Back to Journals » Journal of Pain Research » Volume 11
Upregulation of the high-affinity choline transporter in colon relieves stress-induced hyperalgesia
Received 31 January 2018
Accepted for publication 24 July 2018
Published 21 September 2018 Volume 2018:11 Pages 1971—1982
DOI https://doi.org/10.2147/JPR.S164186
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Erica Wegrzyn
Meng-Juan Lin,1,2 Bao-Ping Yu1,2
1Department of Gastroenterology, Renmin Hospital of Wuhan University, Wuhan, Hubei, China; 2Key Laboratory of Hubei Province for Digestive System Disease, Renmin Hospital of Wuhan University, Wuhan, Hubei, China
Background: Irritable bowel syndrome (IBS) is a common disease with hyperalgesia, the mechanisms of which remain elusive. The cholinergic system is known to be involved in pain inhibitory pathways in multiple diseases, and its involvement in IBS is unknown.
Objective: We aimed to determine whether high-affinity choline transporter CHT1, a major determinant of the cholinergic signaling capacity, is involved in regulating intestinal sensations associated with stress-induced visceral pain.
Materials and methods: An IBS rat model was established by chronic water avoidance stress (WAS). Colonic pathologic alterations were detected by H&E staining. Visceral sensations were determined by scoring the abdominal withdrawal reflex (AWR) and visceromotor response (VMR) magnitude of the electromyogram in response to colorectal distension (CRD). Abdominal mechanical hyperalgesia was assessed by counting the number of withdrawal events evoked by applying von Frey filaments. Real-time PCR, Western blot, and immunostaining were performed to identify CHT1 expression in the colon. Acetylcholine (ACh) secretion was determined by ELISA. Effects of MKC-231, a choline uptake enhancer, on visceral pain were examined.
Results: After 10 days of WAS exposure, AWR score and VMR magnitude in response to CRD were significantly enhanced and the number of withdrawal events was elevated. Protein and mRNA levels of CHT1 were considerably increased in the colon after WAS. CHT1 upregulation in the WAS-exposed group was largely abolished by ammonium pyrrolidinedithiocarbamate. The density of CHT1-positive intramuscular cells and enteric neurons in the myenteric plexus was enhanced in WAS-exposed rats. Pharmacologic enhancement of CHT1 activity by MKC-231 gavage could relieve the visceral pain of WAS rats by upregulating CHT1 protein expression and enhancing ACh production.
Conclusion: CHT1 may exert an antinociceptive effect in stress-induced visceral pain by modulating ACh synthesis through nuclear factor kappa B signaling. MKC-231 could be used as a potential drug to treat disorders with hyperalgesia.
Keywords: irritable bowel syndrome, high-affinity choline transporter, myenteric plexus, visceral hyperalgesia
Introduction
Several lines of evidence indicate that the cholinergic system may play a crucial role in certain important physiologic functions such as movement, sensation, learning, neuroimmune, and endocrine processes.1,2 Numerous studies suggest that the cholinergic system also exhibits significant antinociceptive effects in chronic neuropathic pain,3 as well as in visceral pain.4 Muscarinic receptor agonists and acetylcholinesterase inhibitors have long been known to produce analgesia,5 enhance the analgesia of opiates, and attenuate the development of tolerance to morphine.6 However, administration of cholinomimetics can cause various side effects such as dysfunction of gastrointestinal (GI), cardiovascular, and even the central nervous system,7 which greatly limits their clinical use.
The high-affinity choline transporter CHT1, which is the rate-limiting step for acetylcholine (ACh) synthesis, exerts a crucial role in the functioning of the cholinergic system.8 CHT1 is present on the presynaptic terminal of cholinergic neurons and can take up choline hydrolyzed from ACh at the synaptic cleft into the presynaptic nerve terminal.8 This choline uptake that is Na+ and Cl− dependent is sensitive to hemicholinium-3.8 A previous study showed that CHT1 knockout mice failed to synthesize ACh and died within 1 hour after birth.9 However, MKC-231, a choline uptake enhancer, was reported to improve cholinergic hypofunction by enhancing high-affinity choline uptake, subsequently facilitating ACh synthesis and release.10 To date, numerous studies report that altered expression and dysfunction of CHT1 play a key role in the development of neurodegenerative diseases (especially Alzheimer’s disease),11 vascular diseases (carotid atherosclerosis),12 and tumors (such as pancreatic ductal adenocarcinoma).13 Our recent study suggested for the first time that CHT1 may exert an important inhibitory function in the visceral hyperalgesia induced by chronic pancreatitis.14 However, there are as yet no reports on the involvement of CHT1 in pain modulation of irritable bowel syndrome (IBS).
IBS characterized by visceral hyperalgesia and GI dysmotility is one of the most common GI disorders exhibiting a global prevalence of 11.2% according to the Rome III criteria.15 The pathogenesis of IBS is multifactorial with interaction between psychosocial (stress, behavior, illness, and diet) and biologic (infection, gut microbiota, and immune activation) variables.16 Animal models of water avoidance stress (WAS) have been utilized as psychosocial stressors in an attempt to produce the IBS symptomatology and identify the detailed mechanisms of IBS.17 Therefore, the present study was designed to explore the role of CHT1 in visceral hyperalgesia in a rat model of IBS induced by chronic WAS as a possible target of drug therapies.
Materials and methods
Animals
Sixty male Sprague Dawley rats (weight 180–230 g) were purchased from Hunan SJA Laboratory Animal Co. Ltd. The animals were housed under climate-controlled conditions (22°C±2°C with a 12-hour light/dark cycle and a relative humidity of 40%–60%), and had ad libitum access to food and water. All experiments were approved by the Institutional Animal Care and Use Committee of Wuhan University and were conducted in accordance with the Declaration of the National Institutes of Health Guide for Care and Use of Laboratory Animals and the People’s Republic of China animal welfare legislations in order to minimize the number of experimental animals and their suffering.
The rats were randomly divided into four groups (n=15/group): control group, WAS group, WAS + ammonium pyrrolidinedithiocarbamate (PDTC) group, and WAS + MKC-231 group. The WAS procedure was performed to induce stress-induced hyperalgesia as described previously with minor modifications.18 Briefly, rats were placed on a platform (10×8×8 cm; length × width × height) in the center of a water-filled (25°C) tank (45×25×35 cm; length × width × height) for 1 hour daily, for 10 consecutive days. The water level in the tank was kept at 1 cm below the platform. The animals from the WAS + PDTC group were administered an intraperitoneal injection of PDTC (100 mg/kg) (ammonium pyrrolidinedithiocarbamate, Beyotime, Shanghai, China), a selective nuclear factor kappa B (NF-κB) antagonist, in saline, 1 hour before WAS for 10 days. Rats in the WAS + MKC-231 group were gavaged with MKC-231 (2 mg/kg) (MedChemExpress, Monmouth Junction, NJ, USA) 1 hour before WAS for 10 days. After 48 hours, subsequent experiments such as assessment of visceral pain, colonic histologic examination, and molecular biology tests were conducted. The distal colon was resected and studied.
Observation of abdominal withdrawal reflex (AWR)
To evaluate the visceral hyperalgesia, a colorectal distension (CRD) test was performed by inserting a balloon into the anus for about 4 cm. The polyethylene cannula with the balloon was lightly tied to the root of the rat tail to prevent it from sliding out. The rat was then placed in a small transparent cubicle (20×8×8 cm) on a platform and allowed to adapt for 20 minutes. The AWR was recorded when the pressure of balloon was maintained at 20, 40, 60, and 80 mmHg. A stethoscope-modified three-way device was used to monitor the pressure. AWR in response to graded CRD stimulations was observed and recorded as semiquantitative AWR scores according to the scale of Al-Chaer et al.19 The AWR score was determined based on the behavioral response (head, abdominal, or whole-body movement) to CRD. The scoring criteria are as follows: 0, no behavioral response to CRD; 1, brief head movement followed by immobility; 2, contraction of abdominal muscles; 3, lifting of abdomen; 4, body arching and lifting of pelvic structures. To obtain more accurate scores, each graded CRD was repeated three times and the average value of scores was taken for analysis.
