Back to Journals » Drug Design, Development and Therapy » Volume 12
Upregulation of vascular endothelial growth factor receptor-1 contributes to sevoflurane preconditioning–mediated cardioprotection
Authors Qian B, Yang Y, Yao Y ![]() , Liao Y
, Liao Y ![]() , Lin Y
, Lin Y
Received 15 January 2018
Accepted for publication 1 March 2018
Published 6 April 2018 Volume 2018:12 Pages 769—776
DOI https://doi.org/10.2147/DDDT.S162577
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tuo Deng
Bin Qian,1 Yang Yang,2 Yusheng Yao,3 Yanling Liao,3 Ying Lin3
1Department of Anesthesiology, People’s Hospital Affiliated to Fujian University of Traditional Chinese Medicine, Fuzhou, Fujian, China; 2Department of Anesthesiology, West China Hospital, Sichuan University, Chengdu, Sichuan, China; 3Department of Anesthesiology, The Shengli Clinical Medical College, Fujian Medical University, Fuzhou, Fujian, China
Purpose: Sevoflurane preconditioning (SPC) can provide myocardial protective effects similar to ischemic preconditioning. However, the exact mechanism of SPC remains unclear. Previous studies indicate that vascular endothelial growth factor receptor 1 (VEGFR-1) is involved in ischemic preconditioning-mediated cardioprotection. This study was designed to determine the significance of VEGFR-1 signaling in SPC-mediated cardioprotection.
Materials and methods: Myocardial ischemia–reperfusion (I/R) rat model was established using the Langendorff isolated heart perfusion apparatus. Additionally, after 15 min of baseline equilibration, the isolated hearts were pretreated with 2.5% sevoflurane, 2.5% sevoflurane+MF1 10 µmol/L, or 2.5% sevoflurane+placental growth factor 10 µmol/L, and then subjected to 30 min of global ischemia and 120 min of reperfusion. The changes in hemodynamic parameters, myocardial infarct size, and the levels of creatine kinase-MB, lactate dehydrogenase, cardiac troponin-I, tumor necrosis factor-α, and interleukin 6 in the myocardium were evaluated.
Results: Compared to the I/R group, pretreatment with 2.5% sevoflurane significantly improved the cardiac function, limited myocardial infarct size, reduced cardiac enzyme release, upregulated VEGFR-1 expression, and decreased inflammation. In addition, the selective VEGFR-1 agonist, placental growth factor, did not enhance the cardioprotection and anti-inflammation effects of sevoflurane, while the specific VEGFR-1 inhibitor, MF1, completely reversed these effects.
Conclusion: Our data have demonstrated that 2.5% sevoflurane preconditioning alleviates heart I/R injury, which is probably mediated by the anti-inflammatory property and upregulation of VEGFR-1.
Keywords: sevoflurane, preconditioning, ischemic–reperfusion injury, vascular endothelial growth factor receptor 1, anti-inflammatory
Introduction
Despite substantial advances in prevention and treatment, ischemic cardiac injury and the ventricular dysfunction can provoke major causes of morbidity and mortality throughout the world.1 Accumulated evidence has indicated that sevoflurane, one of the most commonly used volatile anesthetics in clinical practice, is superior to many general anesthetics in its cardiovascular stability and rapid recovery pharmacologic properties, which is ideal for patients with high risk of perioperative cardiac events.2–4 Sevoflurane preconditioning (SPC)-induced heart ischemic tolerance might be associated with downregulation of apoptosis,5 reduction of oxidative stress and the inflammatory cytokines,6 activation of mitochondrial KATP channels,7 attenuation of nuclear factor-κB activation,8 and so forth. However, the mechanism is not fully elucidated, mainly because the pharmacology, metabolic responses, and physiologies are significantly different and cardioprotective methods from the laboratory cannot be directly translated into the clinic.9
Vascular endothelial growth factors (VEGFs) and their receptors including VEGFR-1, VEGFR-2, and VEGFR-3 form a regulatory system crucial for normal development and pathologic angiogenesis.10 Recent progress in the study of the VEGF–VEGFR system showed that it is deeply involved in several pathologic conditions related not only to angiogenesis but also to malignancy, ischemic diseases, and inflammation.11 Although a lot of studies have focused on studies of VEGF expression and its function in myocardial ischemia or hypoxia, relatively little is known regarding the VEGF receptors. An array of studies in preclinical models of ischemia–reperfusion (I/R) injury have provided key insights regarding the role of VEGFR-1 involved in ischemic preconditioning-mediated cardioprotection.12,13 Thus, VEGFR-1 signaling represents a promising target for therapy of I/R injury, and identifying the novel pathologic role of VEGFR-1 is very important for clinical translational research. Therefore, we designed this study to identify the role of VEGFR-1 in SPC-mediated cardioprotection.
