Back to Journals » Therapeutics and Clinical Risk Management » Volume 11
USP18 downregulation in peripheral blood mononuclear cells predicts nonresponse to interferon-based triple therapy in patients with chronic hepatitis C, genotype 1: a pilot study
Authors Frankova S, Jirsa M ![]() , Merta D, Neroldova M
, Merta D, Neroldova M ![]() , Urbanek P, Senkerikova R
, Urbanek P, Senkerikova R ![]() , Spicak J, Sperl J
, Spicak J, Sperl J ![]()
Received 8 August 2015
Accepted for publication 15 October 2015
Published 17 December 2015 Volume 2015:11 Pages 1853—1861
DOI https://doi.org/10.2147/TCRM.S94010
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Garry Walsh
Sona Frankova,1 Milan Jirsa,2 Dusan Merta,3 Magdalena Neroldova,2 Petr Urbanek,4 Renata Senkerikova,1 Julius Spicak,1 Jan Sperl1
1Department of Hepatogastroenterology, 2Laboratory of Experimental Hepatology, 3Department of Anesthesiology, Resuscitation and Intensive Care, Institute for Clinical and Experimental Medicine, 4Department of Internal Medicine, Central Military Hospital, First Medical School, Prague, Czech Republic
Background and aims: Patients with advanced liver fibrosis owing to chronic hepatitis C virus genotype 1 represent a difficult-to-treat group even if a protease inhibitor is added to pegylated interferon alpha and ribavirin. Therefore, only patients with a high chance of cure should be treated with interferon-based treatment.
Patients and methods: Expression of IFNG, IFNLR1, and interferon-sensitive genes CXCL9, IFI16, IFI27, ISG15, and USP18 in peripheral blood mononuclear cells was assessed before and during the initial 12 weeks of treatment. The studied group consisted of 26 treatment-experienced patients of average age of 50 years with advanced liver fibrosis compared to seven healthy volunteers. Fourteen patients were treated with pegylated interferon alpha 2b, ribavirin, and boceprevir and 12 patients with telaprevir. The overall sustained virological response (SVR) rate was 69% (18/26).
Results: A significant difference in the initial expression (median, interquartile range [IQR]) of CXCL9 2.9×, IQR: 1.7–12.4 vs 1.2×, IQR: 0.5–1.8; (P=0.01) IFNG 7.3×, IQR: 1.7–32.6 vs 0.7×, IQR: 0.4–1.3; P=0.002 and USP18 3.7×, IQR: 2.1–7.7 vs 1.4×, IQR: 0.9–1.6; (P=0.03) was found between the SVR and non-SVR groups. Expression of all analyzed genes was progressively increasing during the first 12 weeks of therapy, but a significant difference between SVR and non-SVR group was found only in USP18 expression at week 12 (P=0.001). Initial expression of four genes predicted SVR in univariate analysis (CXCL9 [OR: 12.00, 95% CI: 1.21–118.89], IFI27 [OR: 12.00, 95% CI: 1.21–118.89], IFNG [OR: 10.50, 95% CI: 1.50–73.67], USP18 [OR: 21.00, 95% CI: 2.05–215.18]). In multivariate analysis, only the initial expression of USP18 was identified as a predictor of SVR (P=0.047).
Conclusion: Initial expression of USP18 and the course of its activation could be a reliable predictor of SVR achievement.
Keywords: chronic hepatitis C, interferon-sensitive gene, USP18, protease inhibitor, virological response
Introduction
Hepatitis C virus (HCV) infection represents a leading cause of liver disease.1 Interferon alpha-based therapies, later co-administered with ribavirin (RBV), have been standard-of-care since the beginning of the 1990s, with their nonspecific immunomodulatory effect inducing the clearance of the virus. In the last 4 years, new direct-acting antivirals (DAAs), inhibiting directly the viral enzymes crucial for its replication, have entered into clinical practice, together with pegylated interferon alpha and RBV (P/R) or in different combinations as interferon-free regimens. Nevertheless, interferon-based therapies have remained standard-of-care in many countries and represent a huge proportion of therapies administered worldwide.