Von Frey filament (VFF) test
VFF test was conducted to assess the abdominal mechanical hyperalgesia by counting the number of withdrawal events (either abdominal or whole-body withdrawal) evoked by the application of VFFs in a blinded manner.20 The rats were placed in a plastic box with a mesh floor and acclimated for 30 minutes. Filaments of six different strengths (0.4, 0.6, 1, 1.4, 2, and 4 g; North Coast Medical Morgan Hill, CA, USA) were applied to the abdominal area from underneath the mesh floor in ascending order 10 times. Each application lasted for 1–2 seconds with a 10-second interval between applications. The data were expressed as the number of withdrawal events during the 10 applications in each trial.
Electromyogram (EMG) recording
To evaluate the visceral hyperalgesia, we recorded the EMG signal of abdominal oblique musculature. For EMG measurements, animals were initially anesthetized with isoflurane inhalation, keeping a mild and stable anesthesia throughout the experiment. After anesthesia, the rat was fixed in a supine position. A pair of electrodes was implanted into the external oblique muscle of the rats. The electrodes were connected to a Bio Amp (AD Instruments Pty Ltd, Bella Vista, Australia), which was connected to a Power Lab (AD Instruments) as an EMG acquisition system. CRD stimulation was given in a manner similar to that for behavioral tests after 20 minutes of adaptation. Each recording progression consisted of a 5-minute predistention baseline activity measurement, a 20-second CRD-evoked response (20, 40, 60, and 80 mmHg), and a 3-minute postdistention activity measurement, followed by a 3-minute rest between two CRD episodes. The EMG signals expressed by the visceromotor response (VMR), and the area under the curve (AUC) in response to the CRD stimuli were collected and analyzed using Lab Chart 7 software (AD Instruments). The analytic period was 40 seconds (20 seconds during and 20 seconds after each CRD). The net value for each CRD was calculated by subtracting the AUC of the baseline (40 seconds interval) before each CRD.21
Western blot
Total proteins were extracted using RIPA lysis buffer (Beyotime) and subsequently subjected to centrifugation at 12,000 rpm, 4°C, for 30 minutes. Supernatants were then collected and protein concentrations were determined using the BCA protein assay kit (Beyotime). Samples were mixed with 5× loading buffer and heated at 100°C for 5 minutes to denature the proteins. Thirty micrograms of total proteins were loaded on 10% SDS-PAGE gels and electrophoresed. The separated proteins were transferred to polyvinylidene difluoride membranes (Millipore, Darmstadt, Germany), and the membranes were incubated in 5% skimmed milk at room temperature for 2 hours, to block nonspecific binding. The blots were then incubated overnight at 4°C with the primary antibody against CHT1 (Santa Cruz Biotechnology, Dallas, TX, USA) and β-actin (Beyotime, Shanghai, China). After washing three times with TBST for 10 minutes, the corresponding secondary antibody conjugated to horseradish peroxidase (Boster, Wuhan, China) was applied for 1 hour at room temperature, followed by three washes of TBST for 10 minutes. Specific protein bands were visualized using the ECL kit (Thermo Fisher Scientific, Waltham, MA, USA) and an X-ray film (Kodak, Xiamen, China). The OD of the bands was analyzed using Band Scan 5.0 software (Alpha Innotech Corp. San Leandro, CA, USA). This experiment was repeated six times.
Quantitative real-time PCR (RT-PCR)
Total RNA was extracted from the colon that was frozen in liquid nitrogen with TRIzol reagent (Invitrogen, Carlsbad, CA, USA), following the manufacturer’s instructions. Next, 1.5 µg of total RNA from each sample was used for cDNA synthesis in a total volume of 20 µL, with oligo (dT)18 and Hiscript Reverse Transcriptase (VAZYME, Nanjing, China) included in the reverse transcription system. Quantitative RT-PCR was performed in 20 µL wells with SYBR Green PCR master mix (VAZYME) on the viiA 7 RT-PCR system (ABI, Carlsbad, CA, USA). After incubation at 95°C for 10 minutes as the initiation of thermal cycling, 40 cycles of 95°C for 30 seconds and 60°C for 30 seconds were performed. Each reaction was performed in triplicate. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as a loading control to normalize each sample. The PCR primers used are as follows: CHT1 forward: 5′-GACTGTGTATGGGCTCTGGT-3′, reverse: 5′-TGGCTCTCCTCCGGTAATTC-3′; GAPDH forward: 5′-ACAGCAACAGGGTGGTGGAC-3′, reverse: 5′-TTT GAGGGTGCAGCGAACTT-3′. Specificity of the PCR products was monitored by melting curve analysis. The relative expression of CHT1 mRNA was quantified by the 2−DDct method. This experiment was repeated six times.
Immunohistochemistry
Immunohistochemistry was performed on paraffin-embedded sections. Fresh colonic specimens were fixed in 4% paraformaldehyde solution, embedded in paraffin, and cut longitudinally into 5 µm thick sections. The sections were incubated with rabbit anti-rat polyclonal antibody raised against CHT1 (Millipore, Darmstadt, Germany) at room temperature for 24 hours, followed by incubation with the appropriate secondary antibodies. Subsequently, the sections were stained with diaminobenzidine and counterstained with hematoxylin. Six slices were prepared in each group. Stained sections were examined with an Olympus BX53 microscope (Olympus, Tokyo, Japan). The results were evaluated by average optical density (AOD) using Image Pro Plus software version 6.0 (Media Cybernetics, Silver Spring, MD, USA). AOD = positive area × OD/total area.
Immunofluorescence
Immunofluorescence analysis for CHT1 was performed both on the whole-mounts of colonic myenteric plexus (MP) and longitudinal sections of the colon. Immunofluorescence staining for CHT1 on whole-mounts of colonic MP was performed as follows. Colonic specimens were fixed in 4% paraformaldehyde solution at 4°C for 6–8 hours. Subsequently, the mucosa was removed with sharp forceps under a stereoscopic microscope. The circular muscle was then stripped off carefully at certain intervals. Whole-mount preparations were then blocked with 10% goat serum containing 0.3% Triton X-100 for 1 hour at room temperature. Next, the samples were incubated overnight at 4°C with mouse anti-rat polyclonal CHT1 antibody. After washing in PBS with Tween-20 (PBST), the pinned tissues were incubated overnight at 4°C with rabbit anti-rat polyclonal PGP9.5 antibody (Abcam, Cambridge, England). After washing thrice with PBST for 5 minutes, the tissues were incubated with tetramethylrhodamine-conjugated rabbit anti-mouse secondary antibody (Boster) and FITC-conjugated goat anti-rabbit secondary antibody (Boster) for 1 hour at room temperature. Six slices were prepared in each group. The stained samples were imaged using an Olympus BX53 microscope (Olympus) after they were mounted on a slide with a coverslip, and sealed with glycerol. The results were analyzed using Image Pro Plus software version 6.0. The CHT1-IR neurons were quantified as a relative percentage considering the total number of PGP9.5-IR neurons. Immunofluorescence of colonic longitudinal sections was conducted in a similar manner using the above procedure.
ELISA
ELISA was used to detect ACh release from the colon. For detecting ACh production, fresh colonic tissues were homogenized on ice after dilution and subsequently centrifuged at 2500–3500 rpm for 10 minutes. Supernatants were then collected and ACh content was determined using the ACh ELISA Kit (Jiancheng, Nanjing, China) according to the manufacturer’s instructions. Each sample was assayed in duplicate. Absorbance was read at 550 nm and the concentrations calculated according to the standard curve.
Statistical analysis
Statistical analyses were performed using SPSS version 21.0 (IBM Co, Armonk, NY, USA). Continuous variables were presented as mean ± SD and compared using independent sample t-test and variance analysis. Differences among different groups were analyzed by two-way repeated-measures ANOVA with distention pressure and filament force as the repeated measure. S-N-K post hoc test was used where appropriate. A two-sided P-value <0.05 was regarded as statistically significant.