Materials and methods
All experiment protocols were approved by the Institutional Animal Care and Use Committee of Fujian Medical University and performed in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No 85-23, revised 1996).
Experiment protocols
Male Sprague Dawley rats (8–10 weeks, weighing 270–300 g) were supplied by the Animal Center of the Fujian Medical University. The rats were anesthetized with intraperitoneal injection of 3% sodium pentobarbital (50 mg/kg) and intravenously heparinized (50 IU). All the hearts were rapidly excised and mounted in a Langendorff apparatus (Radnoti, Monrovia, CA, USA) and perfused with 37°C Krebs–Henseleit buffer (KHB) at a constant pressure (75 mmHg). After equilibrating with KHB for 15 min, the hearts were randomly divided into five groups: 1) sham group: the hearts were subjected to 195 min of perfusion without I/R injury; 2) I/R group: the hearts were subjected to 30 min of global ischemia followed by 120 min of reperfusion; 3) SPC group: after preconditioning with 2.5% sevoflurane (Abbott Laboratories, Shanghai, China) for 15 min, the hearts were exposed to 15 min of washout prior to 30 min of global ischemia; 4) SPC+MF1 group: the hearts were exposed to 2.5% sevoflurane and 10 μmol/L MF1 (ImClone, Somerville, NJ, USA), the selective VEGFR-1 inhibitor, for 15 min followed by 15 min washout period before ischemia; and 5) SPC+placental growth factor (PIGF) group: the hearts were exposed to 2.5% sevoflurane and 10 μmol/L PIGF (Abcom, Cambridge, UK), the specific agonist of VEGFR-1, for 15 min followed by 15 min washout period before global ischemia. Our experimental protocol is briefly illustrated in Figure 1 and available online at https://dx.doi.org/10.17504/protocols.io.mmrc456. To implement SPC, the KHB was pre-equilibrated with sevoflurane using a separate reservoir with a Vapor 2000 (Drager Medical AG & Co, Lubeck, Germany).
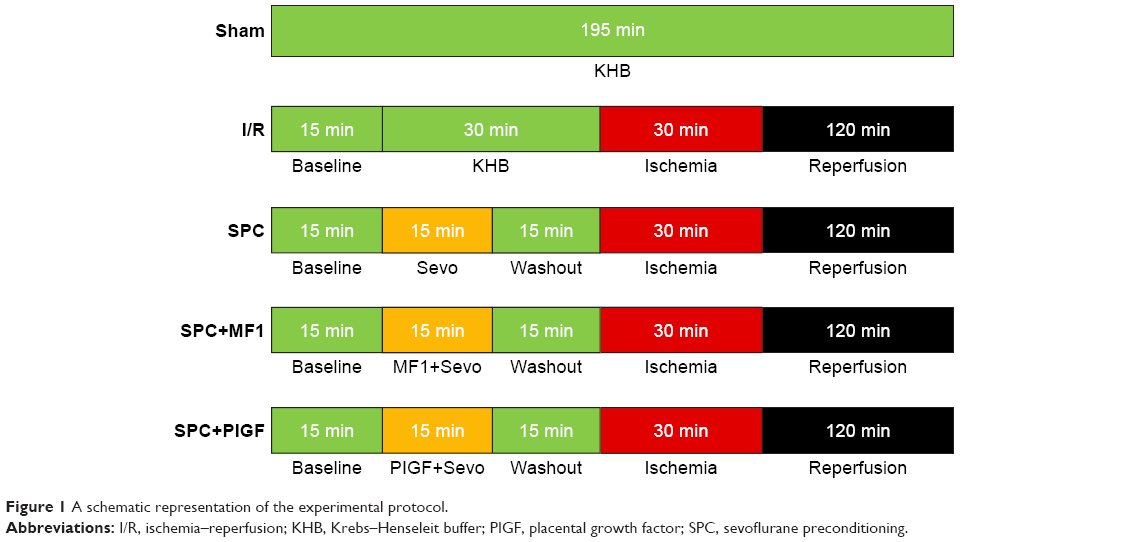 | Figure 1 A schematic representation of the experimental protocol. |
Cardiac function measurement
A distilled water-filled latex balloon linked with a pressure transducer (Model SP844; MEMSCAP, Durham, NC, USA) was inserted into the left ventricle (LV) through the mitral valve immediately after the heart was mounted in Langendorff equipment. Then, the cardiac function parameters, including heart rate, left ventricular developed pressure (LVDP), left ventricular end-diastolic pressure (LVEDP), maximum LVDP increase rate (+dp/dt), and maximum LVDP decrease rate (-dp/dt), were collected via a PowerLab data acquisition system (AD Instruments, Bella Vista, Australia).
