Back to Journals » International Journal of General Medicine » Volume 13
Usefulness of Circulating Methylated p16 as a Noninvasive Molecular Biomarker for Hepatitis C-Related Hepatocellular Carcinoma with Normal Serum Alpha-Fetoprotein Levels
Authors Elsewify WAE ![]() , Hassan EA
, Hassan EA ![]() , Mekky MA
, Mekky MA ![]() , Abd El-Rehim ASED
, Abd El-Rehim ASED ![]() , Sayed ZEAA
, Sayed ZEAA ![]() , Abdel Malek MO, ElMelegy TTH
, Abdel Malek MO, ElMelegy TTH ![]() , Sabry A
, Sabry A ![]()
Received 26 February 2020
Accepted for publication 25 March 2020
Published 30 April 2020 Volume 2020:13 Pages 147—155
DOI https://doi.org/10.2147/IJGM.S249272
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Wael Abd Elgwad Elsewify,1 Elham Ahmed Hassan,2 Mohamed A Mekky,2 Abeer Sharaf El-Din Abd El-Rehim,2 Zain El-Abdeen Ahmed Sayed,3 Mohamed Omar Abdel Malek,2 Tarek TH ElMelegy,4 Abeer Sabry5
1Department of Internal Medicine, Faculty of Medicine, Aswan University, Aswan, Egypt; 2Department of Gastroenterology and Tropical Medicine, Faculty of Medicine, Assiut University, Assiut, Egypt; 3Department of Internal Medicine, Faculty of Medicine, Assiut University, Assiut, Egypt; 4Department of Clinical Pathology, Faculty of Medicine, Assiut University, Assiut, Egypt; 5Department of Internal Medicine, Faculty of Medicine, Helwan University, Cairo, Egypt
Correspondence: Wael Abd Elgwad Elsewify
Department of Internal Medicine, Faculty of Medicine, Aswan University, Aswan 81528, Egypt
Tel +20 1001657295
Fax +20 973480449
Email [email protected]
Background: Screening of hepatocellular carcinoma (HCC) is challenged especially in patients with normal alpha-fetoprotein (AFP) levels. Aberrant p16 methylation has been implicated in HCC.
Objectives and Aims: This study aimed to assess serum methylated p16 (MP16) expression levels and to evaluate MP16 diagnostic performance in HCC detection among HCV-infected Egyptian patients with normal AFP levels.
Methods: MP16 levels were quantified using real-time PCR in 230 serum samples (30 healthy controls, 95 with HCV-HCC, 40 with chronic hepatitis C “CHC” and 65 with HCV cirrhosis). Diagnostic performance of MP16 for diagnosis of HCC was done using receiver operator characteristic curve analysis.
Results: Serum MP16 levels were significantly higher in HCC than CHC, cirrhosis, and healthy subjects and significantly higher in HCC with normal AFP levels than those with higher AFP. ROC curves revealed promising diagnostic performance for MP16 in discriminating HCC with normal AFP levels from non-HCC cases. This predictive ability improved by combining MP16 and AFP (AUC of 0.872 with 100% sensitivity, 76.5% specificity, 79.1% positive predictive value, 100% negative predictive value, and 87.5% accuracy).
Conclusion: MP16 can be a potential noninvasive molecular biomarker for HCC detection in patients with hepatic mass(es) and normal AFP levels especially in those where liver biopsy and radiological imaging cannot be done.
Keywords: methylated p16, hepatitis C virus, hepatocellular carcinoma, alpha-fetoprotein, quantitative real-time PCR
Introduction
Hepatocellular carcinoma (HCC) is the fifth most frequent human malignancies and the third most common cause of death caused by cancer, globally.1 Even though advances in the diagnostic and therapeutic modalities of HCC, there is still increasing in its incidence and lowering of the 5-year survival rate.2 It has been revealed that different carcinogens activate or inhibit certain pathways during HCC development and progression. It is well known that risk factors as chronic viral hepatitis and cirrhosis are associated with HCC and it is also revealed that hepatitis C virus (HCV) infection accelerates the methylation process in HCC.3
Both genetic and epigenetic alterations are being increasingly identified for a better understanding of the patho-hepatic carcinogenesis.4,5 Genetic alterations entail the irreversible DNA sequence changes, hence cumulative oncogene activation and tumour suppressor genes (TSGs) inactivation.6 Furthermore, epigenetic alterations that denote the reversible and heritable modifications in gene expression without any DNA sequence alterations have been also concerned in HCC development and progression.4 DNA methylation is one of the major epigenetic modifications in eukaryotic genome. It plays an essential role in the signalling pathways including apoptosis, cell proliferation, adherence, DNA repair, cell-cycle control, and during cancer development.7 Moreover, aberrant DNA methylation includes genome-wide hypomethylation as well as promoter CpG island hypermethylation.8 Methylation of the promoter CpG islands is present in the precursor lesions of a variety of cancers, including HCC and has been proved to be an early event in carcinogenesis where it inhibits the transcriptional initiation causing permanent silence of the downstream genes.9 Multiple studies have documented certain cancer-related genes to be frequently methylated in HCC such as p16, CDKN2A, RASSF1A, and GSTP1.10–12
Many potential noninvasive biomarkers may help for the early detection of HCC in high-risk population hand in hand or even earlier to the currently available screening tests as alpha-fetoprotein (AFP) analysis and ultrasound.10–14 These biomarkers include the detection of tumour-derived genetic and epigenetic alterations as point mutations, microsatellite instabilities, losses of heterozygosity and DNA hypermethylation in plasma or serum which identically reflects the tumour origin of altered cell-free DNA.15
Previous studies have reported the detection of frequencies or sequences of gene P16 methylation [p16 (INK4A)] which is a cyclin-dependent kinase inhibitor gene located on chromosome 9p21 in HCC patients.6–18
A third of HCC cases have normal AFP levels at the time of HCC diagnosis and usually remain low (<400–500 ng/mL), even with advanced HCC, in addition, a third of non-HCC cases have abnormal AFP.13 Because of both false positives and false negatives, the accuracy of the AFP is challenged and its ongoing use for HCC screening test is still a matter of debate. So, we aimed to quantitatively estimate serum levels of methylated p16 (MP16) and to evaluate its diagnostic performance in the detection of HCC among HCV-infected patients, especially with normal serum AFP levels.
