Back to Journals » The Application of Clinical Genetics » Volume 15
Utility of Measuring Fetal Cavum Septum Pellucidum (CSP) Width During Routine Obstetrical Ultrasound for Improving Diagnosis of 22q11.2 Deletion Syndrome: A Case-Control Study
Authors Pylypjuk CL ![]() , Memon SF
, Memon SF ![]() , Chodirker BN
, Chodirker BN
Received 1 March 2022
Accepted for publication 17 June 2022
Published 26 July 2022 Volume 2022:15 Pages 87—95
DOI https://doi.org/10.2147/TACG.S364543
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Prof. Dr. Martin Maurer
Christy L Pylypjuk,1 Shiza F Memon,2 Bernard N Chodirker3
1Department of Obstetrics, Gynecology and Reproductive Sciences (Section of Maternal-Fetal Medicine), Children’s Hospital Research Institute of Manitoba, University of Manitoba, Winnipeg, MB, Canada; 2Department of Obstetrics, Gynecology & Reproductive Sciences, University of Manitoba, Winnipeg, MB, Canada; 3Departments of Pediatrics and Child Health (Section of Genetics and Metabolism) & Biochemistry and Medical Genetics, University of Manitoba, Winnipeg, MB, Canada
Correspondence: Christy L Pylypjuk, WN5002, HSC Women’s Hospital, 820 Sherbrook Street, Winnipeg, MB, R3A 1R9, Canada, Tel +1 204 787-4821, Fax +1 204 787-2920, Email [email protected]
Objective: To evaluate the utility of measuring fetal cavum septum pellucidum (CSP) width during routine, mid-pregnancy ultrasound for improving diagnosis of 22q11.2 deletion syndrome amongst fetuses with and without conotruncal anomalies.
Patients and Methods: This was a retrospective case-control study (2005– 2016). Fetuses and newborns with 22q11.2 deletion and/or conotruncal cardiac anomalies were identified using a regional, clinical database. A control group was assembled in a 2:1 ratio to create three groups for comparison: i) 22q11.2 deletion syndrome; ii) isolated conotruncal anomalies; and iii) controls. Eligibility was restricted to those with stored ultrasound images between 18– 22 weeks’ gestation and a minimum biparietal diameter of 40 mm. Post-processing measurement of CSP width was performed in a standardized fashion by two blinded and independent study personnel. Descriptive and inferential statistics, regression modeling, and receiver operator curves (ROC) were used to compare outcomes between groups and evaluate sensitivity/specificity of CSP width as a marker of 22q11.2 deletion syndrome.
Results: Twenty-nine cases of 22q11.2 deletion and 64 cases of isolated conotruncal anomalies were matched to 186 healthy controls. Cases with 22q11.2 deletion syndrome had significantly larger CSP widths (5.36 mm; SD=1.2) compared to those with isolated conotruncal anomalies (3.75 mm; SD=1.11) and healthy controls (2.93 mm; SD=0.57; p< 0.0001). There was no difference in CSP width amongst those with 22q11.2 deletion irrespective of the presence/absence of a conotruncal anomaly (p=0.362), or by type of conotruncal anomaly (p=0.211). Using a CSP width cutoff > 4.3 mm, fetuses with 22q11.2 deletion can be accurately identified with good sensitivity (89.7%) and specificity (84%).
Conclusion: Fetuses with 22q11.2 deletion syndrome have dilated CSPs when compared to those with isolated conotruncal anomalies or controls. Because CSP dilation can be evaluated during routine mid-pregnancy ultrasound using standard images of the fetal head, measurement could easily be incorporated to enhance prenatal diagnosis of this phenotypically diverse condition.