Results
Evaluation of animal model
No animals died during the experiment. H&E staining revealed that colon specimens of the control and WAS groups were intact, simultaneously without obvious infiltration of inflammatory cells (Figure 1A). However, the AWR score of WAS rats was significantly higher than that of the control group at the pressure of 40, 60, and 80 mmHg (20 mg: control 0.83±0.12, WAS 1.20±0.13, P>0.05; 40 mmHg: control 1.40±0.17, WAS 2.20±0.15, P<0.05; 60 mmHg: control 2.30±0.14, WAS 3.12±0.11, P<0.05; 80 mmHg: control 2.92±0.16, WAS 3.70±0.12, P<0.05; control n=6 rats, WAS n=6 rats) (Figure 1B). Furthermore, the number of withdrawal events in WAS-exposed rats was considerably enhanced compared to the control group in the VFF test (0.4 g: control 0.21±0.15, WAS 2.12±0.23, P<0.05; 0.6 g: control 1.19±0.23, WAS 3.27±0.31, P<0.05; 1 g: control 1.91±0.28, WAS 5.07±0.33, P<0.05; 1.4 g: control 2.79±0.34, WAS 7.10±0.30, P<0.05; 2 g: control 4.31±0.37, WAS 8.22±0.42, P<0.05; 4 g: control 4.98±0.31, WAS 8.90±0.41, P<0.05; control n=6 rats, WAS n=6 rats) (Figure 1C). In addition, we detected that the VMR amplitude of the WAS group was higher than that in control rats (20 mg: control 72.1±7.9 μv s, WAS 105.0±8.1 μv s, P<0.05; 40 mmHg: control 114.7±8.6 μv s, WAS 168.4±8.3 μv s, P<0.05; 60 mmHg: control 172.4±9.6 μv s, WAS 218.8±9.3 μv s, P<0.05; 80 mmHg: control 211.1±9.1 μv s, WAS 289.7±10.4 μv s, P<0.05; control n=6 rats, WAS n=6 rats) (Figure 1D).
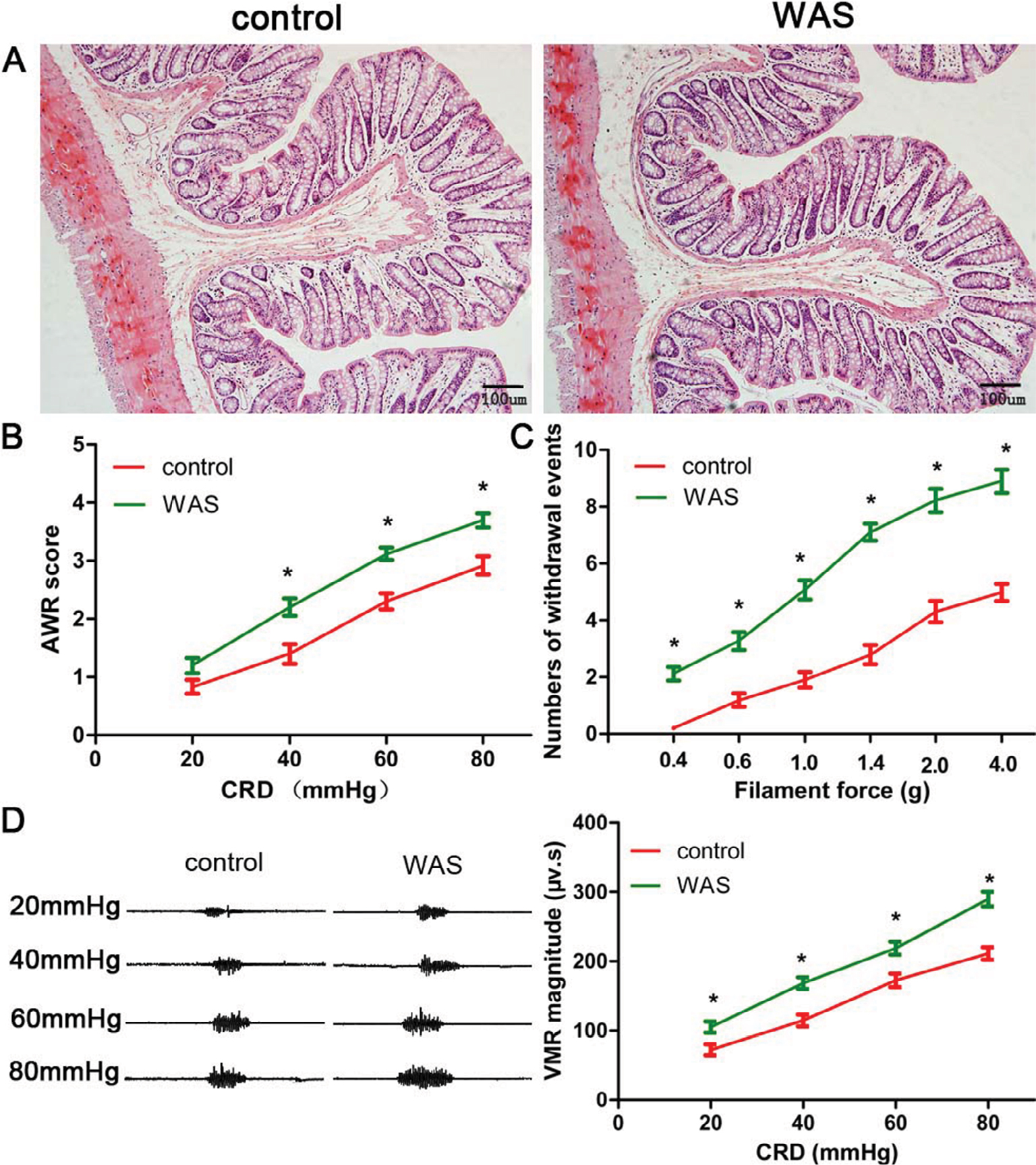 | Figure 1 Evaluation of animal model. Notes: (A) H&E staining (200×) shows that the colon specimens of control (left) and WAS (right) groups are intact, without congestion and obvious inflammatory cell infiltration. (B) The AWR score of WAS rats is significantly higher than that of the control group. (C) The withdrawal events of the WAS group in VFF test are enhanced compared to the control rats. (D) VMR cure (left) and summary data (right) for raw VMR responses to CRD at different pressures in control and WAS rats. VMR amplitude of the WAS group is higher than that of the control rats. Bars: (A) = 100 µm; n of each group = 6; *P<0.05, compared to control. Abbreviations: AWR, abdominal withdrawal reflex; CRD, colorectal distension; VFF, Von Frey filament; VMR, visceromotor response; WAS, water avoidance stress. |
These data suggested that the animal model of chronic stress-induced hyperalgesia, exhibiting the hallmark of IBS, was successfully established after 10 days of WAS exposure.
Enhanced CHT1 expression in the colon of WAS rats
Western blot analysis showed that the CHT1 protein level in the colon was elevated after 10 days of WAS (Figure 2A). The ratios of CHT1 to β-actin obtained in the control and WAS groups were 0.356±0.0312 and 0.675±0.0331 (P<0.05), respectively. Quantitative RT-PCR analysis further confirmed CHT1 upregulation in the colon under chronic stress at the transcriptional level (Figure 2B). The mean 2−ΔΔCt values of CHT1 mRNA levels in the control and WAS groups were 1.000±0.0000 and 1.780±0.0475 (P<0.05), respectively.
