Back to Journals » International Journal of General Medicine » Volume 14
Using Integrated Bioinformatics Analysis to Identify Abnormally Methylated Differentially Expressed Genes in Hepatocellular Carcinoma
Authors Chen QL, Yan Q, Feng KL ![]() , Xie CF
, Xie CF ![]() , Fang CK, Wang JN, Liu LH, Li Y, Zhong C
, Fang CK, Wang JN, Liu LH, Li Y, Zhong C ![]()
Received 1 December 2020
Accepted for publication 19 February 2021
Published 10 March 2021 Volume 2021:14 Pages 805—823
DOI https://doi.org/10.2147/IJGM.S294505
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Qing-Lian Chen,1,2,* Qian Yan,1,* Kun-Liang Feng,1,2,* Chun-Feng Xie,1,2,* Chong-Kai Fang,1,2 Ji-Nan Wang,1,2 Li-Hua Liu,2,3 Ya Li,3 Chong Zhong2,3
1Guangzhou University of Chinese Medicine, Guangzhou, 510405, People’s Republic of China; 2Department of Hepatobiliary Surgery, The First Affiliated Hospital of Guangzhou University of Chinese Medicine, Guangzhou, 510405, People’s Republic of China; 3Lingnan Medical Research Center, Guangzhou University of Chinese Medicine, Guangzhou, 510405, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Chong Zhong
Department of Hepatobiliary Surgery, The First Affiliated Hospital of Guangzhou University of Chinese Medicine, 16 Airport Road, Guangzhou, 510405, People’s Republic of China
Tel +86-20-36596301
Email [email protected]
Objective: For the identification of abnormally methylated differentially expressed genes (MDEGs) in hepatocellular carcinoma (HCC), this study integrated four microarray datasets to investigate the fundamental mechanisms of tumorigenesis.
Methods: We obtained the expression (GSE76427, GSE57957) and methylation (GSE89852, GSE54503) profiles from Gene Expression Omnibus (GEO). The abnormally MDEGs were identified by using R software. We used the clusterProfiler package for the functional and pathway enrichment analysis. The String database was used to build the protein–protein interaction (PPI) network and visualize it in Cytoscape. MCODE was employed in the module analysis. Additionally, Gene Expression Profiling Interactive Analysis (GEPIA) and The Cancer Genome Atlas (TCGA) were employed to validate results. Lastly, we used cBioPortal software to examine the hub genetic alterations.
Results: We identified 162 hypermethylated, down-regulated genes and 190 hypomethylated, up-regulated genes. Up-regulated genes with low methylation were enriched in biological processes, such as keratinocyte proliferation, and calcium homeostasis. Pathway analysis was enriched in the AMPK and PI3K-Akt signaling pathways. The PPI network identified PTK2, VWF, and ITGA2 as hypomethylated, high-expressing hub genes. Down-regulated genes with high methylation were related to responses to peptide hormones and estradiol, multi-multicellular organism process. Pathway analysis indicated enrichment in camp, oxytocin signaling pathways. The PPI network identified CFTR, ESR1, and CXCL12 as hypermethylated, low-expressing hub genes. Upon verification in TCGA databases, we found that the expression and methylation statuses of the hub genes changed significantly, and it was consistent with our results.
Conclusion: The novel abnormally MDEGs and pathways in HCC were identified. These results helped us further understand the molecular mechanisms underlying HCC invasion, metastasis, and development. Hub genes can serve as biomarkers for an accurate diagnosis and treatment of HCC, and PTK2, VWF, ITGA2, CFTR, ESR1, and CXCL12 are included.
Keywords: methylation, hepatocellular carcinoma, hub genes, gene expression, bioinformatics analysis
Background
As the second main cause of death for both men and women, cancer also ranks first or second in causing deaths in each age group among women.1 Hepatocellular carcinoma (HCC), the major form of the primary liver cancer, has been the second principal cause of deaths related to cancer, with annual cases of 800,000 globally,2 and involves many risk factors. Substantial studies have indicated that the risk factors influencing HCC comprise liver cirrhosis, hepatitis B virus (HBV), hepatitis C virus (HCV), diabetes, aflatoxins, alcohol, non-alcoholic fatty liver disease (NAFLD), and certain immune-related factors.3–5 HCC is treated differently depending on the tumor stage, as well as liver function and performance. Early-stage radiofrequency ablation (RFA), liver transplantation (LT), and surgical resection are the three common treatments for early liver cancer, which are considered potentially curative.6,7 In addition, primary liver tumors have high metastatic potential and are chemotherapy-resistant, so the HCC prognosis is inaccurate, and the postoperative recurrence is high.8 According to the Global Cancer Statistics in 2018, liver cancer is predicted to be the fourth leading cause of cancer death, and approximately 782,000 patients die.9 Therefore, screening for early HCC and treatment for advanced HCC are crucial for reducing the mortality of HCC.
Mechanisms of epigenetic regulation are critical for coordinating cellular processes as well as functions, including methylation of deoxyribonucleic acid (DNA), non-coding ribonucleic acid (RNA), and histone acetylation.10 As a key factor influencing the epigenetic modification, DNA methylation plays a vital role in the regulation of gene expression as well as strengthening its connection with chromatin remodeling and histone modification.11 At the precancerous stage, the epigenetic abnormalities accumulate in the liver and thereby lead to tumorigenesis by altering hepatocyte differentiation and survival.12 Many studies have reported changes in DNA methylation specific in HCC, especially at the complete genome level.12–14 At present, changes in DNA methylation can be easily detected quantitatively from fixed tissues; thus, using the DNA methylation spectrum as an alternative biomarker is still an active field in clinical cancer research. At the same time, the detection of abnormally methylated expression genes and the in-depth understanding of their characteristics may help us understand the molecular mechanism of HCC, thus, providing new ideas for the liver cancer treatment.
With the large-scale application of sequencing technology in recent decades and the reduction of its cost, more studies on genomic liver cancer have provided new ideas for the diagnosis and treatment of hepatic carcinoma.15,16 Under this context, microarrays based on the high-throughput platforms have become a potential new tool to identify target genes and epigenetic variations, offering new approaches for the HCC treatment.17 The data for this study were obtained from the GEO database, an international public repository, freely available, with a wide variety of microarray data from different researchers, including different species regions.18 In this study, we used bioinformatics methods to synthesize data from the gene expression profiling microarrays (GSE57957, GSE76427) and gene methylation-spectral microarrays (GSE89852, GSE54503). Target genes were identified using R software to explore the interaction networks and signaling pathways associated with liver cancer. Our aim is to find new abnormally methylated genes that can serve as biomarkers for an early liver cancer diagnosis in the future. The in-depth study of key signaling pathways can provide innovative ideas for the precise treatment of liver tumors.