Keywords: DiGeorge syndrome, 22q11 microdeletion, cavum septum pellucidum, prenatal diagnosis, fetal ultrasound, neurosonography
Introduction
22q11.2 deletion syndrome is a genetic condition resulting from a deletion in the small arm of chromosome 22.1 Also known as “DiGeorge syndrome”, microdeletion in this region contributes to defective development of the pharyngeal pouch system in early embryonic life and, as such, there are a wide range of clinical phenotypes associated with 22q11.2 deletion syndrome.1–4 Structural abnormalities commonly associated with this condition include conotruncal cardiac defects, thymic hypoplasia, craniofacial abnormalities (including cleft palate), gastrointestinal and genitourinary malformations, poor growth, and even problems with thyroid and parathyroid development.1–3 Postnatally, those with 22q11.2 deletion syndrome can also present with hypocalcemia, immunodeficiency, and failure to thrive.1 Long-term, it is estimated that up to 30% of patients with 22q11.2 deletion are diagnosed with schizophrenia or other mental health concerns by late adolescence/early adulthood.4 There are also described associations with developmental delay, specifically cognitive and language deficits.4,5 22q11.2 microdeletion is present in approximately 1 per 4,000 liveborn infants, however it can be difficult to diagnose prior to birth.1
Almost one-third of patients with 22q11.2 deletion syndrome have an underlying conotruncal cardiac defect such as interrupted aortic arch, truncus arteriosus, tetralogy of Fallot, vascular rings, atrial septal defect (ASD) and ventricular septal defect (VSD), among others.1,2 Because of this association, prenatal diagnosis of 22q11.2 deletion syndrome has relied on testing of amniotic fluid sampled from invasive amniocentesis of fetuses with conotruncal anomalies: molecular testing for 22q11.2 microdeletion (and now microarray) has not traditionally been offered in the absence of conotruncal heart anomalies.6,7 Because the other facial-type anomalies can be difficult to visualize by fetal ultrasound, and the non-anomalous features entirely absent antenatally, it can make prenatal diagnosis of 22q11.2 deletion syndrome very challenging.
A recent study by Chaoui et al8 demonstrated that there is a significant increase in width of the cavum septum pellucidum (CSP) in patients with cardiac anomalies and 22q11.2 deletion syndrome compared to healthy controls. The study was the first to suggest that non-invasive screening for 22q11.2 deletion syndrome could be achieved using obstetrical ultrasound.8 The CSP is a naturally occurring space between the fetal brain hemispheres which is filled by cerebrospinal fluid.9,10 It is visible by 16 weeks of gestation and normally regresses after birth, typically by 3 months of age.9,10 The CSP is standardly imaged during routine, mid-trimester obstetrical ultrasound between 18–22 weeks’ gestation as part of screening for intracranial anomalies.10,11 Evaluation of the CSP by obstetrical ultrasound is limited to its presence or absence – it is not routinely measured. There is some emerging evidence that adults with schizophrenia show a persistence of the CSP into adulthood.12,13 The relationship between fetal CSP and the schizophrenic-phenotype of 22q11.2 deletion syndrome remains unknown, as does the relationship between CSP width and 22q11 microdeletion for fetuses without cardiac anomalies.
The purpose of this study was to evaluate the utility of measuring CSP width during routine obstetrical ultrasound for improving prenatal diagnosis of 22q11.2 deletion syndrome amongst fetuses with and without conotruncal cardiac anomalies. If a relationship exists between CSP width and 22q11.2 deletion syndrome at mid-pregnancy – particularly in the absence of conotruncal anomalies – it would improve prenatal diagnosis of 22q11.2 deletion syndrome without conferring any additional costs or resources for the healthcare system. Such a noninvasive marker would also improve diagnosis for those families opting out of antenatal genetic testing and assist healthcare providers with postnatal management of affected newborns.14
Materials and Methods
This was a retrospective case-control study at a tertiary level hospital in Winnipeg, Canada between January 2005 and December 2016. The hospital serves as a regional referral site for all genetics and pediatric cardiology consultations for a population of over 1.3 million individuals and a geographical region composed of rural, urban, and northern/remote communities. Cases of conotruncal anomalies and 22q11.2 deletion within the region are referred to a centralized clinical genetics program located at the study site. Obstetrical ultrasound images performed within the region since 2005 are stored centrally and available for review electronically. Research ethics approval was obtained from the University of Manitoba Health Research Ethics Board. Patient confidentiality was maintained in compliance with institutional standards and in accordance with the Declaration of Helsinki: because this project was retrospective in nature and did not require any direct patient contact, nor did it influence patient management or outcomes, informed consent forms were not required by our institution. Individual patient consent was obtained for use of the stored ultrasound image of the fetal CSP.