Myocardial infarct size assessment
At the end of the experiments, the hearts were frozen at −20°C and then sliced into 2 mm thick sections parallel to the atrioventricular groove. The slices were incubated in a 1% solution of 2,3,5-triphenyl tetrazolium chloride (Sigma, St Louis, MO, USA) for 15 min at 37°C and then fixed in a 4% paraformaldehyde phosphate buffer for 24 h. The infarct size (unstained by 2,3,5-triphenyl tetrazolium chloride) was measured by computerized planimetry using Image-Pro plus 5.0 software (Media Cybernetics, Silver Spring, MD, USA) and expressed as a percentage of the total heart volume.
Determination of myocardial enzyme release
The release of creatine kinase-MB (CK-MB) and lactate dehydrogenase (LDH) in the coronary effluent was measured using an auto-analyzer (Cobas C501; Roche, Basel, Switzerland). Cardiac troponin-I (cTnI) was cardiac measured by troponin-I enzyme-linked immunosorbent assay kit (Life Diagnostics, West Chester, PA, USA).
VEGFR-1 expression was evaluated by Western blot
A portion of the left anterior ventricular tissue was sampled from hearts at the time point of reperfusion 120 min. Samples were immediately frozen in liquid nitrogen and stored at −80°C. The freeze-dried samples were homogenized with lysis buffer. The homogenates were vortexed for 20 s and then centrifuged (14,000×g for 10 min) at 4°C. Protein concentrations were measured by the Bradford method using bovine serum albumin. Equal amounts of protein were electrophoresed on a 12.5% sodium dodecyl sulfate-polyacrylamide gel, then transferred to a nitrocellulose membrane, and probed with polyclonal primary antibodies specific for total VEGFR-1. The Western blot was performed with the rabbit anti-VEGF receptor 1 antibody (1:1,000; Abcom). Appropriate secondary horseradish peroxidase-conjugated goat anti-rabbit antibody (1:2,000; Pierce, Rockford, IL, USA) was used. Bands were developed using an enhanced chemiluminescent detection system (Thermo Fisher Scientific, Waltham, MA, USA), and ImageJ software (National Institutes of Health, Bethesda, MD, USA) was used to quantified the gray value of bands.
Assay of myocardium inflammation
After 120 min of reperfusion, the hearts were immediately harvested and stored at −80°C for inflammation analysis. The frozen ventricle samples were crushed into powder using a liquid nitrogen chilled tissue pulverizer. The amount of frozen ventricle tissues was homogenized in the appropriate buffer for tissue analysis. The levels of tumor necrosis factor (TNF)-α and interleukin (IL)-6 were spectrophotometrically analyzed by a highly sensitive enzyme-linked immunosorbent assay (R&D Systems, Minneapolis, MO, USA).
Statistical analysis
SPSS 18.0 for windows (SPSS Inc., Chicago, IL, USA) was used to conduct statistical analyses. All values are presented as mean ± standard error of the mean. One-way analysis of variance was used to compare the differences among five groups, followed by post hoc test with Bonferroni procedure. The time point effects were detected with repeated-measures analysis of variance. A P-value of <0.05 was considered statistically significant.
Results
SPC improves cardiac LV function recovery after I/R
The Langendorff isolated heart was used to assess the effect of SPC on in vitro cardiac I/R injury. As shown in Table 1, all the hemodynamic parameters at baseline were not different among the groups. During reperfusion, the I/R group hearts exhibited significantly decreased LVDP (Figure 2A) and ±dp/dt (Figure 2B and C), as well as remarkably increased LVEDP (Figure 2D) in comparison to the Sham group (P<0.05). SPC caused a clear cardioprotective effect, as evidenced by improved LVDP and ±dp/dt, and markedly decreased LVEDP (all P<0.05 vs the I/R group; Figure 2A–D).
