Back to Archived Journals » Metalloproteinases In Medicine » Volume 2
Using the laws of thermodynamics to understand how matrix metalloproteinases coordinate the myocardial response to injury
Authors Iyer R, Jung M, Lindsey M
Received 2 June 2015
Accepted for publication 2 September 2015
Published 30 October 2015 Volume 2015:2 Pages 75—82
DOI https://doi.org/10.2147/MNM.S74093
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yoshifumi Itoh
Rugmani Padmanabhan Iyer,1 Mira Jung,1 Merry L Lindsey1,2
1Mississippi Center for Heart Research, Department of Physiology and Biophysics, School of Medicine, University of Mississippi Medical Center, University of Mississippi, 2Research Service, GV (Sonny) Montgomery Veterans Affairs Medical Center, Jackson, MS, USA
Abstract: Following myocardial infarction (MI), the left ventricle (LV) undergoes a series of molecular, cellular, and functional alterations that are both part of the wound healing response to form a scar in the infarct region and the consequence of that response. Using the laws of thermodynamics as an analogy, we present here three laws for categorizing the post-MI LV remodeling process. The first law is that the LV will attempt to maintain equilibrium and compensate as a way to maximize function, the second law is that remodeling is progressive and unidirectional, and the third law is that the final goal is (ideally, but not always achievable) a stable, equilibrated scar. This comparison helps to define the boundaries of the system, whether it be the infarct zone, the LV, the heart, or the entire body. This review provides an overview for those not directly in the field and establishes a framework to help prioritize future research directions.
Keywords: MMP, remodeling, left ventricle, inflammation, fibrosis, extracellular matrix, proteomics
Introduction
Thermodynamics is the branch of physics that deals with the energy and work of a system, and there are four principal laws of thermodynamics that define the properties of a physical system. In this review, we have used the laws of thermodynamics as a framework analogy to explain how the left ventricle (LV) remodels after myocardial infarction (MI), focusing on the roles matrix metalloproteinases (MMPs) play. This schema applies thermodynamics laws to remodeling of the LV post-MI as an example scenario to illustrate the general mechanisms involved in the cardiac system response to injury. We define the laws of MI thermodynamics as an iterative process the myocardium takes in its attempt to maintain LV function and equilibrium in the setting of excessive myocyte loss. We include in our review how departure from these laws may be beneficial or detrimental to drive the remodeling response, which serves to identify potential areas for therapeutic emphasis. While we use MI as an example of cardiac injury, this review applies to other cardiac injuries (eg, pressure and volume overload) as well as other wound injury models (eg, dermal wounds).
Zeroth law of MI thermodynamics: in the absence of pathology, the cardiac system remains in equilibrium
The zeroth law of thermodynamics states that a system is in equilibrium when there is no net change to the system state over time. Our analogous law with respect to LV dynamics is that in the absence of pathology, the factors that maintain normal function of the cardiac system will remain in equilibrium with each other. Specifically, the extracellular matrix (ECM) and the cellular constituents of the myocardium maintain LV equilibrium to ensure normal LV function. MMPs and fibroblasts coordinate ECM turnover and maintain a balanced ECM. This law explains the young (non-aged) myocardium and is not applicable to the post-MI response. An important consideration for this law is that, by its very definition, it is not sustainable. As a system ages, there will be a net change in cardiac structure and function.1–5 Other exceptions to the law include exercise or pregnancy, both of which induce changes to the cardiac system in the absence of pathology.6,7
First law of MI thermodynamics: following MI, the cardiac system remodels in an attempt to maintain maximum LV function
The first law of thermodynamics states that the total energy of an isolated system is constant; energy can be transformed from one form to another, but cannot be created or destroyed. This law applies the conservation of energy principle to heat and thermodynamic processes. In the case of MI, the LV undergoes an orchestrated sequence of cellular and extracellular events to heal the infarcted myocardium in a way that seeks to optimize LV structure and function. MI yields an acute energy crisis due to myocyte loss, and the LV response begins immediately. Within minutes of ischemia, cardiomyocyte death occurs and is a driving force to trigger an influx of inflammatory leukocytes into the infarcted myocardium.8 The MI response time course can be divided into three overlapping phases: inflammatory, proliferative, and maturation phases.8
The initial inflammatory phase involves the robust infiltration of leukocytes (primarily neutrophils and macrophages, with lymphocytes also contributing) as well as the upregulation of chemokine and cytokine signaling. The influx of neutrophils yields MMP release and activation, which leads to net ECM breakdown that is necessary for removal of necrotic tissue debris to prepare the infarct area for scar formation. Neutrophils stimulate an influx of macrophages, which phagocytose apoptotic neutrophils to resolve the pro-inflammatory response. The proliferative phase is dominated by ECM synthesis to form a collagen-based scar and neovessel formation, coordinated by macrophage regulation of myofibroblast differentiation and endothelial cell activation. The third phase involves scar maturation and an attempt to return to equilibrium. Of note, this post-MI equilibrium is not the same homeostasis that occurred prior to MI, but rather is a reset of homeostasis at a new level. The goal at this stage, in the absence of an effective myocyte cell replacement strategy, is to prevent the further decline in function, which would result in the progression to heart failure.