Materials and Methods
Study Design
This case–control study was done at Assiut University Hospital, Assiut, Egypt, from July 2018 to November 2019. The study was approved by the Local Ethics Committee of Assiut University Hospital and was conducted in accordance with the provisions of the Declaration of Helsinki. A written informed consent was obtained from all the participants before enrolment.
Study Population
The study group included 200 naïve chronic HCV-infected patients and they were divided into 95 patients with HCC and 105 patients with benign liver diseases “BLD” including 40 had chronic hepatitis C “CHC” and 65 had liver cirrhosis. Diagnosis of chronic liver disease was based on clinical, biochemical, and imaging findings. The severity of liver cirrhosis was measured by Child-Pugh and MELD scores.19,20 The diagnosis of HCC was based on triphasic computed tomography scan according to the guidelines for the diagnosis and treatment of HCC.21 Thirty volunteers who were sex- and age-matched, had negative markers for HBV and HCV infections, and apparently healthy based on clinical and laboratory examination were served as normal controls.
Those patients were consecutively selected from outpatient clinics and inpatient wards of The Tropical Medicine and Gastroenterology, and Internal Medicine Departments, Assiut University Hospital, Assiut, Egypt, while controls were selected randomly from outpatient clinic and the patients’ relatives. Patients with evidence of non-HCV related liver diseases, HCV/HBV co-infection, HIV co-infection, hepatic metastasis, or receiving antiviral therapy were excluded.
Medical history and clinical examination were performed and measurement of liver function tests, serum creatinine, AFP, and serum methylated p16 (MP16) levels was undertaken.
Collection and Processing of Blood Specimens
Under aseptic precautions, 3 ml of blood was collected before therapy from each subject into plain polypropylene tube. Blood was allowed to clot in the tube and then centrifuged for 10 min at 4000 rpm. Sera were inspected to ensure it is clear and non-haemolyzed and were carefully transferred into plain polypropylene tubes. Part of collected serum was used for chemical investigations and remaining part was stored at −20°C until further processing. Extraction of DNA from sera, bisulfite conversion, and quantitative real-time PCR (qPCR) amplification were done in the Molecular Unit, Laboratory of Clinical Immunology, Assiut University Hospital, Egypt. The laboratory specialist was blinded to patient diagnosis and patient disease category.
Serum creatinine and liver function tests were done on the automated chemistry analyzer Dimension RxL Max (Siemens, USA), while serum α-fetoprotein was measured using Architect AFP reagent kit (REF 3P36, Abbott, Ireland) on Architect i1000 (Abbott, USA). This was performed according to manufacturers’ instructions.
DNA Extraction from Sera
DNA was extracted from serum using QIAamp DNA Mini Kit (catalog no. 51304, Qiagen, Hilden, Germany) using the blood and body fluids protocol according to manufacturer’s instructions, a final elution volume of 50 µl was used.
DNA Bisulfite Conversion
The principle of DNA bisulfite conversion is that treating DNA with bisulfite would result in the conversion of unmethylated cytosine residues into uracil. Methylated cytosine residues, on the other hand, would remain unchanged. Thus, the DNA sequences of methylated and unmethylated genomic regions after bisulfite conversion would differ and can be differentially amplified by sequence-specific PCR primers. DNA Bisulfite conversion was carried out using EpiTect Fast DNA Bisulfite Kit (catalog no. 59824, Qiagen, Hilden, Germany) as follows:
- The bisulfite reaction mixture was prepared following the manufacturer’s recommended setup using 25 µl of DNA elutes.
- Reaction tubes were loaded into Veriti 96 well-thermal cycler (Applied Biosystems, USA) programmed for the following cycling conditions: 2 cycles (5 min denaturation at 95°C, then 20 min incubation at 60ºC) followed by a hold step at 20ºC.
- Cleanup of converted DNA was done according to manufacturer’s instructions using carrier RNA in final concentration of 10 μg/mL in buffer BL.
- Bisulfite converted DNA was finally eluted using 12 µl of buffer BE.
Real-Time PCR Amplification
For relative quantitation of MP16, amplification of endogenous control (GAPDH) was performed to normalize levels of methylated p16 towards differences in the amount of DNA loaded into PCR reaction tubes. For each sample, each of MP16 and GAPDH was amplified in a separate PCR tube. All amplification/quantification experiments included a No-Template Control (containing all the components of the reaction except for the DNA template) to detect carry-over contamination.