The interferon-induced HCV clearance is a complex, immune-mediated process.2 A detailed clarification of the mechanism of viral elimination is still lacking, nevertheless, previous studies described the induction of many interferon-sensitive genes (ISG) in hepatocytes and other cells following interferon alpha administration.3 Chen et al4 first described the differences in ISG activation in the liver in subjects prone to HCV elimination in comparison with nonresponders to therapy. HCV infection modifies the baseline expression profiles of ISG compared with healthy subjects both in hepatocytes and peripheral blood mononuclear cells (PBMC),4–7 and the course of the ISG expression profiles is most likely unfavorable with persistence of the chronic HCV infection. Clinical data are in accordance with this hypothesis; the capacity to eliminate the virus with interferon alpha and RBV is reduced in the course of the disease. It decreases with the age of the patient and with fibrosis progression, and both these factors are influenced by the duration of the infection.8 In the livers of nonresponders, long-term exposed to the virus, ISGs are upregulated and are not prone to further stimulation by interferon alpha.9 Asselah et al5 proved a significantly higher baseline liver ISG activity in nonresponders in comparison with responders to therapy. Based on the activity of selected ISGs in the liver, they were able to predict sustained virological response (SVR) with 94% probability. Chen et al10 investigated the ISG baseline expression profile in hepatocytes and liver macrophages in the same individuals, and found a difference. The group of nonresponders was characterized by higher ISG expression in hepatocytes and lower in macrophages when compared with responders to P/R therapy. Furthermore, they confirmed the principal importance of ISG15 and USP18 in the specific activation of interferon alpha anti-HCV pathway. Hou et al6 described a reliable prediction of elimination of the virus based on the expression profiles of 18 ISGs investigated in peripheral blood. On the contrary, Taylor et al11 investigated relative changes in PBMC ISGs after stimulation with pegylated interferon alpha and did not find any significant changes between responders and nonresponders. Taylor’s results describe upregulation of ISGs in PBMC, contrary to MacParland et al, who, when comparing pretreatment ISG expression levels to healthy volunteers, found upregulated as well as downregulated genes in PBMC prior to interferon therapy.12
The DAAs inhibit replication of HCV directly by targeting the virus replication cycle, their mechanism of action is not immune-mediated. The first DAAs, first-generation protease inhibitors (boceprevir [BOC] and telaprevir [TVR]), must be administered in combination with P/R. This triple therapy is more effective compared with P/R combination, but its efficacy in the patients with advanced fibrosis and cirrhosis is still unsatisfactory.
The aim of our study was to clarify whether irresponsiveness to interferon-based triple therapy in patients with advanced liver disease depends on the unfavorable pretreatment ISG expression profile and whether we can predict SVR achievement based on the pretreatment expression levels of ISG, IFNG, and IFNLR1 or on the expression variation of these genes during the first 12 weeks of therapy.
With respect to the above-described data, PBMC were used to investigate gene expression because their acquisition was considered easier and safer than the acquisition of liver tissue, especially when analyzing the expression profile in different time points during treatment.
Patients and methods
Study design and eligibility of patients
A total of 26 patients with advanced liver fibrosis (Metavir score ≥ F3) were treated for chronic hepatitis C in two outpatient specialty clinics in Prague, Czech Republic, from December 2011 to April 2014. The cohort consisted of 17 males and 9 females of average age of 50 years (range 30–62). All patients were Caucasians infected with genotype 1 (24 patients with subtype 1b, 2 patients with subtype 1a) and treatment-experienced (all had been treated previously at least once with P/R, 16 were nonresponders and 10 relapsers), distribution of IL28B genotypes was as follows: CC 2/26, CT 16/26, and TT 8/26. Pretreatment liver biopsy was performed in all patients, out of whom eleven had fibrosis F3 and 15 had fibrosis F4 according to the Metavir score. All patients had compensated liver disease with no signs of proteosynthetic dysfunction (normal albumin, bilirubin, and prothrombin time values), ascites or encephalopathy. Patients with history of liver disease decompensation, hepatitis B infection or HIV co-infection, and patients receiving any immunosuppressive or immunomodulation therapy at the time of treatment initiation were not included in the study.
Fourteen patients were treated with once weekly subcutaneously administered pegylated interferon alpha 2b together with weight-adjusted RBV 1,000–1,200 mg daily, BOC (total daily dose 2,400 mg) was added at week 4 after a lead-in phase. Twelve patients were treated with once weekly subcutaneously administered pegylated interferon alpha 2a together with weight-adjusted RBV 1,000–1,200 mg daily and TVR (total daily dose 2,250 mg). The anticipated treatment duration in both groups was 48 weeks in all patients. Virological futility rules were applied at treatment weeks 4, 8, and 12 according to prescribing information of both DAAs. No patient who discontinued therapy owing to an adverse event was included in the final evaluation. HCV RNA was assessed at weeks 4, 8, 12, 24, 36, and 48. SVR was assessed as HCV RNA negativity 24 weeks posttreatment. Blood draws for PBMC isolation were performed at day 0 and treatment weeks 4, 8, and 12 in both groups.
The control group consisted of seven healthy control subjects (two men, five women) of mean age of 42 years. The control subjects did not have any history of liver disease. All had normal activity of alanine aminotransferase, negative serological markers of hepatitis B infection and negative anti-HCV antibodies and HCV RNA.
All study participants provided written consent approved by the Institutional Review Board of the Institute for Clinical and Experimental Medicine and Thomayer’s Hospital (Prague, Czech Republic), with prospective personal data collection, PBMC isolation, and DNA and RNA sampling prior to the initiation of therapy, and the study was approved by the Institutional Review Board and conformed to the Declaration of Helsinki Ethical Guidelines.