Methods
Data Information of Microarray
We obtained two gene expression profiling datasets (GSE57957, GSE76427) and two gene methylation profiling datasets (GSE89852, GSE54503) through GEO (https://www.ncbi.nlm.nih.gov/geo/). The statement to confirm that all methods were carried out in accordance with relevant guidelines and regulations, and that all experimental protocols were approved by a named institutional and/or licensing committee, and that informed consent was obtained from all subjects or, if subjects are under 18, from a parent and/or legal guardian can be obtained from GEO (SingHealth Centralized Institutional Review Board, PMID: 25093504; SingHealth Institutional Review Board, PMID: 29117471; the Ethics Committees of the National Cancer Center, the National Center for Global Health and Medicine, and Yotsuya Medical Cube, PMID: 28426876; the Institutional Review Board of Columbia University Medical Center (CUMC), PMID: 25861255). The GPL10558 platform (Illumina Human HT-12 V4.0 expression BeadChip) was used to register GSE57957 and GSE76427. GSE57957 obtained 39 tumor samples and 39 adjacent non-tumor samples; whereas, GSE76427 included 115 primary tissue samples and 52 adjacent non-tumor tissue samples derived from 115 HCC patients. GSE89852 and GSE54503 were registered on the GPL13534 platform (Illumina Human Methylation 450 BeadChip). GSE89852 included 37 paired normal and HCC samples from 37 patients; whereas, the samples of 66 HCC tumor and 66 adjacent non-tumor were obtained in GSE54503.
Data Acquisition and Processing
The methylation and expression profiling microarray datasets were analyzed to identify differential expression in the microarray data by using the limma package (v3.12) in R software (v3.6.1) (https://www.r-project.org/). The cut-off standard of P < 0.05 was set to analyze the differentially expressed genes (DEGs). The cut-off values of FDR < 0.05 and β > 0.2 were used to analyze the differentially methylated genes (DMGs). Finally, for the differential expression analysis results, the Venn Diagram package (v1.6.20) in R software was used to identify the overlapping DMGs and DEGs. Overlapping hypermethylated and down-regulated genes generated a Venn diagram of genes with hypermethylation and low expression. Overlapping hypomethylated and up-regulated genes generated a Venn diagram of hypomethylated and highly expressed genes.
Analysis of Functional and Pathway Enrichment
The functional profiles for genes and gene were analyzed and visualized by using the clusters R package software of clusterProfiler (v3.0.4). We investigated the functional profiles of Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene ontology (GO). We set the P value Cutoff to 0.05 and the Q value Cutoff to 0.05.
Generation of PPI Network and Module Analysis
STRING database is a search tool for gene and protein interactions, which helps users easily obtain unique, wide-ranging experiments, and predictive interaction information, and it was used in this study to construct the PPI network of differential genes. The interaction score ≥0.4 was set as a threshold. We then imported the results of the STRING database into Cytoscape software (v3.7.2) and used cytoHubba (v3.18.0) to view the hub genes. To select the six hub genes, we used five ranking methods and overlapped the top ten genes. Betweenness, Closeness, Degree, EcCentricity and Edge percolated component (EPC) methods were used to analyze hypermethylation and down expression genes, while, Betweenness, Bottleneck, Closeness, Radiality and Stress methods were used for hypermethylation and down expression genes. In the Cytoscape software, the MCODE plugin was utilized for module analysis, and the standard values for defining modules were used (MCODE score >3; the number of nodes >5).
Hub Gene Verification
The online database GEPIA (http://gepia.cancer-pku.cn/) is used to interactively analyze gene expression profiles founded on the TCGA database. Customizable features, such as normal/tumor differential expression analysis, tumor type, pathology staging, survival analysis, dimensional analysis, correlation analysis, and similar gene detection, are provided by GEPIA. In this study, we identified differential mRNA expression levels of the hub genes between the liver cancer tumor and non-tumor samples using GEPIA. TCGA comprises comprehensive data of cancer genomic alterations, including mutation, copy number variation, mRNA expression, and methylation data. Besides, the HPA database was employed to validate the translational levels of the hub genes.
Survival Analysis and Genetic Alteration of Hub Genes
The multidimensional cancer genomics are explored, visualized, and analyzed through a resource network provided by cBioPortal (http://www.cbioportal.org/). In this database, large-scale cancer genomic datasets for a variety of cancers are allowed to be visualized, analyzed, and downloaded. In this study, the cBioPortal was used to explore the genetic alterations of the hub genes and the correlation between DNA methylation status and mRNA expression. The GEPIA online tool was employed to assess the prognostic value of the hub genes. We set P < 0.05 as a threshold to indicate the significant difference.
Results
Identification of Abnormally MDEGs in HCC
Figure 1 shows a study design flowchart. By R software, we separately analyzed the data from each microarray to acquire the DEGs or DMGs. In two datasets, 4264 genes were up-regulated (5379 in GSE57957; 6826 in GSE76427) while 2888 were down-regulated (4213 in GSE57957; 6873 in GSE76427). For microarrays of gene methylation, 1780 overlapping hypermethylated genes (7391 in GSE89852; 5116 in GSE54503) together with 4550 overlapping hypomethylated genes (41,046 in GSE89852; 23,579 in GSE54503) were identified. By comparing these two sets of genes, a number of 162 hypermethylated, low-expressing genes and 190 hypomethylated, highly expressed genes were identified (Figure 2). The clustered heat map of GSE57957 and GSE76427 (with the top 50 DEGs) and the top 50 DMGs of GSE89852 and GSE54503 are shown in Figure 3.