Real-time PCR amplification of methylated p16 was done using custom methylated sequence-specific detection primers (catalog no. 4304970, Applied Biosystems, USA), Taqman probe (FAM/TAMRA; catalog no. 450025, Applied Biosystems, USA), and EpiTectMethyLight PCR+ROX™ vial Kit (catalog no. 59496, Qiagen, Hilden, Germany). PCR reaction mixture was prepared following manufacturer recommendations: 10 µl of 2x Epitect Methy Light master mix (w/o ROX), 0.4 µl of 50x ROX dye solution, 0.4 μM of each primer, 0.2 μM probe, and 2.5 µl of bisulfite converted DNA, in a final total volume of 20 µl per reaction. The previously described10,22,23 sequences of methylated p16 primers: 5́-TTATTAGAGGGTGGGGCGGATCGC-3́ and 5́-GACCCCGAACCGCGACCGTAA-3́ and the previously described10 probe sequence: 5́-AGTAGTATGGAGTTTTCGGTTGATTGGTTG-3́ were used.
GAPDH real-time PCR amplification was performed using human GAPDH endogenous control primer/probe set (VIC™/MGB probe, catalog no. 4326317E, Applied Biosystems, USA) and TaqMan® Universal PCR Master Mix II- no UNG (catalog no. 4440043, Applied Biosystems, USA). PCR reaction mixture was prepared according to manufacturer’s instructions: 10 µl of 2x TaqMan Universal PCR Master Mix, 1 µl of 20x GAPDH endogenous control primer/probe, and 5 µl of DNA in a final total volume of 20 µl per reaction.
PCR tubes were loaded into 7500 Fast Real-Time PCR System (Applied Biosystems, USA) with initial denaturation/activation step of 10 min at 95°C followed by 40 cycles (denaturation for 15 s at 95°C and annealing/extension for 60 s at 60ºC).
Interpretation of Results
In each run, raw data were analysed using the automated setting (7500 Fast Software, Version 2.0.5) to assign baseline and threshold. The cycle threshold (Ct) was determined which is the fractional cycle number at which the fluorescence exceeds the given threshold.
The 2-ΔΔCt method24 was used to calculate the relative quantitation of MP16 as follows:
- For each patient/control subject, ΔCt was calculated as the difference between Ct of endogenous control (Ct GAPDH) and Ct of target MP16;
ΔCt sample = Ct MP16 of a sample – Ct GAPDH of the same sample.
- The mean ΔCt of control subjects (ΔCt calibrator) was calculated as the difference between mean Ct of GAPDH assay of the control subjects and mean Ct of the MP16 assay of the control subjects.
- For each sample (patient and control), the ΔΔCt of each target MP16 was calculated as the difference between ΔCt of this sample and ΔCt calibrator (mean ΔCt of control subjects);
ΔΔCt = ΔCt sample – ΔCt calibrator
- Then, the equation 2–ΔΔCt was applied for each sample; Fold difference = 2–ΔΔCt
Accordingly, MP16 assay results were presented as fold changes relative to the mean of control group (healthy subjects) using the 2-ΔΔCt equation.
Statistical Analysis
All statistical analyses were done by using Statistical Package for the Social Sciences (SPSS) for Windows version 16 (SPSS Inc., Chicago, IL, USA) and MedCalc program. The Kolmogorov–Smirnov Test of Normality was used to test the normality of data. The quantitative data were expressed as mean ± standard deviation (SD) or median and the range (minimum–maximum) for normally or abnormally distributed data, respectively. They were compared using Mann–Whitney U-test or The Kruskal–Wallis test for two or more groups of abnormally distributed data, respectively. The qualitative data were expressed as a percentage. Spearman’s rank correlation coefficient (rho) was used to find correlations. The receiver operating characteristic curves (ROC) were plotted to measure the performance of MP16 (with and without AFP) in predicting the presence of HCC and to select its optimal cut-off value at which the sensitivity, specificity, positive (PPV) and negative (NPV) predictive values, and overall accuracy could be calculated. All tests were two-tailed and P-values < 0.05 were considered significant.
Results
Characteristics of the Study Population
The present study was performed on 200 patients with HCV-related CLD (158 males and 42 females with mean age of 59.8 ± 7 years) and 30 healthy subjects (9 females and 21 males with a mean age of 55.5 ± 7.6 years). The baseline demographic and clinical data of the studied population are summarized in Table 1.
