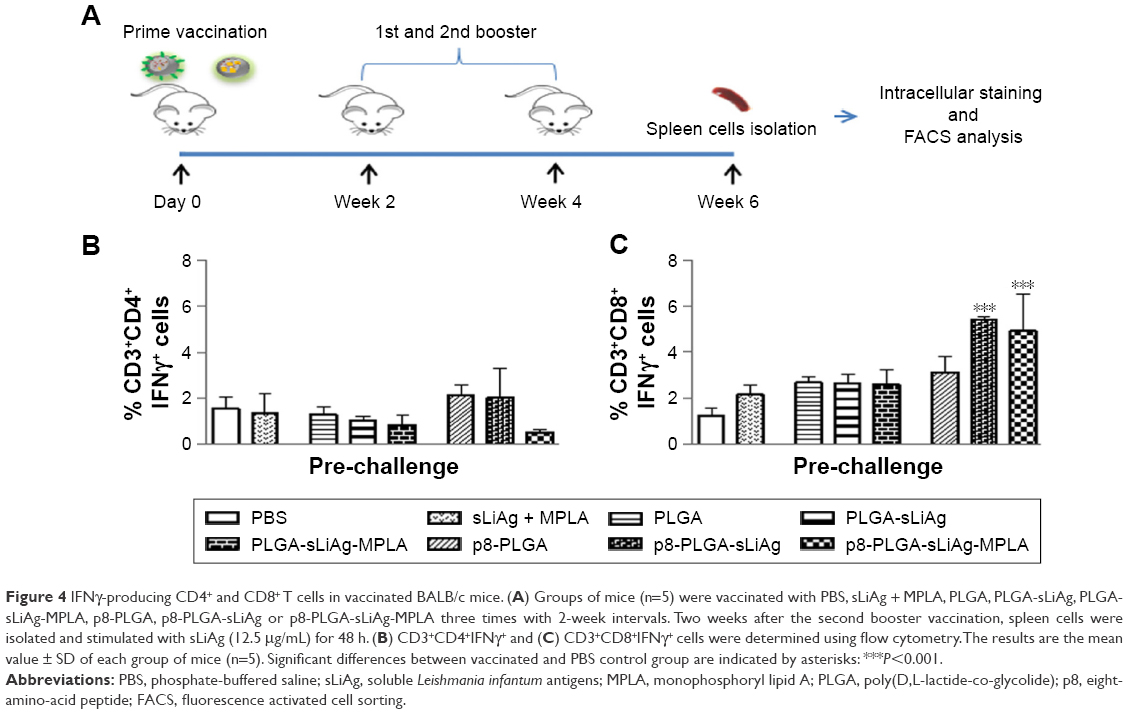 | Figure 4 IFNγ-producing CD4+ and CD8+ T cells in vaccinated BALB/c mice. (A) Groups of mice (n=5) were vaccinated with PBS, sLiAg + MPLA, PLGA, PLGA-sLiAg, PLGA-sLiAg-MPLA, p8-PLGA, p8-PLGA-sLiAg or p8-PLGA-sLiAg-MPLA three times with 2-week intervals. Two weeks after the second booster vaccination, spleen cells were isolated and stimulated with sLiAg (12.5 μg/mL) for 48 h. (B) CD3+CD4+IFNγ+ and (C) CD3+CD8+IFNγ+ cells were determined using flow cytometry. The results are the mean value ± SD of each group of mice (n=5). Significant differences between vaccinated and PBS control group are indicated by asterisks: ***P<0.001. |
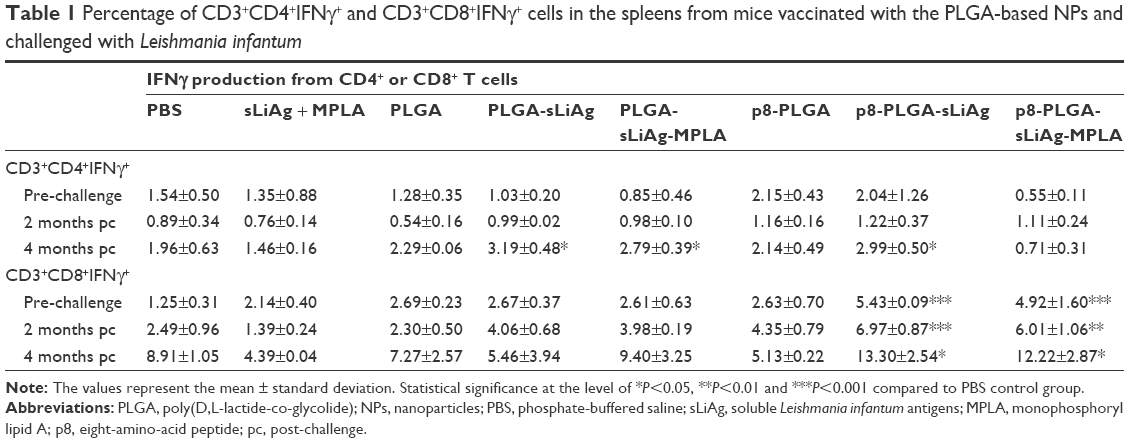 | Table 1 Percentage of CD3+CD4+IFNγ+ and CD3+CD8+IFNγ+ cells in the spleens from mice vaccinated with the PLGA-based NPs and challenged with Leishmania infantum |
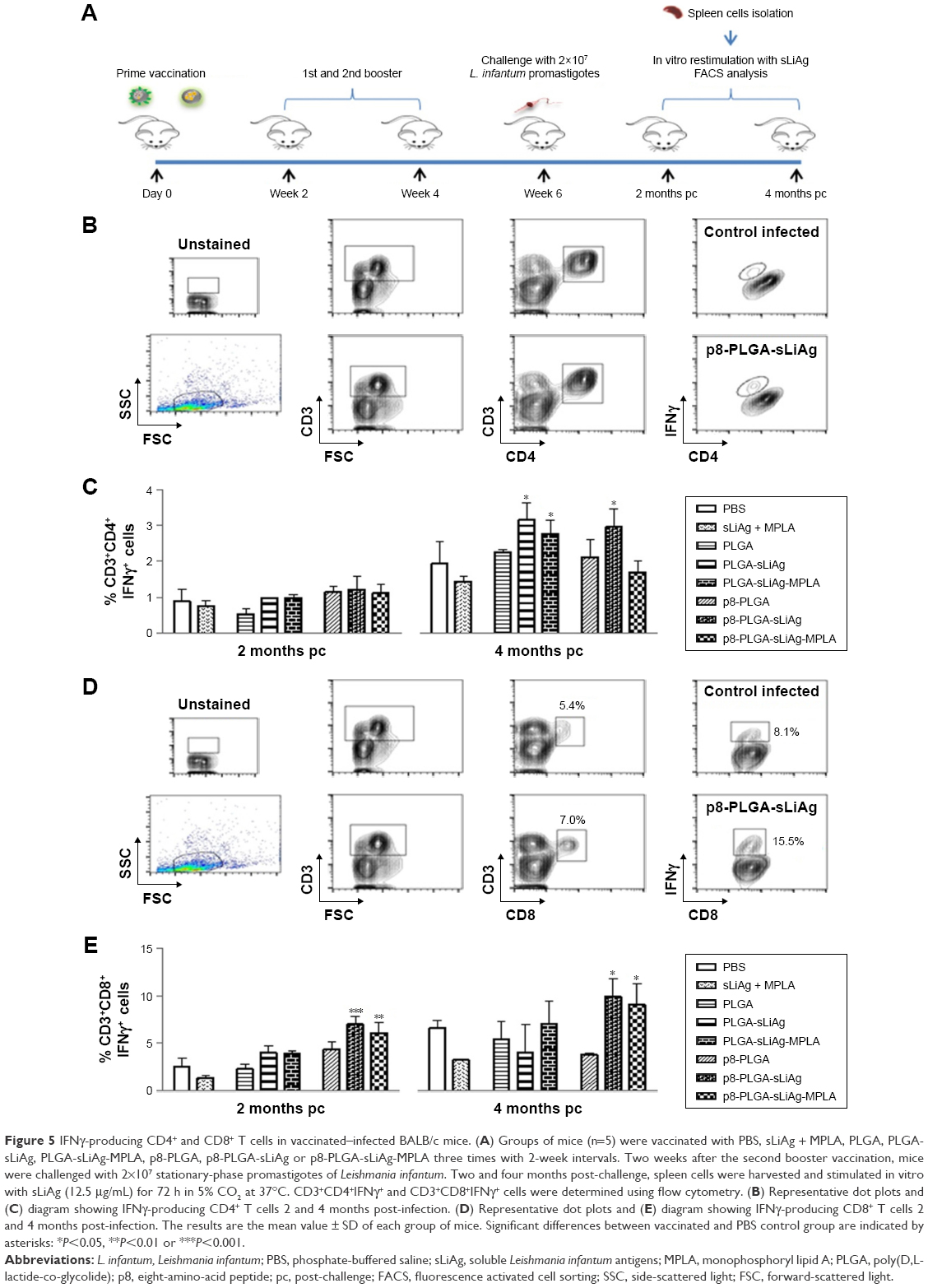 | Figure 5 IFNγ-producing CD4+ and CD8+ T cells in vaccinated–infected BALB/c mice. (A) Groups of mice (n=5) were vaccinated with PBS, sLiAg + MPLA, PLGA, PLGA-sLiAg, PLGA-sLiAg-MPLA, p8-PLGA, p8-PLGA-sLiAg or p8-PLGA-sLiAg-MPLA three times with 2-week intervals. Two weeks after the second booster vaccination, mice were challenged with 2×107 stationary-phase promastigotes of Leishmania infantum. Two and four months post-challenge, spleen cells were harvested and stimulated in vitro with sLiAg (12.5 μg/mL) for 72 h in 5% CO2 at 37°C. CD3+CD4+IFNγ+ and CD3+CD8+IFNγ+ cells were determined using flow cytometry. (B) Representative