HCV RNA assessment
HCV RNA was assessed by the Roche AmpliPrep/COBAS® TaqMan® HCV Test v2.0 (Roche Molecular Systems, Branchburg, NJ, USA). Serum HCV RNA levels were determined at baseline, at weeks 4, 12, 24, 36, 48 of treatment and 12 and 24 weeks after the end of therapy.
IL28B genotyping
Patients were genotyped for IL28B rs12979860 C/T polymorphism by polymerase chain reaction – restriction fragment length polymorphism assay, as described by Fabris et al.13
PBMC and mRNA isolation
Blood was collected in ethylenediaminetetraacetic acid 9 mL tubes, diluted with an equal volume (9 mL) of 1% phosphate-buffer and then layered over a 9 mL Lymphoprep gradient (Axis-Shield, Oslo, Norway). The tubes were centrifuged at 1,400 rpm for 30 minutes at 10°C. The buffy-coat layer was transferred to 15 mL RNAse-free tubes, diluted with phosphate-buffered saline and centrifuged at 1,000 rpm at 10°C. The supernatant was discarded and PBMC were retained and ready for RNA isolation. RNA was extracted from PBMC with the RNeasy Mini Kit (Qiagen, Valencia, CA, USA). RNA purity and concentration were assessed by spectrophotometry. mRNA was reverse-transcribed into sscDNA using the RevertAid Premium First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Waltham, MA, USA).
Gene expression analysis
The expression of a set of seven genes was analyzed in PBMC. The set was selected according to the above-cited literature and consisted of CXCL9, IFI16, IFI27, IFNG, IFNLR1, ISG15, and USP18. The genes were characterized by the association with immune response to HCV infection. Real-time PCR was performed using TaqMan® Assays in 384-Well Microfluidic Cards (Applied Biosystems, Foster City, CA, USA). Custom cards were designed with the Custom TaqMan array configuration tool. Real-time PCR was performed in a 7900HT Real-Time PCR System (Thermo Fisher Scientific) according to the manufacturer’s suggested cycling conditions 50°C 2 minutes and 95°C 20 seconds for the initial cycle and 95°C 1 second and 60°C 20 seconds for the following 40 cycles. All samples were measured in quadruplicates; cDNA was prepared from 400 ng of total RNA and TaqMan Fast Advanced Master Mix (4444963, Thermo Fisher Scientific) were used for each assay. The data were analyzed using the SDS 2.2 software. Each 384-well plate included a set of control reactions quantifying expression of three housekeeping genes HPRT1, ACTB, and GAPDH. Gene expression values (Ct) were normalized to the expression of housekeeping genes present in the card (2−ΔCt). Relative quantification of gene expression was carried out by the 2−ΔΔCt method. Final results were expressed as folds of expression medians compared with median expression levels in a group of seven healthy controls; GAPDH was used as the housekeeping gene in the statistical analysis of the cohort.
Statistical analysis
The data are presented as means and standard deviations, medians and ranges or as frequencies, as appropriate. Mann–Whitney test and chi-square test were used for comparisons of the medians and frequencies, respectively. Paired Mann–Whitney test was used to compare the gene expression changes at different time points. Logistic regression was used to determine significant predictors of SVR. Cutoff points for continuous variables were obtained from Receiver Operating Characteristic analysis. P-value <0.05 was considered statistically significant throughout the study. Statistical analysis was performed using the R programming language version 3.2.0 (www.r-project.org).
Results
Treatment outcomes
The overall SVR rate was 69% (18/26), 64% (9/14) in BOC and 75% (9/12) in TVR group. Of the 26 treated patients, eight did not achieve SVR (four null-responders, two breakthroughs, two relapsers). SVR rates according to IL28B genotypes were as follows: CC 1/2 (50%), CT 13/16 (81%), TT 4/8 (50%). In the further analysis, all the patients who did not achieve an SVR were assessed as non-SVR group. The routinely used pretreatment predictors of SVR, ie, age, sex, response to a previous course of therapy, IL28B genotype, HCV RNA viral load, stage of liver fibrosis according to Metavir score (F3 vs F4), and the treatment administered (BOC vs TVR) did not differ significantly in patients who had achieved SVR in comparison with non-SVR group – the data are shown in Table 1.
 | Table 1 Patients’ characteristics |
Initial gene expression
The initial expression of IFNLR1 was decreased in comparison with the group of healthy volunteers whereas expression of the remaining six genes was slightly upregulated compared with healthy controls (Figure 1). A significant difference in the initial expression of CXCL9, IFNG, and USP18 was found between the groups of SVR and non-SVR patients (Table 2). The initial expression of IFI27 2.2×, interquartile range (IQR): 1.7–3.1 vs 8.3×, IQR: 3.5–43.5; (P=0.02), and USP18 1.4×, IQR: 0.9–1.8 vs 6.5×, IQR: 2.4–9.7; (P=0.005), was significantly lower in IL28B genotype TT-carriers (8/26) than in CT-carriers (16/26). The initial ISG expression was independent of HCV viral load and patients’ age.