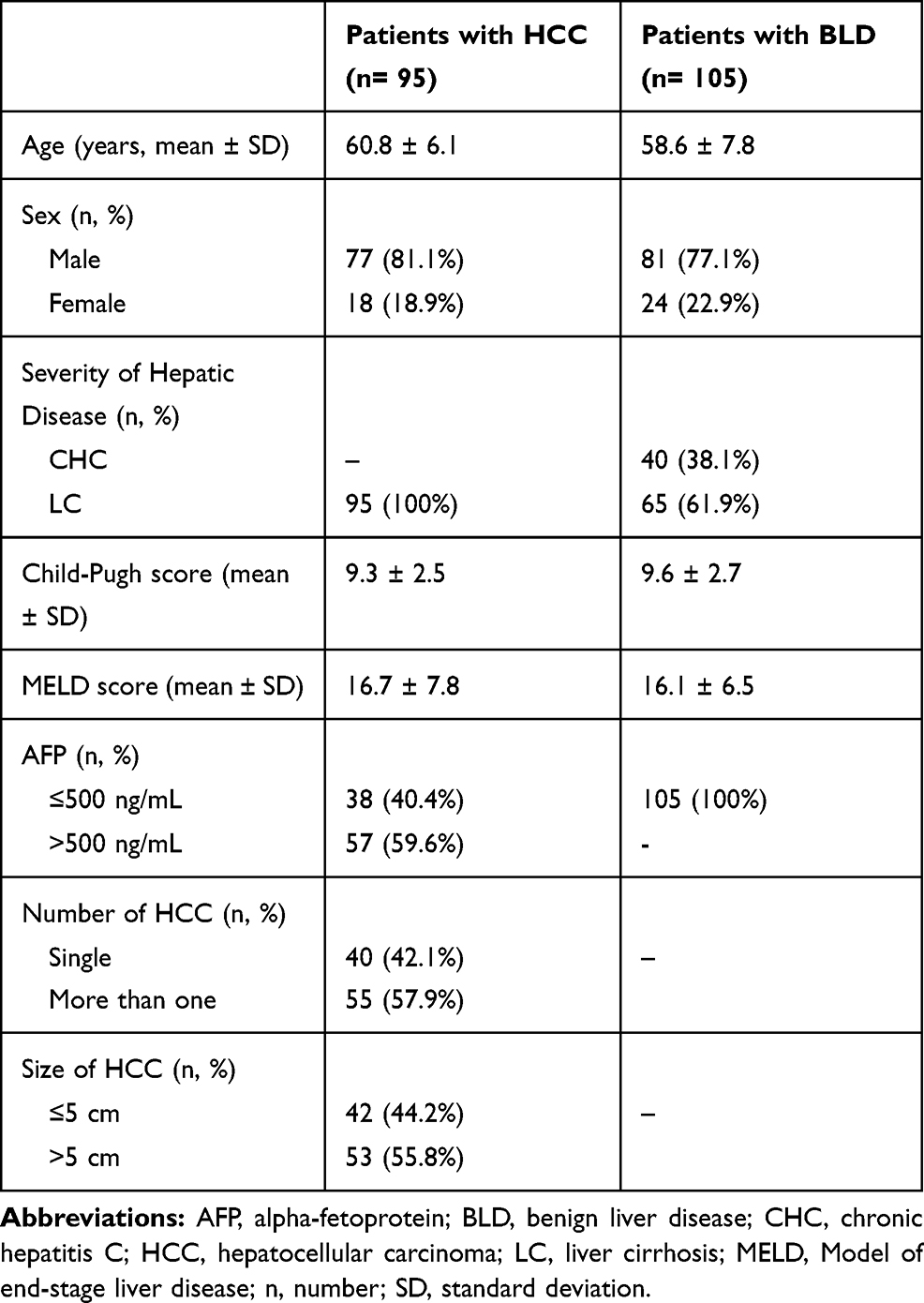 |
Table 1 Baseline Demographic and Clinical Characteristics of Patients with HCV-Related Chronic Liver Disease |
Expression of Methylated p16 in Serum of Patients and Healthy Controls
Serum MP16 expression was expressed in all analysed samples. There were no significant differences in serum MP16 levels with respect to gender and age among patients with HCC, BLD, and healthy controls (P > 0.05). Serum MP16 levels were significantly elevated in HCC cases as compared to BLD cases and normal controls (P < 0.001). Furthermore, MP16 levels were significantly higher in patients with BLD than normal group (P < 0.001). However, among BLD group, no significant difference in MP16 levels between chronic hepatitis C and liver cirrhosis patients [median; 108.3 (0.88–404.5) vs 142.54 (0.75–5.5x105); P = 0.365] (Table 2 and Figure 1).
