Back to Journals » Cancer Management and Research » Volume 12
Upregulated SET Promotes Cell Survival Through Activating Akt/NF-κB Signal in Colorectal Carcinoma
Authors Zhu J, Shi L, Du G, Li L, Liu M
Received 3 April 2020
Accepted for publication 9 June 2020
Published 19 June 2020 Volume 2020:12 Pages 4735—4745
DOI https://doi.org/10.2147/CMAR.S255930
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Antonella D'Anneo
Jianjun Zhu,1 Lihong Shi,2 Genlai Du,1 Li Li,1 Ming Liu1
1Department of Medical Cellular Biology and Genetics, Shanxi Medical University, Taiyuan 030001, People’s Republic of China; 2Department of Human Anatomy, School of Basic Medicine, Shanxi Medical University, Taiyuan 030001, People’s Republic of China
Correspondence: Lihong Shi
Department of Human Anatomy, School of Basic Medicine, Shanxi Medical University, 56 Xinjian South Road, Taiyuan 030001, People’s Republic of China
Tel/ Fax +86-351-3985176
Email [email protected]
Purpose: SET has been proven to be an oncogene, which promotes the initiation and progression in several kinds of malignant carcinomas. However, the expression and its functional roles in colorectal carcinoma (CRC) remained unknown.
Materials and Methods: CRC tissues samples, CRC cell lines and xenograft mouse tumors were used in this study. The mRNA and protein expressions were detected by quantitative real-time polymerase chain reaction (qRT-PCR), immunohistochemistry (IHC), and Western blot (WB), respectively. siRNAs were used to silence the gene expression. Cell viability, cell proliferation, colony formation, and apoptosis were measured by MTS assay, EdU incorporation assay, plated colony formation assay, and flow cytometry, respectively. Western blot was applied to evaluate the levels of Akt, p-Akt, c-Myc and cyclin D1. Xenograft mouse model was performed to observe the role of SET in vivo.
Results: Our results revealed that SET was up-regulated in CRC, and the expression of SET was increased with the development of CRC. SET knockdown in vitro attenuated cell proliferation activity, and increased cell apoptosis in CRC cells. Moreover, the knockdown of SET reduces tumorigenic potential in nude mice. For the mechanism, knockdown of SET promoted the dephosphorylation of Akt, followed by suppressing the translocation of NF-κB to nucleus. In addition, SET knockdown-mediated dephosphorylation of Akt downregulated the expression of c-Myc and Cyclin D1, which inhibited the cell survival in CRC.
Conclusion: Our results indicated that SET promoted cell survival via activating Akt/NF-κB signaling pathway in CRC, which strongly suggested that SET might be a potential therapeutic target in the colorectal carcinoma treatment.
Keywords: SET, colorectal adenocarcinoma, PP2A, Akt, NF-κB
Introduction
Colorectal adenocarcinoma is the second leading cause of malignancy-associated mortality worldwide, and its prevalence is increasing in recent years.1,2 The carcinogenesis of colorectal adenocarcinoma is a multi-step process which involves a number of genomic alterations.3 Therefore, it is urgent to investigate the roles of these suppressor genes/oncogenes in colorectal adenocarcinoma for cancer prevention, early diagnosis and therapeutic development.
SET is a multifunctional oncoprotein, involved in apoptosis, transcription, nucleosome assembly and histone binding, which ultimately promotes the initiation and progression of cancer.4 The SET protein has two isoforms, which were named as isoform 1 and isoform 2, and both were potent inhibitors of protein phosphatase 2A (PP2A).5 Overexpression of SET was characterized as being tumor-specific and was associated with the worse clinical outcomes in many human malignant carcinomas.4,6 Moreover, SET had been proven to be associated with the development of therapeutic resistance in several kinds of carcinomas.7 Mody et al reported that SET isoform 2 was overexpressed in aggressive pancreatic cancer cell lines and overexpressing SET isoform 2 promoted cellular proliferation, migration, invasion, and colony formation in pancreatic cancer.6 Jiang et al reported that SET mRNA expression was up-regulated in colorectal carcinoma, and SET promoted cellular proliferation and inhibited cell apoptosis in colorectal adenocarcinoma cells and oral squamous cells.4
However, the potential roles of SET remained unclear in colorectal carcinoma. In this study, we comprehensively investigated the expression of SET and its functional roles in the progression of colorectal carcinoma both in vitro and in vivo. More importantly, the molecular mechanisms underlying the oncogenic role of SET in promoting the cell survival were deeply explained in colorectal carcinoma.