 | Table 1 The basal hemodynamics of isolated hearts |
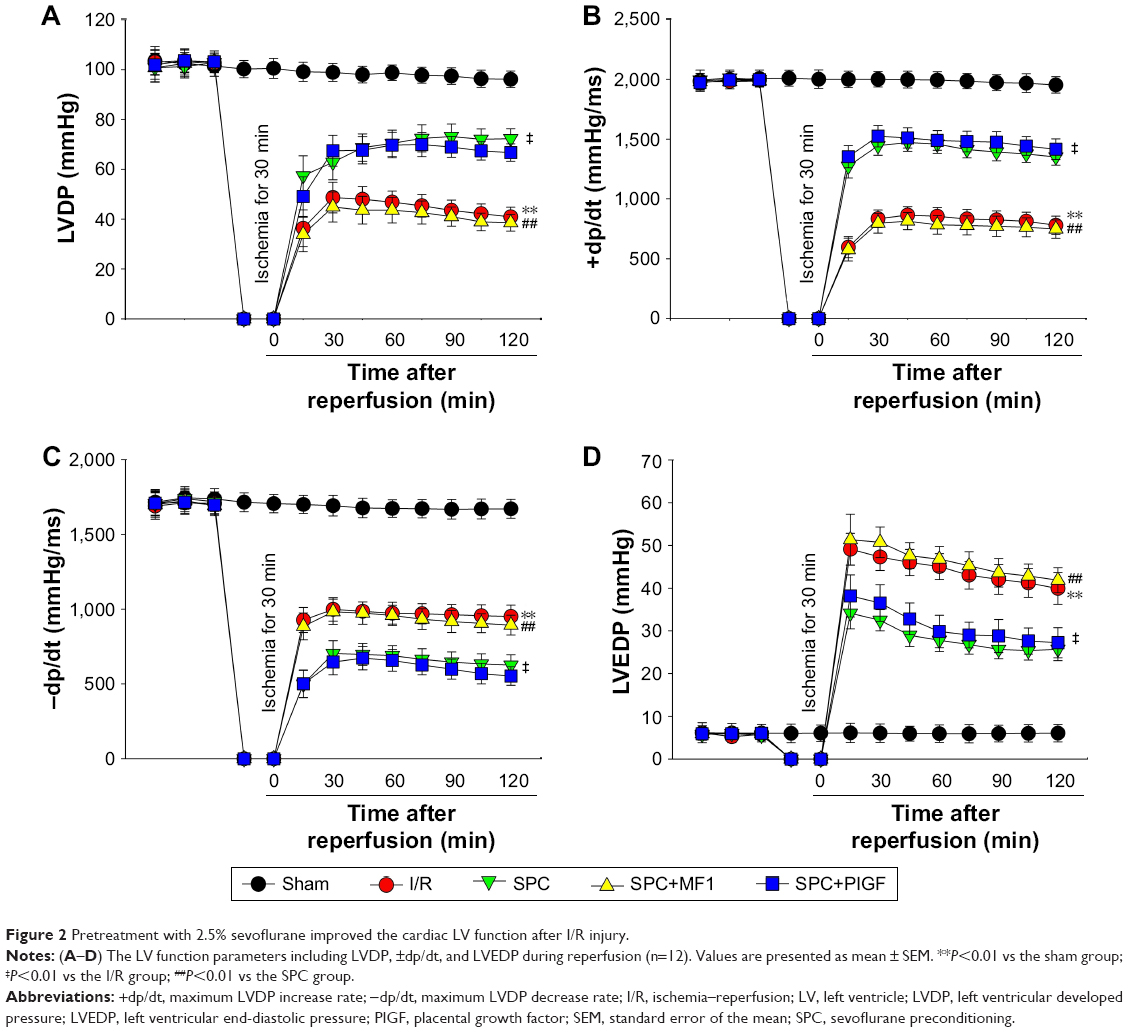 | Figure 2 Pretreatment with 2.5% sevoflurane improved the cardiac LV function after I/R injury. |
SPC limits myocardial infarct size and reduces myocardial necrosis
Global ischemia followed by KHB reperfusion caused increased myocardial infarct size (37.68%±4.85% in the I/R group vs 5.32%±1.11% in the sham group, P<0.01). Such an effect was significantly attenuated by SPC (17.5%±3.4% in the SPC group, P<0.01 vs the I/R group; Figure 3A and B). As markers of myocardial necrosis, the levels of CK-MB (1.03±0.14 IU/L), LDH (6.25±0.84 IU/L), and cTnI (37.68±4.85 ng/mL) were greatly increased in the I/R group, whereas lesser cardiac enzyme release was observed in the SPC group (all P<0.01 vs the I/R group; Figure 3C–E).