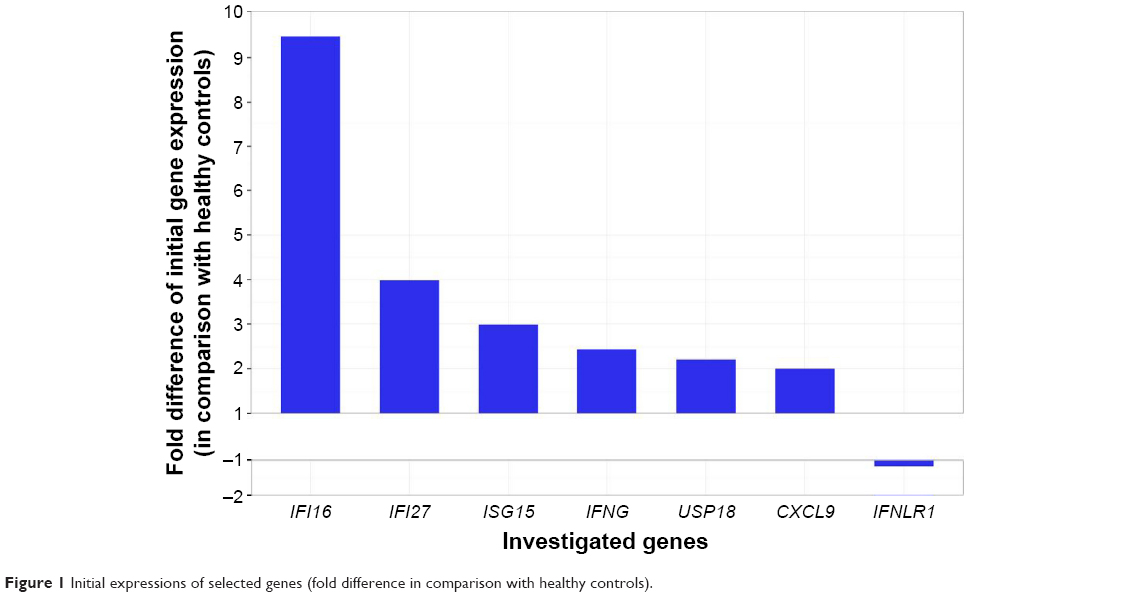 | Figure 1 Initial expressions of selected genes (fold difference in comparison with healthy controls). |
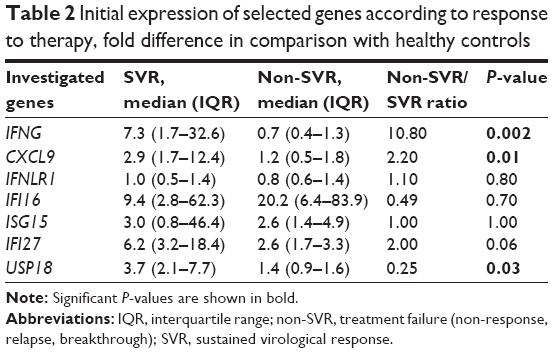 | Table 2 Initial expression of selected genes according to response to therapy, fold difference in comparison with healthy controls |
Course of ISG activation
Expression of all analyzed genes was progressively increasing during the first 12 weeks of interferon therapy. At weeks 4, 8, and 12, gene expression profiles did not differ between BOC and TVR groups. When comparing their activation between SVR and non-SVR group, a significant difference was found only in USP18 expression at week 12 (P=0.001), as shown in Figure 2. Its expression rose continuously until week 12 in the SVR group, while in the non-SVR group a decrease at week 12 was observed (Figure S1). The course of gene expressions at different time points did not correlate with HCV RNA levels.