Materials and Methods
Collection of Tissue Samples and Cell Culture
The present study was approved by the Medical Research Ethics Committee of Shanxi Medical University (Taiyuan, China), and written informed consents were obtained from all the patients who participated in this study. 87 pairs of surgically resected CRC and adjacent colorectal tissues (>5 cm away from the tumor edges) were collected from the Affiliated Hospital of Shanxi Medical University. All of samples were formalin fixed and paraffin embedded for subsequent IHC examinations. Another 20 fresh tissues were collected and frozen in liquid nitrogen, then stored in −80°C refrigerator for mRNA analysis. The Ls174T and DLD-1 cell lines were purchased from Shanghai Cell Bank of the Chinese Academy of Sciences (Shanghai, China). Ls174T and DLD-1 cells were routinely cultured.
Public Dataset Collection
TCGA public datasets mRNA expression in CRC tissues were downloaded from UALCAN online database (http://ualcan.path.uab.edu/analysis.html).8
Immunohistochemistry (IHC)
For immunohistochemistry (IHC), tissues were fixed in 10% of formalin and embedded in paraffin. Immunohistochemistry was performed on four-micrometer paraffin sections. The level of SET was independently evaluated by two pathologists according to the proportion and intensity of positive cells within five microscopic visual fields per slide as previously described.9 Each sample was assigned a score based on the proportion and the intensity of positive cells (For the proportion, 0≤10%, 1=10%-25%, 2=26%-50%, 3=51%-75%, 4≥75%; For the intensity, 0=negative, 1=weak, 2=moderate, 3=strong). The score of the sample ranged from 0 to 12. Primary antibodies used in this study were listed in Supplementary Table S1.
Quantitative Reverse Transcription PCR (qRT-PCR)
RNA extraction, cDNA synthesis, and qPCR reactions were performed as described previously.3 Briefly, total RNA was extracted using the TRIzol regent (Invitrogen). Reverse transcription reaction were performed according to the manufacturer’s instructions. Target genes were amplified using SYBR Green PCR Kit (Takara). β-actin was used as internal control. Primer sequences were synthesized by Sangon Biotech (Shanghai, China) and were provided in Supplementary Table S2.
Western Blot Assay
Western blot assay was performed as previously described.3 Briefly, protein samples loaded into the 10% PAGE were separated and transferred to PVDF membrane (Invitrogen). After blocking the PVDF membrane, the PVDF membrane was incubated with the primary antibody at 4°C overnight, followed by incubating with the secondary antibody (1:10,000) at room temperature for 1h. In the end, the protein bands were quantified by Quantity One Software. Antibodies used in this study were listed in Supplementary Table S1.
RNA Interference (RNAi)
Small interfering RNAs (siRNA) were synthesized by Ribobio (Guangzhou, China). The control siRNA fragment was designed not to silence any genes. siRNA fragment was transfected by using Lipofectamine 2000 (Invitrogen, CA, USA) according to the manufacturer’s protocol. Briefly, a mixture composed of siRNA and Lipofectamine 2000 were transfected into the cells. Six hours later, the cells were routinely cultured. 48h later, the mRNA and protein expression of target molecules were verified by qRT-PCR and Western Blot, respectively.
Cell Apoptosis Assay
Cell apoptosis was determined by Annexin V-FITC detection Kit (BestBio, Shanghai, China) according to the manufacturer’s protocol. Briefly, cells resuspended in 400 µL Binding buffer were stained with 5µL Annexin V at 4°C for 15 min protected from light. Then, 10µL PI was added into the cell suspension, followed by incubating at 4°C for 10 min. The cell apoptosis were analyzed by flow cytometry (Beckman, Fullerton, CA).
MTS Assay and Colony Formation Assay
Cells plated into 96-well plates were routinely cultured. After adding 20 μL MTS reagent into the well, the cells were incubated at 37°C for 2 hour protected from light. The plate was read at the absorbance wavelength of 490 nm by a microplate reader (Bio-Rad, Hercules, CA, USA). To evaluate the colony formation ability, 1000 cells in logarithmic growth phase were seeded into 25 cm2 plate. After 14 days of routine incubation, the cells were washed with PBS buffer, and the colonies were methanol fixed and stained with crystal violet.
Ethynyl Deoxyuridine (EdU) Incorporation Assay
The proliferation activity of cells was analyzed using EdU incorporation assay kit (Ribobio, Guangdong, China) according to the manufacturer’s instructions. Briefly, cells were incubated with 5 μM EdU in normal medium at 37°C for 2 h, followed by fixing with 4% formaldehyde for 30 min at room temperature and permeating with 0.5% Triton X-100 for 20 min. Then, the cells were incubated with 1 × Apollo® reaction cocktail for 30 min at room temperature. Finally, the nuclear DNA were stained with Hoechst 33342 for 30 min and visualized under a fluorescent microscope.