dot plots and (C) diagram showing IFNγ-producing CD4+ T cells 2 and 4 months post-infection. (D) Representative dot plots and (E) diagram showing IFNγ-producing CD8+ T cells 2 and 4 months post-infection. The results are the mean value ± SD of each group of mice. Significant differences between vaccinated and PBS control group are indicated by asterisks: *P<0.05, **P<0.01 or ***P<0.001. |
It is well known that Th1 immunity is marked by IFNγ secretion, while Th2 cytokines like IL-4 promote humoral responses. Moreover, the dominant production of IFNγ against IL-4 and/or IL-10 plays an important role in the induction of immunity against leishmaniasis. Thus, we compared the levels of the above cytokines in the supernatants of spleen cells restimulated with sLiAg 4 months post-infection in order to gain insight into the cellular mechanisms present at this time point, which are characterized by significant reduction of parasite load in vaccinated mice. sLiAg-recall assays revealed that, in mice vaccinated with PLGA-sLiAg, levels of secreted IFNγ were increased by 38.86% compared to PBS control mice. Moreover, IFNγ production was significantly higher in mice vaccinated with PLGA-sLiAg-MPLA, compared to PBS control mice (134.00±9.53 vs 62.29±25.03 pg/mL, P<0.001). Importantly, the surface modification of PLGA-sLiAg or PLGA-sLiAg-MPLA NPs with the p8 peptide further increased IFNγ production compared to non-modified NPs (p8-PLGA-sLiAg: 175.5±47.4 vs 103.6±7.07 pg/mL and p8-PLGA-sLiAg-MPLA: 171.20±18.16 vs 134.00±9.53 pg/mL, P<0.05) (Table 2).