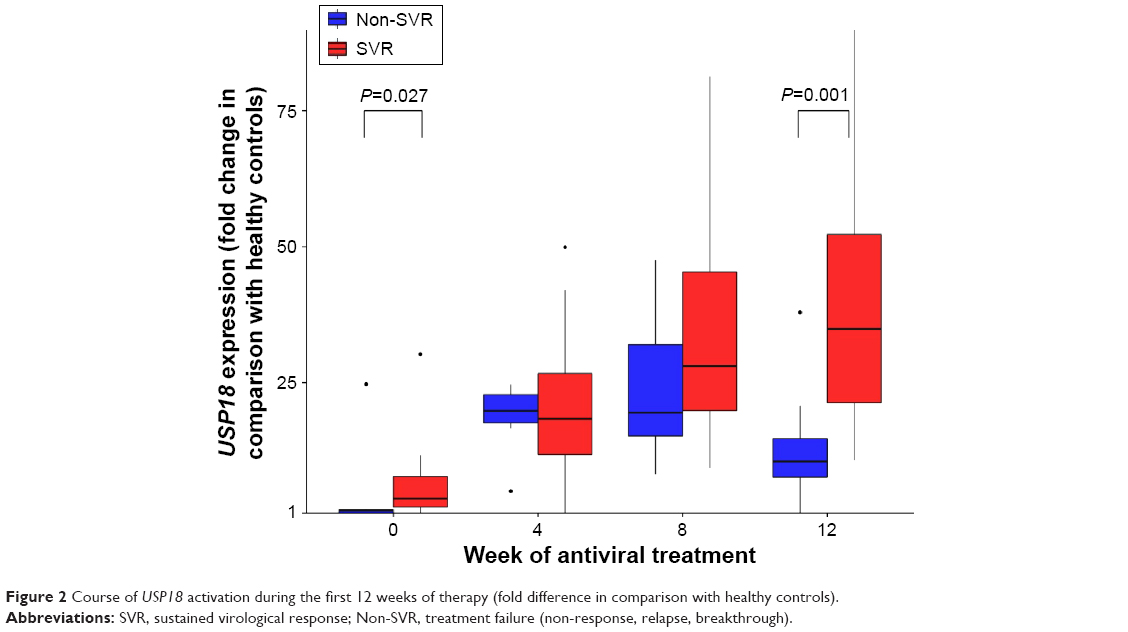 | Figure 2 Course of USP18 activation during the first 12 weeks of therapy (fold difference in comparison with healthy controls). |
Prediction of SVR
None of the above-described standard clinical pretreatment SVR predictors had predictive value in our group of patients with advanced liver disease (Figure 3A). Out of seven selected genes, the initial expression of four genes was found to be a predictor of SVR achievement in univariate analysis (USP18, IFNG, IFI27, and CXCL9), as shows Table 3 and Figure 3B. In multivariate analysis, the initial expression of USP18 was identified as a predictor of SVR (P=0.047).
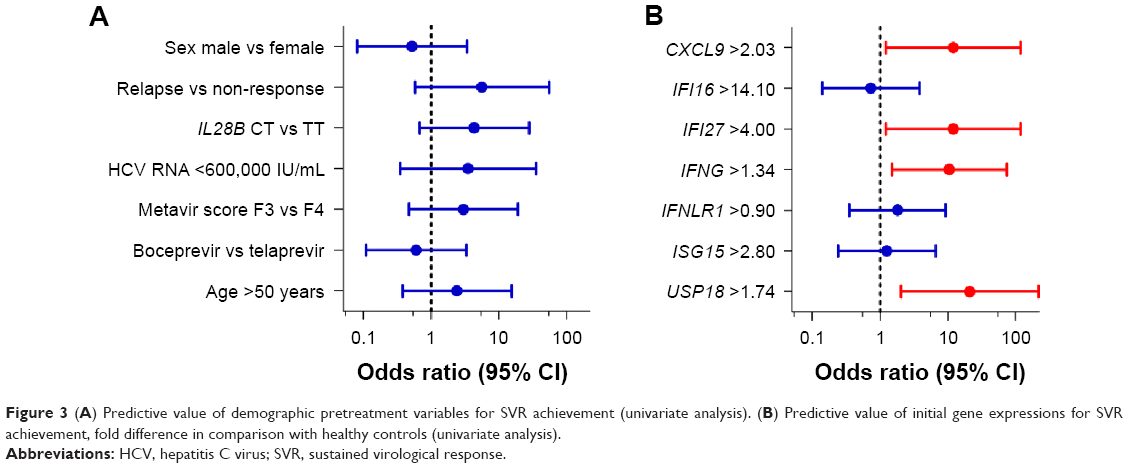 | Figure 3 (A) Predictive value of demographic pretreatment variables for SVR achievement (univariate analysis). (B) Predictive value of initial gene expressions for SVR achievement, fold difference in comparison with healthy controls (univariate analysis). |
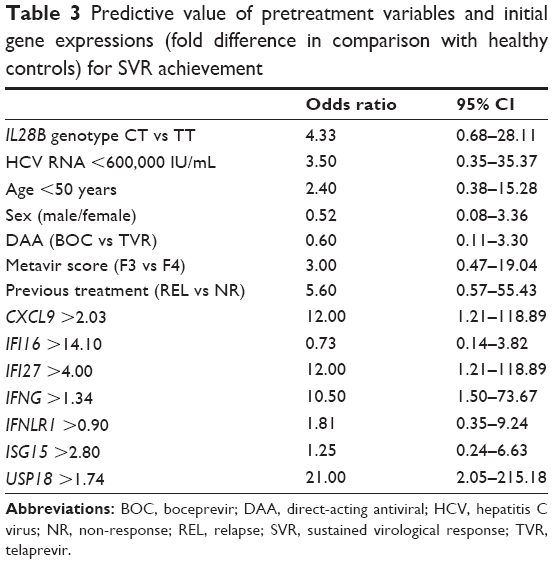 | Table 3 Predictive value of pretreatment variables and initial gene expressions (fold difference in comparison with healthy controls) for SVR achievement |
Discussion
Our cohort represents a homogenous group of difficult-to-treat individuals of older age with long duration of the disease and with advanced liver fibrosis (stage F3 and F4 according to Metavir score). All had failed in the previous course of P/R therapy. This explains the fact that the expression of most studied genes did not differ between the patient groups (SVR and non-SVR) and did not increase significantly during the retreatment with triple therapy with P/R + DAAs. Nonetheless, we found in this cohort significantly higher initial expression levels of IFNG, CXCL9, and USP18 in patients who had achieved SVR than in those in whom the treatment had failed. Another significant difference was observed in USP18 expression levels at week 12 between the same patient groups. The pretreatment IFNG mRNA expression levels in PBMCs are already known to be lower in non-SVR patients treated with P/R without DAAs.14 Since CXCL9 is an interferon gamma-sensitive gene, one may expect the same. However, during treatment, expression of both IFNG and CXCL9 did not differ between the SVR and non-SVR patients and it was not upregulated compared with the initial values found in the non-SVR group. We suppose that the observed inability to further upregulate IFNG and CXCL9 and also USP18 in the non-SVR group may result from the previously reported exhaustion of the immune system owing to longer duration of HCV infection.15 Exhaustion of the immune system in long-persistent HCV infection has been explained by different mechanisms, one of the mechanisms being the impact of disrupted tetherin signalization on protein kinase R activity.16 Another mechanism to be considered is the proteolytic degradation of the molecules involved in interferon regulating factor 3 (IRF3) signaling by NS3/4A protease, a nonstructural HCV protein crucial for its replication.2 Our data show that in patients without response to interferon stimulation treated with P/R in combination even with the NS3/4A protease inhibitor, the interferon-irresponsiveness was not broken through. This indicates that even the pharmacological inhibition of the NS3/4A protease does not lead to full restoration of sensitivity to interferon.