Nude Mice Xenograft Model
Four-week-old male BALB/c nude mice with the average body weight of 18–22g were chosen, and the xenografts were initiated by subcutaneous injection of Ls174T cells into the back of nude mice. One week later, siSET group mice were injected with siR-vivo™ siSET (50 mg/Kg) every 3 days via the tumor regional injection. siCtrl group mice were injected with siR-vivo™ control siRNA. One month later, the nude mice were sacrificed. The tumor volume (mm3) was calculated by the formula (length ×wide2)/2. This study was approved by the ethics committee of the Shanxi Medical University. All processes were performed according to the Council of Agriculture Guidebook for the Care and Use of Laboratory Animals.
TUNEL Assay
For analysis of apoptosis in vivo, terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) assay (Roche Applied Science, Rotkreuz, Switzerland) was employed according to the manufacturer’s instructions. Briefly, tissue sections were incubated with Proteinase K (Sigma, Munich, Germany) at 37 °C for 30 min, followed by incubating with TUNEL buffer at 37 °C for 1 hour in the dark. Then, the cell nucleus was stained with DAPI (Beyotime). Images of TUNEL/DAPI-stained sections were collected by a fluorescence microscope (DM5000B, Leica, Heerbrugg, Switzerland).
Statistical Analysis
SPSS 17.0 software (SPSS, Chicago, IL) was used for all statistical analyses and P values less than 0.05 were considered to be statistically significant. Data were represented as mean ± SD. t-tests were used for comparisons between two groups. One-way ANOVA were used for comparisons among three or more groups.
Results
SET Is Up-Regulated in Human CRC Tissues
To study the functional roles of SET in the progression of colorectal carcinoma, both the mRNA expression of SET were determined by qRT-PCR in 20 pairs of colorectal carcinoma tissues. Our results showed that the mRNA expression of SET was up-regulated in 15 out of 20 (75%) colorectal carcinoma tissues when compared with adjacent normal colorectal tissues (p=0.0006) (Figure 1A). The above findings were further supported by the bioinformatic analysis based on TCGA public mRNA expression datasets from CRC and normal tissue samples (Figure 1B). To further validate this result, SET expression was investigated in 87 pairs of human CRC and adjacent normal colorectal tissues by IHC. Our results indicated that SET expression was mainly localized to cell nucleus. In total, 63 out of 87 (72.14%) CRC tissues displayed high SET protein expression levels when compared with adjacent normal tissues (p<0.01) (Figure 1C).
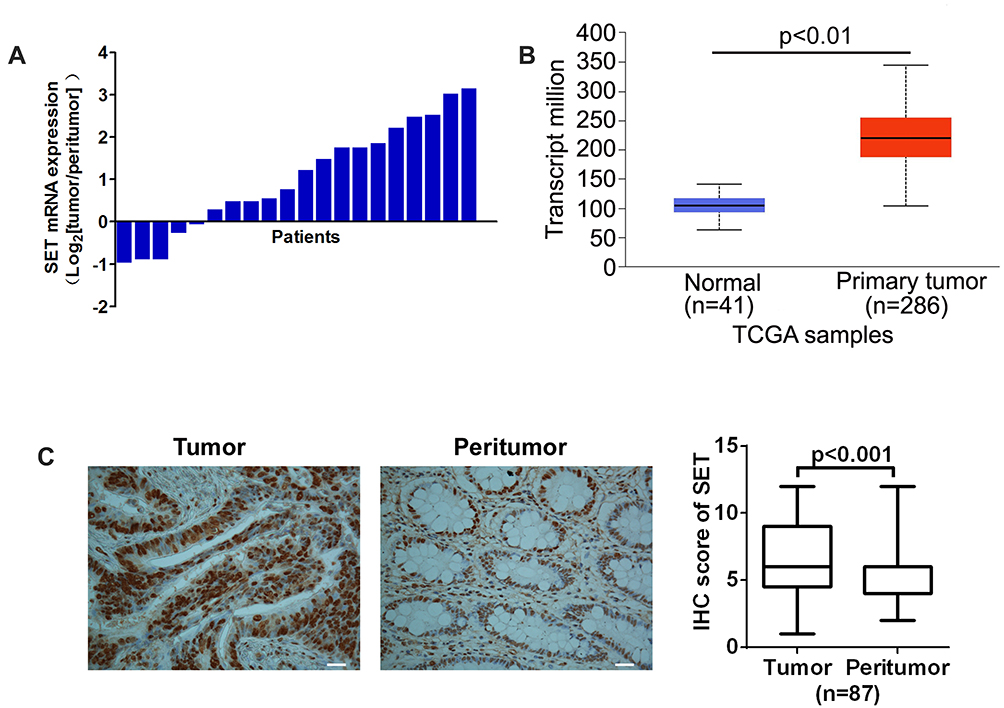 |
Figure 1 SET is over-expressed in colorectal carcinoma. (A) The relative mRNA expression ratio (Log2 transformed) of tumor/peritumor for SET examined by qPCR in 20 pairs of CRC tissues. (B) The relative mRNA expression levels of tumor and peritumor of SET were analyzed in public data TCGA downloaded from UALCA online database. (C) Representative IHC staining image (Left) and IHC score (Right) of SET in 87 paired CRC tissues (tumor and peritumor). Data were expressed as mean ± SD. Scale bar, 50 μm. |
In order to further investigate whether the upregulation of SET was associated with CRC progression, we analyzed the relationship between the SET expression and the pathological characteristics of CRC patients. Although no significant correlations were observed between SET expression and gender, age or differentiation (p=0.276, p=0.481, p=0.283), a statistically significant correlation between SET expression and Dukes’ stage of CRC was identified (p=0.003) (Table 1). Altogether, these results indicate that SET is up-regulated in colorectal carcinoma, which promotes the progression of CRC.