Using a genetics clinical database, all antenatal (fetal) and postnatal (infant) cases of conotruncal cardiac anomalies and/or 22q11.2 deletion syndrome during the study period were ascertained. The database was sequentially searched for 1) fetuses identified on amniocentesis to have a 22q deletion based on either FISH or microarray testing without any evidence of another chromosome anomaly or microdeletion/microduplication, 2) infants diagnosed with a 22q deletion based on either FISH or microarray testing without any evidence of another chromosome anomaly or microdeletion/microduplication, 3) fetuses diagnosed with a conotruncal heart defect (tetralogy of Fallot, interrupted aortic arch, truncus arteriosus, pulmonary atresia, double outlet right ventricle, aberrant right subclavian artery, vascular ring, right sided aortic arch), and 4) infants diagnosed with a conotruncal heart defect. Those designated as “isolated” conotruncal anomalies had no evidence of 22q deletion by either FISH or microarray testing, and had no evidence of another chromosome anomaly by QF-PCR, karyotype or microarray. Cases were reviewed to exclude duplications. The database was then reviewed to ascertain a group of structurally normal, euploid controls matched 2:1 by year of ascertainment (ie, patients who had an amniocentesis resulting in a normal fetal karyotype, QF-PCR, or microarray). Clinic charts were then cross-referenced to ensure accuracy of database information and case selection. Cases were also restricted to singleton pregnancies, and excluded for incorrect dating, lack of stored images, and/or the presence of other anomalies. This resulted in formation of three study groups: i) 22q11.2 deletion syndrome; ii) isolated conotruncal anomalies; and iii) controls. The final list of cases and controls was randomly assorted to ensure blinding of independent observers was maintained during the review of ultrasound images.
Linkage to stored obstetrical ultrasound images was then performed. In keeping with existing literature and the known relationship between CSP width and biparietal diameter, eligibility was restricted to those patients with stored images between 18–22 weeks’ gestation and a minimum biparietal diameter of 40 mm.10 A post-processing review of stored obstetrical ultrasound images and measurements of CSP width was performed by two experienced, independent and blinded study personnel in a standardized fashion as previously described.10 Using standardly obtained fetal head views in the transverse plane at the level of the thalami and symmetric view of both hemispheres, the CSP width was measured by placing the calipers on the inner edges of the lateral borders of the CSP (Figure 1).10 CSP measurements between observers were averaged if within 10% of each other: cases with >10% difference between measurements were planned to be resolved by a third observer, but this was not required. To further evaluate interobserver reliability, CSP widths of controls were compared to previously published normal ranges of CSP during mid-pregnancy scan.10 The gestational age at time of scan and fetal biometry were also collected from stored ultrasound reports and entered into a standardized data collection form.
 |
Figure 1 Fetal cavum septum pellucidum (CSP) with caliper measurements (in yellow) at 20 week ultrasound. |
Statistical analysis was performed using Stata v.14.2 (Stata Corp, College Station, TX) software. Descriptive statistics were used to present study findings. Continuous variables were described as means with standard deviations (SD) or medians with interquartile ranges (IQR) depending on distribution. Dichotomous/categorical variables were described as proportions. CSP width was primarily described as a mean with standard deviation. However, to account for the small sample size per group, CSP widths were also presented graphically using box-and-whisker plots of median and IQR as well. Chi-square, analysis of variance, and Kruskal-Wallis tests were used to compare the three study groups and across subgroups of conotruncal anomalies. Linear regression was used to evaluate the relationship between CSP width and BPD. A receiver operator curve (ROC) was generated to determine the sensitivity and specificity of CSP width in relation to 22q11.2 deletion syndrome status. CSP width was also evaluated as a dichotomous variable separately, by using: i) >95th percentile by biparietal diameter; and ii) the optimal cut-off modeled from the ROC curve. Interobserver reliability was evaluated using the Spearman correlation coefficient.