 |
Figure 1 The flow chart of this study. Abbreviations: DEGs, differentially expressed genes; DMGs, differentially methylated genes; Hyper-LGs, hypermethylation and low expression genes; Hpo-HGs, hypomethylation and high expression genes. |
 |
Figure 2 Identification of abnormally methylated-differentially expressed genes (MDEGs) in mRNA expression profiling datasets (GSE57957, GSE76427) and gene methylation profiling datasets (GSE89852, GSE54503). (A) Hypermethylation and low expression genes. (B) Hypomethylation and high expression genes. |
 |
Figure 3 Clustered heat map of the top 50 differentially expressed genes (DEGs) and differentially methylated genes (DMGs). Red: upregulated genes; Blue: downregulated genes. (A and B) The heat map of top 50 DEGs in GSE57959 and GSE76427; (C and D) The heat map of top 50 DMGs in GSE89852 and GSE54503. |
Analysis of GO Functional and KEGG Pathway Enrichment
We used the clusterProfiler R package to perform GO functional annotation and KEGG pathway enrichment analysis. The results of the significant enrichment analyses are shown in Table 1. For hypomethylated and highly expressed genes, biological process (BP) analysis results were mainly enriched in calcium homeostasis, keratinocyte proliferation, and collagen-activation signaling pathways. The analysis results of Molecular function (MF) recommended that these genes are mainly involved in growth factor binding, PDZ domain binding, insulin-like growth factor binding, histone serine kinase activity, and adenosine monophosphate (AMP)-activated protein kinase activity. In addition, cellular component (CC) analysis primarily indicated cell-matrix junctions, focal adhesions, cell-matrix adhesion junctions, and membrane anchoring components. For the hypermethylated and low-expressing genes, the BP analysis was mainly enriched in peptide hormone responses, multicellular biological processes, female pregnancy, and regulation of estradiol. MF enrichment indicated proximal promoter sequence-specific DNA binding, organic anion transmembrane transporter, RNA polymerase II proximal promoter sequence-specific DNA binding, and peptidoglycan receptor activity. In addition, CC analysis results were mainly enriched in the basolateral plasma membrane, multivesicular body, filamentous actin, and astrocyte projection.
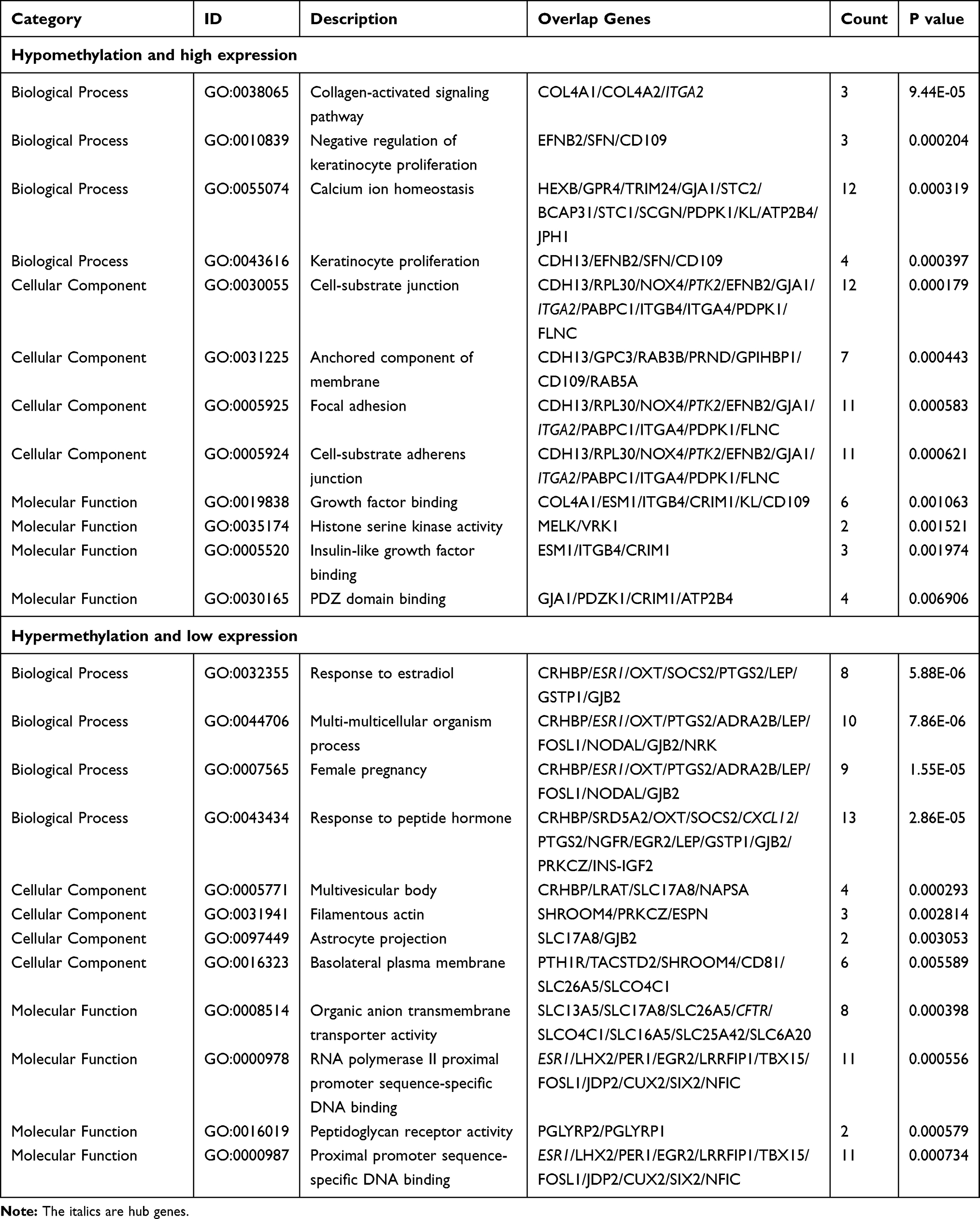 |
Table 1 Gene Ontology Analysis of Aberrantly Methylated Differentially Expressed Genes in Hepatocellular Carcinoma |
Analysis of KEGG Pathway
It can be seen from the KEGG pathway analysis that the hypomethylated, highly expressed genes were remarkably enriched in extracellular matrix (ECM)-receptor interaction, focal adhesion, and phosphatidylinositol 3-kinase (PI3K)-Akt signaling pathways. Table 2 shows that Hypermethylated and low-expressing genes were enriched in axon guidance, cyclic adenosine monophosphate (cAMP) signaling, and Wnt signaling pathway.