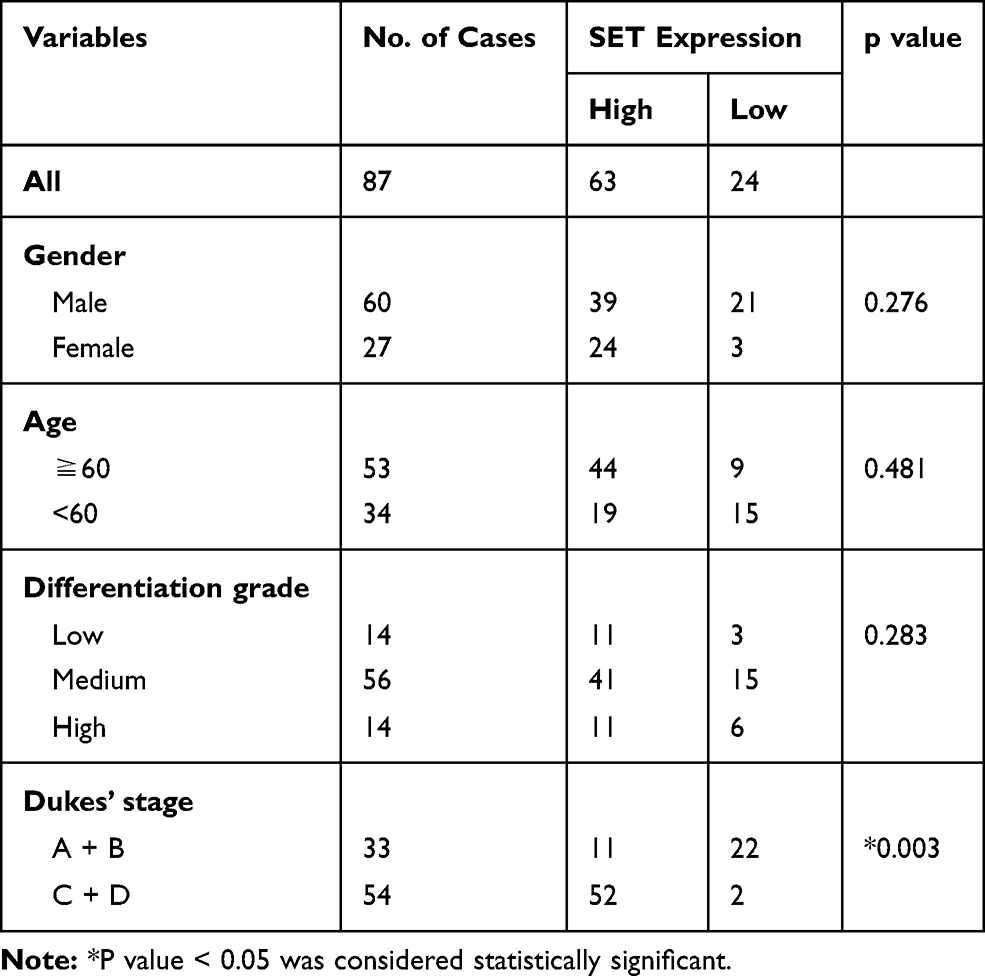 |
Table 1 Relationship Between Tumor SET Expression and Clinicopathologic Features of Colorectal Carcinoma Patients |
Knockdown of SET Enhances CRC Cell Apoptosis
In order to confirm whether the up-regulated SET played a functional role in the process of cell apoptosis in CRC, siRNA targeted to silence SET was employed to perform this assay. As shown in Figure 2A, the mRNA expressions of SET in Ls174T and DLD-1 cell lines were significantly decreased upon silencing the SET gene. Furthermore, the protein expressions of SET were also significantly decreased upon silencing the SET gene (Figure 2B). In addition, flow cytometry analysis showed that the cell apoptosis rates were significantly increased after knockdown of SET in Ls174T and DLD-1 cells, when compared with that in the control group (Figure 2C). Moreover, the expressions of the cleaved caspase 3 remarkably increased after knockdown of SET in Ls174T and DLD-1 cells (Figure 2D). Altogether, these results indicate that SET promotes the development of colorectal carcinoma through inhibiting cell apoptosis.