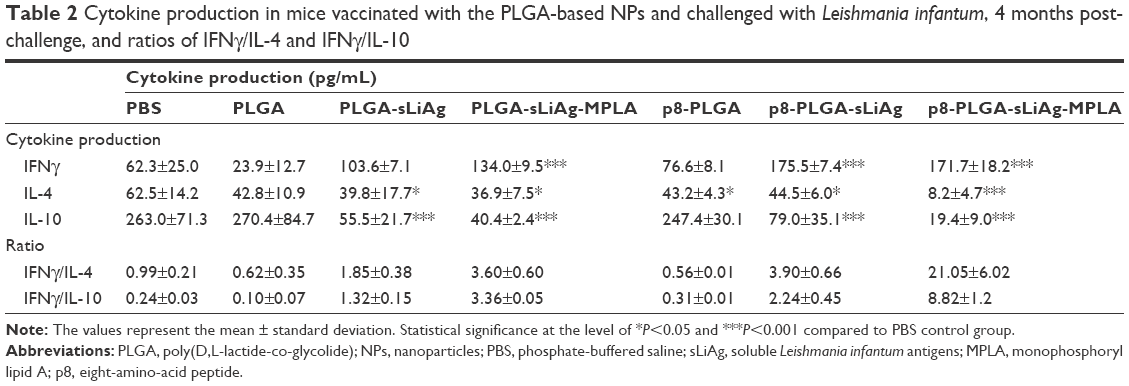 | Table 2 Cytokine production in mice vaccinated with the PLGA-based NPs and challenged with Leishmania infantum, 4 months post-challenge, and ratios of IFNγ/IL-4 and IFNγ/IL-10 |
Furthermore, the spleen cells obtained from mice vaccinated with PLGA-sLiAg, PLGA-sLiAg-MPLA and p8-PLGA-sLiAg showed a decrease in their capacity to produce IL-4 compared to PBS control group, a phenomenon that was enhanced in the spleen cells obtained from p8-PLGA-sLiAg-MPLA-vaccinated mice (PLGA-sLiAg: 39.8±17.7 vs 62.5±14.2 pg/mL, P<0.05, PLGA-sLiAg-MPLA: 37.0±7.5 vs 62.5±14.2 pg/mL, P<0.05, p8-PLGA-sLiAg: 44.5±6.0 vs 62.5±14.2 pg/mL, P<0.05 and p8-PLGA-sLiAg-MPLA: 8.2±4.7 vs 62.5±14.2 pg/mL, P<0.001) (Table 2). Moreover, all PLGA-sLiAg-vaccinated mice groups produced significantly lower IL-10 levels (PLGA-sLiAg: 55.5±21.7 vs 263.0±71.3 pg/mL, P<0.001, PLGA-sLiAg-MPLA: 40.4±2.4 vs 263.0±71.3 pg/mL, P<0.001, p8-PLGA-sLiAg: 79.00±35.1 vs 263.0±71.3 pg/mL, P<0.001 and p8-PLGA-sLiAg-MPLA: 19.4±9.0 vs 263.0±71.3 pg/mL, P<0.001) compared to PBS control mice, irrespective of their modification (Table 2).
Collectively, it could be said that the enhanced production of IFNγ in mice vaccinated with PLGA-sLiAg, PLGA-sLiAg-MPLA, p8-PLGA-sLiAg and p8-PLGA-sLiAg-MPLA outbalanced both IL-4 and IL-10 production compared to control infected mice (Table 2), demonstrating the development of protective cellular immune responses associated with the existence of protective sLiAg-specific CD4+ Th1 and/or CD8+ T cells, followed by secondary specific sero-responses (Figure 2).
Discussion
The induction of sustained immune responses for the lifelong protection of individuals from a variety of diseases is the primary goal of a successful vaccine. Several attempts have been undertaken to develop such a vaccine against leishmaniasis, but there is still no licensed vaccine for humans. To date, deliberate inoculation of live parasites (leishmanization) is the only vaccination strategy capable of achieving long-term protection against the disease. However, observations of adverse side effects30,31 have shifted the focus of vaccine development to subunit vaccines. These vaccines are safer than live vaccines, but they lack strong immunogenicity and thus require the use of an adjuvant.32 Therefore, microparticulate and nanoparticulate formulations are currently considered as the ideal vaccine delivery systems. Although several antigens have been tested for vaccine development, soluble leishmanial antigens are still part of vaccine development research due to their proven immunogenicity, as already mentioned. Immunization of BALB/c mice with sLiAg entrapped in positively or negatively charged liposomal carriers has elicited protection against VL.33–35
In a previous study of ours, we have found that PLGA-based nanoformulations loaded with soluble antigens from late log-phase L. infantum promastigotes, the causative agent of VL, in combination with MPLA as an adjuvant or surface modified with p8 mimicking the TNFα docking region can be taken up more efficiently by DCs, expressing a mature and fully functionalized phenotype. These fully functionalized DCs could present sLiAg and induce the development of antigen-specific Th1 cells.26 The development of the above in vitro system for evaluating the synthesized nanoformulations prompted us to investigate PLGA NPs encapsulating sLiAg and MPLA and surface modified with the p8 peptide for their protective efficacy against experimental VL.