 |
Table 2 KEGG Pathway Analysis of Aberrantly Methylated Differentially Expressed Genes in Hepatocellular Carcinoma |
Construction of PPI Network and cytoHubba Analysis
We constructed 80 nodes and 106 edges from the hypomethylated, highly expressed genes, and 108 nodes and 166 edges from the hypermethylated, low-expressing genes. The PPI networks for hypomethylated, highly expressed genes and hypermethylated, low-expressing genes are shown in Figures 4 and 5, respectively. The hub genes were selected by using cytoHubba. By overlapping the top ten genes based on five ranking methods in cytoHubba, three down-regulated, hyper methylated genes (CFTR, ESR1, and CXCL12) and three up-regulated, hypomethylated genes (PTK2, VWF, and ITGA2) were identified (Table 3).
 |
Figure 4 Protein–protein interaction (PPI) network of the hypomethylation and high expression genes. |
 |
Figure 5 Protein–protein interaction (PPI) network of the hypermethylation and low expression genes. |
 |
Table 3 Hub Genes Among the Aberrantly Methylated Differentially Expressed Genes Ranked in cytoHubba |
Module Analysis
Figure 6 shows that two modules in the hypomethylated, highly expressed genes network, and one module in the hypermethylated, low-expressing genes network were statistically significant. In Table 4, we analyzed the GO terms and KEGG pathways. The results indicated that the hypomethylated, highly expressed gene was enriched in pathways significantly concerning ECM-receptor interaction, focal adhesion, and PI3K-Akt signaling pathway. Hypermethylated, low-expressing genes showed enrichment in Estrogen signaling pathway and cytokine-cytokine receptor interaction.
 |
Figure 6 Module analysis of abnormally methylated differentially expressed genes (MDEGs). (A) The hypomethylation and high expression genes in Module 1; (B) the hypomethylation and high expression genes in Module 2; (C) the hypermethylation and low expression genes in Module 1. |
 |
Table 4 Module Analysis of the Protein–Protein Interaction Network |
Identification and Validation of Six Selected Genes
The GEPIA database was employed to investigate the six-hub gene expression, contributing to the further validation of the results. We found that the expression of the three up-regulated, hypomethylated genes and the three down-regulated, hypermethylated genes were dramatically different between tumor and normal tissues (Figure 7). However, the expression of CFTR and ITGA2 was not significantly different between tumor and normal samples. Further experiments are required to confirm this result. Nevertheless, data for the rest four genes were in agreement with our data. Next, TCGA was employed to confirm the results, and the outcome is shown in Table 5. It is found that agreed with our findings, the methylation and expression statuses were altered in TGCA data significantly. Furthermore, the HPA database was used to detect the hub gene expression at the translation level. For up-regulated, hypomethylated genes, ITGA2 and PTK2 were expressed higher in the liver cancer tissue (Figure 8). The difference in VWF between normal liver and cancer samples was not distinct. For down-regulated, hypermethylated genes, the lower expression in the liver cancer was demonstrated through the immuno-histochemical ESR1 results. The differences between ESR1 and CFTR in normal liver and cancer samples were not apparent. To determine if the identified genes are translationally relevant to the liver cancer, further verifications from more clinical samples are required.
 |
Table 5 Validation of the Hub Genes in TCGA |
 |
Figure 7 The expression of the six genes in mRNA expression, using data from the TCGA database in GEPIA. |
 |
Figure 8 Validation of the six hub genes on a translational level using the Human Protein Atlas database. |
Genetic Information of the Six Genes
The connection between six genes and prognosis was evaluated by conducting a survival analysis with GEPIA. We discovered that ITGA2 and ESR1 were tightly related to the overall survival (OS; Figure 9A). For the up-regulated, hypomethylated oncogene ITGA2, its upregulation led to shorter OS. However, downregulated expression of the down-regulated, hypermethylated oncogene ESR1 resulted in worse OS in HCC patients. As for the rest four genes, the trends were close to our prognosis, but not statistically significant. By using the cBioPortal tool, a hub gene network was constructed to find the corresponding drug and the potential therapeutic target. A network made up of six hub genes and 50 most frequently altered neighboring genes of them are illustrated in Figure 9B. The drug targets network showed that five genes, ESR1, CFTR, CXCL12, VWF, and PTK2, are the most popular drug targets nowadays (Figure 9C). We surmise that in the future, ITGA2 could find application as a new drug target. Figure 10A and B exhibits the information on the hub gene alteration. It can be seen that the six hub genes were varied in 277 (51%) in the 442 sequenced cases/patients. Meanwhile, PTK2 and ITGA were altered most often (46% and 8%), including deep deletion, amplification, and missense. The correlation between mRNA expression and DNA methylation of the hub genes in the HCC patient dataset of TCGA is shown in Figure 10C. A negative correlation, which indicates that the methylation regulates the genes’ mRNA expression (except for ESR1), was found and demonstrated the important role of methylation in expressing these genes.
 |
Figure 9 Overall survival analyses of ESR1 and ITGA2 in HCC patient. (A) Downregulation of ESR1 was closely associated with worse overall survival; (B) downregulation of ITGA2 resulted in longer overall survival; (C) the network contained our six hub genes and the 50 most frequently altered neighbor genes. The relationship between hub genes and drugs is also exhibited. |
 |
Figure 10 Genetic alterations connected with the six genes and the relationship between mRNA expression and DNA methylation in the TCGA HCC study. (A) A visual summary of alteration based on a query of the six hub genes, which were altered in 277 (51%) in the 442 sequenced cases/patients; (B) alteration frequency of hub genes; (C) the correlation between mRNA expression and DNA methylation of the six hub genes in the HCC patient dataset of TCGA. |
Discussion
In the development of HCC, abnormal methylation of genes related to tumor not only occurs in the terminal tumor, but is also a common early event. In addition, the frequency of promoter abnormal methylation develops from a precancerous lesion to HCC.19 Related studies have demonstrated that epigenetically silenced tumor suppressor genes can be re-expressed by the use of demethylation and histone modifiers.20,21 It could be a routine treatment similar to other malignancies in the coming years. Therefore, epigenetic changes in tumors can not only serve as indicators or “biomarkers” for screening patients at high stake for HCC, but also provide innovative ideas for the treatment of HCC. In our research, we identified three hypomethylated, highly expressed genes, and three hypermethylated, low-expressing genes using bioinformatics analysis. We clearly verified the functional and enrichment analysis of the hub genes. The interaction network results revealed that, with the development of HCC, relevant genes may involve important pathways related to molecular regulation.