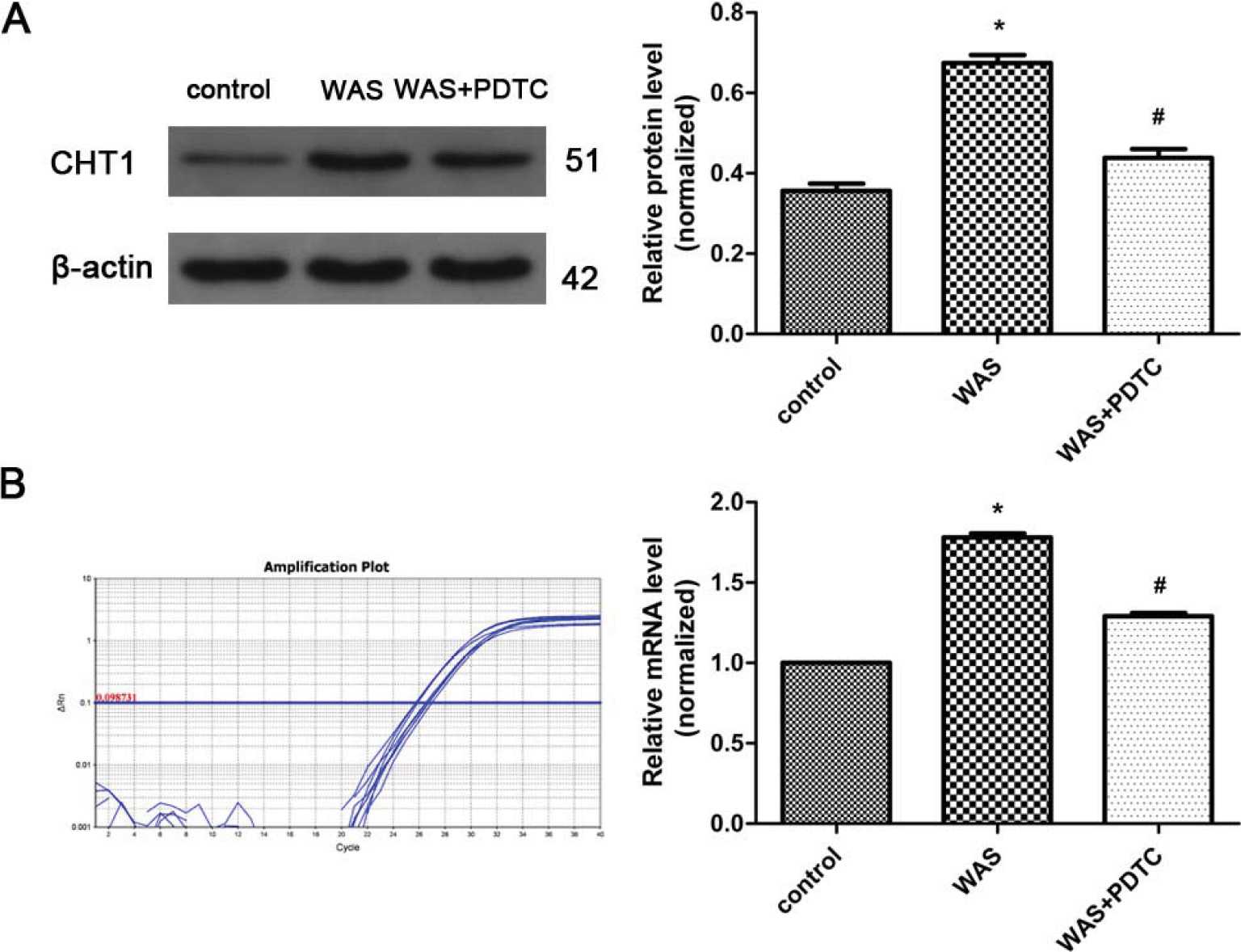 | Figure 2 Protein and mRNA levels of CHT1 in the colon. Notes: (A) Western blot analysis reveals that CHT1 protein expression is enhanced in WAS rats compared to control rats, whereas the elevated CHT1 expression is partly abolished after treatment with PDTC. (B) CHT1 mRNA levels in WAS-exposed rats are higher than those in control rats, and are considerably declined after treatment with PDTC. n of each group =6; *P<0.05, compared to control; #P<0.05, compared to the WAS group. Data are presented as mean ± SD and analyzed with the S-N-K post hoc test. Abbreviations: PDTC, ammonium pyrrolidinedithiocarbamate; WAS, water avoidance stress. |
PDTC was administered to explore the mechanism of CHT1 upregulation in WAS-exposed rats. Western blot and quantitative RT-PCR analyses revealed that both CHT1 protein (0.675±0.0331 in WAS group and 0.439±0.0370 in WAS + PDTC group, P<0.05) and mRNA (1.780±0.0475 in WAS group and 1.293±0.0326 in the WAS + PDTC group, P<0.05) levels in the PDTC-treated group were significantly declined compared to those in the WAS-exposed rats (Figure 2A, B), indicating that upregulation of CHT1 protein expression in WAS-exposed group was largely abolished by PDTC.
Taken together, these results indicated that CHT1 expression in the colon was considerably upregulated after 10 consecutive days of WAS exposure, partly mediated by the NF-κB signaling pathway.
Immunostaining of CHT1 in the colon
Immunohistochemical staining of longitudinal paraffin sections revealed that CHT1 was robustly expressed in the colon, both in the mucosal and muscular layer. Furthermore, we clearly observed two populations of CHT1-positive cells in the muscular layer (Figure 3A). One population was mainly fusiform in shape and was found within the circular muscle, exhibiting the morphologic characteristics of interstitial cells of Cajal (ICC) and smooth muscle cells. The other population was predominantly round in shape located between the circular muscle and longitudinal muscle, showing the hallmark of MP neurons. Upon statistical analysis, we found that the AOD of CHT1 in the muscular layer of WAS-exposed rats was elevated (0.562±0.0371 in control group and 0.786±0.0394 in WAS group, P<0.05).
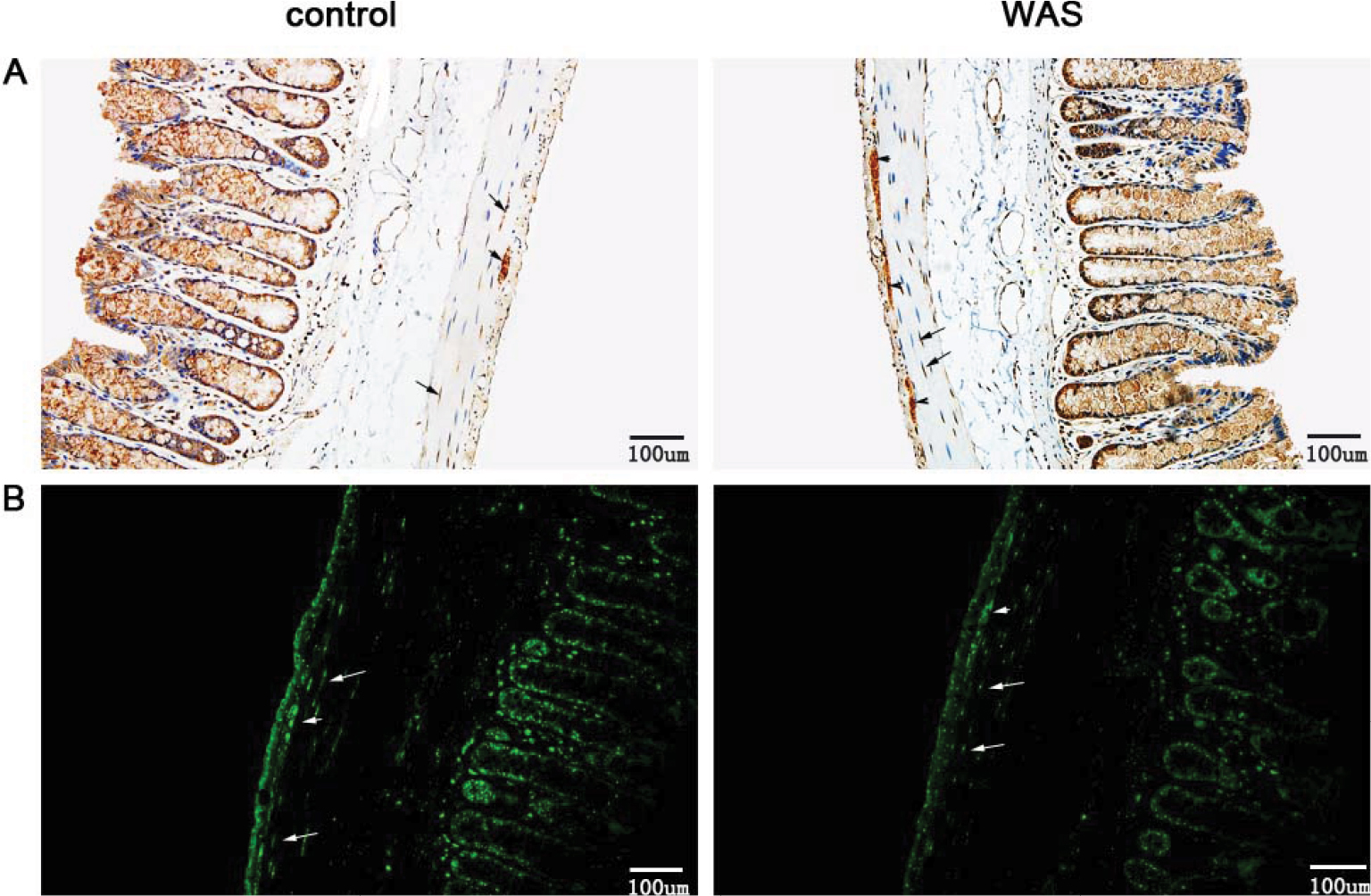 | Figure 3 Immunostaining of CHT1 in the colon. Notes: (A) Immunohistochemical staining for CHT1 reveals that the AOD of CHT1 in the muscular layer of WAS rats (right) is increased compared to that of control rats (left) (200×). (B) Immunofluorescence staining for CHT1 shows that the density of CHT1 in the muscular layer is significantly elevated after 10 days of WAS (200×). Both the black and white arrows indicate CHT1-positive cells that are mainly fusiform in shape and found within the circular muscle, exhibiting the morphologic characteristics of ICC and smooth muscle cells. Both black and white arrowheads indicate CHT1-positive cells that are predominantly round in shape, located between the circular muscle and longitudinal muscle, showing the hallmark of MP neurons. Bars: (A) = 100 µm, (B) = 100 µm; n of each group = 6. Abbreviations: AOD, average optical density; ICC, interstitial cells of Cajal; MP, myenteric plexus; WAS, water avoidance stress. |
Immunofluorescence staining clearly reveals the above two populations of CHT1-positive cells in the muscular layer (Figure 3B). These results support the immunohistochemistry findings. Furthermore, the density of CHT1-postive cells in the muscular layer was significantly enhanced after 10 days of WAS.