L. infantum parasites multiply faster in the liver during the first 4 weeks of infection; however, self-control of the infection is observed in this organ. By contrast, parasites multiply slower in the spleen; however, infection is persistent in this organ.36 The fact that PLGA and p8-PLGA NPs are taken up by spleen cells after subcutaneous administration26 strengthens the possible role of these NPs as effective vaccine carriers against VL. Indeed, subcutaneous vaccination of susceptible BALB/c mice with PLGA-sLiAg, PLGA-sLiAg-MPLA, p8-PLGA-sLiAg or p8-PLGA-sLiAg-MPLA resulted in significant reduction of the parasitic load 2 and 4 months post-infection both in the liver and spleen. Notably, 4 months post-infection, the level of protection in the liver ranged from 64% to 92%, speeding up the phenomenon of self-healing observed in this organ, while vaccination with PLGA nanoformulations resulted in significant parasite reduction, also, in the spleen, ranging from 44% to 99% (Figure 1). Encapsulation of MPLA in PLGA-sLiAg NPs resulted in significant reduction of parasitic load in accordance with previous studies demonstrating the necessity for an adjuvant for experimental vaccines against leishmaniasis36–40 but also against other intracellular pathogens.41,42 The conjugation of the p8 peptide to PLGA-sLiAg NPs resulted in protection levels similar to those induced by PLGA-sLiAg-MPLA vaccination. Interestingly, vaccination with PLGA-sLiAg-MPLA or p8-PLGA-sLiAg resulted in almost sterile protection in the spleen 4 months post-challenge infection indicating an important role of MPLA encapsulation, as well as for p8 conjugation to nanoformulations.
Higher IgG2a/IgG1 is associated with resistance to leishmaniasis, as it is considered an indicator of enhanced Th1 vs Th2 immune response.15 According to our results, vaccination only with PLGA-sLiAg or PLGA-sLiAg-MPLA raised specific anti-sLiAg IgG1 antibodies (Figure 2), a result which is in accordance with the absence of IFNγ production from CD4+ T cells. Although high IgG1 titers, signature of Th2 response, are associated with susceptibility in VL, previously published results support that early development of Th2 cells holds an important role in the generation of CD8+ T cell memory and contributes to protection against infection in VL models.43,44 Moreover, vaccination with PLGA-sLiAg-MPLA resulted in significantly higher levels of IgG2a antibodies compared to control infected mice, indicating the importance of MPLA in enhancing Th1-type responses (Figure 2). Although protection against leishmaniasis depends more on the cellular than on the humoral immune response, it is worth noting that the skewing IgG subtypes observed at 4 months post-challenge are strong surrogates of protection,45 which indicate the decrease of parasite load.46–48
Protection against leishmaniasis is believed to be dependent upon IL-12 driven production of IFNγ, which drives the immune response towards a Th1-type phenotype. This process is suppressed during infection, and a successful immunization protocol should be able to activate the signal between antigen-presenting cells and T cells. IFNγ, from CD4+ Th1 cells or CD8+ T cells as part of the acquired immune response, mediates macrophage activation, nitric oxide production and parasite killing.49
In different models of leishmaniasis, CD4+ T cell production of IFNγ has been found to be necessary and sufficient for inducing protection,50,51 and some of the earlier studies reported that CD8+ T cells were ineffective in providing efficient control against challenge infection with L. major.52 However, the present perception of CD8+ T cells has changed with significant observations of failure of CD8+ T cell-deficient mice to control parasitic growth suggesting that CD8+ T cells play an important role in immunity against infection with L. major.53–55 There are also data that strongly support the crucial role of CD8+ T cells not only in primary response to infection in experimental VL but also as major mediators of resistance upon reinfection.56–59
In order to analyze the relative contribution of CD4+ and CD8+ IFNγ-producing T cells in protection elicited in our experimental model, the percentage of these populations in spleen cells of vaccinated and infected mice was determined. Vaccination with p8-PLGA-sLiAg or p8-PLGA-sLiAg-MPLA significantly enhanced CD3+CD8+IFNγ+ cells compared to control group. The activation of CD8+ T cells was long-lasting in terms of mice vaccinated with p8-PLGA-sLiAg or p8-PLGA-sLiAg-MPLA contributing to high levels of protection and almost elimination of the parasite. This result is in accordance with previous studies showing that CD8+ T cells play an important role in protection against VL in mice vaccinated with sLiAg encapsulated in liposomes.34,60
PLGA particles loaded with the model antigen OVA promote cross-presentation and the development of CD8+ T cell population.61,62 Additionally, our observations are supported by in vivo studies showing that antigen cross-presentation and the subsequent generation of cytotoxic T cell responses is correlated with PLGA NPs63,64 and particularly with those with size of 300 nm, similar to the size of nanoformulations used in the present study.65 It is believed that PLGA NPs promote cross-presentation of antigens through the mechanism of endosomal escape, that is, NPs’ cytosolic delivery following endocytosis.66 Acidic enviroment in endosomal compartment results in positive charge of PLGA NPs, which possibly facilitates endosomal escape.67 However, even if a particular type of NP promotes a certain pathway of antigen processing, it does not mean the exclusion of the other alternative pathway of antigen presentation.68 Consequently, PLGA-based NPs used in the present study may have presented encapsulated sLiAg through major histocompatibility complex (MHC) class I and II molecules, as shown by both CD4+ and CD8+ T cells activation.