The GO enrichment analysis showed that the BP of genes with hypomethylation and high expression mainly focused on calcium ion homeostasis and keratinocyte proliferation. It is reported that intracellular calcium homeostasis can be adjusted from autophagy, which is closely linked to the emergence and development of tumor formation and autophagy.22,23 Among the genes with high methylation and low expression, BP mainly concentrates in the response to peptide hormones, multi-multicellular organism processes, and female pregnancy. Previous studies have shown that atrial natriuretic peptides (ANP) can eliminate pancreatic adenocarcinoma in up to 80% of thymus-free mice.24 Vasoactive intestinal peptide (VIP), as a regulator of the inflammatory response, is expressed in many types of tumors. Studies have shown that VIP can suppress the apoptosis of HCC cells through the cAMP/Bcl-xl pathway to prevent the progression of HCC.25 In terms of CC, genes with hypomethylation and high expression are mainly concerned with cell-substrate junctions, focal adhesions, and cell-substrate adherences junctions; whereas, genes with hypermethylation and low expression are mainly concerned with the composition of the multivesicular body, basolateral plasma membrane, and filamentous actin. The adhesion and capture of tumor cells in the microcirculation are the primary conditions for tumor vascular metastasis. Therefore, overexpression of cell adhesion molecules can enhance the interaction between focal adhesion and ECM receptors, and accelerate the metastasis of liver cancer cells in the blood vessels and settlement at the metastasis site.26–28 Leda-1/Pianp is a transmembrane protein of type I located in the basolateral membrane of polarized epithelial cells and enhances the tumor cells’ resistance to unstable junctions and the tightness and stability of intercellular junctions.29 In terms of MF, the main function of genes with hypomethylation and high expression is binding to growth factors, PDZ domain binding, and insulin-like growth factor binding. However, the MF of genes with hypermethylation and low expression is mainly for RNA polymerase II proximal promoter sequence-specific DNA binding and organic anion transmembrane transporter activity. For example, the insulin-like growth factor-I receptor (IGF-IR) in most of the transformed cells exhibits potential cell-survival and antiapoptotic activities. Additionally, its expression is negatively regulated by p53, BRCA1, and WT1.30 Studies based on animal models, as well as in vitro, have demonstrated that IGF plays a key role in the cell cycle, proliferation, survival, migration, apoptosis inhibition, protein synthesis, and cell growth of HCC.31
From the KEGG pathway enrichment analysis, hypomethylated, highly expressed genes were found to be significantly enriched in HPV infection, ECM-receptor interactions, focal adhesions, the PI3K-Akt signaling pathway, and the AMPK signaling pathway. Studies show that by inhibiting the PI3K/Akt pathway, petroleum ether (PE) may induce the apoptosis of hepatocellular carcinoma cells, thus producing an anti-tumor effect.32 Hou et al indicated that inhibition of the PI3K/Akt pathway could significantly reduce the phosphorylation level of FOXO3a, accompanied by an increase in the total level of FOXO3a, and significantly inhibited the proliferation of tumor cells.33 Stearoyl-CoA desaturase 1 (SCD1) regulates autophagy through the AMPK pathway and has a great influence on HCC.34 At the same time, we found that interaction of neuroactive ligand-receptor is the most abundant enrichment pathway with low expression of hypermethylated genes. In addition, concentration is also found in the cAMP pathway, oxytocin signaling pathway, axon guidance, and Wnt signaling pathway. Liu et al proposed a Bayesian method to demonstrate that mitogen-activated protein kinase (MAPK) signaling pathway and neuroactive ligand–receptor interaction are closely related to HCC.35 Tao et al found that the upstream KEGG pathway of methylated genes in individual hepatocytes in HBV-associated hepatocellular carcinoma is a “neuroactive ligand–receptor interaction”.36 These results show that the neuroactive ligand-receptor pathway greatly affects the pathogenesis of liver cancer.
The results based on PPI analysis showed that three hypomethylated and highly expressed hub genes were PTK2, VWF, and ITGA2. PTK2 activates Wnt/β-catenin signaling by fostering the beta-catenin nuclear accumulation in HCC cells, which is connected with OS, marker expression of liver cancer stem cell, and recurrence-free survival (RFS) in HCC patients.37 In addition, increased PTK2 expression can improve the prognosis of chronic lymphocytic leukemia (CLL) patients treated with cyclophosphamide and rituximab-cyclophosphamide. PTK2 expression may be a useful biomarker in future clinical trials.38 In our study, PTK2 expression in tumor samples was higher than that in normal tissues, and there was a statistically dramatic difference between these two groups. At the same time, our data showed a negative correlation between PTK2 mRNA expression and DNA methylation. The PTK2 mutation rate was found to be up to 46%, suggesting that abnormal methylation of DNA may result in mutations and can potentially be a target for future drug therapy. As the largest plasma protein in humans, VWF is an adhesive polysaccharide protein that intervenes the adhesion of platelets to the endothelial surface and subendothelial matrix. It is also the carrier of clotting factor VIII in the blood.39 A review of elevated VWF levels concerned with increased risk of myocardial infarction, stroke, venous thrombosis, and diabetes suggests that the VWF gene plays a crucial role in the regulation of the blood clotting and bleeding balance.40 In addition to its important role in hemostasis, an increasing amount of literature has shown that VWF has additional anti-tumor effects, mainly through negative regulation of angiogenesis and apoptosis.41 Liu et al demonstrated that VWF is connected to the pathogenesis of HBV infection and duplication, and is also related to the clinicopathological staging of HBV-infected HCC patients.42 The results showed that the VWF expression in normal and tumor tissues was dramatically different, and that the VWF expression in liver cancer samples was apparently higher than that in normal tissues. However, data obtained from HPA showed no obvious difference in VWF gene expression between normal liver tissues and tumor tissues. The mutation rate of VWF was up to 7%, and the expression of mRNA was negatively correlated with methylation. This may be the result of abnormal methylation, which needs to be confirmed by further experiments. It has been reported that ITGA2 is essential in cell survival, invasion, migration, and angiogenesis.43,44 Overexpression of ITGA2, which is methylation-independent, is connected with inaccurate prognosis in acute myeloid leukemia (AML).45 In addition, ADAR1 can enhance HCC metastasis by increasing ITGA2 expression and promoting tumor cell adhesion to the ECM.44 In our study, we found that highly expressed ITGA2 is closely related to OS and RFS. In the verification of HPA data, the expression of ITGA2 in liver cancer tissues was obviously higher than that in normal samples. The mutation rate of ITGA2 was 8%, suggesting that an analogous mechanism might exist in HCC tumorigenesis. At the same time, we found that the expression of ITGA2 was negatively correlated with the methylation state. Therefore, we can speculate that ITGA2 gene mutation may lead to its hypomethylation; thus, contributing to the emergence and development of liver cancer. The three hypomethylated and highly expressed genes screened in this study were all highly expressed in the liver cancer tissues, and the mRNA expression levels were negatively correlated with the methylation state. This suggests that abnormal methylation of these three genes may accelerate the development of liver cancer.