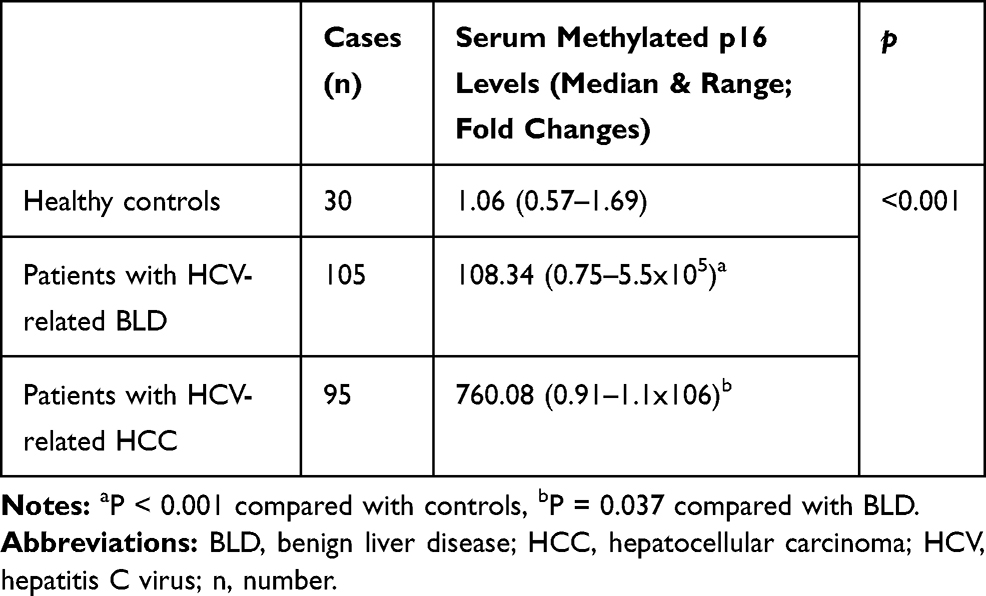 |
Table 2 Distribution of Serum Methylated P16 Levels Among Healthy Controls and Cases |
 |
Figure 1 Comparison between serum methylated p16 levels among different groups (healthy controls and HCV-related chronic liver disease). Abbreviations: CHC, chronic hepatitis C; HCC, hepatocellular carcinoma; LC, liver cirrhosis. |
Relationship Between Serum MP16 Expression and Clinical Characteristics in HCC Patients
No correlations were found between serum MP16 levels and age (rho = 0.110, P = 0.304), severity of liver cirrhosis based on Child-Pugh (rho = 0.042, P = 0.694) and MELD (rho = 0.059, P = 0.578) scores and AFP levels (rho = 0.240, P = 0.161). However, cases of HCC with normal AFP levels had significantly higher MP16 levels [median; 2091 (4.69–6.1×105) fold changes] when compared to HCC cases with higher AFP levels [median; 190.9 (1.07–1.5×106) fold changes, P = 0.028] and non-HCC cases [median; 142.54 (0.75–5.5×105) fold changes P = 0.004] as shown in Figure 2. In addition, serum MP16 levels were not associated with gender [median MP16 levels in males; 174.85 (0.75–1.1×106) fold changes vs those in females; 820.3 (1.07–5.5×105) fold changes, P = 0.111] and tumour characteristics; tumour size [median MP16 levels in those with HCC of ≤5 cm; 2091 (1.07–1.1×106) fold changes vs those with HCC > 5 cm; 2047 (0.91–8×105) fold changes, P = 0.111] and number [median MP16 levels in those with one lesion; 1962 (1.07–1.1×106) fold changes vs those with more than one lesion; 2202.3 (0.91–8×105) fold changes, P = 0.409].
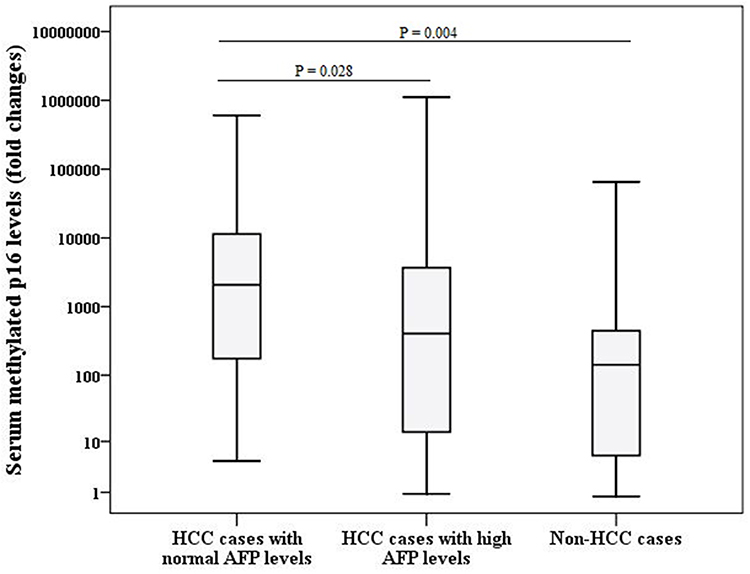 |
Figure 2 Comparison between serum methylated p16 levels among different groups (HCC cases and non-HCC cases). Abbreviations: AFP, alpha-fetoprotein; HCC, hepatocellular carcinoma. |
Diagnostic Performance of Methylated p16 for Prediction of HCC
As shown in Figure 2, ROC curves were developed to evaluate the diagnostic potential of MP16 as a noninvasive biomarker candidate. For diagnosis of HCC vs non-HCC cases, ROC curve analysis showed an area under the curve (AUC): 0.7 (95% CI: 0.580–0.772) with a sensitivity, specificity, PPV, NPV, and overall accuracy of 51.1%, 80.4%, 70.2%, 64.5%, 65% and 66.5%, respectively, at optimal cut-off value 464.65 fold changes (Figure 3A). With the combination of MP16 and AFP, the AUC was increased to 0.872 (95% CI: 0.742–0.951) with 100% sensitivity, 76.5% specificity, 79.1% PPV, 100% NPV, and 87.5% accuracy for discriminating HCC (Figure 3B). Furthermore, ROC analysis also showed that serum MP16 expression levels to distinguish patients with HCC and normal AFP levels from those without HCC had an AUC of 0.823 (95% CI: 0.694–0.942) with 55% sensitivity, 95.7% specificity, 75% PPV, 90.1% NPV, and 88% overall accuracy at optimal cut-off value of >824.14 (Figure 3C).