In contrast to observations in murine CL, where a polarized response is sufficient for protection69 and a concomitant Th2 abrogates even strong Th1 function,70,71 a mixed Th1/Th2 response is essential for protection against VL.43,72,73 Studies in mice74 and humans75 indicated that IL-10 and not IL-4 is the major immunosuppressive cytokine in VL and the absence of IL-10 is associated with resistance to infection.76,77 In accordance with the above studies, we showed that in mice vaccinated with PLGA-sLiAg, PLGA-sLiAg-MPLA, p8-PLGA-sLiAg and p8-PLGA-sLiAg-MPLA, significantly lower production of IL-10, compared to control infected mice, was observed proving the important role of this cytokine in protection from VL.
In this context, determination of cytokine levels secreted from spleen cell cultures of vaccinated and infected mice restimulated with sLiAg was conducted. The modification of PLGA-sLiAg and PLGA-sLiAg-MPLA NPs with the p8 resulted in higher production of IFNγ compared to mice vaccinated with non-modified NPs. Along with IFNγ production, a co-existence of IL-4 was observed. However, the ratio of IFNγ/IL-4 production was higher in vaccinated compared to non-vaccinated mice (Table 2) indicating that the balance of Th1/Th2 was in favor of protective responses.
Conclusion
The present study demonstrates that vaccination with the multifunctionalized PLGA NPs encapsulating sLiAg and/or MPLA and/or surface modified with the p8 peptide resulted in activation of IFNγ-producing CD4+ and CD8+ T cells. However, only PLGA-sLiAg-MPLA and p8-PLGA-sLiAg NPs could provide almost sterile protection against infection with L. infantum. The protection observed was attributed to increased production of IFNγ followed by suppression of IL-4 and IL-10 production. It seems that PLGA-sLiAg-MPLA and p8-PLGA-sLiAg can achieve the same result through probably different mechanisms, since solely p8-modified NPs were able to activate CD8+ T cells. This could be an explanation of why MPLA encapsulation in already p8-modified nanoformulations did not further enhance the protection effect. Collectively, the data presented here suggest that the above nanoformulations hold promise for future vaccination strategies against VL.
Acknowledgments
This work was supported by NanoLeish project (09SYN-14-643) within the Cooperation Action of the General Secretariat for Research and Technology, funded by the government of Greece, and the European Regional Development Fund of the European Union under the Operational Programme for Competitiveness and Entrepreneurship, National Strategic Reference Framework 2007–2013, awarded to EK. MM was awarded a fellowship from Hellenic Pasteur Institute.
Disclosure
The authors report no conflicts of interest in this work.
References
Colmenares M, Kar S, Goldsmith-Pestana K, McMahon-Pratt D. Mechanisms of pathogenesis: differences amongst Leishmania species. Trans R Soc Trop Med Hyg. 2002;96 Suppl 1:S3–S7. | ||
Alvar J, Vélez ID, Bern C, et al; WHO Leishmaniasis Control Team. Leishmaniasis worldwide and global estimates of its incidence. PLoS One. 2012;7(5):e35671. | ||
Croft SL, Sundar S, Fairlamb AH. Drug resistance in leishmaniasis. Clin Microbiol Rev. 2006;19(1):111–126. | ||
Santos DO, Coutinho CE, Madeira MF, et al. Leishmaniasis treatment – a challenge that remains: a review. Parasitol Res. 2008;103(1):1–10. | ||
de Oliveira CI, Nascimento IP, Barral A, Soto M, Barral-Netto M. Challenges and perspectives in vaccination against leishmaniasis. Parasitol Int. 2009;58(4):319–324. | ||
Okwor I, Uzonna J. Vaccines and vaccination strategies against human cutaneous leishmaniasis. Hum Vaccin. 2009;5(5):291–301. | ||
Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Préat V. PLGA-based nanoparticles: an overview of biomedical applications. J Control Release. 2012;161(2):505–522. | ||
Hamdy S, Haddadi A, Hung RW, Lavasanifar A. Targeting dendritic cells with nano-particulate PLGA cancer vaccine formulations. Adv Drug Deliv Rev. 2011;63(10–11):943–955. | ||
Jiang W, Gupta RK, Deshpande MC, Schwendeman SP. Biodegradable poly(lactic-co-glycolic acid) microparticles for injectable delivery of vaccine antigens. Adv Drug Deliv Rev. 2005;57(3):391–410. | ||
Goforth R, Salem AK, Zhu X, et al. Immune stimulatory antigen loaded particles combined with depletion of regulatory T-cells induce potent tumor specific immunity in a mouse model of melanoma. Cancer Immunol Immunother. 2009;58(4):517–530. | ||
Ma W, Chen M, Kaushal S, et al. PLGA nanoparticle-mediated delivery of tumor antigenic peptides elicits effective immune responses. Int J Nanomedicine. 2012;7:1475–1487. | ||