During scar formation, there is a balance between too little scar formation and excessive infarct expansion (which can cause rupture) and excessive scar formation and infarct stiffening (which can cause heart failure).9 Remodeling events therefore, must be orchestrated during wound healing for optimal responses. This includes the timely influx of leukocytes to regulate cytokine synthesis and oversee the removal of tissue debris, followed by their timely removal to ensure optimum resolution of inflammation and generation of a collagen-rich scar. Based on the well-known energy equation, E = mc2 (where E = energy, m = mass, and c = speed of light), we propose a first law of MI thermodynamics equation:

Equation 1 indicates that, in order to optimize LV function preservation post-MI, the factors most modifiable within the infarct zone are the inflammation and ECM components. Two assumptions of this equation are that the time of infarction has proceeded past the point of myocyte salvage and regeneration therapies are not optimal, which are currently valid assumptions for infarcts that are not reperfused. While the first law of thermodynamics has several theoretical and philosophical considerations, the relatively simple essence of the law is conservation of energy. In this review, we have used conservation of LV function as a platform to define the first law of MI thermodynamics, where the post-MI cardiac system limits the extent of LV dysfunction by modifying the inflammation response.
Scar formation is variable and is highly dependent on the inflammation component. Modulating the inflammatory response by regulating leukocyte functions, modulating ECM turnover by regulating MMPs (ECM breakdown) or fibroblast differentiation (ECM synthesis), or modulating neovascularization by regulating endothelial cell activation all feed forward to alter scar quality and consequently LV function. How an LV proceeds along the remodeling process continuum is a major deciding factor of long-term post-MI complications (eg, development of heart failure or sudden cardiac death). Altering inflammation or ECM turnover can improve or worsen LV function, depending on the perturbation. We propose that interventions altering variables within the first law of MI thermodynamics may have the largest effects on outcomes.
Second law of MI thermodynamics: following MI, the LV undergoes unidirectional modifications that lead to irreversible transformation in geometry and function
The second law of thermodynamics maintains that the state of entropy (disorder) of the universe, as a closed isolated system, will always increase over time and that changes in entropy can never be negative. In the case of MI, the analogy would be that post-MI remodeling events proceed in one direction, culminating in irreversible changes to LV structure and function. This law indicates that the direction of LV remodeling progresses toward scar formation. Each upstream event of the remodeling process regulates downstream events, leading to an overall effect on scar quality and cardiac function. For example, the magnitude of neutrophil infiltration feeds forward to regulate macrophage entry into infarct zone. Through intercellular communication, factors released from neutrophils regulate macrophages and factors released from macrophages regulate fibroblasts and endothelial cells. Based on the entropy equation, ΔS = ΔQ/T (where S = entropy, Q = heat transfer, and T = temperature), we propose a second law of MI thermodynamics equation:
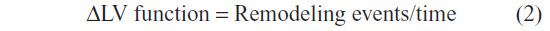
In this equation, remodeling events include leukocyte infiltration, necrotic cardiomyocyte removal, ECM degradation, macrophage polarization, neutrophil apoptosis, myofibroblast differentiation, resolution of inflammation, and collagen synthesis. Remodeling post-MI is time dependent in a similar manner as entropy is temperature dependent. Therefore, for the second law of MI thermodynamics equation, we have substituted the temperature variable with a time variable. In the entropy equation, dividing by temperature means that the effect of a specific level of heat transfer is diminished at higher temperatures; analogously, the effect of specific remodeling events may diminish if they occur at later time points along the remodeling continuum. Therefore, the time element is a crucial component when considering therapeutic strategies.