 |
Figure 3 Area under the receiver operating characteristic curves (AUC) of (A) methylated p16 [AUC= 0.7; 95% CI (0.580–0.772)] for predicting HCC versus non-HCC, (B) combination of methylated p16 and AFP [AUC= 0.872; 95% CI (0.742–0.951)] for predicting HCC versus non-HCC, and (C) methylated p16 [AUC= 0.823; 95% CI (0.694–0.942)] for predicting HCC in patients with hepatic mass and normal AFP versus non-HCC cases. Abbreviations: AFP, alpha-fetoprotein; HCC, hepatocellular carcinoma. |
Discussion
Hypermethylation of gene promoters has been demonstrated as an early event in hepatocellular carcinogenesis.25 In this study, we quantified the circulating MP16 levels using Rt-PCR in patients with HCV-related CLD for screening and early detection of HCC. These levels were significantly higher in patients with HCC as compared to patients with BLD or normal controls. Previous studies explored the detection of frequencies or sequences of gene P16 methylation in HCC, whereas higher frequencies of MP16 (65% and 96%) were observed in HCC cases.9,16-18 While, Wong et al16 showed that quantities of MP16 sequences were detected in peripheral circulation of 80% of HCC patients.
We found that MP16 levels were significantly higher in patients with BLD than normal controls but these levels were not significantly different between liver cirrhosis and CHC patients. These results were matched with Chu et al,26 who informed that 17% of cirrhosis patients had serum DNA with aberrant p16 methylation. Furthermore, Narimatsu et al17 had comparable results suggesting that hepatitis viruses may induce p16 methylation in liver tissues with chronic inflammation before the appearance of HCC. However, Wong et al10 reported that p16 hypermethylation was not detected in the plasma/serum of patients with either liver cirrhosis or hepatitis. This discrepancy indicates that this correlation is still controversial.27
P16 gene is an important tumour suppressor gene located on chromosome 9p21 and it is one of the most common altered genes observed in various human tumours.28 Earlier studies demonstrated that p 16 (p16INK4A) gene methylation might play an imperative role in p16 gene inactivation which leads to p16-mediated cell-cycle control disruption and therefore participate in a role in hepatocarcinogenesis.29,30 Several studies have indicated that these epigenetic changes might deceive “addict” cancer cells to signal transduction pathway changes during the early tumour stages.31,32 The appearance of these serum/plasma tumour-derived DNA altered genes may be due to their release from the tumour during cell turnover, cellular necrosis, or apoptosis.33
In the current study, serum MP16 levels were not significantly influenced by the tumour characters (number and size) which was in accordance with earlier series.18,34 However, Zhang et al9 found that p16 methylation may be implicated in tumour progression.
Similar to previous studies,34,35 we found no significant correlation between serum MP16 levels and AFP levels. However, MP16 levels were significantly higher in those with normal levels of AFP. Nevertheless, Wong et al36 demonstrated that circulating MP l6 sequences were detected in all individuals with higher serum AFP of >45 µg/L, whereas they were found in 57% of cases who had lower AFP concentrations of <45µg/L. This indicates its importance in the screening of HCC in high-risk individuals with normal AFP levels.
Despite elevated MP16 levels in HCC cases, MP16 had low performance in prediction of HCC, where AUC was 0.7 with 51.1% sensitivity and 80.4% specificity and 66.5% overall accuracy at the optimal cut-off value of 464.65-fold changes. Even though this performance got better with adding AFP (AUC was 0.872 with 100% sensitivity, 76.5% specificity, and 87.5% accuracy), the utility of using MP16 in the presence of high AFP for HCC diagnosis is questionable. On the other hand, in this study, about 40% of patients with HCC had normal AFP levels (<400–500 ng/mL) where their serum MP16 levels were significantly higher than patients without HCC. Furthermore, MP16 had a good diagnostic performance in discriminating HCC cases with normal AFP levels from non-HCC cases with an AUC of 0.823, high specificity and NPV (95.7% and 90.1%), and reasonable sensitivity and PPV (55% and 75%) at cut-off value of 824.14-fold changes suggesting their potential value for HCC detection. Owing to the previously mentioned findings, assessment of serum MP16 expression levels may be restricted for cases with hepatic mass(es) and normal AFP levels as a potential noninvasive biomarker for HCC diagnosis especially in cirrhotic patients where liver biopsy (eg, those with severely impaired prothrombin time or low platelet count) and/or radiological imaging (eg, renal impairment, high cost, or less availability) cannot be done.
Many resources have been triggered for the development of different epigenetic therapeutic approaches because of their reversible alterations. They have emerged as attractive targets for therapeutic intervention.37 These epigenetic drugs are able to reexpress silenced genes, either by demethylation of methylated promoter regions (demethylating agents) or by histone acetylation.38 Several compounds have been preclinically tested and showed promising results in other cancers and HCC as 5-azacytidine (Vidaza), 5-aza-dC (decitabine), MS-275, and Valproic Acid.39 Therefore, we recommend performing quantitative analysis of methylated gene p16 in large cohorts with liver cirrhosis especially for high-risk patients who had suggestive clinical manifestations of HCC as loss of weight, repeatedly hepatic encephalopathy without evidence of predisposing factors or rapid accumulating ascites even without focal lesion(s) in imaging. Furthermore, those who had MP16 levels > 464.65-fold changes should be subjected for meticulous laboratory and careful imaging follow-up for early HCC detection and hence a trial of epigenetic therapeutic interventions.