Moon JJ, Suh H, Polhemus ME, Ockenhouse CF, Yadava A, Irvine DJ. Antigen-displaying lipid-enveloped PLGA nanoparticles as delivery agents for a Plasmodium vivax malaria vaccine. PLoS One. 2012;7(2):e31472. | ||
Pandit S, Cevher E, Zariwala MG, Somavarapu S, Alpar HO. Enhancement of immune response of HBsAg loaded poly (L-lactic acid) microspheres against hepatitis B through incorporation of alum and chitosan. J Microencapsul. 2007;24(6):539–552. | ||
Thomas C, Rawat A, Hope-Weeks L, Ahsan F. Aerosolized PLA and PLGA nanoparticles enhance humoral, mucosal and cytokine responses to hepatitis B vaccine. Mol Pharm. 2011;8(2):405–415. | ||
Danesh-Bahreini MA, Shokri J, Samiei A, Kamali-Sarvestani E, Barzegar-Jalali M, Mohammadi-Samani S. Nanovaccine for leishmaniasis: preparation of chitosan nanoparticles containing Leishmania superoxide dismutase and evaluation of its immunogenicity in BALB/c mice. Int J Nanomedicine. 2011;6:835–842. | ||
Eskandari F, Talesh GA, Parooie M, et al. Immunoliposomes containing Soluble Leishmania Antigens (SLA) as a novel antigen delivery system in murine model of leishmaniasis. Exp Parasitol. 2014;146:78–86. | ||
Tafaghodi M, Eskandari M, Kharazizadeh M, Khamesipour A, Jaafari MR. Immunization against leishmaniasis by PLGA nanospheres loaded with an experimental autoclaved Leishmania major (ALM) and Quillaja saponins. Trop Biomed. 2010;27(3):639–650. | ||
Tafaghodi M, Khamesipour A, Jaafari MR. Immunization against leishmaniasis by PLGA nanospheres encapsulated with autoclaved Leishmania major (ALM) and CpG-ODN. Parasitol Res. 2011;108(5):1265–1273. | ||
Santos DM, Carneiro MW, de Moura TR, et al. Towards development of novel immunization strategies against leishmaniasis using PLGA nanoparticles loaded with kinetoplastid membrane protein-11. Int J Nanomedicine. 2012;7:2115–2127. | ||
Kaye P, Scott P. Leishmaniasis: complexity at the host-pathogen interface. Nat Rev Microbiol. 2011;9(8):604–615. | ||
Holaday BJ. Role of CD8+ T cells in endogenous interleukin-10 secretion associated with visceral leishmaniasis. Mem Inst Oswaldo Cruz. 2000;95(2):217–220. | ||
McMahon-Pratt D, Alexander J. Does the Leishmania major paradigm of pathogenesis and protection hold for New World cutaneous leishmaniases or the visceral disease? Immunol Rev. 2004;201:206–224. | ||
Murray HW, Berman JD, Davies CR, Saravia NG. Advances in leishmaniasis. Lancet. 2005;366(9496):1561–1577. | ||
Kaye PM, Curry AJ, Blackwell JM. Differential production of Th1- and Th2-derived cytokines does not determine the genetically controlled or vaccine-induced rate of cure in murine visceral leishmaniasis. J Immunol. 1991;146(8):2763–2770. | ||
Melby PC, Yang J, Zhao W, Perez LE, Cheng J. Leishmania donovani p36(LACK) DNA vaccine is highly immunogenic but not protective against experimental visceral leishmaniasis. Infect Immun. 2001;69(8):4719–4725. | ||
Margaroni M, Agallou M, Kontonikola K, et al. PLGA nanoparticles modified with a TNFα mimicking peptide, soluble Leishmania antigens and MPLA induce T cell priming in vitro via dendritic cell functional differentiation. Eur J Pharm Biopharm. 2016;105:18–31. | ||
Gouzelou E, Haralambous C, Antoniou M, et al. Genetic diversity and structure in Leishmania infantum populations from southeastern Europe revealed by microsatellite analysis. Parasit Vectors. 2013;6:342. | ||
Agallou M, Margaroni M, Karagouni E. Cellular vaccination with bone marrow-derived dendritic cells pulsed with a peptide of Leishmania infantum KMP-11 and CpG oligonucleotides induces protection in a murine model of visceral leishmaniasis. Vaccine. 2011;29(31):5053–5064. | ||
Tellevik MG, Muller KE, Løkken KR, Nerland AH. Detection of a broad range of Leishmania species and determination of parasite load of infected mouse by real-time PCR targeting the arginine permease gene AAP3. Acta Trop. 2014;137:99–104. | ||
Coler RN, Reed SG. Second-generation vaccines against leishmaniasis. Trends Parasitol. 2005;21(5):244–249. | ||
Handman E. Leishmaniasis: current status of vaccine development. Clin Microbiol Rev. 2001;14(2):229–243. | ||
Coler RN, Duthie MS, Hofmeyer KA, et al. From mouse to man: safety, immunogenicity and efficacy of a candidate leishmaniasis vaccine LEISH-F3+GLA-SE. Clin Transl Immunology. 2015;4(4):e35. | ||
Bhowmick S, Ravindran R, Ali N. Leishmanial antigens in liposomes promote protective immunity and provide immunotherapy against visceral leishmaniasis via polarized Th1 response. Vaccine. 2007;25(35):6544–6556. | ||