This law indicates that in the post-MI setting, LV remodeling regulated within a specific time frame may be necessary to attain maximum benefit to conserving LV geometry and function. Remodeling events can be regulated by several methods, including regulating 1) the inflammatory response (eg, neutrophil or macrophage phenotypes and cytokine production or secretion), 2) ECM turnover (eg, breakdown by MMPs or synthesis by fibroblasts), 3) cellular crosstalk, or 4) the timing or quality of each event (Figure 1).
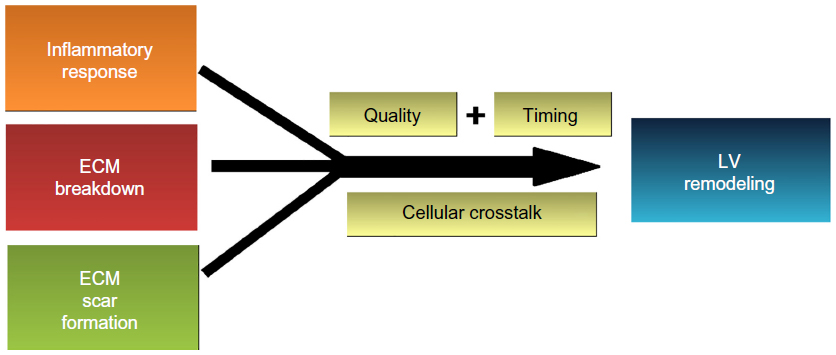 | Figure 1 Remodeling of the left ventricle following myocardial infarction is the product of the inflammatory response and turnover of the extracellular matrix to remove necrotic myocytes and generate an infarct scar. |
Third law of MI thermodynamics: following MI, the infarct approaches scar equilibrium over time
The third law of thermodynamics states that the entropy of a system approaches a constant value as the temperature approaches absolute zero. When time is substituted for temperature, the infarct reaches a state of mature scar over several weeks to months post-MI. Inflammatory signals are downregulated, and ECM turnover reduces from a state of rapid growth to a state of maintenance at this late phase.10–12 Scar tissue provides structural stability to the LV and provides tensile strength to the infarct.13,14 Therefore, effective scar formation, which includes scar contraction, is a crucial process in determining long-term outcomes. The mechanically most advantageous scenario is the development of a thick scar with a minimal loss in the length to width ratio. Importantly, the third law is not actually possible to achieve but is merely a mathematical reflection to convey the limits of the universe and describe an optimal situation. Similarly, in the MI setting, attaining a sturdy scar with stable collagen structure and maximally conserved cardiac function is an optimum goal. The chronic maturation phase is not a permanently stable condition, as both the scar and remote regions continue to remodel, and the LV can accelerate off a course of stability and spiral into a state of heart failure progression.14 The myofibroblast in the cardiac infarct scar is responsible for wound contraction, as well as scar maintenance, and this cell persists many years post-MI.15–17
Using the laws to understand how interventions improve LV remodeling
While there are numerous examples of interventions that either beneficially or negatively contribute to LV remodeling, we will focus the discussion here on how altering MMP activity can impact LV remodeling outcomes. MMP expression and activity increases post-MI; in particular, MMP-1, MMP-2, MMP-7, MMP-8, MMP-9, MMP-12, MMP-13, MMP-14, and MMP-28 have been shown to increase during the acute post-MI phase.18–20 Null and transgenic mouse models or MMP inhibitors (MMPi) reveal extensive MMP roles in LV remodeling, including effects on all remodeling components.18 Later, we provide examples of how the MI thermodynamics laws can explain responses to MMP targeted therapeutic intervention (summarized in Table 1).