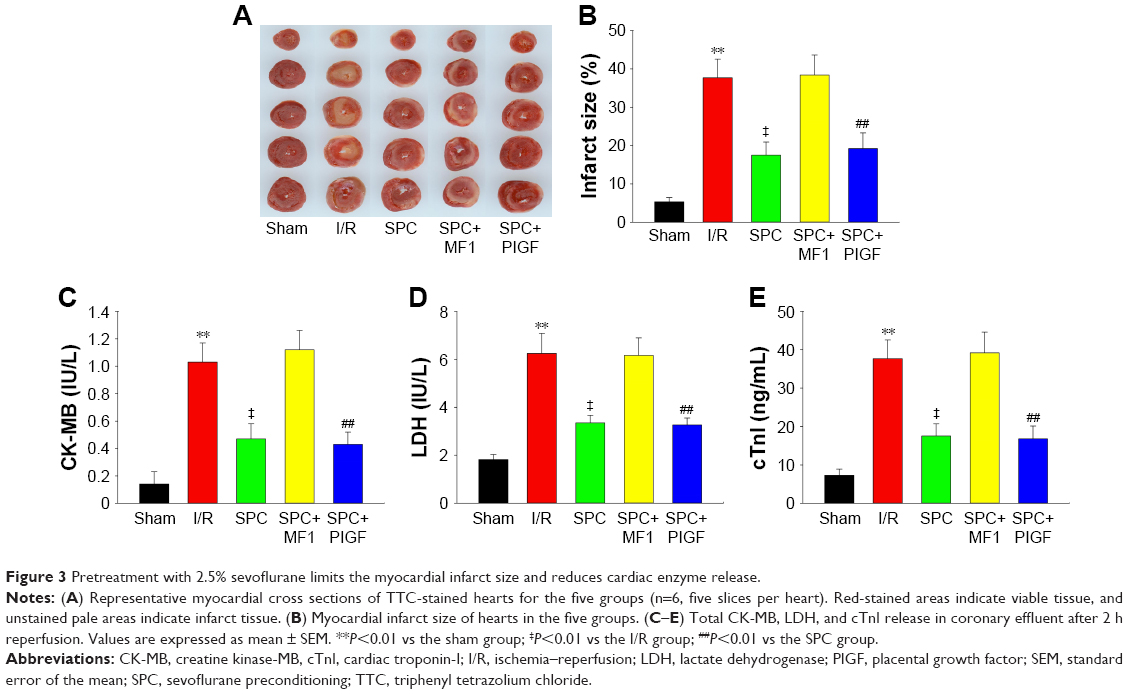 | Figure 3 Pretreatment with 2.5% sevoflurane limits the myocardial infarct size and reduces cardiac enzyme release. |
Inhibition of VEGFR-1 by MF1 reverses SPC-mediated cardioprotection
Compared with the SPC group, administration of pharmacologic inhibitor MF1 at 10 μmol/L (SPC+MF1 group) significantly attenuated SPC-mediated cardioprotection, as evidenced by decreased LV cardiac function (Figure 2) and increased myocardial infarct size (P<0.01; Figure 3A and B). Consistently, the release of CK-MB (1.12±0.14 IU/L), LDH (6.17±0.73 IU/L), and cTnI (39.2±5.4 ng/mL) was greatly increased than that (CK-MB, 0.47±0.11 IU/L; LDH, 3.35±0.32 IU/L; cTnI, 17.5±3.2 ng/mL) in the SPC group (all P<0.01; Figure 3C–E). There was no difference between the I/R group and the SPC+MF1 group (all P>0.05). These data provide mechanistic evidence that upregulation of VEGFR-1 may contribute to SPC-induced cardioprotection.
Activation of VEGFR-1 by PIGF does not enhance SPC-mediated cardioprotection
SPC alleviated I/R-induced cardiac function impairment, as shown by increased LVDP and ±dp/dt, and decreased LVEDP compared to the I/R group. Administration of pharmacologic agonist PIGF at 10 μmol/L (SPC+PIGF group) did not enhance SPC-mediated cardioprotective effect. These hemodynamic parameters during recovery were not different from the SPC group (all P<0.01; Figure 2). As shown in Figure 3A and B, lesser myocardial infarct size was observed in the SPC group, whereas PIGF did not further reduce myocardial infarct size (19.2%±4.1% in the SPC+PIGF group vs 17.5%±3.2% in the SPC group, P>0.05). In addition, the levels of CK-MB, LDH, and cTnI were greatly reduced in the SPC+PIGF group as compared to the I/R group and were not different from the SPC group (all P>0.05; Figure 3C–E).
SPC increased the expression of VEGFR-1
We measured the expression of VEGFR-1 in the LV tissue of isolated hearts using Western blot. Of note, VEGFR-1 was markedly increased nearly 2.6-fold in the SPC group compared with the I/R group (P<0.01; Figure 4). There was no difference between the sham group and the I/R group (P>0.05).