Ravindran R, Maji M, Ali N. Vaccination with liposomal leishmanial antigens adjuvanted with monophosphoryl lipid-trehalose dicorynomycolate (MPL-TDM) confers long-term protection against visceral leishmaniasis through a human administrable route. Mol Pharm. 2012;9(1):59–70. | ||
Thakur A, Kaur H, Kaur S. Studies on the protective efficacy of freeze thawed promastigote antigen of Leishmania donovani along with various adjuvants against visceral leishmaniasis infection in mice. Immunobiology. 2015;220(9):1031–1038. | ||
Bhardwaj S, Vasishta RK, Arora SK. Vaccination with a novel recombinant Leishmania antigen plus MPL provides partial protection against L. donovani challenge in experimental model of visceral leishmaniasis. Exp Parasitol. 2009;121(1):29–37. | ||
Gurunathan S, Wu CY, Freidag BL, Seder RA. DNA vaccines: a key for inducing long-term cellular immunity. Curr Opin Immunol. 2000;12(4):442–447. | ||
Jaafari MR, Badiee A, Khamesipour A, et al. The role of CpG ODN in enhancement of immune response and protection in BALB/c mice immunized with recombinant major surface glycoprotein of Leishmania (rgp63) encapsulated in cationic liposome. Vaccine. 2007;25(32):6107–6117. | ||
Nagill R, Kaur S. Enhanced efficacy and immunogenicity of 78kDa antigen formulated in various adjuvants against murine visceral leishmaniasis. Vaccine. 2010;28(23):4002–4012. | ||
Agallou M, Margaroni M, Athanasiou E, et al. Identification of BALB/c immune markers correlated with a partial protection to Leishmania infantum after vaccination with a rationally designed multi-epitope cysteine protease a peptide-based nanovaccine. PLoS Negl Trop Dis. 2017;11(1):e0005311. | ||
Hensel MT, Marshall JD, Dorwart MR, et al. Prophylactic herpes simplex virus 2 (HSV-2) vaccines adjuvanted with stable emulsion and toll-like receptor 9 agonist induce a robust HSV-2-specific cell-mediated immune response, protect against symptomatic disease, and reduce the latent viral reservoir. J Virol. 2017;91(9).pii:e02257-16. | ||
Song P, He S, Zhou A, et al. Vaccination with toxofilin DNA in combination with an alum-monophosphoryl lipid A mixed adjuvant induces significant protective immunity against Toxoplasma gondii. BMC Infect Dis. 2017;17(1):19. | ||
Stäger S, Smith DF, Kaye PM. Immunization with a recombinant stage-regulated surface protein from Leishmania donovani induces protection against visceral leishmaniasis. J Immunol. 2000;165(12):7064–7071. | ||
Agallou M, Smirlis D, Soteriadou KP, Karagouni E. Vaccination with Leishmania histone H1-pulsed dendritic cells confers protection in murine visceral leishmaniasis. Vaccine. 2012;30(34):5086–5093. | ||
Plotkin SA. Vaccines: correlates of vaccine-induced immunity. Clin Infect Dis. 2008;47(3):401–409. | ||
Nico D, Claser C, Borja-Cabrera GP, et al. Adaptive immunity against Leishmania nucleoside hydrolase maps its c-terminal domain as the target of the CD4+ T cell-driven protective response. PLoS Negl Trop Dis. 2010;4(11):e866. | ||
Nico D, Gomes DC, Alves-Silva MV, et al. Cross-protective immunity to Leishmania amazonensis is mediated by CD4+ and CD8+ epitopes of Leishmania donovani nucleoside hydrolase terminal domains. Front Immunol. 2014;5:189. | ||
Nico D, Gomes DC, Palatnik-de-Sousa I, Morrot A, Palatnik M, Palatnik-de-Sousa CB. Leishmania donovani nucleoside hydrolase terminal domains in cross-protective immunotherapy against Leishmania amazonensis murine infection. Front Immunol. 2014;5:273. | ||
Cunningham AC. Parasitic adaptive mechanisms in infection by leishmania. Exp Mol Pathol. 2002;72(2):132–141. | ||
Heinzel FP, Sadick MD, Mutha SS, Locksley RM. Production of interferon gamma, interleukin 2, interleukin 4, and interleukin 10 by CD4+ lymphocytes in vivo during healing and progressive murine leishmaniasis. Proc Natl Acad Sci U S A. 1991;88(16):7011–7015. | ||
Moll H, Scollay R, Mitchell GF. Resistance to cutaneous leishmaniasis in nude mice injected with L3T4+ T cells but not with Ly-2+ T cells. Immunol Cell Biol. 1988;66(Pt 1):57–63. | ||
Huber M, Timms E, Mak TW, Röllinghoff M, Lohoff M. Effective and long-lasting immunity against the parasite Leishmania major in CD8-deficient mice. Infect Immun. 1998;66(8):3968–3970. | ||
Belkaid Y, Von Stebut E, Mendez S, et al. CD8+ T cells are required for primary immunity in C57BL/6 mice following low-dose, intradermal challenge with Leishmania major. J Immunol. 2002;168(8):3992–4000. | ||
Ruiz JH, Becker I. CD8 cytotoxic T cells in cutaneous leishmaniasis. Parasite Immunol. 2007;29(12):671–678. | ||
Uzonna JE, Joyce KL, Scott P. Low dose Leishmania major promotes a transient T helper cell type 2 response that is down-regulated by interferon gamma-producing CD8+ T cells. J Exp Med. 2004;199(11):1559–1566. | ||