 |
Figure 2 Knockdown of SET promotes CRC cell apoptosis. (A) qPCR and (B) Western blot analysis of SET level in Ls174T and DLD-1 cells transiently transfected with siRNA. (C) Apoptosis analysis by flow cytometry in Ls174T and DLD-1 cells treated as indicated. (D) Western blot analysis of cleaved caspase 3 level in Ls174T and DLD-1 cells treated as indicated. siSET: siRNA specially target to silence SET; si Ctrl: control siRNA. Data were expressed as mean ± SD. **P < 0.01. |
Knockdown of SET Attenuates CRC Cell Growth in vitro
Next, we investigated the roles of SET in CRC cell growth. As shown in Figure 3A, cell viability were significantly reduced upon silencing SET expression in Ls174T and DLD-1 cells, when compared with that in the control group, as determined by MTS assay. Moreover, the cell proliferation activity was also significantly decreased after knockdown of SET in Ls174T and DLD-1 cells, as determined by EdU assay (Figure 3B). Furthermore, the colony formation ability was also significantly decreased after knockdown of SET in Ls174T and DLD-1 cells, as determined by plate colony formation assay (Figure 3C). Altogether, our results indicate that SET functions to promote the cell growth in the progression of CRC.
 |
Figure 3 Knockdown of SET inhibits CRC cell growth in vitro. (A) Cell viability detected by CCK8 assay after transfected with siRNA in Ls174T and DLD-1 cells. (B) Cell proliferation activity detected by EdU assay after transfected with siRNA in Ls174T and DLD-1 cells. Scale bar, 50 μm. (C) Colony formation assay for CRC cells treated as indicated. Data were expressed as mean ± SD. **P < 0.01. |
Knockdown of SET Attenuates CRC Growth in vivo
Our previous data demonstrated that up-regulated SET promoted colorectal carcinoma cells growth in vitro, but the functional roles of SET in vivo remained unknown. Here, our data showed that the growth capacity of xenograft tumors injected with siR-vivo™ si SET was significant decreased, compared to that in the control group (Figure 4A and B). Next, we investigated the effects of SET on the cell apoptosis and proliferation activity in vivo using TUNEL assay and immunohistochemical staining of Ki67. Our data showed that knockdown of SET increased the cell apoptosis in vivo, and knockdown of SET attenuated the proliferation activity of tumor cell in vivo (Figure 4C and D). Taken together, our results in vivo indicate the functional roles of SET in promoting the CRC growth.
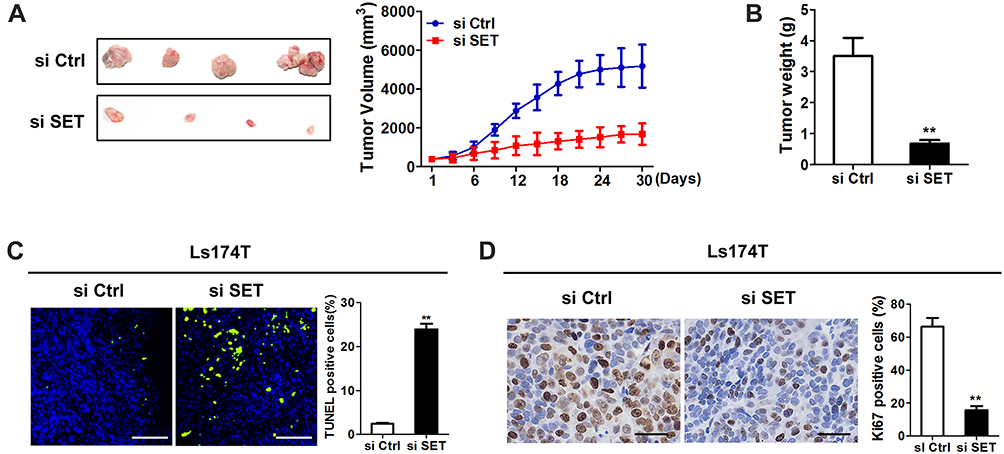 |
Figure 4 Knockdown of SET inhibits CRC tumor growth in vivo. (A) Tumor growth curves of subcutaneous xenograft tumor model (Right); dissected tumors (Left) from sacrificed mice were shown. (B) Tumor weight were calculated and shown. (C) TUNEL staining in tumor tissues of nude mice xenograft model with treatment as indicated. Blue: Hochest; Green: TUNEL positive nucleus. Scale bar, 50 μm. (D) Representative IHC staining images of Ki67 in xenograft tumor treated as indicated. Scale bar, 20 μm. Data shown were the mean ± SD from three independent experiments. **P < 0.01. |
Knockdown of SET Inhibits CRC Cell Survival Through Enhancing the Dephosphorylation of Akt
Although our previous data demonstrated that SET knockdown inhibited CRC growth both in vitro and in vivo, the mechanism underlying role of SET knockdown in inhibiting the progression of colorectal carcinoma remained unclear. SET is the natural inhibitor of PP2A, and PP2A is a phosphatase with relatively poor specificity and functions in many cellular pathways through controlled phosphorylate of various substrates, such as Akt.10 We first tested the effect of SET knockdown on Akt phosphorylation. As shown in Figure 5A, SET depletion in CRC cells leaded to a significantly increased of the activity of PP2A. In addition, SET knockdown had no effect on the total protein expression of