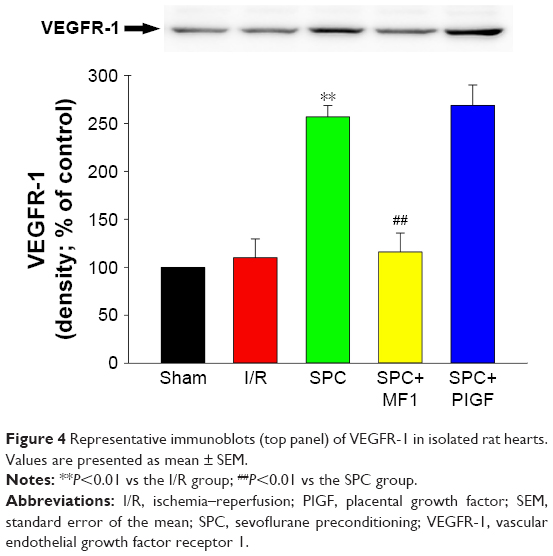 | Figure 4 Representative immunoblots (top panel) of VEGFR-1 in isolated rat hearts. Values are presented as mean ± SEM. |
SPC decreases inflammation in the myocardium
After I/R, the levels of TNF-α (251.3±19.7 pg/mL) and IL-6 (86.5±7.4 pg/mL) were greatly increased in the I/R group compared to those (63.6±5.9 and 27.8±3.5 pg/mL, respectively) in the sham group (P<0.01, respectively; Figure 5A and B). Sevoflurane pretreatment significantly inhibited this I/R-induced myocardial inflammation, as evidenced by markedly reduced levels of TNF-α (125.4±9.8 pg/mL) and IL-6 (42.9±5.2 pg/mL) in the SPC group (all P<0.01 vs the I/R group; Figure 5A and B). PIGF did not enhance the anti-inflammation effect of sevoflurane (113.5±6.2 and 39.4±4.8 pg/mL in the SPC+PIGF group, respectively, all P>0.05 vs the SPC group), while MF1 reversed this effect (267.5±17.8 and 92.4±9.2 pg/mL in the SPC+MF1 group, respectively, all P<0.01 vs the SPC group).
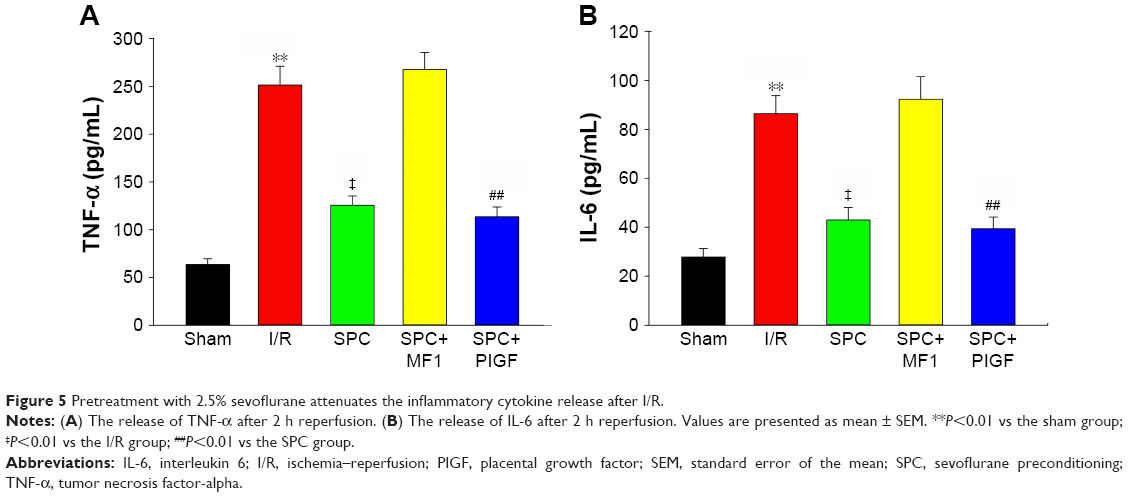 | Figure 5 Pretreatment with 2.5% sevoflurane attenuates the inflammatory cytokine release after I/R. |
Discussion
This study suggests that 2.5% SPC significantly improved cardiac function and limited myocardial infarct size, thus exhibiting profound protection against myocardial I/R injury. This was further demonstrated by a marked reduction in myocardial enzyme. To elucidate the underlying mechanism, we also investigated the status of VEGFR-1 and inflammatory reactions in these hearts. Our results clearly show that SPC increased the expression of VEGFR-1 and decreased the levels of TNF-α and IL-6. In addition, the selective VEGFR-1 agonist (PIGF) did not alter the cardioprotection and anti-inflammation of sevoflurane. But MF1, the specific blocker of VEGFR-1 completely abolished these beneficial effects, indicating VEGFR-1 plays an important role in SPC-induced cardioprotection. Taken together, our data suggest that VEGFR-1 is a candidate target for attenuating cardiac I/R injury.