Results
From the 409 fetuses and infants initially identified in the clinical repository, 134 were excluded after review due to the following factors: incorrect diagnosis, twins/higher-order multiples, inadequate head views to assess CSP, ultrasound performed at gestational age beyond 18–22 weeks’ gestation, biparietal diameter less than 40 mm, or presence of multiple anomalies (Figure 2). Of the remaining 275 cases, three groups were assembled: i) 22q11.2 deletion syndrome (n=29); ii) isolated conotruncal anomalies (n=64); and iii) controls (n=182) (Figure 2). The mean maternal age was 31.6 years (SD=5.6) and similar across all three groups (p=0.603). Most patients (52.7%) were multiparous. There were no obvious differences between groups with respect to mean gestational age at time of ultrasound (p=0.455) or biparietal diameter (p=0.362). One-third of cases of 22q11 deletion syndrome (9/20) had structurally normal hearts.
 |
Figure 2 Flow diagram of study subjects. Abbreviations: GA, gestational age; US, ultrasound. |
The mean CSP width was significantly larger amongst fetuses with 22q11.2 deletion syndrome (5.36 mm, SD=1.2), compared to those with isolated conotruncal cardiac defects (3.75 mm, SD=1.11) and controls (2.93 mm; SD=0.57; p<0.0001) (Figure 3). These differences persisted amongst fetuses with 22q11.2 deletion syndrome irrespective of the presence (or absence) of a conotruncal defect (5.22 mm; SD=1.4 versus 5.7 mm; SD=1.5; p=0.362) (Figure 4). All fetuses with 22q11.2 deletion syndrome – with or without conotruncal anomalies – had significantly larger CSPs than fetuses with any type of isolated conotruncal anomalies. For fetuses with isolated conotruncal anomalies, CSP widths were similar by subtype of conotruncal anomaly: tetralogy of Fallot 3.6 mm (SD=0.91); double outlet right ventricle 3.96 mm (SD=1.2); other conotruncal lesions 4 mm (SD=1.5); p=0.211) (Figure 4). CSP widths of control patients were comparable to those of previously published CSP widths at ~20 weeks GA.10
 |
Figure 3 Box-and-whisker plot comparison of median CSP width between groups. Abbreviation: CSP, cavum septum pellucidum. |
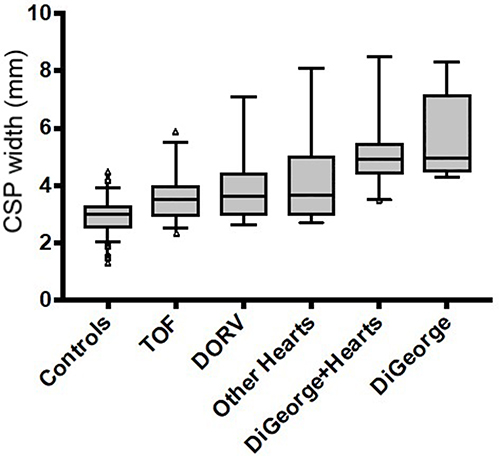 |
Figure 4 Subgroup comparison of median CSP width with box-and-whisker plots. Abbreviations: CSP, cavum septum pellucidum; TOF, tetralogy of Fallot; DORV, double outlet right ventricle. Note: Triangles indicate outliers. |
It was possible to measure CSP width in all 275 study subjects with stored images using standardly obtained views between 18–22 weeks’ gestation, with good interobserver agreement (intraclass correlation=0.92; p<0.0001). There was a positive linear correlation between CSP width and biparietal diameter (r2=0.43; p<0.0001). Using a CSP width cut-off >4.3 mm as modeled from the optimal ROC (area under the curve=0.9181), accurate identification of fetuses with 22q11.2 deletion syndrome is possible with 89.7% sensitivity and 84.4% specificity (Figure 5). Using a cut-off of >95th percentile of CSP width per biparietal diameter, accurate identification of fetuses with 22q11.2 deletion syndrome is possible with sensitivity and specificity of 79.3% and 94.7%, respectively.