The present data indicated that the enhanced expression of CHT1 in the colon of WAS rats was restricted to the muscular layer.
MKC-231 alleviated hyperalgesia of WAS-exposed rats
MKC-231, a choline uptake enhancer, was administered to explore the role of CHT1 in stress-induced hyperalgesia. Western blot analysis showed that the ratios of CHT1 to β-actin in WAS rats were considerably increased after 10 consecutive days of MKC-231 treatment (0.634±0.0317 in WAS group and 0.781±0.0361 in WAS + MKC-231 group, P<0.05) (Figure 4A), indicating that CHT1 protein expression in the colon was upregulated by MKC-231. ELISA revealed that the ACh content in the colon of WAS-exposed rats was significantly increased in contrast to that in the control rats (2.417±0.247 µg/mg in the control group and 3.574±0.221 µg/mg in the WAS group, P<0.05). Furthermore, the ACh content of WAS rats was considerably elevated after treatment with MKC-231 (3.574±0.221 µg/mg in the WAS group and 4.258±0.244 µg/mg in the WAS + MKC-231 group, P<0.05) (Figure 4B).
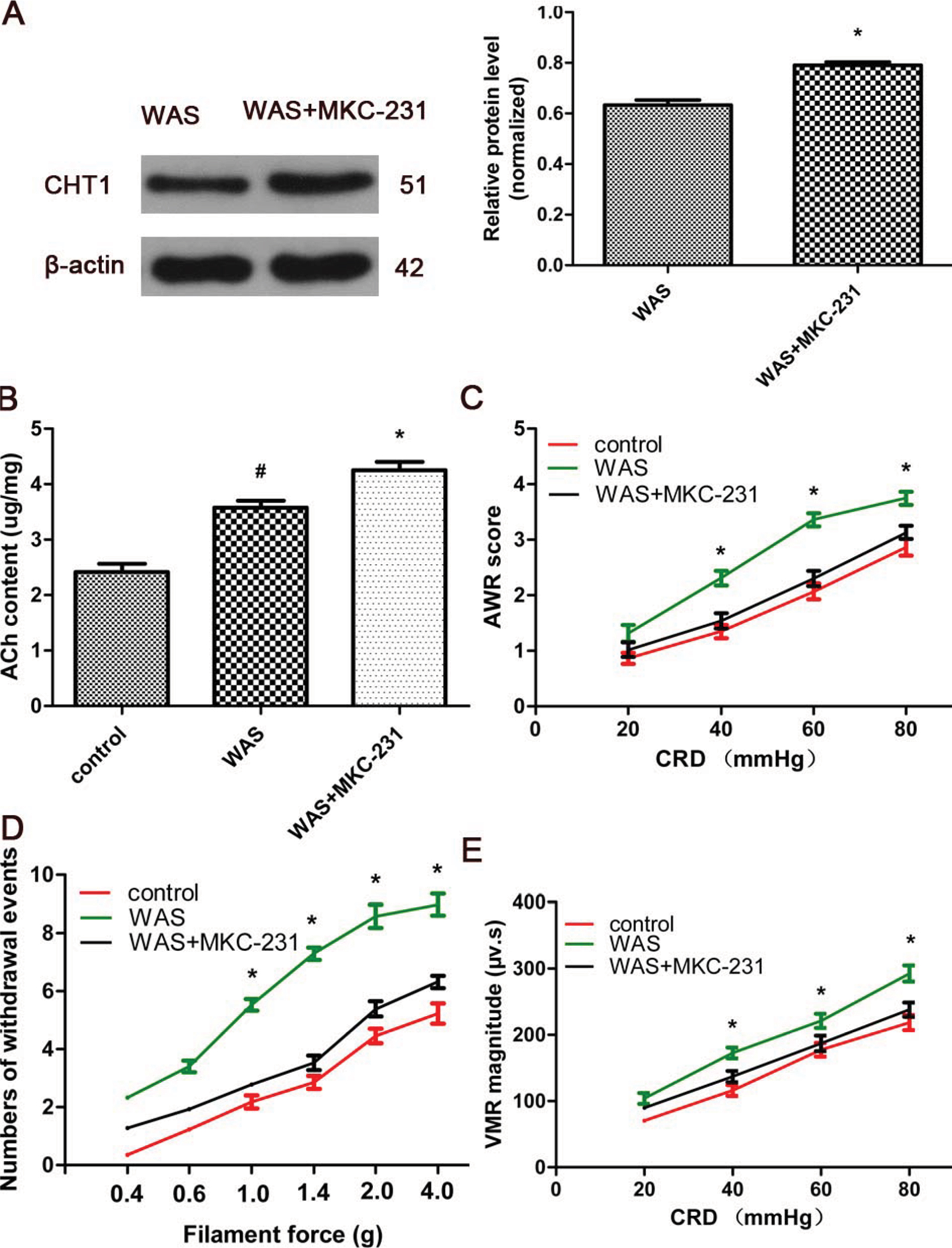 | Figure 4 Effect of MKC-231 on pain threshold and CHT1 expression in WAS rats. Notes: (A) Immunoblots (left) and a histogram (right) for CHT1 in the colon of WAS and WAS-MKC-231-treated rats reveal that the ratios of CHT1 to β-actin expression in WAS rats are considerably increased after 10 consecutive days of MKC-231 treatment. (B) The ACh content of WAS rats is higher than that of the control rats, but lower than that of the MKC-231-treated rats. (C) The AWR score of WAS + MKC-231 rats is significantly reduced in contrast to the WAS group. (D) Withdrawal events for the WAS + MKC-231 group in the VFF test are declined compared to those in the WAS group. (E) VMR amplitude of the WAS + MKC-231 group is lower than that of the WAS rats. n of each group = 6; *P<0.05, compared to the WAS group. Data are presented as mean ± SD and analyzed by two-way repeated-measures ANOVA with distention pressure and filament force as the repeated measures. Abbreviations: AWR, abdominal withdrawal reflex; CRD, colorectal distension; VFF, Von Frey filament; VMR, visceromotor response; WAS, water avoidance stress. |
In our study, different means were used to assess hyperalgesia. We detected that the AWR score of MKC-231-treated rats was reduced when compared with the WAS group at the pressure of 40, 60, and 80 mmHg (20 mg: control 0.86±0.10, WAS 1.32±0.15, WAS + MKC-231 1.02±0.13, P>0.05; 40 mmHg: control 1.35±0.12, WAS 2.31±0.13, WAS + MKC-231 1.54±0.14, P<0.05; 60 mmHg: control 2.07±0.14, WAS 3.36±0.12, WAS + MKC-231 2.30±0.14, P<0.05; 80 mmHg: control 2.86±0.15, WAS 3.75±0.14, WAS + MKC-231 3.13±0.12, P<0.05; control n=6 rats, WAS n=6 rats, WAS + MKC-231 n=6 rats) (Figure 4C). In the VFF test, number of withdrawal events in MKC-231-exposed rats was considerably declined compared to that in the WAS group (0.4 g: control 0.36±0.12, WAS 2.32±0.15, WAS + MKC-231 1.27±0.14, P>0.05; 0.6 g: control 1.22±0.13, WAS 3.41±0.20, WAS + MKC-231 1.94±0.12, P>0.05; 1 g: control 2.18±0.23, WAS 5.53±0.21, WAS + MKC-231 2.77±0.16, P<0.05; 1.4 g: control 2.86±0.27, WAS 7.30±0.21, WAS + MKC-231 3.54±0.25, P<0.05; 2 g: control 4.46±0.25, WAS 8.58±0.40, WAS + MKC-231 5.39±0.26, P<0.05; 4 g: control 5.23±0.34, WAS 8.98±0.38, WAS + MKC-231 6.32±0.21, P<0.05; control n=6 rats, WAS n=6 rats, WAS + MKC-231 n=6 rats) (Figure 4D). Furthermore, we noticed that the VMR amplitude of the WAS + MKC-231 group was significantly lower than that of WAS rats (20 mg: control 70.6±7.4 μv s, WAS 104.0±8.1 μv s, WAS + MKC-231 89.7±7.6 μv s, P>0.05; 40 mmHg: control 116.0±8.3 μv s, WAS 172.7±8.1 μv s, WAS + MKC-231 137.1±8.5 μv s, P<0.05; 60 mmHg: control 177.5±10.3 μv s, WAS 221.2±10.7 μv s, WAS + MKC-231 187.1±11.6 μv s, P<0.05; 80 mmHg: control 218.4±11.4 μv s, WAS 292.6±12.2 μv s, WAS + MKC-231 238.0±10.7 μv s, P<0.05; control n=6 rats, WAS n=6 rats, WAS + MKC-231 n=6 rats) (Figure 4E).