VEGF is one of the most important angiogenic cytokines and has proinflammatory properties.14 Actually, VEGFR-1 is not only significantly expressed in vascular endothelial cells but also widely expressed on the membrane of macrophage lineage cells such as monocytes, transducing a vital signal for the migration and chemokine/cytokine production of these cells. According to previous studies, VEGFR-1 exerts a negative regulating effect on VEGF signaling.10 A wealth of evidence has demonstrated that upregulation of VEGF and VEGFR-1 promotes vascular remodeling and reendothelialization.15,16 Besides, as a negative component in VEGF signaling, upregulation of VEGFR-1 will lead to an anti-inflammatory effect. From the data of this study, sevoflurane pretreatment was found to cause upregulation of the expression of VEGFR-1, which is consistent with previously reported results.17,18
Inflammation, which plays a key role in many pathologic states including I/R injury, may promote the expression of adhesive molecules in the vasculature, resulting in the infiltration of a larger amount of monocytes, neutrophils, and macrophages.19 Accumulated evidence has indicated that I/R induces vigorous inflammatory reactions, which is verified by significantly increased levels of TNF-α and IL-6.20,21 The increased levels of proinflammatory cytokines from activated leukocytes can lead to myocardial tissue damage. To evaluate the relationship between the anti-inflammatory properties and sevoflurane pretreatment-induced cardioprotection, we conducted this experiment to determine whether or not this extract affected the I/R-induced changes in TNF-α and IL-6. Our data showed that I/R increased the levels of both TNF-α and IL-6, whereas SPC greatly reduced the levels of these cytokines. In addition, blockade of VEGFR-1 function by a specific pharmacologic inhibitor (MF1) reverses SPC-mediated cardioprotection and anti-inflammation. Overall, SPC may reduce myocardial I/R injury via upregulation of VEGFR-1 and anti-inflammatory activities.
We have documented in this study for the first time that pretreatment with sevoflurane mediated cardioprotection, which is associated with upregulation of VEGFR-1. To provide more conclusive evidence of how VEGFR-1 is involved in sevoflurane pretreatment-mediated cardioprotection will require further researches with several types of gene knockout mice and assessment with pharmacologic inhibitors. For example, the flt-1TK−/- mice specifically lack signaling from VEGFR-1 tyrosine kinase, which appear to be useful for exploring the role of VEGFR-1 signals in various pathologic conditions, especially ischemic diseases and inflammation, but this is beyond the scope of our study. Targeting this signaling with an antibody or related ligand should improve the quality of life of patients. Antitumor drugs targeting VEGF–VEGFRs, such as VEGFR tyrosine kinase inhibitors, were successfully developed and clinically used. However, drugs used to treat ischemic diseases related to VEGFR-1 are not yet available. Therefore, further translational studies at the wider range are necessary.
Conclusion
In summary, using an isolated rat heart model, we have demonstrated that 2.5% sevoflurane pretreatment exerts a profound cardioprotective effect against I/R injury, which seems to be associated with upregulation of VEGFR-1 and anti-inflammation. Our results reveal that VEGFR-1 may be a promising candidate for the treatment of myocardial I/R injury.
Acknowledgments
We thank Prof Yangqin Chen and Dr Xiaodan Wu from Fujian Medical University for their support and cooperation. This study was supported by the following grants: Natural Science Foundation of Fujian Province (2015J01373), Young and Middle-aged Backbone Talents Training Project of Fujian Province (2016-ZQN-7), and Fujian Medical Innovation Project (2015-CXB-24).
Disclosure
The authors report no conflicts of interest in this work.