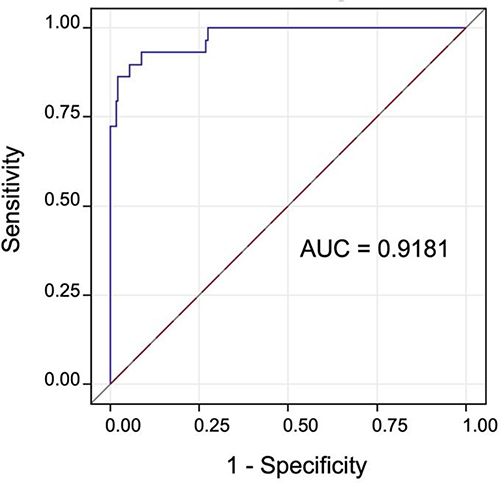 |
Figure 5 ROC curve modeled to represent the diagnostic accuracy of CSP width for 22q11.2 deletion syndrome. Abbreviations: AUC, area under the curve; CSP, cavum septum pellucidum. |
Discussion
Improved prenatal diagnosis of 22q11.2 deletion syndrome is possible. Our study illustrates that by simply adding measurement calipers to standardly obtained images at the time of routine, mid-pregnancy obstetrical ultrasound – and without any additional healthcare costs or resources – accurate identification of fetuses with underlying 22q11 microdeletion can be achieved with good sensitivity and specificity. As mean CSP widths were similar in fetuses that had 22q11.2 deletion syndrome with or without co-existing conotruncal anomalies, the results of this study will be particularly transformative in rethinking the possibility of fetal diagnosis of 22q11.2 deletion syndrome in the absence of anomalies. With the creation of two separate control groups, our study showed that increases in CSP width were independently associated with 22q11 microdeletion and not attributable simply to the presence of an underlying conotruncal anomaly and/or any potential effects of shunting deoxygenated blood on the developing fetal brain.15 It was also interesting to observe that there appeared no difference in CSP widths by type of cardiac anomaly amongst those fetuses with conotruncal defects, which had not before been described in the literature.
The mean CSP width of our healthy controls and positive linear relationship between CSP and biparietal diameter are consistent with previously published studies – both findings attesting to the reliability, reproducibility, and case selection of our study. Similarly to the study published by Chauoi et al,8 CSP width was greater amongst fetuses with 22q11.2 deletion syndrome compared to controls; however, their group did not include evaluation of fetuses with isolated conotruncal defects. In that study, the CSP width amongst fetuses with 22q11.2 deletion syndrome (all of whom had co-existing cardiac anomalies) averaged 5.6 mm and thus was similar with the fetal cases of 22q11.2 deletion syndrome in our study (5.36 mm). However, in that study all cases of 22q11.2 deletion syndrome had conotruncal anomalies and there was no description of CSP width for patients with 22q11.2 deletion syndrome and structurally normal hearts. Additionally, ultrasounds of normal controls spanned 16–34 weeks’ gestation and did not evaluate the utility of CSP measurement specifically at the time of routine obstetrical ultrasound, ~18–22 weeks’ gestation.8 Interestingly, the Chaoui group described how CSP dilation was more obvious and occurred more frequently at later gestational ages beyond 22 weeks in cases of 22q11.2 deletion syndrome (65.7% of fetuses <22 weeks versus 85.7% of fetuses >22–34 weeks).8 Persistence of CSP dilation or relative change in width over time may represent another sonographic marker to enhance fetal diagnosis of 22q11.2 deletion syndrome and warrants further study.
Because only one-third of those affected by 22q11.2 deletion syndrome actually have a cardiac anomaly, it means that two-thirds of cases are being missed by current prenatal screening algorithms. One recent study illustrated that ~95% of prenatally diagnosed cases of 22q11.2 deletion syndrome occur in the setting of cardiac anomalies.16 Historically, confirmatory diagnosis of 22q11.2 deletion syndrome antenatally centered around molecular testing of amniotic fluid obtained by amniocentesis: more recently, chromosomal microarray has further expanded prenatal detection of 22q11 microdeletion.6,7 With modern advances in non-invasive prenatal testing (NIPT) using cell-free fetal DNA, screening for 22q11 microdeletion is now possible using a peripheral maternal blood sample.17 However, many families decline invasive testing by amniocentesis, and others still decline any type of genetic testing antenatally (including NIPT), thus highlighting the need for potential sonographic markers to improve prenatal diagnosis of this condition. For families who prefer to defer genetic testing to the postnatal period, there remains a delay in diagnosis of 22q11.2 microdeletion at a crucial time where neonatal therapy may be important.3 Because 22q11.2 deletion syndrome can be associated with critical hypocalcemia, any delay in recognizing underlying 22q11 microdeletion – especially in those newborns without anomalies who are discharged from hospital early – could be life-threatening.1,3 One study from France emphasized the magnitude of the issue of under-diagnosis of 22q11.2 deletion syndrome.14 That paper described a mean age of diagnosis of 22q11.2 deletion syndrome over 9 years of age.14 Many of those patients were eventually diagnosed due to minor congenital abnormalities, dysmorphisms, hypocalcemia, and intellectual delay. Not surprisingly, patients with structural anomalies were diagnosed earlier (average age 2.5 years old) compared to those without any anomalies, further underscoring the need for non-invasive markers that could be used for screening in the general population.14 More than one in five parents were diagnosed in adulthood following a diagnosis of 22q11.2 deletion syndrome in their offspring.14 In addition to CSP width, there are other fetal ultrasound markers that could be used to improve prenatal diagnosis of 22q11.2 deletion syndrome.16,18