These results suggested that MKC-231 could relieve hyperalgesia in WAS rats via upregulation of CHT1 protein expression and enhancing ACh production.
Enhanced percentage of CHT1-positive neurons in the MP
MP neurons were immunohistochemically identified using the anti-PGP9.5 antibody. The neuronal network-like structure of MP was clearly observed on colonic MP whole-mounts after immunostaining. Compared to control rats (Figure 5A), the percentages of CHT1-IR neurons in the MP of WAS-exposed rats (Figure 5B) were significantly increased (46% ± 4% in the control group and 55% ± 7% in the WAS group, P<0.05). In addition, the percentage of CHT1-positive neurons in the WAS + MKC-231 group was significantly enhanced compared to that in the WAS group (55% ± 7% in the WAS group and 62% ± 8% in the WAS + MKC-231 group, P<0.05) (Figure 5C).
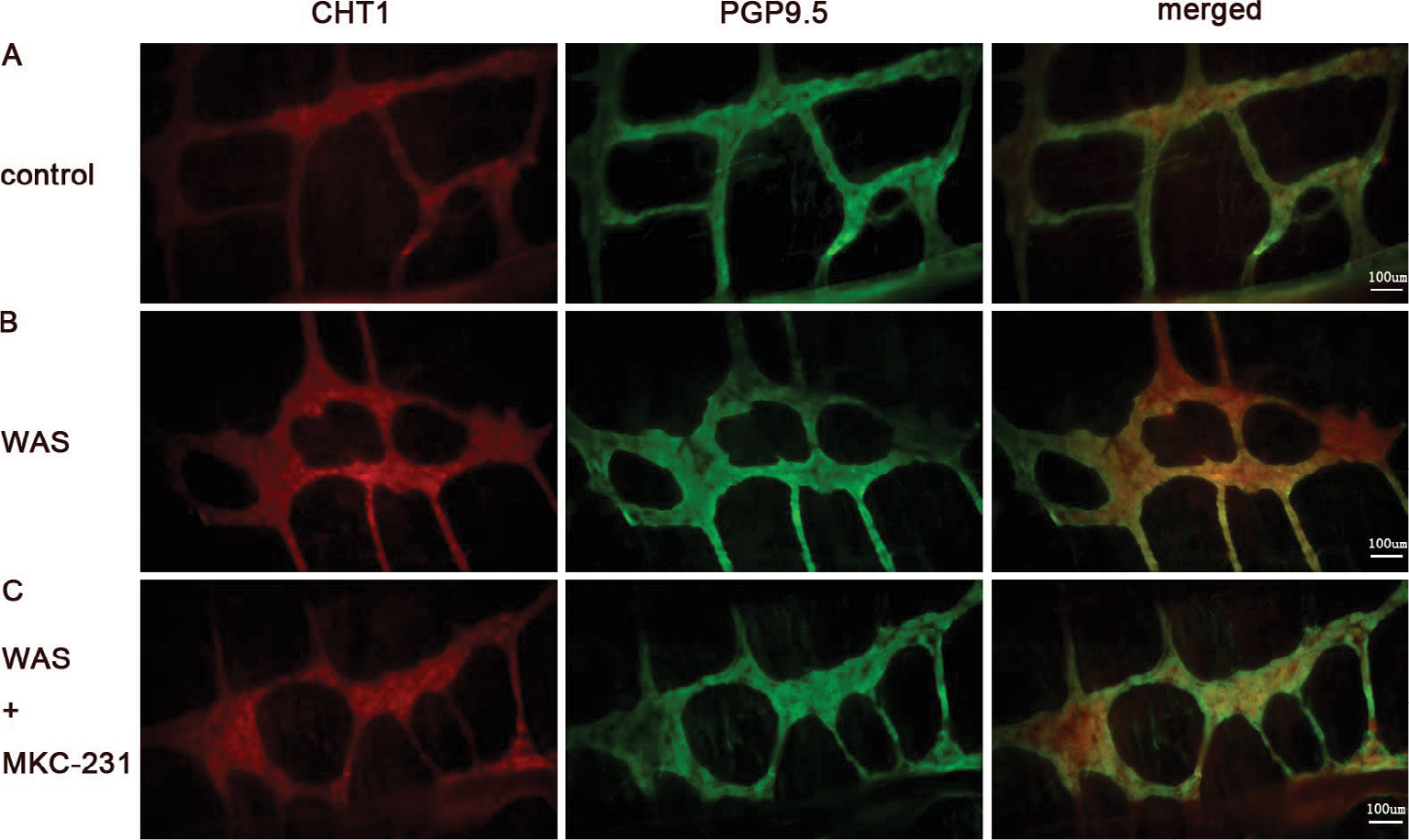 | Figure 5 Distribution of CHT1 in the MP of colon. Notes: Immunofluorescence for CHT1, PGP9.5, and merged picture on whole-mount preparations of colonic MP from control (A), WAS (B), and WAS + MKC-231 rats (C) (200×) was displayed. The percentage of CHT1-positive neurons in the MP of WAS is significantly higher than that in the control group. In addition, the percentage of CHT1-positive neurons in the WAS + MKC-231 group is enhanced compared to that in the WAS group. Bars = 100 µm; n of each group = 6. Abbreviations: MP, myenteric plexus; PGP9.5, protein gene product 9.5; WAS, water avoidance stress. |
These findings indicated that percentage of CHT1-positive neurons in MP was significantly enhanced after 10 days of WAS. Furthermore, MKC-231 could increase the CHT1-IR neuronal subpopulation in MP.
Discussion
Presently, the detailed mechanism of IBS remains elusive. However, the pathogenesis of IBS is known to be multifactorial including a disrupted gut–brain axis,22 visceral hypersensitivity,23 altered gut microbiota,24 and immune activation.25 Animal models of IBS are pivotal in clarifying its pathogenesis. To date, repeated WAS has been developed to establish an animal model of stress-induced IBS with visceral hyperalgesia, motility impairment, anxiety, and colonic immune activation,10 which can persist for the following 20 days. The WAS model, which is optimum for studies on IBS, is a brain–gut interaction model and mimics some clinical and pathophysiologic characteristics of IBS-diarrhea. In the present study, elevated visceral hyperalgesia in response to CRD and enhanced abdominal mechanical hyperalgesia to VFF were observed after 10 days of WAS exposure, indicating that the animal model of stress-induced hyperalgesia was successfully established in our research.