 | Table 1 MMP Interventions that improve post-MI outcomes |
While the promising effects of MMP inhibition on LV remodeling and cardiac function in animal models encouraged the use of MMP inhibitors (MMPi) as a potential therapy for prevention of development of heart failure post-MI, these findings have not translated to the human scenario. The reasons for this not only include selectivity and specificity issues in inhibitors developed to date, but also include our lack of understanding of the full range of MMP functions.18 For example, the substrate list for MMP-9 is extensive and includes collagen and fibronectin (ECM), as well as interleukin-1 and plasminogen (cytokines and growth factors) and citrate synthase (intracellular components).21,22
Inhibition using broad-spectrum MMPi decreases early LV dilation post-MI in mice and rabbits.23,24 Animals treated with the broad-spectrum MMPi CP-471,474 showed significantly smaller post-MI end-systolic and end-diastolic dimensions. In pigs treated with another broad MMPi, PD166793, post-MI LV end-diastolic dimensions were reduced, while tissue inhibitor of metalloproteinase (TIMP)-1 concentrations were increased.25 When the broad-spectrum MMPi, PGE-530742, was given at a dose that inhibited MMP-1 and MMP-7, administering 3 days pre- or 3 days post-MI, it had no effect on infarct area and did not attenuate LV dilation.26 Treatment did, however, increase collagen deposition in the border zone and decrease deposition in the remote zone when given pre-MI, indicating selectivity and timing are critical factors. Another MMPi, 2R-2-[5-[4-[ethyl-methylamino]phenyl] thiophene-2-sulfonylamino]-3-methylbutyric acid (TISAM) given at a dose that inhibited MMP-2, MMP-9, and MMP-14 but not MMP-1, MMP-3, or MMP-7 showed both beneficial and detrimental effects post-MI, including not only increased survival rates due to reduced cardiac rupture rates but also reduced macrophage infiltration.27 These studies illustrate that MMP inhibition modulates both ECM and inflammatory factors and follow the first and second laws where changes to early remodeling events regulate LV geometry. The examples highlight the importance of treatment timing. Additionally, these studies underscore that MMPs do not regulate all variables equally, and which MMPs are being inhibited and the downstream consequences of this inhibition need to be considered. These studies also highlight that outcome variables being measured to assess MMPi efficacy need to take short-term (inflammation and ECM turnover) and long-term (LV function) effects into consideration.
As an example of how the MI laws can be used to understand remodeling, MMP-9 perturbation has been used to control the remodeling response (Table 2).22,28–32 Post-MI, MMP-9 null mice demonstrate reduced macrophage infiltration, collagen accumulation, and LV dilation over the 1st weeks post-MI.29,31 The first law can be used to explain these findings, as changes in inflammation and ECM variables simultaneously draw the LV toward attenuated dilation and improved LV function. Likewise, transgenic overexpression of MMP-9 only in macrophages attenuates the inflammatory response and improves LV function early post-MI, indicating that MMP-9 deriving from the macrophage has different net functions compared to MMP-9 deriving from all cell types, including neutrophils and other post-MI cell sources.30 Both of these scenarios demonstrate that the remodeling flow is unidirectional and time dependent.