To our knowledge, this is one of the first studies to evaluate quantitatively MP16 expression and to evaluate its diagnostic performance in the detection of HCC among HCV-infected patients especially in with normal AFP in our community. However, some limitations as small sample-sized and a single-centre study were considered. The quantitative assessment of MP16 in liver tissue and its correlation with serum levels was not accessible. This is a preliminary study so, phased validation with large-scale samples is needed to confirm these findings, to demonstrate whether it can be incorporated into routine clinical practice in patients with hepatic masses and normal AFP and early hepatocarcinogenesis detection and to assess its ability of monitoring tumour progression, response to treatment and recurrence.
Conclusion
Serum MP16 expression levels were higher in HCC than non-HCC patients with HCV-related chronic liver disease. Furthermore, MP16 may be a potential molecular biomarker for diagnosis of HCC in HCV-infected patients with hepatic mass(es) and normal AFP levels with high specificity and reasonable sensitivity especially in those where liver biopsy and radiological imaging cannot be done. Understanding roles of MP16 in modulating the signalling pathways for HCC might be helpful in lowering the incidence and progression of HCC through epigenetic therapeutic interventions.
Acknowledgment
This work was supported by a grant from the Grant Office, Faculty of Medicine, Assiut University, Egypt (Grant No. R31-2015).
Disclosure
The authors declare that they have no competing interests.
References
1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. doi:10.3322/caac.20107
2. Huang Z, Hu Y, Hua D, Wu Y, Song M, Cheng Z. Quantitative analysis of multiple methylated genes in plasma for the diagnosis and prognosis of hepatocellular carcinoma. Exp Mol Pathol. 2011;91:702–707. doi:10.1016/j.yexmp.2011.08.004
3. Nishida N, Nagasaka T, Nishimura T, Ikai I, Boland CR, Goel A. Aberrant methylation of multiple tumor suppressor genes in aging liver, chronic hepatitis, and hepatocellular carcinoma. Hepatology. 2008;47:908–998. doi:10.1002/hep.22110
4. Wong IH, Johnson PJ, Lai PB, Lau WY, Lo YM. Tumor-derived epigenetic changes in the plasma and serum of liver cancer patients: implications for cancer detection and monitoring. Ann N Y Acad Sci. 2000;906:102–105. doi:10.1111/j.1749-6632.2000.tb06598.x
5. Toh TB, Lim JJ, Chow EK. Epigenetics of hepatocellular carcinoma. Clin Transl Med. 2019;8:13. doi:10.1186/s40169-019-0230-0
6. Saintigny P, Zhang L, Fan YH, et al. Gene expression profiling predicts the development of oral cancer. Cancer Prev Res (Phila). 2011;4:218–229. doi:10.1158/1940-6207.CAPR-10-0155
7. Li XQ, Guo YY, De W. DNA methylation and microRNAs in cancer. World J Gastroenterol. 2012;18:882–888. doi:10.3748/wjg.v18.i9.882
8. Calvisi DF, Ladu S, Gorden A, et al. Mechanistic and prognostic significance of aberrant methylation in the molecular pathogenesis of human hepatocellular carcinoma. J Clin Invest. 2007;117:2713–2722. doi:10.1172/JCI31457
9. Zhang YJ, Wu HC, Shen J, et al. Predicting hepatocellular carcinoma by detection of aberrant promoter methylation in serum DNA. Clin Cancer Res. 2007;13:2378–2384. doi:10.1158/1078-0432.CCR-06-1900
10. Wong IHN, Lo YMD, Zhang J, et al. Detection of aberrant p16 methylation in the plasma and serum of liver cancer patients. Cancer Res. 1999;59:71–73.
11. Yeo W, Wong N, Wong WL, Lai PB, Zhong S, Johnson PJ. High frequency of promoter hypermethylation of RASSF1A in tumor and plasma of patients with hepatocellular carcinoma. Liver Int. 2005;25:266–272. doi:10.1111/j.1478-3231.2005.01084.x
12. Wang J, Qin Y, Li B, Sun Z, Yang B. Detection of aberrant promoter methylation of GSTP1 in the tumor and serum of Chinese human primary hepatocellular carcinoma patients. Clin Biochem. 2006;39(4):344–348. doi:10.1016/j.clinbiochem.2006.01.008
13. Colombo M. Screening for cancer in viral hepatitis. Clin Liver Dis. 2001;5(1):109–122. doi:10.1016/S1089-3261(05)70156-2
14. Li L, Zhao H, Chen B, et al. Noninvasive identification of immune-related biomarkers in hepatocellular carcinoma. J Oncol. 2019;2019:2531932. doi:10.1155/2019/2531932
15. Li L, Choi JY, Lee KM, et al. DNA methylation in peripheral blood: a potential biomarker for cancer molecular epidemiology. J Epidemiol. 2012;22:384–394. doi:10.2188/jea.JE20120003
16. Wong IH, Zhang J, Lai PB, Lau WY, Lo YM. Quantitative analysis of tumor-derived methylated p16INK4a sequences in plasma, serum, and blood cells of hepatocellular carcinoma patients. Clin Cancer Res. 2003;9:1047–1052.