The antinociceptive effect of the cholinergic system in both neuropathic pain3 and visceral pain4 has been known for long. The latest evidence suggested that degeneration of basal forebrain cholinergic neurons may be relevant to hyperalgesia in patients with Alzheimer’s disease.26 The mechanism underlying the antinociceptive action of the cholinergic system is not yet fully understood. However, an increasing body of evidence indicates that the antinociceptive effects may be associated with the cholinergic anti-inflammatory pathway,27 interaction with opioid systems,6 increase in the release of inhibitory neurotransmitters, such as glycine28 and gamma-aminobutyric acid,29 and decrease in the release of excitatory transmitters such as glutamate29 through ACh and its receptors, mainly muscarinic receptors. Recent studies reporting that the high-affinity choline transporter CHT1 functions as the rate-limiting step for ACh synthesis have ignited significant interest in this family of choline transporters.5 The slc5a7 gene that encodes CHT1 was cloned in 2000.8 CHT1 is mainly present in neuronal cytoplasm and the presynaptic membrane and is widely distributed in both peripheral and central nervous systems.30 In addition, CHT1 is cholinergic-specific according to the results of in situ hybridization and immunohistochemical experiments.8 To date, only a minority of evidence has demonstrated the presence of CHT1 immunoreactivity in the enteric nervous system.31 In the current study, we observed that CHT1 was expressed robustly in the colon, especially in the mucosa and muscular layer. Furthermore, we detected enhanced expression of CHT1 protein and mRNA in the colon after 10 consecutive days of WAS exposure, indicating that colonic CHT1 expression could be directly regulated by chronic stress. Further findings suggested that altered CHT1 expression in the colon of WAS-exposed rats was partly mediated by NF-κB signaling and that CHT1 might affect the GI sensation by affecting ACh production. A growing body of evidence has demonstrated that activation of NF-κB signaling is involved in pain regulation in inflammatory diseases,32 cancer,33 and autoimmune diseases,34 via the release of inflammatory cytokines such as IL-1β, IL-6, and TNF-α. The current study suggests that NF-κB signaling could also modulate the hyperalgesia originating from psychological stress, partly by affecting CHT1 expression. Painful stimuli are known to increase ACh release in the spinal cord through the descending inhibitory serotonergic and noradrenergic pain modulatory pathways.35 However, our data suggest that the elevated ACh content may be directly attributed to CHT1 upregulation.
Previous studies indicated that intestinal pathologic alterations might contribute to the visceral hyperalgesia in IBS.36 Colonic mucosal biopsies of IBS patients have revealed an increase in activated lymphocytes, mast cells, and neutrophilic37,38 and eosinophilic granulocytes.39 In the present study, we observed that the density of CHT1-IR cells in the colonic muscular layer of rats with stress-induced hyperalgesia was enhanced. Further immunofluorescence staining of colonic MP whole-mounts demonstrated that the percentage of CHT1-IR neurons in MP was significantly increased after 10 days of WAS. Given that CHT1 is exclusively present in cholinergic nerve fibers and represents another biomarker of cholinergic nerves,31 we can safely conclude that the cholinergic neurons in colonic MP may be directly modulated by chronic stress and be relevant to stress-induced hyperalgesia.
MKC-231, known as a choline uptake enhancer, has been reported to take CHT1 as its principal target.40,41 Animal experiments have demonstrated that oral administration of MKC-231 significantly improved the learning and memory deficit associated with cholinergic hypofunction by directly affecting the trafficking of CHT1 and increasing the number of transporters.40,41 Furthermore, MKC-231 could antagonize phencyclidine-induced behavioral deficits and reduction in septal cholinergic neurons in rats.42 These reports suggest that MKC-231 could be a therapeutic drug for the treatment of Alzheimer’s disease and schizophrenia, but without any significant side effects.35 Here, we demonstrate that MKC-231 could alleviate visceral pain in WAS rats by upregulation of CHT1 protein expression and enhancing ACh production. In turn, we could safely conclude that enhanced CHT1 expression might play a key role in inhibiting stress-induced visceral pain. Therefore, upregulation of CHT1 or enhanced CHT1 activity may be used to treat disorders with hyperalgesia.
CHT1 dysfunction is closely associated with multiple diseases, such as atherosclerosis,13 depression,43 and Alzheimer’s disease.11 Recently, our team demonstrated for the first time that CHT1 in the dorsal root ganglion may be involved in regulating chronic pain in chronic pancreatitis.14 The current work further demonstrates that CHT1 may play an important inhibitory role in stress-induced visceral hyperalgesia. Upregulation of CHT1 was observed after establishment of stress-induced visceral pain, possibly as a response to the noxious stimulus. Simultaneously, nociceptive mechanisms (such as corticotropin-releasing factor44) were increased and were greater than the inhibitory effect, resulting in constant pain due to stress. These findings laid the foundation for research regarding the role of CHT1 in pain.
However, our present study inevitably has certain limitations. First of all, in terms of the experimental design, the baseline behavioral performance of the experimental animals was not evaluated, making it impossible for the same cohort to conduct self-comparison before and after treatment. Secondly, the functional alterations of CHT1 in rats with stress-induced hyperalgesia were not examined. However, choline uptake, an important property of CHT1, plays a critical role in physiologic and pathologic conditions. Thirdly, whether and how CHT1 interacted with other pain signaling, such as serotonin signaling45 and transient receptor potential vanilloid type-1 pathway,46 was not explored, resulting in the impossibility to exclude the epiphenomenon. Last but not the least, our findings come from the animal experiments. In view of distinct species differences, it is hard to translate these findings to the clinical application of IBS. Therefore, further studies regarding the role of CHT1 in hyperalgesia induced by chronic stress are warranted. First, to what extent the analgesic effect of MKC-231 is attributed to CHT1 will be tested via blockade of choline uptake by coadministering a CHT1 inhibitor, such as hemicholinium-3. Moreover, we will focus our future research on the functional alterations in choline uptake and the relationship between CHT1 and other pain signaling in rodents with hyperalgesia. In addition, our future research will be extended to the patients with IBS. We anticipate that pharmacologic modulation of CHT1 can provide a new therapeutic target for the treatment of a number of GI disorders, including IBS.
Conclusion
In conclusion, our data demonstrate that CHT1 is involved in the regulation of stress-induced visceral pain and that MKC-231 could alleviate visceral pain in rats with IBS. These findings identify new mechanisms underlying visceral hyperalgesia associated with enhanced stress responsiveness and may pave the way for novel treatment strategies for IBS and related disorders.
Acknowledgment
This study was supported by National Natural Science Foundation of China (NSFC) (Nos. 81170351 and 81770638).