 | Table 2 Examples of applying the laws of MI thermodynamics to post-MI inflammation and extracellular matrix responses following matrix metalloproteinase-9 modulation |
Indirect MMP inhibition can also be achieved by blocking upstream activators as an intervention strategy. For example, the endothelin receptor blocker sitaxsentan decreases MMP-1, MMP-2, and MMP-9 and increases TIMP-1 and prevents LV dilation post-MI.33 Doxycycline is a well-known example; this antibiotic has been shown to suppress activities of MMP-2, MMP-8, MMP-9, MMP-12, TIMP-1, and TIMP-2 and to have beneficial effects on LV dysfunction and deposition of type I collagen post-MI.34,35 Another study in MI patients using angiotensin-converting enzyme inhibitors showed reduced collagenolytic activity by reducing MMP-1 levels that slowed the progressive enlarging of LV dimensions post-MI.26,36 Angiotensin-converting enzyme inhibitors have also been shown to be selective MMP-9 inhibitors.37–39
Deletion of TIMPs in mice also leads to adverse remodeling post-MI, both through increasing MMP activity as well as regulating MMP-independent functions. TIMP-1 deletion aggravates LV remodeling post-MI.40,41 TIMP-2 null mice show increased infarct expansion and inflammatory response leading to exacerbated LV dysfunction post-MI.42 TIMP-3 and TIMP-4 deficiency caused increased cardiac rupture.43,44
One concept highlighted by these examples is that understanding the complete phenotypic effect of MMP or TIMP modulation is important for understanding its role in the post-MI response. In addition to tissue level changes in LV structure and function and cellular level changes in inflammatory or endothelial cell numbers and phenotypes, using broad stroke methods such as genomic or proteomic screens to capture molecular level alterations is also needed. Earlier studies evaluating specific MMP null mouse models missed the complexity of effects by limiting screens to obvious ECM changes in scar structure and changes in mortality rates due to rupture.
Using the laws to understand how interventions worsen LV remodeling
While the above examples demonstrate how the laws can be applied to multiple cases where improvements are seen, there are instances where interventions lead to worsened remodeling (Table 3). Strategies that delay inflammation resolution increase LV dilation and dysfunction at 7 days post-MI. For example, the MMP-12 inhibitor, RXP 470.1, given at 3 hours post-MI suppressed neutrophil apoptosis to prolong inflammation, resulting in exacerbated LV dilation.19 A prolonged inflammatory response impairs collagen deposition leading to formation of a weak scar, thus increasing the probability of chamber dilation.45 Cardiac-restricted overexpression of the membrane type (MT) 1-MMP (MMP-14) resulted in adverse remodeling and reduced survival post-MI.30 Consistent with the second law, MMP-14 increases early post-MI to influence downstream cardiac function.18,46 MMP-28 deletion inhibited M2 anti-inflammatory macrophage activation, which also exacerbated LV dysfunction and increased cardiac rupture rates.20 These studies highlight that remodeling is a unidirectional process and inhibition of an early step can affect the overall outcome. These studies are exceptions to the laws in that reducing components of the inflammatory process do not always have a beneficial effect on LV remodeling.20 These studies also highlight that we need to delineate individual MMP roles to identify which MMPs follow the laws and under which specific conditions or times.
 | Table 3 MMP interventions that worsen post-MI outcomes |
Concluding remarks
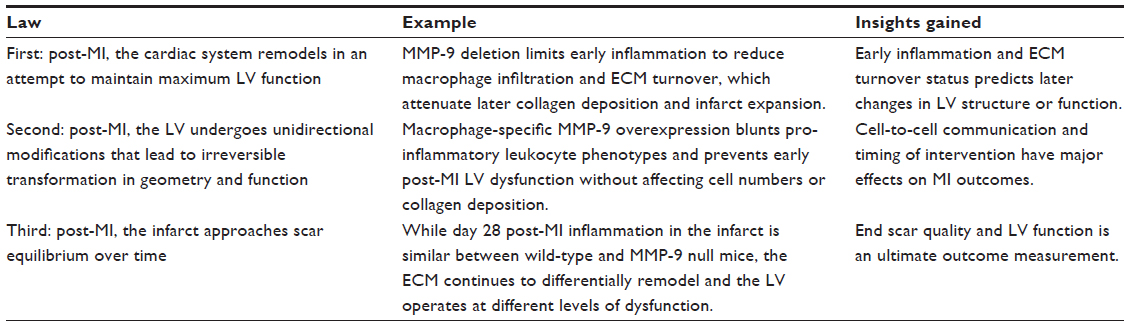
In conclusion, we have discussed how translating the laws of thermodynamics into an MI laws analogy can help to explain the response of the LV to MI. Introducing entropy, energy, and dynamics as post-MI concepts may provide depth and new insights to better understand the remodeling process. LV remodeling post-MI is complicated and involves molecular, cellular, and tissue level components in continual flux over time. Based on the first law, changes that occur to the inflammatory and ECM turnover patterns will predict changes in LV structure and function. Based on the second law, consideration should be given for cell-to-cell communication and timing of the intervention tested. Based on the third law, focusing on the end scar quality is an ultimate outcome measurement. When using interventions, such as one that modulates an MMP activity directly or indirectly, a more detailed evaluation would include how the intervention affects inflammatory and ECM components as well as tissue level structure and function. By suggesting a set of conditions to establish cause and effect relationships between LV remodeling events and MMP actions, our laws can help to delineate the mechanisms of the MMP field generally and the MI field specifically.