Akt, but the level of p-Akt was significantly decreased after knockdown of SET in CRC cells (Figure 5B). Given the above, we speculated that SET might act as its oncogenic role by altering the phosphorylation of Akt. To prove this, we treated Ls174T cells with siRNA-PP2A and SC79, a highly Akt activator. The results showed that inhibitory effect of SET knockdown on cell growth was effectively reversed upon siRNA-PP2A or SC79 treatment (Figure 5C and D), as supported by EdU incorporation assay (Figure 5E). Collectively, these data indicate that SET acts as an oncogenic function in CRC through altering Akt activity.
 |
Figure 5 Knockdown of SET inhibits cell survival of CRC through enhancing the dephosphorylation of Akt (A) The activity of intracellular PP2A was determined by enzyme assays in CRC cells. (B) Western blot analysis of Akt and p-Akt level in Ls174T cells transiently transfected with siRNA. (C and D) CCK-8 cell viability assay in Ls174T cells treated as indicated. (E) EdU incorporation assay in LS174T cells treated as indicated. SC79: 20 μM SC79. siPP2A: siRNA specially target to silence PP2A. Data were shown as the mean ± SD from three independent experiments. *P<0.05, **P < 0.01. |
Knockdown of SET Decreases the Expressions of c-Myc and Cyclin D1 by Regulating Akt Signal Pathway
Akt functions in many cellular pathways through activating NF-κB.11 Thus, we speculated that SET/Akt/NF-κB signaling axis may act on some key transcription factors, thereby contributing to malignant growth of CRC cells. As shown in Figure 6A, knockdown of SET clearly decreased the level of NF-κB in nucleus from Ls174 cells, indicating that SET knockdown inhibited the translocation of NF-κB to nucleus in Ls174T cells, while this effect could be effectively reversed by SC79, the inhibitor of Akt. Evidences reporting that NF-κB translocates to the nucleus, and promotes gene expression, including c-Myc, Cyclin D1, which function in promoting the malignant growth of tumor cells.12 As shown in Figure 6B, SET knockdown leaded to a significant decrease of the protein levels of c-Myc and cyclin D in Ls174T cells, while this effect could also be effectively reversed by SC79. This was also supported by the results of qRT-PCR (Figure 6C). The above findings indicate that SET can activate c-Myc and Cyclin D1 through modulating Akt/NF-κB signals, thereby contributing to malignant growth of CRC cells.
 |
Figure 6 Knockdown of SET decreases the levels of c-Myc and cyclin D1 by regulating Akt. (A) Western blot analysis of NF-κB in Ls174T cells treated as indicated. (B) Western blot analysis of c-Myc and Cyclin D1 in Ls174T cells treated as indicated. (C) qPCR analysis of c-Myc and Cyclin D1 in Ls174T cells treated as indicated. SC79: 20 μM SC79. Data were shown as the mean ± SD from three independent experiments. **P < 0.01. |
Discussion
In the majority of studies to date, SET has been reported to be overexpressed in different cancers, and up-regulated SET is correlated with poor prognosis in several human malignancies, including pancreatic, renal, esophageal and colon cancer.13–15 Sobral et al reported that SET was a multifunctional protein, which was up-regulated in heads and neck squamous cell carcinoma.16 Consistent with previous studies, our present results revealed that the expression of SET was significantly up-regulated in human colorectal adenocarcinoma tissues, compared with that in the adjacent normal colorectal tissues. Furthermore, the expression of SET was well positively correlated with the development of colorectal carcinoma. However, in the present study, no significant correlation was observed between SET expression and CRC patients’ other characteristics, including gender, age or tumor differentiation. Together, these results strongly suggest that upregulated SET promotes the progression of colorectal carcinoma.