Authors contribution
All authors conceived and designed the experiments. M-JL conducted the experiments. All authors contributed toward data analysis, drafting and critically revising the paper, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Hurst R, Rollema H, Bertrand D. Nicotinic acetylcholine receptors: from basic science to therapeutics. Pharmacol Ther. 2013;137(1):22–54. | ||
Ehlert FJ, Pak KJ, Griffin MT. Muscarinic agonists and antagonists: effects on gastrointestinal function. Handb Exp Pharmacol. 2012;208(208):343–374. | ||
Freitas K, Ghosh S, Ivy Carroll F, Lichtman AH, Damaj MI. Effects of α7 positive allosteric modulators in murine inflammatory and chronic neuropathic pain models. Neuropharmacology. 2013;65:156–164. | ||
Joshi SK, Mikusa JP, Weaver B, Honore P. Morphine and ABT-594 (a nicotinic acetylcholine agonist) exert centrally mediated antinociception in the rat cyclophosphamide cystitis model of visceral pain. J Pain. 2008;9(2):146–156. | ||
Eisenach JC. Muscarinic-mediated analgesia. Life Sci. 1999;64(6–7):549–554. | ||
Gawel K, Gibula-Bruzda E, Dziedzic M, et al. Cholinergic activation affects the acute and chronic antinociceptive effects of morphine. Physiol Behav. 2017;169:22–32. | ||
Jones PG, Dunlop J. Targeting the cholinergic system as a therapeutic strategy for the treatment of pain. Neuropharmacology. 2007;53(2):197–206. | ||
Okuda T, Haga T. Functional characterization of the human high-affinity choline transporter. FEBS Lett. 2000;484(2):92–97. | ||
Ferguson SM, Bazalakova M, Savchenko V, et al. Lethal impairment of cholinergic neurotransmission in hemicholinium-3-sensitive choline transporter knockout mice. Proc Natl Acad Sci U S A. 2004;101(23):8762–8767. | ||
Takashina K, Bessho T, Mori R, Eguchi J, Saito K-I. MKC-231, a choline uptake enhancer: (2) effect on synthesis and release of acetylcholine in AF64A-treated rats. J Neural Transm. 2008;115(7):1027–1035. | ||
Wang Z, Yang L, Zheng H. Role of APP and Aβ in synaptic physiology. Curr Alzheimer Res. 2012;9(2):217–226. | ||
Neumann SA, Linder KJ, Muldoon MF, et al. Polymorphic variation in choline transporter gene (CHT1) is associated with early, subclinical measures of carotid atherosclerosis in humans. Int J Cardiovasc Imaging. 2012;28(2):243–250. | ||
Penet MF, Shah T, Bharti S, et al. Metabolic imaging of pancreatic ductal adenocarcinoma detects altered choline metabolism. Clin Cancer Res. 2015;21(2):386–395. | ||
Luo D, Chen L, Yu B. Inhibition of the high affinity choline transporter enhances hyperalgesia in a rat model of chronic pancreatitis. Biochem Biophys Res Commun. 2017;488(1):204–210. | ||
Lovell RM, Ford AC. Global prevalence of and risk factors for irritable bowel syndrome: a meta-analysis. Clin Gastroenterol Hepatol. 2012;10(7):712–721. | ||
Gwee KA, Ghoshal UC, Chen M. Irritable bowel syndrome in Asia: pathogenesis, natural history, epidemiology, and management. J Gastroenterol Hepatol. 2018;33(1):99–110. | ||
Rojas-Macías V, Rodríguez-Fandiño O, Jiménez-Ponce F, et al. External validity of a relevant model for irritable bowel syndrome (IBS) using chronic stress by water avoidance in Wistar rats. Rev Gastroenterol Mex. 2010;75(4):421–426. | ||
Tang D, Qian AH, Song DD, et al. Role of the potassium chloride cotransporter isoform 2-mediated spinal chloride homeostasis in a rat model of visceral hypersensitivity. Am J Physiol Gastrointest Liver Physiol. 2015;308(9):G767–G778. | ||
Al-Chaer ED, Kawasaki M, Pasricha PJ. A new model of chronic visceral hypersensitivity in adult rats induced by colon irritation during postnatal development. Gastroenterology. 2000;119(5):1276–1285. | ||
Winston JH, He ZJ, Shenoy M, Xiao SY, Pasricha PJ. Molecular and behavioral changes in nociception in a novel rat model of chronic pancreatitis for the study of pain. Pain. 2005;117(1–2):214–222. | ||
Qi D, Wu S, Zhang Y, Li W. Electroacupuncture analgesia with different frequencies is mediated via different opioid pathways in acute visceral hyperalgesia rats. Life Sci. 2016;160:64–71. | ||
Mach T. The brain-gut axis in irritable bowel syndrome--clinical aspects. Med Sci Monit. 2004;10(6):RA125–131. | ||
Zhou Q, Verne GN. New insights into visceral hypersensitivity--clinical implications in IBS. Nat Rev Gastroenterol Hepatol. 2011;8(6):349–355. | ||
Bhattarai Y, Muniz Pedrogo DA, Kashyap PC. Irritable bowel syndrome: a gut microbiota-related disorder? Am J Physiol Gastrointest Liver Physiol. 2017;312(1):G52–G62. | ||
Hyland NP, Quigley EM, Brint E. Microbiota-host interactions in irritable bowel syndrome: epithelial barrier, immune regulation and brain-gut interactions. World J Gastroenterol. 2014;20(27):8859–8866. | ||
Vierck CJ, Yezierski RP, Wiley RG. Pain sensitivity following loss of cholinergic basal forebrain (CBF) neurons in the rat. Neuroscience. 2016;319:23–34. | ||
Tracey KJ. Physiology and immunology of the cholinergic antiinflammatory pathway. J Clin Invest. 2007;117(2):289–296. | ||
Wang XL, Zhang HM, Li DP, Chen SR, Pan HL. Dynamic regulation of glycinergic input to spinal dorsal horn neurones by muscarinic receptor subtypes in rats. J Physiol. 2006;571(Pt 2):403–413. | ||
Li DP, Chen SR, Pan YZ, Levey AI, Pan HL. Role of presynaptic muscarinic and GABA(B) receptors in spinal glutamate release and cholinergic analgesia in rats. J Physiol. 2002;543(Pt 3):807–818. | ||
Misawa H, Nakata K, Matsuura J, et al. Distribution of the high-affinity choline transporter in the central nervous system of the rat. Neuroscience. 2001;105(1):87–98. | ||
Harrington AM, Lee M, Ong SY, et al. Immunoreactivity for high-affinity choline transporter colocalises with VAChT in human enteric nervous system. Cell Tissue Res. 2010;341(1):33–48. | ||
Khalid S, Ullah MZ, Khan AU, et al. Antihyperalgesic properties of Honokiol in inflammatory pain models by targeting of NF-κB and Nrf2 signaling. Front Pharmacol. 2018;9:140. | ||
Jian-Min T, Jun L, Sheng-Yang C, et al. Tashinone II A-sulfoacid-natrum elevates the pain threshold through inhibiting nuclear factor kappa B pathway in neuropathic cancer pain. Indian J Cancer. 2015;52(Suppl 3): E179–181. | ||
Xia ZB, Yuan YJ, Zhang QH, et al. Salvianolic acid B suppresses inflammatory mediator levels by downregulating NF-κB in a rat model of rheumatoid arthritis. Med Sci Moit. 2018;24:2524–2532. | ||
Eisenach JC, Detweiler DJ, Tong C, D’Angelo R, Hood DD. Cerebrospinal fluid norepinephrine and acetylcholine concentrations during acute pain. Anesth Analg. 1996;82(3):621–626. | ||
Stasi C, Bellini M, Bassotti G, Blandizzi C, Milani S. Serotonin receptors and their role in the pathophysiology and therapy of irritable bowel syndrome. Tech Coloproctol. 2014;18(7):613–621. | ||
Chadwick VS, Chen W, Shu D, et al. Activation of the mucosal immune system in irritable bowel syndrome. Gastroenterology. 2002;122(7):1778–1783. | ||
O’Sullivan M, Clayton N, Breslin NP, et al. Increased mast cells in the irritable bowel syndrome. Neurogastroenterol Motil. 2000;12(5):449–457. | ||
Walker MM, Warwick A, Ung C, Talley NJ. The role of eosinophils and mast cells in intestinal functional disease. Curr Gastroenterol Rep. 2011;13(4):323–330. | ||
Bessho T, Takashina K, Tabata R, et al. Effect of the novel high affinity choline uptake enhancer 2-(2-oxopyrrolidin-1-yl)-N-(2,3-dimethyl-5,6,7,8-tetrahydrofuro[2,3-b] quinolin-4-yl)acetoamide on deficits of water maze learning in rats. Arzneimittelforschung. 1996;46(4):369–373. | ||
Takashina K, Bessho T, Mori R, et al. MKC-231, a choline uptake enhancer: (3) mode of action of MKC-231 in the enhancement of high-affinity choline uptake. J Neural Transm. 2008;115(7):1037–1046. | ||
Shirayama Y, Yamamoto A, Nishimura T, Katayama S, Kawahara R. Subsequent exposure to the choline uptake enhancer MKC-231 antagonizes phencyclidine-induced behavioral deficits and reduction in septal cholinergic neurons in rats. Eur Neuropsychopharmacol. 2007;17(9):616–626. | ||
Hahn MK, Blackford JU, Haman K, et al. Multivariate permutation analysis associates multiple polymorphisms with subphenotypes of major depression. Genes Brain Behav. 2008;7(4):487–495. | ||
Fukudo S. Role of corticotropin-releasing hormone in irritable bowel syndrome and intestinal inflammation. J Gastroenterol. 2007;42(Suppl 17):48–51. | ||
Mawe GM, Coates MD, Moses PL. Review article: intestinal serotonin signalling in irritable bowel syndrome. Aliment Pharmacol Ther. 2006;23(8):1067–1076. | ||
Akbar A, Yiangou Y, Facer P, et al. Increased capsaicin receptor TRPV1-expressing sensory fibres in irritable bowel syndrome and their correlation with abdominal pain. Gut. 2008;57(7):923–929. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.