It is well known that individuals with 22q11.2 deletion syndrome are at increased risk of developing schizophrenia and other neuropsychiatric complications in late adolescence and adulthood.4,12–14 In fact, almost one-third of patients with 22q11 microdeletion will develop schizophrenia during their lifetime.4,20 Like the other features of 22q11.2 deletion syndrome, the neuropsychiatric phenotype is also highly variable.19–22 The psychiatric manifestations of 22q11.2 deletion syndrome are also known to change across the lifespan of those individuals affected.20,21 According to Trzesniak et al, adults with schizophrenic phenotype of 22q11.2 deletion syndrome have a persistent and widened CSP.12 However, counseling about longer term neurodevelopmental outcomes is less frequently incorporated into discussion with families at the time of a prenatal diagnosis of 22q11.2 deletion syndrome.23–25 In one survey of parents of children with 22q11.2 deletion syndrome, most parents recalled receiving information about the medical risks of 22q11 microdeletion but none remembered receiving any information about psychiatric risk;21 yet parents in that study almost universally stated that caring for their child with 22q11.2 deletion syndrome and psychotic illness was their greatest source of anxiety and stress.21,24,25 Educational achievement is also highly variable amongst children with 22q11.2 deletion syndrome.5,22 It seems that most attend mainstream school in early years, but academic struggles tend to start in middle years. Later in life, only about one-third of individuals with 22q11.2 deletion syndrome will be employed in the open market versus ~40% unemployed and the remainder receiving employment assistance.22 Comprehensive counseling about all aspects of 22q11.2 deletion syndrome along with better predictors of long-term outcome and functioning (like CSP width) would aide families in decision-making and improve access to educational and mental health supports to improve developmental trajectories earlier.21,23–25 The correlation between CSP width at mid-pregnancy and long-term neuropsychiatric development is not presently known, and individualized counseling about neurodevelopmental outcomes after cardiac anomalies remains challenging.26
Our study has many important strengths. It is the first one in the literature to include structurally normal fetuses with 22q11.2 deletion syndrome to evaluate CSP width independently from co-existence of a cardiac defect. Our study also highlights that CSP measurement at midpregnancy could improve prenatal diagnosis of 22q11.2 deletion syndrome, particularly in the absence of cardiac anomalies or other structural defects during fetal ultrasound. The potential utility of CSP measurement is further attested to by the strong interobserver reliability between two separate and blinded observers. As a case-control study, there are some inherent limitations. Given the relative rarity with which 22q11.2 deletion syndrome is diagnosed during pregnancy or early infancy – especially in the absence of anomalies – our number of cases is small despite the 10-year study period and use of a regional database. There also exists the possibility of known or unknown covariates confounding results. Another intrinsic limitation of case-control studies pertains to selection bias in sampling. This risk should be minimized by use of a regional database which serves as a central intake for all cases of 22q11.2 deletion syndrome in the catchment area and should reflect as close to a true population mean as possible. While it is the local standard of practice for children with conotruncal cardiac defects to be referred for genetic testing (including 22q11 microdeletion), cases where a formal referral was never made – however unlikely – would be missed by this review. The consistency of CSP widths measured amongst cases of 22q11.2 deletion syndrome in our study with those of other studies provides further confidence in our selection strategy. Future multicenter studies should be conducted to evaluate CSP width and 22q11.2 deletion. Research is also needed to develop and evaluate performance of other ultrasound markers of 22q11.2 deletion syndrome for screening in the general population. Correlation between CSP width and other markers with long-term psychosocial functioning is also needed to improve counseling of families and coordinating community resources to improve outcomes.