A number of studies reported that SET increased the proliferation ability and inhibited cell apoptosis in kinds of malignant tumors.6,9,16 Sobral et al reported that SET promoted cell proliferation, survival and resistance to cell death in heads and neck squamous cell carcinoma.16 Liu et al reported that knockdown of SET in NSCLC cells resulted in attenuated proliferative and invasive ability.9 Consist with the above studies, our present study demonstrated that the colorectal carcinoma cell growth ability was significantly decreased in vitro, and the tumorigenic potential of colorectal carcinoma cells in vivo was remarkably reduced after knockdown of SET. Altogether, our results strongly indicate that SET promotes the progression of colorectal carcinoma through increasing cancer cell proliferation activity.
SET, also named as I2PP2A, is a biological inhibitor of the cellular serine/threonine protein phosphatase 2A (PP2A). Liu et al reported that the biological effect of SET on proliferation was mediated by the inhibition of the PP2A, and their results showed that restoration of PP2A using SET antagonist FTY720 impaired proliferative and invasive potential in vitro, as well as inhibited tumor growth in vivo.9 In the present study, our results indicated that the activity of PP2A was significantly increased after knockdown of SET in colorectal carcinoma cells.
Meanwhile, a number of study demonstrated that PP2A was an important and ubiquitously expressed serine threonine phosphatase and functioned through dephosphorylating many critical cellular molecules, including Akt, p53, c-Myc and β-catenin.10,17 Akt mediated important cellular decisions involved in growth, survival and metabolism.11 Previous studies identified that PP2A, PP1 and the PHLPP can dephosphorylate the T308 and S473 sites on Akt.18 O’Shaughnessy et al reported that Akt was dephosphorylated through PP2A.19 Consist with the previous studies, our present study demonstrated that SET silencing-mediated increased activity of PP2A promoted the dephosphorylation of Akt, strongly suggesting that the phosphorylation of Akt was participated into the process of SET-induced cell survival in CRC.
Akt activates IKK, which ultimately leads to NF-κB activation and cell survival.11 Majority studies demonstrated that activation of IKK finally caused the phosphorylation and degradation of IκB, enabling NF-κB translocate to the nucleus and promotes new gene expression, including c-Myc, Cyclin D1, Karin et al12 c-Myc is a multifunctional transcription factor that has been linked to a diverse range of cellular functions, including cell cycle regulation, proliferation, growth, differentiation and metabolism.20 Junttila reported that the increased stability of c-Myc was often observed in human cancer specimens and cell lines.21 Moreover, other studies reported that c-Myc was stabilized and activated by phosphorylation at serine 62 in malignant cancer, and PP2A was a critical negative regulator of c-Myc by dephosphorylating serine 62 of c-Myc.22 Farrell et al reported that decreased expression of PP2A in human pancreatic cancer contributed to overexpression and stabilization of c-Myc, which led to an increased proliferation ability of malignant tumor, whereas increased PP2A activity increased c-Myc degradation.23 Consist with previous studies, our present study indicated that SET activated Akt, followed by the translocation of NF-κB to nucleus, and increased the expression of c-Myc and Cyclin D1, strongly suggesting that SET promotes the cell survival by activating Akt/NF-κB signaling pathway in CRC.
In summary, our results provided the strong evidence to support the notion that up-regulated SET promoted the cell survival through inhibiting PP2A to activate Akt/NF-κB signaling pathway, suggested that SET might be a potential target in CRC treatment.
Ethical Approval
This study was ethically approved by the Medical Research Ethics Committee of Shanxi Medical University (Taiyuan, China). All study participants provided informed consent. All procedures performed in studies involving animals were in accordance with the ethical standards of the animal experimental committee of Shanxi Medical University (Taiyuan, China).
Disclosure
The authors declare no conflicts of interest for this article.