The IFNL4 ss469415590 genotype, strongly linked with the IL28B rs12979860 variant, is the most reliable genetic predictor of SVR in interferon-based therapies.17 The IFNL4 variant ss469415590 ΔG converts the inactive IFNL4 pseudogene into an active gene producing interferon-lambda 4, which is likely to suppress signaling by other interferons essential for HCV clearance. ISG expression in the liver is strongly associated with IL28B/IFNL4 genotype.18,19 The data concerning the association between ISG expression in PBMC and IL28B/IFNL4 genotype are controversial. In our cohort, we found a significant difference in the initial expression of two genes (IFI27 and USP18) between IL28B CT and TT-carriers. Unfortunately, we were not able to evaluate CC group because of a low number of patients (only two in our cohort). The expression of IFNL4 gene was described only in infected hepatocytes,20 it has not been studied in PBMC so far. However, the results of other authors support our findings; Bordi et al found an association between favorable IL28B/IFNL4 genotype and IFNAR-1 transcription.21 By this mechanism, IL28B/IFNL4 genotype would be responsible for different levels of ISG expression in the PBMC. In contrast, Rallon et al did not find an association between ISG expression profile, assessed in PBMC, and IL28B genotype in a cohort of HCV–HIV co-infected patients.22 The absence of an association described by this group could be explained by impaired immunoreactivity in HIV/HCV co-infected patients.
We did not find any difference between patients treated with BOC or TVR in the course of ISG activation, although we had anticipated a difference in this point because BOC is added to P/R after a leading-phase of 4 weeks, whereas TVR therapy is initiated at day 1. These data suggest that the addition of a protease inhibitor to the therapy does not influence interferon-induced ISG activation. First-generation protease inhibitors (BOC and TVR) have recently been replaced by simeprevir. The advantage of simeprevir is its better tolerability, but the virological efficacy in patients with advanced liver fibrosis is similar to that of BOC and TVR.23 Therefore, we assume that our data could be applicable to patients treated with simeprevir, but a validation study of our data is needed before the routine use of this predictor in clinical practice. Simeprevir-containing triple therapy still remains a standard-of-care in many countries, but the patients with genotype 1 and advanced liver fibrosis should rather be candidates for interferon-free regimens. Therefore, in the regions with limited access to the interferon-free treatments, there is a need for the selection of patients. Selection according to ISG expression, especially USP18 in PBMC, may represent a suitable and reliable approach.