Conclusion
Prenatal diagnosis of 22q11.2 deletion syndrome is challenging in the general population, particularly in the absence of fetal cardiac anomalies. Measurement of CSP width using standardly obtained obstetrical ultrasound images can accurately detect fetuses with 22q11.2 deletion with great specificity and sensitivity. Beyond diagnosis, future studies are needed to evaluate the relationship between fetal CSP width and neurodevelopment/psychiatric outcomes to better prognosticate long-term outcomes in this highly phenotypically-variable condition.
Acknowledgments
Thank you to Marie Hadfield for assistance in data collection and the staff in our local fetal assessment units for obtaining the quality ultrasound images that supported this project.
Disclosure
The authors report no conflicts of interest in this work. Dr. Christy Pylypjuk has received grants from the Manitoba Medical Services Foundation and Children’s Hospital Research Institute of Manitoba, Health Sciences Centre Foundation, University of Manitoba, and the Winnipeg Foundation Martha Donovan Women’s Leadership Award, as well as transportation and lodging to speak at the annual conference of the Society of Obstetricians and Gynecologists of Canada (no direct payments were received), outside of the submitted work.
References
1. McDonald-McGinn DM, Sullivan KE, Marino B, et al. 22q11.2 deletion syndrome. Nat Rev Dis Primers. 2015;1:15071. doi:10.1038/nrdp.2015.71
2. Ryan AK, Goodship JA, Wilson DI, et al. Spectrum of clinical features associated with interstitial chromosome 22q11 deletions: a European collaborative study. J Med Genet. 1997;34(10):798–804. doi:10.1136/jmg.34.10.798
3. Bassett AS, McDonald-McGinn DM, Devriendt K, et al. Practical guidelines for managing patients with 22q11.2 deletion syndrome. J Pediatr. 2011;159(2):332–9.e1. doi:10.1016/j.jpeds.2011.02.039
4. Qin X, Chen J, Zhou T. 22q11.2 deletion syndrome and schizophrenia. Acta Biochim Biophys Sin. 2020;52(11):1181–1190. doi:10.1093/abbs/gmaa113
5. Duijff SN, Klaassen PW, de Veye HF, Beemer FA, Sinnema G, Vorstman JA. Cognitive development in children with 22q11.2 deletion syndrome. Br J Psychiatry. 2012;200(6):462–468. PMID: 22661678. doi:10.1192/bjp.bp.111.097139
6. Wapner RJ, Martin CL, Levy B, et al. Chromosomal microarray versus karyotyping for prenatal diagnosis. N Engl J Med. 2012;367:2175–2184. doi:10.1056/NEJMoa1203382
7. Grati FR, Molina Gomes D, Ferreira JC, et al. Prevalence of recurrent pathogenic microdeletions and microduplications in over 9500 pregnancies. Prenat Diagn. 2015;35:801–809. doi:10.1002/pd.4613
8. Chaoui R, Heling K, Zhao Y, et al. Dilated cavum septi pellucidi in fetuses with microdeletion 22q11. Prenatal Diagn. 2016;36:911–915. doi:10.1002/pd.4911
9. Winter TC, Kennedy AM, Byrne J, Woodward PJ. The cavum septi pellucidi: why is it important? J Ultrasound Med. 2010;29(3):427–444. PMID: 20194938.. doi:10.7863/jum.2010.29.3.427
10. Abele H, Babiy-Pachomow O, Sonek J, et al. The cavum septi pellucidi in euploid and aneuploid fetuses. Ultrasound Obstet Gynecol. 2013;42:156–160. doi:10.1002/uog.12393
11. Salomon LJ, Alfirevic Z, Berghella C, et al. Practice guidelines for performance of the routine mid-trimester fetal ultrasound scan. ISUOG. 2010. doi:10.1002/uog.8831
12. Trzesniak C, Oliveira IR, Kempton MJ, et al. Are cavum septum pellucidum abnormalities more common in schizophrenia spectrum disorders? A systematic review and meta-analysis. Schizophr Res. 2011;125:1–12. doi:10.1016/j.schres.2010.09.016