With regard to the hypermethylated and low-expressing genes, three genes were identified: CFTR, ESR1, and CXCL12. CFTR is a large glycoprotein located on chromosome 7, belonging to the adenosine triphosphate (ATP)-binding superfamily of proteins. CFTR is expressed in various cell types and is concerned with epithelial cells in the airway, sinuses, gastrointestinal tract, sweat glands, and genital and nervous systems.46 At present, there are no reports on the impact of CFTR on the occurrence and development of liver cancer. Further experiments are needed to confirm its function in HCC. In both males and females, the estrogen receptor (ER), ESR1, is the main receptor in the liver and mediates the liver’s response to estrogen.47 As an important sex hormone, estrogen functions in various processes such as cell metabolism, cell differentiation, and tissue development.48 Villa et al proved that the transcript status of ER in patients with HCC has a novel value in prognosis.49 Moreover, in inoperable patients with HCC, ER transcription was the largest negative predictor of survival. Naugler et al found a protective effect of ER alpha in liver cancer.50 In males, Kupffer cells (KCs) can produce estrogen-mediated inhibitions of interleukin (IL)-6 to reduce the risk of liver cancer, and these outcomes may be useful for the prevention of HCC. In this research, we discovered that the ESR1 expression in tumor tissues was lower than that in normal tissues, and the difference between the two tissues was statistically significant. This finding was consistent with the HPA validation data. Highly expressed ERS1 is associated with better OS and RFS. The mutation rate of the ESR1 gene was 6%, and the mRNA expression was positively correlated with the methylation state. This conclusion requires further experimental confirmation. Chemokines are potential targets for anti-tumor angiogenesis therapy, and the chemokine, CXCL12, promotes tumor metastasis and angiogenesis mainly through the receptor, CXCR4.51 A meta-analysis investigating the relationship between CXCL12 expression and various cancer survivals indicated that CXCL12 plays a crucial role in cancer occurrence.52 However, its role in liver cancer needs to be further verified. From our results, we found that the expression of CXCL12 was indeed low in HCC samples from multiple datasets. The gene mutation rate of CXCL12 was 5%, and its expression was negatively correlated with the methylation state. We speculate that the mutation of CXCL12 may lead to hypermethylation of the gene and decreased mRNA expression, thus, contributing to the emergence and development of liver cancer. Compared with non-tumor tissues, the three differentially expressed genes screened in this study were down-regulated in liver cancer; however, in liver cancer, their action mechanism of remains unclear. Therefore, we surmise that the abnormal methylation of these three genes may promote the development of liver cancer through other aspects besides expression.
Module analysis of PPI network for hypermethylated and low-expressing genes indicated that the estrogen signaling pathway, cAMP signaling pathway, and interaction of neuroactive ligand-receptor, might be involved in the HCC progression. As discussed above, the neuroactive ligand-receptor interaction pathway plays a critical role in liver cancer, which is highly consistent with our conclusion. Although the liver is not a classic target organ for estrogen, studies have shown that ER exhibits a rapid hepatocyte response to estrogen, suggesting that estrogen signaling contributes to rapid occurrence and progression of liver cancer. Meanwhile, studies have shown that estrogen can regulate the HCC malignant degree in vivo by depressing the invasion of tumor cells, impeding cell cycle progression, and facilitating apoptosis.53 Cyclic Adenosine monophosphate (cAMP) is a key intracellular second messenger that can affect plenty of cellular processes.54 Hara et al pointed out that VIP can impede the HCC progression through apoptosis via the cAMP/Bcl-xL pathway.25 More research may be needed to verify our results. The PPI network module analysis for hypomethylated and highly expressed genes demonstrated that the focal adhesion, ECM-receptor interaction, human papillomavirus infection, and proteasome might be connected with HCC progression. The focal adhesion and ECM-receptor interaction are crucial cellular processes in the development of cancer. However, the roles of HPV infection and the proteasome in HCC, together with the impact of abnormal methylation, remain unknown. In HCC occurrence and progression, future investigations are needed to explain how abnormal methylation influences the functional roles of these pathways.
There are some limitations to be noted in our research. First, there is a lack of analysis of clinical data in our study. Secondly, there are inconsistencies in the conclusions obtained from data obtained from different platforms; and, further experiments are required to verify our conclusions. Thirdly, HCC is closely related to HBV, HBC, chronic alcoholism, smoking, aflatoxins, and other factors. No relevant data sets were selected for analysis in this study. In general, our conclusions are based on bioinformatics analysis, which has certain reference value; however, to further verify our results, complementary molecular experiments are encouraged.
Conclusions
In summary, new abnormally methylated genes and HCC-related pathways were discovered in this study. Moreover, we have verified the six selected genes with abnormal methylation from multiple aspects, and these outcomes could provide us with a greater insight into the development and progression of HCC. Based on abnormal methylation, hub genes, including PTK2, VWF, ITGA2, CFTR, ESR1, and CXCL12, could serve as biomarkers for an accurate HCC diagnosis and treatment. It is hoped that these findings can attract more attention to the molecular mechanisms of cancer in the future.