USP18, a member of the ubiquitin-specific protease family, has been shown to cleave protein conjugates with ISG15 (ISGylated proteins) in mice.24 However, the primary role of USP18 seems to be different: the protein acts as a negative regulator of type I interferon signaling.25 Expression of ISG including USP18 and ISG15 is increased in hepatocytes in treatment-naïve HCV patients nonresponding to P/R therapy in comparison with responders to the same treatment and expression of these genes increases less in nonresponders than in responders during the therapy. By contrast, higher pretreatment expression levels of USP18 and ISG15 have been found in Kupffer cells in responders in comparison with nonresponders.10
In our cohort, the initial USP18 expression in PBMC turned out to be the only factor predicting SVR achievement in multivariate analysis. This finding supports previously published data on the relevance of USP18 and ISG15 in induction of antiviral immunity by interferon alpha.26,27 Furthermore, USP18 was one of the genes whose initial expression was associated with IL28B genotype in our cohort.
Acknowledgment
The study was supported by the grant of Internal Grant Agency of Ministry of Health of the Czech Republic (No NT/11235-5).
Disclosure
The authors report no conflicts of interest in this work.
References
Cooke GS, Lemoine M, Thursz M, et al. Viral hepatitis and the Global Burden of Disease: a need to regroup. J Viral Hepat. 2013;20(9):600–601. | ||
Thimme R, Binder M, Bartenschlager R. Failure of innate and adaptive immune responses in controlling hepatitis C virus infection. FEMS Microbiol Rev. 2012;36(3):663–683. | ||
Bolen CR, Robek MD, Brodsky L, et al. The blood transcriptional signature of chronic hepatitis C virus is consistent with an ongoing interferon-mediated antiviral response. J Interferon Cytokine Res. 2013;33(1):15–23. | ||
Chen L, Borozan I, Feld J, et al. Hepatic gene expression discriminates responders and nonresponders in treatment of chronic hepatitis C viral infection. Gastroenterology. 2005;128(5):1437–1444. | ||
Asselah T, Bieche I, Narguet S, et al. Liver gene expression signature to predict response to pegylated interferon plus ribavirin combination therapy in patients with chronic hepatitis C. Gut. 2008;57(4):516–524. | ||
Hou J, van Oord G, Groothuismink ZM, et al. Gene expression profiling to predict and assess the consequences of therapy-induced virus eradication in chronic hepatitis C virus infection. J Virol. 2014;88(21):12254–12264. | ||
Ji X, Cheung R, Cooper S, Li Q, Greenberg HB, He XS. Interferon alfa regulated gene expression in patients initiating interferon treatment for chronic hepatitis C. Hepatology. 2003;37(3):610–621. | ||
McHutchison JG, Lawitz EJ, Shiffman ML, et al. Peginterferon alfa-2b or alfa-2a with ribavirin for treatment of hepatitis C infection. N Engl J Med. 2009;361(6):580–593. | ||
Sarasin-Filipowicz M, Oakeley EJ, Duong FH, et al. Interferon signaling and treatment outcome in chronic hepatitis C. Proc Natl Acad Sci U S A. 2008;105(19):7034–7039. | ||
Chen L, Borozan I, Sun J, et al. Cell-type specific gene expression signature in liver underlies response to interferon therapy in chronic hepatitis C infection. Gastroenterology. 2010;138(3):1123–1133, e1121–e1123. | ||
Taylor MW, Tsukahara T, McClintick JN, Edenberg HJ, Kwo P. Cyclic changes in gene expression induced by Peg-interferon alfa-2b plus ribavirin in peripheral blood monocytes (PBMC) of hepatitis C patients during the first 10 weeks of treatment. J Transl Med. 2008;6:66. | ||