17. Narimatsu T, Tamori A, Koh N, et al. p16 promoter hypermethylation in human hepatocellular carcinoma with or without hepatitis virus infection. Intervirology. 2004;47:26–31. doi:10.1159/000076639
18. Katoh H, Shibata T, Kokubu A, et al. Epigenetic instability and chromosomal instability in hepatocellular carcinoma. Am J Pathol. 2006;168:1375–1384. doi:10.2353/ajpath.2006.050989
19. Pugh RN, Murray-Lyon IM, Dawson JL, Pietroni MC, Williams R. Transection of the oesophagus for bleeding oesophageal varices. Br J Surg. 1973;60:646–649. doi:10.1002/bjs.1800600817
20. Wiesner RH, Edwards E, Freeman R, et al. Model for end-stage liver disease (MELD) and allocation of donor livers. Gastroenterology. 2003;124:91–96. doi:10.1053/gast.2003.50016
21. Bruix J, Sherman M. Management of hepatocellular carcinoma: an update. Hepatology. 2011;53(3):1020–1022. doi:10.1002/hep.24199
22. Herman JG, Graff JG, Myohanen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA. 1996;93:9821–9826. doi:10.1073/pnas.93.18.9821
23. Wong IHN, Lo YMD, Yeo W, Lau WY, Johnson PJ. Frequent p15 promoter methylation in tumor and peripheral blood from hepatocellular carcinoma patients. Clin Cancer Res. 2000;6:3516–3521.
24. Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative CT method. Nat Protoc. 2008;3(6):1101–1108. doi:10.1038/nprot.2008.73
25. Wang M, Ye Q, Mao D, Li H. Research progress in liver-regenerating microenvironment and DNA methylation in hepatocellular carcinoma: the role of traditional Chinese medicine. Med Sci Monit. 2020;26:e920310. doi:10.12659/MSM.920310
26. Chu HJ, Heo J, Seo SB, et al. Detection of aberrant p16INK4A methylation in sera of patients with liver cirrhosis and hepatocellular carcinoma. J Korean Med Sci. 2004;19:83–86. doi:10.3346/jkms.2004.19.1.83
27. Shim YH, Yoon GS, Choi HJ, Chung YH, Yu E. p16 Hypermethylation in the early stage of hepatitis B virus-associated hepatocarcinogenesis. Cancer Lett. 2003;190:213–219. doi:10.1016/S0304-3835(02)00613-4
28. Sherr CJ. G1 phase progression: cycling on cue. Cell. 1994;79(4):551–555. doi:10.1016/0092-8674(94)90540-1
29. Zang JJ, Xie F, Xu JF, et al. P16 gene hypermethylation and hepatocellular carcinoma: a systematic review and meta-analysis. World J Gastroenterol. 2011;17:3043–3048. doi:10.3748/wjg.v17.i25.3043
30. Zheng Y, Hlady RA, Joyce BT, et al. DNA methylation of individual repetitive elements in hepatitis C virus infection-induced hepatocellular carcinoma. Clin Epigenetics. 2019;11(1):145. doi:10.1186/s13148-019-0733-y
31. Baylin SB, Ohm JE. Epigenetic gene silencing in cancer - a mechanism for early oncogenic pathway addiction? Nat Rev Cancer. 2006;6:107–163. doi:10.1038/nrc1799
32. Toyota M, Issa JP. Epigenetic changes in solid and hematopoietic tumors. Semin Oncol. 2005;32:521–530. doi:10.1053/j.seminoncol.2005.07.003
33. Jahr S, Hentze H, Englisch S, et al. DNA fragments in the blood plasma of cancer patients: quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001;61:1659–1665.
34. Lou C, Du Z, Yang B, Gao Y, Wang Y, Fang S. Aberrant DNA methylation profile of hepatocellular carcinoma and surgically resected margin. Cancer Sci. 2009;100:996–1004. doi:10.1111/j.1349-7006.2009.01138.x
35. Li X, Hui AM, Sun L, et al. p16INK4A hypermethylation is associated with hepatitis virus infection, age, and gender in hepatocellular carcinoma. Clin Cancer Res. 2004;10:7484–7489. doi:10.1158/1078-0432.CCR-04-1715
36. Wong IH, Lo YM, Lai PB, Johnson PJ. Relationship of p16 methylation status and serum alpha-fetoprotein concentration in hepatocellular carcinoma patients. Clin Chem. 2000;46:1420–1422. doi:10.1093/clinchem/46.9.1420
37. Herath NI, Leggett BA, MacDonald GA. Review of genetic and epigenetic alterations in hepatocarcinogenesis. J Gastroenterol Hepatol. 2006;21:15–21. doi:10.1111/j.1440-1746.2005.04043.x
38. Ganesan A, Arimondo PB, Rots MG, Jeronimo C, Berdasco M. The timeline of epigenetic drug discovery: from reality to dreams. Clin Epigenetics. 2019;11:174. doi:10.1186/s13148-019-0776-0
39. Herceg Z, Paliwal A. Epigenetic mechanisms in hepatocellular carcinoma: how environmental factors influence the epigenome. Mutat Res. 2011;727(3):55–61. doi:10.1016/j.mrrev.2011.04.001
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.