Abbreviations
AML, acute myeloid leukemia; AMP, adenosine monophosphate; AMPK, adenosine monophosphate-activated protein kinase; ATP, adenosine triphosphate; BP, biological process; cAMP, cyclic adenosine monophosphate; CC, cellular component; CLL, chronic lymphocytic leukemia; DEGs, differentially expressed genes; DMGs, differentially methylated genes; DNA, deoxyribonucleic acid; ER, estrogen receptor; GEO, gene expression omnibus; GEPIA, gene expression profiling interactive analysis; GO, gene ontology; HBV, hepatitis B virus; HCC, hepatocellular carcinoma; HCV, hepatitis C virus; HPV, human papillomavirus; IGF-IR, insulin-like growth factor-I receptor; KCs, kupffer cells; KEGG, Kyoto encyclopedia of genes and genomes; LT, liver transplantation; MDEGs, methylated differentially expressed genes; MF, molecular function; MAPK, mitogen-activated protein kinase; NAFLD, non-alcoholic fatty liver disease; PE, petroleum ether; PPI, protein–protein interaction; RFA, radiofrequency ablation; RFS, recurrence-free survival; RNA, ribonucleic acid; TACE, transcatheter arterial chemoembolization; TCGA, the cancer genome atlas; VIP, vasoactive intestinal peptide.
Data Sharing Statement
The datasets used and/or analyzed in the current study are available from the corresponding author on reasonable request.
Author Contributions
CZ carried out the study design. QLC and QY carried out data acquisition and functional analysis, KLF, CFX performed pathway enrichment analysis. KLF, CFX, CKF, JNW, LHL, and YL were involved in data analysis. QLC, KLF and CZ wrote and revised the manuscript. All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Funding
Authors would like to thank the National Natural Science Foundation of China (Grant No.81873303), Natural Science Foundation of Guangdong Province, China (2019A1515011013), Key Projects of Educational Commission of Guangdong Province, China (2019KZDXM045), and Medical innovation project of the First Affiliated Hospital of Guangzhou University of Chinese Medicine (2019IIT18) for financial support.
Disclosure
The authors declare that have no competing interests in this work.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics. CA Cancer J Clin. 2019;69(1):7–34. doi:10.3322/caac.21551
2. Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–E386. doi:10.1002/ijc.29210
3. Gomaa AI, Khan SA, Toledano MB, Waked I, Taylor-Robinson SD. Hepatocellular carcinoma: epidemiology, risk factors and pathogenesis. World J Gastroenterol. 2008;14(27):4300–4308. doi:10.3748/wjg.14.4300
4. Parikh S, Hyman D. Hepatocellular cancer: a guide for the internist. Am J Med. 2007;120(3):194–202. doi:10.1016/j.amjmed.2006.11.020
5. Singal AG, El-Serag HB. Hepatocellular carcinoma from epidemiology to prevention: translating knowledge into practice. Clin Gastroenterol Hepatol. 2015;13(12):2140–2151. doi:10.1016/j.cgh.2015.08.014
6. Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet. 2003;362(9399):1907–1917. doi:10.1016/s0140-6736(03)14964-1
7. Lopez PM, Villanueva A, Llovet JM. Systematic review: evidence-based management of hepatocellular carcinoma – an updated analysis of randomized controlled trials. Aliment Pharmacol Ther. 2006;23(11):1535–1547. doi:10.1111/j.1365-2036.2006.02932.x
8. Bruix J, Gores GJ, Mazzaferro V. Hepatocellular carcinoma: clinical frontiers and perspectives. Gut. 2014;63(5):844–855. doi:10.1136/gutjnl-2013-306627
9. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
10. Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33(Suppl):245–254. doi:10.1038/ng1089
11. Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci. 2006;31(2):89–97. doi:10.1016/j.tibs.2005.12.008
12. Farazi PA, DePinho RA. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat Rev Cancer. 2006;6(9):674–687. doi:10.1038/nrc1934
13. Sceusi EL, Loose DS, Wray CJ. Clinical implications of DNA methylation in hepatocellular carcinoma. HPB. 2011;13(6):369–376. doi:10.1111/j.1477-2574.2011.00303.x
14. Song MA, Tiirikainen M, Kwee S, Okimoto G, Yu H, Wong LL. Elucidating the landscape of aberrant DNA methylation in hepatocellular carcinoma. PLoS One. 2013;8(2):e55761. doi:10.1371/journal.pone.0055761
15. Goodwin S, McPherson JD, McCombie WR. Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet. 2016;17(6):333–351. doi:10.1038/nrg.2016.49
16. Huang W, Skanderup AJ, Lee CG. Advances in genomic hepatocellular carcinoma research. Gigascience. 2018;7(11). doi:10.1093/gigascience/giy135
17. Nakagawa H, Shibata T. Comprehensive genome sequencing of the liver cancer genome. Cancer Lett. 2013;340(2):234–240. doi:10.1016/j.canlet.2012.10.035
18. Edgar R, Domrachev M, Lash AE. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30(1):207–210. doi:10.1093/nar/30.1.207
19. Tischoff I, Tannapfe A. DNA methylation in hepatocellular carcinoma. World J Gastroenterol. 2008;14(11):1741–1748. doi:10.3748/wjg.14.1741
20. Gailhouste L, Liew LC, Yasukawa K, et al. Differentiation therapy by epigenetic reconditioning exerts antitumor effects on liver cancer cells. Mol Ther. 2018;26(7):1840–1854. doi:10.1016/j.ymthe.2018.04.018
21. Raggi C, Factor VM, Seo D, et al. Epigenetic reprogramming modulates malignant properties of human liver cancer. Hepatology. 2014;59(6):2251–2262. doi:10.1002/hep.27026
22. Engedal N, Torgersen ML, Guldvik IJ, et al. Modulation of intracellular calcium homeostasis blocks autophagosome formation. Autophagy. 2013;9(10):1475–1490. doi:10.4161/auto.25900
23. White E, Mehnert JM, Chan CS. Autophagy, metabolism, and cancer. Clin Cancer Res. 2015;21(22):5037–5046. doi:10.1158/1078-0432.CCR-15-0490
24. Vesely DL, Elchelbrum EJ, Sun Y, et al. Elimination of up to 80% of human pancreatic adenocarcinomas in athymic mice by cardiac hormones. In Vivo. 2007;21:445–452.