MacParland SA, Corkum CP, Burgess C, Karwowska S, Kroll W, Michalak TI. Differential expression of interferon alpha inducible genes in peripheral blood mononuclear cells from patients chronically infected with hepatitis C virus and healthy donors. Int Immunopharmacol. 2015;25(2):545–552. | ||
Fabris C, Falleti E, Cussigh A, et al. IL-28B rs12979860 C/T allele distribution in patients with liver cirrhosis: role in the course of chronic viral hepatitis and the development of HCC. J Hepatol. 2011;54(4):716–722. | ||
He Q, Graham CS, Durante Mangoni E, Koziel MJ. Differential expression of toll-like receptor mRNA in treatment non-responders and sustained virologic responders at baseline in patients with chronic hepatitis C. Liver Int. 2006;26(9):1100–1110. | ||
Rehermann B. Chronic infections with hepatotropic viruses: mechanisms of impairment of cellular immune responses. Semin Liver Dis. 2007;27(2):152–160. | ||
Hotter D, Sauter D, Kirchhoff F. Emerging role of the host restriction factor tetherin in viral immune sensing. J Mol Biol. 2013;425(24):4956–4964. | ||
Prokunina-Olsson L, Muchmore B, Tang W, et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nature Genet. 2013;45(2):164–171. | ||
Honda M, Sakai A, Yamashita T, et al. Hepatic ISG expression is associated with genetic variation in interleukin 28B and the outcome of IFN therapy for chronic hepatitis C. Gastroenterology. 2010;139(2):499–509. | ||
Dill MT, Duong FH, Vogt JE, et al. Interferon-induced gene expression is a stronger predictor of treatment response than IL28B genotype in patients with hepatitis C. Gastroenterology. 2011;140(3):1021–1031. | ||
Amanzada A, Kopp W, Spengler U, Ramadori G, Mihm S. Interferon-lambda 4 (IFNL4) transcript expression in human liver tissue samples. PLoS One. 2013;8(12):e84026. | ||
Bordi L, Caglioti C, Garbuglia AR, et al. IFNL4 and IFNL3 associated polymorphisms strongly influence the spontaneous IFN-alpha receptor-1 expression in HCV-infected patients. PLoS One. 2015;10(2):e0117397. | ||
Rallon NI, Lopez-Fernandez LA, Garcia MI, et al. Interferon-stimulated genes are associated with peginterferon/ribavirin treatment response regardless of IL28B alleles in hepatitis C virus/HIV-coinfected patients. Aids. 2013;27(5):687–696. | ||
Reddy KR, Zeuzem S, Zoulim F, et al. Simeprevir versus telaprevir with peginterferon and ribavirin in previous null or partial responders with chronic hepatitis C virus genotype 1 infection (ATTAIN): a randomised, double-blind, non-inferiority phase 3 trial. Lancet Infect Dis. 2015;15(1):27–35. | ||
Malakhova OA, Yan M, Malakhov MP, et al. Protein ISGylation modulates the JAK-STAT signaling pathway. Genes Dev. 2003;17(4):455–460. | ||
Malakhova OA, Kim KI, Luo JK, et al. UBP43 is a novel regulator of interferon signaling independent of its ISG15 isopeptidase activity. EMBO J. 2006;25(11):2358–2367. | ||
Randall G, Chen L, Panis M, et al. Silencing of USP18 potentiates the antiviral activity of interferon against hepatitis C virus infection. Gastroenterology. 2006;131(5):1584–1591. | ||
Chua PK, McCown MF, Rajyaguru S, et al. Modulation of alpha interferon anti-hepatitis C virus activity by ISG15. J Gen Virol. 2009;90(Pt 12):2929–2939. |
Supplementary material
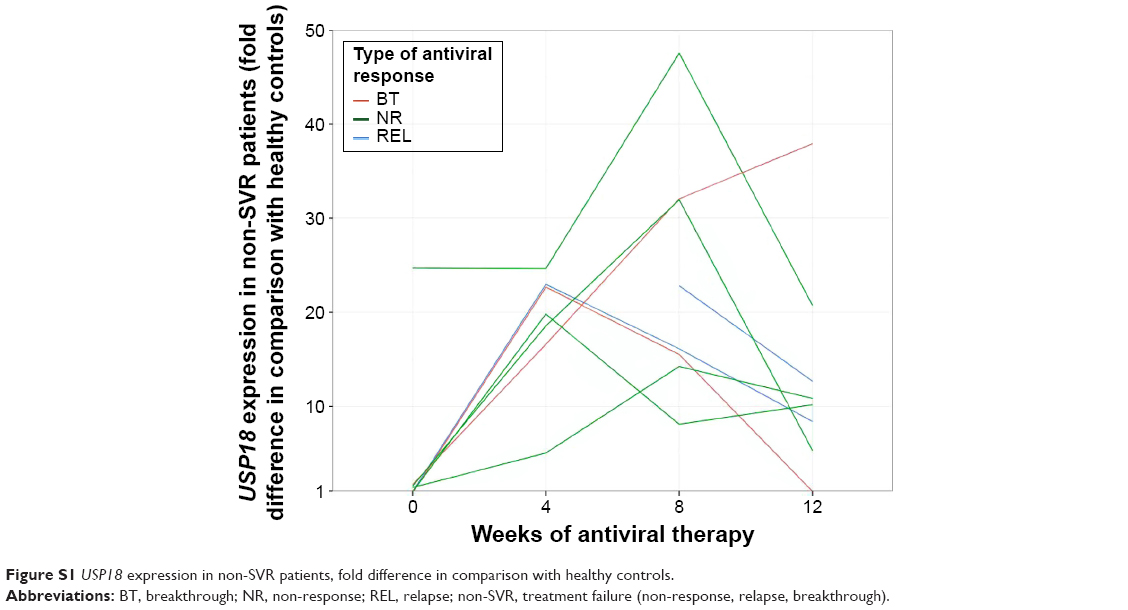 | Figure S1 USP18 expression in non-SVR patients, fold difference in comparison with healthy controls. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.