13. Murphy KC, Jones LA, Owen MJ. High rates of schizophrenia in adults with velocardio-facial syndrome. Arch Gen Psychiatry. 1999;56(10):940–945. doi:10.1001/archpsyc.56.10.940
14. Ingrao T, Lambert L, Valduga M, et al. Syndrome de microdélétion 22q11.2: analyse du parcours patient avant le diagnostic génétique [22q11.2 microdeletion syndrome: analysis of the care pathway before the genetic diagnosis]. Arch Pediatr. 2017;24(11):1067–1075. French. doi:10.1016/j.arcped.2017.08.017
15. Peyvandi S, Donofrio MT. Circulatory changes and cerebral blood flow and oxygegnation during transition in newborns with congenital heart disease. Semin Pediatr Neurol. 2018;28:38–47. doi:10.1016/j.spen.2018.05.005
16. Schindewolf E, Khalek N, Johnson MP, et al. Expanding the fetal phenotype: prenatal sonographic findings and perinatal outcomes in a cohort of patients with a confirmed 22q11.2 deletion syndrome. Am J Med Genet A. 2018;176(8):1735–1741. PMID: 30055034; PMCID: PMC6467263. doi:10.1002/ajmg.a.38665
17. Dugoff L, Mennuti MT, McDonald-McGinn DM. The benefits and limitations of cell-free DNA screening for 22q11.2 deletion syndrome. Prenat Diagn. 2017;37(1):53–60. PMID: 27329064. doi:10.1002/pd.4864
18. Chaoui R, Heling KS, Lopez AS, Thiel G, Karl K. The thymic-thoracic ratio in fetal heart defects: a simple way to identify fetuses at high risk for microdeletion 22q11. Ultrasound Obstet Gynecol. 2011;37:397–403. PMID: 21308838. doi:10.1002/uog.8952
19. Fiksinski AM, Schneider M, Murphy CM, et al. Understanding the pediatric psychiatric phenotype of 22q11.2 deletion syndrome. Am J Med Genet A. 2018;176(10):2182–2191. PMID: 30194907; PMCID: PMC6209526. doi:10.1002/ajmg.a.40387
20. Jonas RK, Montojo CA, Bearden CE. The 22q11.2 deletion syndrome as a window into complex neuropsychiatric disorders over the lifespan. Biol Psychiatry. 2014;75(5):351–360. PMID: 23992925; PMCID: PMC3875621. doi:10.1016/j.biopsych.2013.07.019
21. Hercher L, Bruenner G. Living with a child at risk for psychotic illness: the experience of parents coping with 22q11 deletion syndrome: an exploratory study. Am J Med Genet A. 2008;146A(18):2355–2360. PMID: 18698620. doi:10.1002/ajmg.a.32466
22. Mosheva M, Pouillard V, Fishman Y, et al. Education and employment trajectories from childhood to adulthood in individuals with 22q11.2 deletion syndrome. Eur Child Adolesc Psychiatry. 2019;28(1):31–42. PMID: 29934817. doi:10.1007/s00787-018-1184-2
23. Karas DJ, Costain G, Chow EW, et al. Perceived burden and neuropsychiatric morbidities in adults with 22q11.2 deletion syndrome. J Intellect Disabil Res. 2014;58:198–210. doi:10.1111/j.1365-2788.2012.01639.x
24. Bassett AS, Costain G, Marshall CR. Neuropsychiatric aspects of 22q11.2 deletion syndrome: considerations in the prenatal setting. Prenat Diagn. 2017;37(1):61–69. PMID: 27718271; PMCID: PMC5243851. doi:10.1002/pd.4935
25. Martin N, Mikhaelian M, Cytrynbaum C, et al. 22q11.2 deletion syndrome: attitudes towards disclosing the risk of psychiatric illness. J Genet Couns. 2012;21:825–834. doi:10.1007/s10897-012-9517-7
26. Paladini D, Alfirevic Z, Carvalho J, et al. Prenatal counseling for neurodevelopmental delay in congenital heart disease: results of a worldwide survey of experts’ attitudes advise caution. Ultrasound Obstet Gynecol. 2016;47:667–671. doi:10.1002/uog.15852
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.