25. Hara M, Takeba Y, Iiri T, et al. Vasoactive intestinal peptide increases apoptosis of hepatocellular carcinoma by inhibiting the cAMP/Bcl-xL pathway. Cancer Sci. 2019;110(1):235–244. doi:10.1111/cas.13861
26. Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. 2002;2(8):563–572. doi:10.1038/nrc865
27. Yilmaz M, Christofori G, Lehembre F. Distinct mechanisms of tumor invasion and metastasis. Trends Mol Med. 2007;13(12):535–541. doi:10.1016/j.molmed.2007.10.004
28. Zhang C, Peng L, Zhang Y, et al. The identification of key genes and pathways in hepatocellular carcinoma by bioinformatics analysis of high-throughput data. Med Oncol. 2017;34(6):101. doi:10.1007/s12032-017-0963-9
29. Evdokimov K, Biswas S, Schledzewski K, et al. Leda-1/Pianp is targeted to the basolateral plasma membrane by a distinct intracellular juxtamembrane region and modulates barrier properties and E-Cadherin processing. Biochem Biophys Res Commun. 2016;475(4):342–349. doi:10.1016/j.bbrc.2016.05.092
30. Werner H, Maor S. The insulin-like growth factor-I receptor gene: a downstream target for oncogene and tumor suppressor action. Trends Endocrinol Metab. 2006;17(6):236–242. doi:10.1016/j.tem.2006.06.007
31. Adamek A, Kasprzak A. Insulin-Like Growth Factor (IGF) system in liver diseases. Int J Mol Sci. 2018;19(5). doi:10.3390/ijms19051308
32. Hui F, Qin X, Zhang Q, et al. Alpinia oxyphylla oil induces apoptosis of hepatocellular carcinoma cells via PI3K/Akt pathway in vitro and in vivo. Biomed Pharmacother. 2019;109:2365–2374. doi:10.1016/j.biopha.2018.11.124
33. Hou Y, Sun G, Jiang X, Zhu Z, Yang J. Nuclear forkhead box O3a accumulation contributing to the proliferative suppression in liver cancer cells by PI3K/Akt signaling pathway. J Cancer Res Ther. 2018;1998–4138. doi:10.4103/0973-1482.204891
34. Huang GM, Jiang QH, Cai C, Qu M, Shen W. SCD1 negatively regulates autophagy-induced cell death in human hepatocellular carcinoma through inactivation of the AMPK signaling pathway. Cancer Lett. 2015;358(2):180–190. doi:10.1016/j.canlet.2014.12.036
35. Liu Z, Gartenhaus RB, Tan M, Jiang F, Jiao X. Gene and pathway identification with Lp penalized Bayesian logistic regression. BMC Bioinform. 2008;9:412. doi:10.1186/1471-2105-9-412
36. Tao R, Li J, Xin J, et al. Methylation profile of single hepatocytes derived from hepatitis B virus-related hepatocellular carcinoma. PLoS One. 2011;6(5):e19862. doi:10.1371/journal.pone.0019862
37. Fan Z, Duan J, Wang L, et al. PTK2 promotes cancer stem cell traits in hepatocellular carcinoma by activating Wnt/beta-catenin signaling. Cancer Lett. 2019;450:132–143. doi:10.1016/j.canlet.2019.02.040
38. Weisser M, Yeh RF, Duchateau-Nguyen G, et al. PTK2 expression and immunochemotherapy outcome in chronic lymphocytic leukemia. Blood. 2014;124(3):420–425. doi:10.1182/blood-2013-12-538975
39. Franchini M, Lippi G. The role of von Willebrand factor in hemorrhagic and thrombotic disorders. Crit Rev Clin Lab Sci. 2007;44(2):115–149. doi:10.1080/10408360600966753
40. Xiang Y, Hwa J. Regulation of VWF expression, and secretion in health and disease. Curr Opin Hematol. 2016;23(3):288–293. doi:10.1097/MOH.0000000000000230
41. Franchini M, Frattini F, Crestani S, Bonfanti C, Lippi G. von Willebrand factor and cancer: a renewed interest. Thromb Res. 2013;131(4):290–292. doi:10.1016/j.thromres.2013.01.015
42. Liu Y, Wang X, Li S, et al. The role of von Willebrand factor as a biomarker of tumor development in hepatitis B virus-associated human hepatocellular carcinoma: a quantitative proteomic based study. J Proteomics. 2014;106:99–112. doi:10.1016/j.jprot.2014.04.021
43. San Antonio JD, Zoeller JJ, Habursky K, et al. A key role for the integrin alpha2beta1 in experimental and developmental angiogenesis. Am J Pathol. 2009;175(3):1338–1347. doi:10.2353/ajpath.2009.090234
44. Zhang Z, Ramirez NE, Yankeelov TE, et al. Alpha2beta1 integrin expression in the tumor microenvironment enhances tumor angiogenesis in a tumor cell-specific manner. Blood. 2008;111(4):1980–1988. doi:10.1182/blood-2007-06-094680
45. Lian XY, Zhang W, Wu DH, et al. Methylation-independent ITGA2 overexpression is associated with poor prognosis in de novo acute myeloid leukemia. J Cell Physiol. 2018;233(12):9584–9593. doi:10.1002/jcp.26866
46. Elborn JS. Cystic fibrosis. Lancet. 2016;388(10059):2519–2531. doi:10.1016/s0140-6736(16)00576-6
47. Khristi V, Ratri A, Ghosh S, et al. Disruption of ESR1 alters the expression of genes regulating hepatic lipid and carbohydrate metabolism in male rats. Mol Cell Endocrinol. 2019;490:47–56. doi:10.1016/j.mce.2019.04.005
48. Deroo BJ, Buensuceso AV. Minireview: estrogen receptor-beta: mechanistic insights from recent studies. Mol Endocrinol. 2010;24(9):1703–1714. doi:10.1210/me.2009-0288
49. Villa E, Moles A, Ferretti I, et al. Natural history of inoperable hepatocellular carcinoma: estrogen receptors’ status in the tumor is the strongest prognostic factor for survival. Hepatology. 2000;32(2):233–238. doi:10.1053/jhep.2000.9603
50. Naugler WE, Sakurai T, Kim S, et al. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2008;317:121–124. doi:10.1126/science.1140485
51. Èller AM, Homey B, Soto H, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410(6824):50–56.
52. Samarendra H, Jones K, Petrinic T, et al. A meta-analysis of CXCL12 expression for cancer prognosis. Br J Cancer. 2017;117(1):124–135. doi:10.1038/bjc.2017.134
53. Xu H, Wei Y, Zhang Y, et al. Oestrogen attenuates tumour progression in hepatocellular carcinoma. J Pathol. 2012;228(2):216–229. doi:10.1002/path.4009
54. Beavo JA, Brunton LL. Cyclic nucleotide research — still expanding after half a century. Nat Rev Mol Cell Biol. 2002;3(9):710–718.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.