References
Writing Committee for the VISION Study Investigators; Devereaux PJ, Biccard BM, Sigamani A, et al. Association of postoperative high-sensitivity troponin levels with myocardial injury and 30-day mortality among patients undergoing noncardiac surgery. JAMA. 2017;317(16):1642–1651. | ||
Fradorf J, Huhn R, Weber NC, et al. Sevoflurane-induced preconditioning: impact of protocol and aprotinin administration on infarct size and endothelial nitric-oxide synthase phosphorylation in the rat heart in vivo. Anesthesiology. 2010;113(6):1289–1298. | ||
Zhao J, Wang F, Zhang Y, et al. Sevoflurane preconditioning attenuates myocardial ischemia/reperfusion injury via caveolin-3-dependent cyclooxygenase-2 inhibition. Circulation. 2013;128(11 Suppl 1):S121–S129. | ||
Lu Y, Wang L, Liu N, Dong T, Li R. Sevoflurane preconditioning in on-pump coronary artery bypass grafting: a meta-analysis of randomized controlled trials. J Anesth. 2016;30(6):977–986. | ||
Wei J, Chen S, Xue S, et al. Blockade of inflammation and apoptosis pathways by siRNA prolongs cold preservation time and protects donor hearts in a porcine model. Mol Ther Nucleic Acids. 2017;9:428–439. | ||
Lucchinetti E, Lou PH, Gandhi M, Clanachan AS, Zaugg M. Differential effects of anesthetics and opioid receptor activation on cardioprotection elicited by reactive oxygen species-mediated postconditioning in Sprague-Dawley rat hearts. Anesth Analg. Epub 2017 Dec 15. | ||
Obal D, Dettwiler S, Favoccia C, Scharbatke H, Preckel B, Schlack W. The influence of mitochondrial KATP-channels in the cardioprotection of preconditioning and postconditioning by sevoflurane in the rat in vivo. Anesth Analg. 2005;101(5):1252–1260. | ||
Kunst G, Klein AA. Peri-operative anaesthetic myocardial preconditioning and protection – cellular mechanisms and clinical relevance in cardiac anaesthesia. Anaesthesia. 2015;70(4):467–482. | ||
Guerrero-Orriach JL, Escalona Belmonte JJ, Ramirez Fernandez A, Ramirez Aliaga M, Rubio Navarro M, Cruz Manas J. Cardioprotection with halogenated gases: how does it occur? Drug Des Devel Ther. 2017;11:837–849. | ||
Shibuya M. Structure and function of VEGF/VEGF-receptor system involved in angiogenesis. Cell Struct Funct. 2001;26(1):25–35. | ||
Shibuya M. VEGF-VEGFR system as a target for suppressing inflammation and other diseases. Endocr Metab Immune Disord Drug Targets. 2015;15(2):135–144. | ||
Addya S, Shiroto K, Turoczi T, et al. Ischemic preconditioning-mediated cardioprotection is disrupted in heterozygous Flt-1 (VEGFR-1) knockout mice. J Mol Cell Cardiol. 2005;38(2):345–351. | ||
Thirunavukkarasu M, Juhasz B, Zhan L, et al. VEGFR1 (Flt-1+/−) gene knockout leads to the disruption of VEGF-mediated signaling through the nitric oxide/heme oxygenase pathway in ischemic preconditioned myocardium. Free Radic Biol Med. 2007;42(10):1487–1495. | ||
Carmeliet P, Moons L, Luttun A, et al. Synergism between vascular endothelial growth factor and placental growth factor contributes to angiogenesis and plasma extravasation in pathological conditions. Nat Med. 2001;7(5):575–583. | ||
Plaschke K, Staub J, Ernst E, Marti HH. VEGF overexpression improves mice cognitive abilities after unilateral common carotid artery occlusion. Exp Neurol. 2008;214(2):285–292. | ||
Vidal F, Aragones J, Alfranca A, de Landazuri MO. Up-regulation of vascular endothelial growth factor receptor Flt-1 after endothelial denudation: role of transcription factor Egr-1. Blood. 2000;95(11):3387–3395. | ||
Amano H, Kato S, Ito Y, et al. The role of vascular endothelial growth factor receptor-1 signaling in the recovery from ischemia. PLoS One. 2015;10(7):e0131445. | ||
Wang C, Weihrauch D, Schwabe DA, et al. Extracellular signal-regulated kinases trigger isoflurane preconditioning concomitant with upregulation of hypoxia-inducible factor-1 alpha and vascular endothelial growth factor expression in rats. Anesth Analg. 2006;103(2):281–288. | ||
Ioannou A, Kannan L, Tsokos GC. Platelets, complement and tissue inflammation. Autoimmunity. 2013;46(1):1–5. | ||
Yang J, Fan Z, Yang J, Ding J, Yang C, Chen L. microRNA-22 attenuates myocardial ischemia-reperfusion injury via an anti-inflammatory mechanism in rats. Exp Ther Med. 2016;12(5):3249–3255. | ||
Wang C, Sun H, Song Y, et al. Pterostilbene attenuates inflammation in rat heart subjected to ischemia-reperfusion: role of TLR4/NF-kappaB signaling pathway. Int J Clin Exp Med. 2015;8(2):1737–1746. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.