Acknowledgments
This work was supported by American Heart Association Postdoctoral Grant 14POST18770012 to RPI; the National Institutes of Health HHSN 268201000036C (N01-HV-00244), HL075360, and GM114833 to MLL, P01HL051971, and P20GM104357; and the Biomedical Laboratory Research and Development Service of the Veterans Affairs Office of Research and Development Award 5I01BX000505 to MLL.
Disclosure
The authors report no conflicts of interest in this work.
References
Chiao YA, Dai Q, Zhang J, et al. Multi-analyte profiling reveals matrix metalloproteinase-9 and monocyte chemotactic protein-1 as plasma biomarkers of cardiac aging. Circ Cardiovasc Genet. 2011;4(4):455–462. | |
Ma Y, Chiao YA, Zhang J, Manicone AM, Jin YF, Lindsey ML. Matrix metalloproteinase-28 deletion amplifies inflammatory and extracellular matrix responses to cardiac aging. Microsc Microanal. 2012;18(1):81–90. | |
Yabluchanskiy A, Ma Y, Chiao YA, et al. Cardiac aging is initiated by matrix metalloproteinase-9-mediated endothelial dysfunction. Am J Physiol Heart Circ Physiol. 2014;306(10):H1398–H1407. | |
Chiao YA, Ramirez TA, Zamilpa R, et al. Matrix metalloproteinase-9 deletion attenuates myocardial fibrosis and diastolic dysfunction in ageing mice. Cardiovasc Res. 2012;96(3):444–455. | |
Ma Y, Chiao YA, Clark R, et al. Deriving a cardiac ageing signature to reveal MMP-9-dependent inflammatory signalling in senescence. Cardiovasc Res. 2015;106(3):421–431. | |
Yousef ZR, Redwood SR, Bucknall CA, Sulke AN, Marber MS. Late intervention after anterior myocardial infarction: effects on left ventricular size, function, quality of life, and exercise tolerance: results of the Open Artery Trial (TOAT Study). J Am Coll Cardiol. 2002;40(5):869–876. | |
Mathieu E, Lamirault G, Toquet C, et al. Intramyocardial delivery of mesenchymal stem cell-seeded hydrogel preserves cardiac function and attenuates ventricular remodeling after myocardial infarction. PLoS One. 2012;7(12):e51991. | |
Frangogiannis NG. The inflammatory response in myocardial injury, repair, and remodelling. Nat Rev Cardiol. 2014;11(5):255–265. | |
French BA, Kramer CM. Mechanisms of post-infarct left ventricular remodeling. Drug Discov Today Dis Mech. 2007;4(3):185–196. | |
Vanhoutte D, Schellings M, Pinto Y, Heymans S. Relevance of matrix metalloproteinases and their inhibitors after myocardial infarction: a temporal and spatial window. Cardiovasc Res. 2006;69(3):604–613. | |
Pfeffer MA, Braunwald E. Ventricular remodeling after myocardial infarction. Experimental observations and clinical implications. Circulation. 1990;81(4):1161–1172. | |
St John Sutton M, Pfeffer MA, Moye L, et al. Cardiovascular death and left ventricular remodeling two years after myocardial infarction: baseline predictors and impact of long-term use of captopril: information from the Survival and Ventricular Enlargement (SAVE) trial. Circulation. 1997;96(10):3294–3299. | |
Trueblood NA, Xie Z, Communal C, et al. Exaggerated left ventricular dilation and reduced collagen deposition after myocardial infarction in mice lacking osteopontin. Circ Res. 2001;88(10):1080–1087. | |
Czubryt MP. Common threads in cardiac fibrosis, infarct scar formation, and wound healing. Fibrogenesis Tissue Repair. 2012;5(1):19. | |