Basu R, Bhaumik S, Haldar AK, et al. Hybrid cell vaccination resolves Leishmania donovani infection by eliciting a strong CD8+ cytotoxic T-lymphocyte response with concomitant suppression of interleukin-10 (IL-10) but not IL-4 or IL-13. Infect Immun. 2007;75(12):5956–5966. | ||
McElrath MJ, Murray HW, Cohn ZA. The dynamics of granuloma formation in experimental visceral leishmaniasis. J Exp Med. 1988;167(6):1927–1937. | ||
Polley R, Stager S, Prickett S, et al. Adoptive immunotherapy against experimental visceral leishmaniasis with CD8+ T cells requires the presence of cognate antigen. Infect Immun. 2006;74(1):773–776. | ||
Tsagozis P, Karagouni E, Dotsika E. CD8(+) T cells with parasite-specific cytotoxic activity and a Tc1 profile of cytokine and chemokine secretion develop in experimental visceral leishmaniasis. Parasite Immunol. 2003;25(11–12):569–579. | ||
Sharma SK, Dube A, Nadeem A, et al. Non PC liposome entrapped promastigote antigens elicit parasite specific CD8+ and CD4+ T-cell immune response and protect hamsters against visceral leishmaniasis. Vaccine. 2006;24(11):1800–1810. | ||
Heit A, Schmitz F, Haas T, Busch DH, Wagner H. Antigen co-encapsulated with adjuvants efficiently drive protective T cell immunity. Eur J Immunol. 2007;37(8):2063–2074. | ||
Silva AL, Rosalia RA, Varypataki E, Sibuea S, Ossendorp F, Jiskoot W. Poly-(lactic-co-glycolic-acid)-based particulate vaccines: particle uptake by dendritic cells is a key parameter for immune activation. Vaccine. 2015;33(7):847–854. | ||
Shen H, Ackerman AL, Cody V, et al. Enhanced and prolonged cross-presentation following endosomal escape of exogenous antigens encapsulated in biodegradable nanoparticles. Immunology. 2006;117(1):78–88. | ||
Zwaveling S, Ferreira Mota SC, Nouta J, et al. Established human papillomavirus type 16-expressing tumors are effectively eradicated following vaccination with long peptides. J Immunol. 2002;169(1):350–358. | ||
Joshi VB, Geary SM, Salem AK. Biodegradable particles as vaccine delivery systems: size matters. AAPS J. 2013;15(1):85–94. | ||
Sneh-Edri H, Likhtenshtein D, Stepensky D. Intracellular targeting of PLGA nanoparticles encapsulating antigenic peptide to the endoplasmic reticulum of dendritic cells and its effect on antigen cross-presentation in vitro. Mol Pharm. 2011;8(4):1266–1275. | ||
Stolnik S, Garnett MC, Davies MC, et al. The colloidal properties of surfactant-free biodegradable nanospheres from poly(β-malic acid-co-benzyl malate)s and poly(lactic acid-co-glycolide). Colloids Surf A. 1995;97(3):235–245. | ||
Zupančič E, Curato C, Paisana M, et al. Rational design of nanoparticles towards targeting antigen-presenting cells and improved T cell priming. J Control Release. 2017;258:182–195. | ||
Scott P, Artis D, Uzonna J, Zaph C. The development of effector and memory T cells in cutaneous leishmaniasis: the implications for vaccine development. Immunol Rev. 2004;201:318–338. | ||
Afonso LC, Scharton TM, Vieira LQ, Wysocka M, Trinchieri G, Scott P. The adjuvant effect of interleukin-12 in a vaccine against Leishmania major. Science. 1994;263(5144):235–237. | ||
Sjölander A, Baldwin TM, Curtis JM, Handman E. Induction of a Th1 immune response and simultaneous lack of activation of a Th2 response are required for generation of immunity to leishmaniasis. J Immunol. 1998;160(8):3949–3957. | ||
Ghosh A, Zhang WW, Matlashewski G. Immunization with A2 protein results in a mixed Th1/Th2 and a humoral response which protects mice against Leishmania donovani infections. Vaccine. 2001;20(1–2):59–66. | ||
Mazumdar T, Anam K, Ali N. A mixed Th1/Th2 response elicited by a liposomal formulation of Leishmania vaccine instructs Th1 responses and resistance to Leishmania donovani in susceptible BALB/c mice. Vaccine. 2004;22(9–10):1162–1171. | ||
Murphy ML, Wille U, Villegas EN, Hunter CA, Farrell JP. IL-10 mediates susceptibility to Leishmania donovani infection. Eur J Immunol. 2001;31(10):2848–2856. | ||
Karp CL, el-Safi SH, Wynn TA, et al. In vivo cytokine profiles in patients with kala-azar. Marked elevation of both interleukin-10 and interferon-gamma. J Clin Invest. 1993;91(4):1644–1648. | ||
Belkaid Y, Hoffmann KF, Mendez S, et al. The role of interleukin (IL)-10 in the persistence of Leishmania major in the skin after healing and the therapeutic potential of anti-IL-10 receptor antibody for sterile cure. J Exp Med. 2001;194(10):1497–1506. | ||
Belkaid Y, Piccirillo CA, Mendez S, Shevach EM, Sacks DL. CD4+CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature. 2002;420(6915):502–507. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.