References
1. Nitsche U, Späth C, Müller TC, et al. Colorectal cancer surgery remains effective with rising patient age. Int J Colorectal Dis. 2014;29(8):971–979. doi:10.1007/s00384-014-1914-y
2. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
3. Zhu J, Li K, Dong L, Chen Y. Role of FBXL20 in human colorectal adenocarcinoma. Oncol Rep. 2012;28(6):2290–2298. doi:10.3892/or.2012.2065
4. Jiang Q, Zhang C, Zhu J, Chen Q, Chen Y. The set gene is a potential oncogene in human colorectal adenocarcinoma and oral squamous cell carcinoma. Mol Med Rep. 2011;4(5):993–999. doi:10.3892/mmr.2011.526
5. Macfarlan T, Parker JB, Nagata K, Chakravarti D. Thanatos-associated protein 7 associates with template activating factor-Ibeta and inhibits histone acetylation to repress transcription. Mol Endocrinol. 2006;20(2):335–347. doi:10.1210/me.2005-0248
6. Mody HR, Hung SW, Naidu K, et al. SET contributes to the epithelial-mesenchymal transition of pancreatic cancer. Oncotarget. 2017;8(40):67966–67979. doi:10.18632/oncotarget.19067
7. Hung MH, Chen KF. Reprogramming the oncogenic response: SET protein as a potential therapeutic target in cancer. Expert Opin Ther Targets. 2017;21(7):685–694. doi:10.1080/14728222.2017.1336226
8. Chandrashekar DS, Bashel B, Balasubramanya SAH, et al. UALCAN: A portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia. 2017;19(8):649–658. doi:10.1016/j.neo.2017.05.002
9. Liu H, Gu Y, Wang H, et al. Overexpression of PP2A inhibitor SET oncoprotein is associated with tumor progression and poor prognosis in human non-small cell lung cancer. Oncotarget. 2015;6(17):14913–14925. doi:10.18632/oncotarget.3818
10. Mukhopadhyay A, Tabanor K, Chaguturu R, Aldrich JV. Targeting inhibitor 2 of protein phosphatase 2A as a therapeutic strategy for prostate cancer treatment. Cancer Biol Ther. 2013;14(10):962–972. doi:10.4161/cbt.25943
11. Manning BD, Toker A. AKT/PKB signaling: navigating the network. Cell. 2017;169(3):381–405. doi:10.1016/j.cell.2017.04.001
12. Karin M, Cao Y, Greten FR, Li ZW. NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer. 2002;2(4):301–310. doi:10.1038/nrc780
13. Dong L, Zhu J, Wen X, Jiang T, Chen Y. Involvement of SET in the Wnt signaling pathway and the development of human colorectal cancer. Oncol Lett. 2014;7(4):1203–1208. doi:10.3892/ol.2014.1866
14. Loganathan J, Jiang J, Smith A, et al. The mushroom Ganoderma lucidum suppresses breast-to-lung cancer metastasis through the inhibition of pro-invasive genes. Int J Oncol. 2014;44(6):2009–2015. doi:10.3892/ijo.2014.2375
15. Leopoldino AM, Squarize CH, Garcia CB, et al. Accumulation of the SET protein in HEK293T cells and mild oxidative stress: cell survival or death signaling. Mol Cell Biochem. 2012;363(1–2):65–74. doi:10.1007/s11010-011-1158-x
16. Sobral LM, Sousa LO, Coletta RD, et al. Stable SET knockdown in head and neck squamous cell carcinoma promotes cell invasion and the mesenchymal-like phenotype in vitro, as well as necrosis, cisplatin sensitivity and lymph node metastasis in xenograft tumor models. Mol Cancer. 2014;13:32. doi:10.1186/1476-4598-13-32
17. Rodgers JT, Vogel RO, Puigserver P. Clk2 and B56β mediate insulin-regulated assembly of the PP2A phosphatase holoenzyme complex on Akt. Mol Cell. 2011;41(4):471–479. doi:10.1016/j.molcel.2011.02.007
18. Van Kanegan MJ, Adams DG, Wadzinski BE, Strack S. Distinct protein phosphatase 2A heterotrimers modulate growth factor signaling to extracellular signal-regulated kinases and Akt. J Biol Chem. 2005;280(43):36029–36036. doi:10.1074/jbc.M506986200
19. O’Shaughnessy RF, Welti JC, Sully K, Byrne C. Akt-dependent Pp2a activity is required for epidermal barrier formation during late embryonic development. Development. 2009;136(20):3423–3431. doi:10.1242/dev.037010
20. Shao Q, Kannan A, Lin Z, Stack BC
21. Junttila MR, Westermarck J. Mechanisms of MYC stabilization in human malignancies. Cell Cycle. 2008;7(5):592–596. doi:10.4161/cc.7.5.5492
22. Janghorban M, Farrell AS, Allen-Petersen BL, et al. Targeting c-MYC by antagonizing PP2A inhibitors in breast cancer. Proc Natl Acad Sci U S A. 2014;111(25):9157–9162. doi:10.1073/pnas.1317630111
23. Farrell AS, Allen-Petersen B, Daniel CJ, et al. Targeting inhibitors of the tumor suppressor PP2A for the treatment of pancreatic cancer. Mol Cancer Res. 2014;12(6):924–939. doi:10.1158/1541-7786.MCR-13-0542
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.