Gabbiani G, Ryan GB, Majne G. Presence of modified fibroblasts in granulation tissue and their possible role in wound contraction. Experientia. 1971;27(5):549–550. | |
Vracko R, Thorning D. Contractile cells in rat myocardial scar tissue. Lab Invest. 1991;65(2):214–227. | |
Cleutjens JP, Verluyten MJ, Smiths JF, Daemen MJ. Collagen remodeling after myocardial infarction in the rat heart. Am J Pathol. 1995; 147(2):325–338. | |
Iyer RP, de Castro Bras LE, Jin YF, Lindsey ML. Translating Koch’s postulates to identify matrix metalloproteinase roles in postmyocardial infarction remodeling: cardiac metalloproteinase actions (CarMA) postulates. Circ Res. 2014;114(5):860–871. | |
Iyer RP, Patterson NL, Zouein FA, et al. Early matrix metalloproteinase- 12 inhibition worsens post-myocardial infarction cardiac dysfunction by delaying inflammation resolution. Int J Cardiol. 2015;185:198–208. | |
Ma Y, Halade GV, Zhang J, et al. Matrix metalloproteinase-28 deletion exacerbates cardiac dysfunction and rupture after myocardial infarction in mice by inhibiting M2 macrophage activation. Circ Res. 2013;112(4):675–688. | |
de Castro Brás LE, Cates CA, DeLeon-Pennell KY, et al. Citrate synthase is a novel in vivo matrix metalloproteinase-9 substrate that regulates mitochondrial function in the postmyocardial infarction left ventricle. Antioxid Redox Signal. 2014;21(14):1974–1985. | |
Halade GV, Jin YF, Lindsey ML. Matrix metalloproteinase (MMP)-9: a proximal biomarker for cardiac remodeling and a distal biomarker for inflammation. Pharmacol Ther. 2013;139(1):32–40. | |
Rohde LE, Ducharme A, Arroyo LH, et al. Matrix metalloproteinase inhibition attenuates early left ventricular enlargement after experimental myocardial infarction in mice. Circulation. 1999;99(23):3063–3070. | |
Lindsey ML, Gannon J, Aikawa M, et al. Selective matrix metalloproteinase inhibition reduces left ventricular remodeling but does not inhibit angiogenesis after myocardial infarction. Circulation. 2002;105(6):753–758. | |
Mukherjee R, Brinsa TA, Dowdy KB, et al. Myocardial infarct expansion and matrix metalloproteinase inhibition. Circulation. 2003;107(4):618–625. | |
Yarbrough WM, Mukherjee R, Escobar GP, et al. Selective targeting and timing of matrix metalloproteinase inhibition in post-myocardial infarction remodeling. Circulation. 2003;108(14):1753–1759. | |
Matsumura S, Iwanaga S, Mochizuki S, Okamoto H, Ogawa S, Okada Y. Targeted deletion or pharmacological inhibition of MMP-2 prevents cardiac rupture after myocardial infarction in mice. J Clin Invest. 2005; 115(3):599–609. | |
Heymans S, Luttun A, Nuyens D, et al. Inhibition of plasminogen activators or matrix metalloproteinases prevents cardiac rupture but impairs therapeutic angiogenesis and causes cardiac failure. Nat Med. 1999;5(10):1135–1142. | |
Lindsey ML, Escobar GP, Dobrucki LW, et al. Matrix metalloproteinase- 9 gene deletion facilitates angiogenesis after myocardial infarction. Am J Physiol Heart Circ Physiol. 2006;290(1):H232–H239. | |
Zamilpa R, Ibarra J, de Castro Brás LE, et al. Transgenic overexpression of matrix metalloproteinase-9 in macrophages attenuates the inflammatory response and improves left ventricular function post-myocardial infarction. J Mol Cell Cardiol. 2012;53(5):599–608. | |
Ducharme A, Frantz S, Aikawa M, et al. Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J Clin Invest. 2000;106(1):55–62. | |
Ramirez TA, Iyer RP, Ghasemi O, et al. Aliskiren and valsartan mediate left ventricular remodeling post-myocardial infarction in mice through MMP-9 effects. J Mol Cell Cardiol. 2014;72:326–335. | |
Podesser BK, Siwik DA, Eberli FR, et al. ET(A)-receptor blockade prevents matrix metalloproteinase activation late postmyocardial infarction in the rat. Am J Physiol Heart Circ Physiol. 2001;280(3):H984–H991. | |
Phatharajaree W, Phrommintikul A, Chattipakorn N. Matrix metalloproteinases and myocardial infarction. Can J Cardiol. 2007;23(9):727–733. | |
DeCoux A, Lindsey ML, Villarreal F, Garcia RA, Schulz R. Myocardial matrix metalloproteinase-2: inside out and upside down. J Mol Cell Cardiol. 2014;77:64–72. | |
Papadopoulos DP, Economou EV, Makris TK, et al. Effect of angiotensin-converting enzyme inhibitor on collagenolytic enzyme activity in patients with acute myocardial infarction. Drugs Exp Clin Res. 2004;30(2):55–65. | |
Yamamoto D, Takai S. Pharmacological implications of MMP-9 inhibition by ACE inhibitors. Curr Med Chem. 2009;16(11):1349–1354. | |
Yamamoto D, Takai S, Jin D, Inagaki S, Tanaka K, Miyazaki M. Molecular mechanism of imidapril for cardiovascular protection via inhibition of MMP-9. J Mol Cell Cardiol. 2007;43(6):670–676. | |
Yamamoto D, Takai S, Miyazaki M. Prediction of interaction mode between a typical ACE inhibitor and MMP-9 active site. Biochem Biophys Res Commun. 2007;354(4):981–984. | |
Ikonomidis JS, Hendrick JW, Parkhurst AM, et al. Accelerated LV remodeling after myocardial infarction in TIMP-1-deficient mice: effects of exogenous MMP inhibition. Am J Physiol Heart Circ Physiol. 2005;288(1):H149–H158. | |
Creemers EE, Davis JN, Parkhurst AM, et al. Deficiency of TIMP-1 exacerbates LV remodeling after myocardial infarction in mice. Am J Physiol Heart Circ Physiol. 2003;284(1):H364–H371. | |
Kandalam V, Basu R, Abraham T, et al. TIMP2 deficiency accelerates adverse post-myocardial infarction remodeling because of enhanced MT1-MMP activity despite lack of MMP2 activation. Circ Res. 2010; 106(4):796–808. | |
Kandalam V, Basu R, Abraham T, et al. Early activation of matrix metalloproteinases underlies the exacerbated systolic and diastolic dysfunction in mice lacking TIMP3 following myocardial infarction. Am J Physiol Heart Circ Physiol. 2010;299(4):H1012–H1023. | |
Koskivirta I, Kassiri Z, Rahkonen O, et al. Mice with tissue inhibitor of metalloproteinases 4 (Timp4) deletion succumb to induced myocardial infarction but not to cardiac pressure overload. J Biol Chem. 2010;285(32):24487–24493. | |
Frangogiannis NG. Regulation of the inflammatory response in cardiac repair. Circ Res. 2012;110(1):159–173. | |
Spinale FG, Mukherjee R, Zavadzkas JA, et al. Cardiac restricted overexpression of membrane type-1 matrix metalloproteinase causes adverse myocardial remodeling following myocardial infarction. J Biol Chem. 2010;285(39):30316–30327. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.