Back to Journals » The Application of Clinical Genetics » Volume 14
Updates on Genes and Genetic Mechanisms Implicated in Primary Angle-Closure Glaucoma
Authors Kondkar AA ![]()
Received 8 December 2020
Accepted for publication 18 February 2021
Published 9 March 2021 Volume 2021:14 Pages 89—112
DOI https://doi.org/10.2147/TACG.S274884
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Martin Maurer
Altaf A Kondkar1– 3
1Department of Ophthalmology, College of Medicine, King Saud University, Riyadh, Saudi Arabia; 2Glaucoma Research Chair in Ophthalmology, College of Medicine, King Saud University, Riyadh, Saudi Arabia; 3King Saud University Medical City, King Saud University, Riyadh, Saudi Arabia
Correspondence: Altaf A Kondkar
Department of Ophthalmology, College of Medicine, King Saud University, P. O. Box 245, Riyadh, 11411, Saudi Arabia
Tel +96612825290
Fax +96614775724
Email [email protected]
Abstract: Primary angle-closure glaucoma (PACG) is estimated to affect over 30 million people worldwide by 2040 and is highly prevalent in the Asian population. PACG is more severe and carries three times the higher risk of blindness than primary open-angle glaucoma, thus representing a significant public health concern. High heritability and ethnic-specific predisposition to PACG suggest the involvement of genetic factors in disease development. In the recent past, genetic studies have led to the successful identification of several genes and loci associated with PACG across different ethnicities. The precise cellular and molecular roles of these multiple loci in the development and progression of PACG remains to be elucidated. Nonetheless, these studies have significantly increased our understanding of the emerging cellular processes and biological pathways that might provide more significant insights into the disease’s genetic etiology and may be valuable for future clinical applications. This review aims to summarize and update the current knowledge of PACG genetics analysis research.
Keywords: angle-closure, candidate genes, extracellular matrix, genetics, glaucoma, GWAS, PACG, pathways, polymorphisms, trabecular meshwork
Introduction
Glaucoma is characterized by the degeneration of retinal ganglion cells (RGCs) and progressive damage of the optic nerve axons, most commonly due to elevated intraocular pressure (IOP), leading to irreversible blindness.1 It is estimated that glaucoma may affect 111.8 million people globally by 2040.2 The prevalence of glaucoma varies significantly by geographical regions and ethnic groups. Among the different glaucoma types, the worldwide population affected by primary angle-closure glaucoma (PACG) is estimated to exceed 20 million in 2020 and over 30 million by 2040.2 Based on recent reports, the PACG burden is estimated to be the highest in Asia (0.73%, 95% credible interval 0.18 to 1.96),3 including middle-east compared to the Caucasians and Africans. It is also more common (60%) among women.3,4 The likelihood of severe bilateral visual impairment is three times higher in PACG than primary open-angle glaucoma (POAG) and poses a significant public health concern.5
PACG is clinically characterized by an iridotrabecular contact (≥ 270°), resulting in aqueous outflow obstruction and elevated IOP due to closure of an existing narrow angle of the anterior chamber associated with glaucomatous optic neuropathy and visual field changes.6 Currently, there is no widely accepted classification of PACG for both clinical or research purposes. However, on the basis of clinical phenotypes, anatomic configurations, etiology of angle-closure and natural history, PACG can be classified as a pupillary block, plateau iris, or peripheral iris crowding (non-pupillary block), and multiple mechanism pattern.7,8 Pupillary block is considered to be the principal mechanism in the angle-closure pathogenesis. The PACG eye typically exhibits the following ocular biometric findings: a shallow anterior chamber depth (ACD), increased thickness and more anterior position of the lens, hyperopic refractive error, and short axial length (AL).8 Other anterior segment parameters, such as trabecular to ciliary process distance, iris volume and thickness, anterior chamber area and volume, and lens vault, are also considered significant risk indicators for angle-closure.8 Due to the natural course of the disease, PACG diagnosis is often made at the advanced phenotypic stages involving chronic visual loss or acute angle-closure.1 Unraveling the underlying molecular and cellular mechanisms of PACG etiology or PACG phenotypes might help identify at-risk individuals at the early stage of the disease.
Many different factors affect the progression of the anterior chamber angle from narrow to angle-closure (Figure 1). Advanced aging, female gender, Asian ethnicity, anatomical characteristics, family history, and environmental factors to a certain extent are all considered major predisposing risk factors for PACG.3,4,8–12 The age and sex-adjusted odds of developing angle-closure were 13.6 times higher in affected patients’ siblings.11 Also, the narrow angle risk was seven times higher in first-degree relatives in Singaporean Chinese with overall heritability of PACG of 60%.10 The reports of the familial tendency towards the disease and racial differences in predisposition to PACG imply an underlying genetic basis for the development of PACG.4,9
 |
Figure 1 Schematic representation of risk factors contributing to angle-closure in PACG. The curved arrow from the ciliary epithelium indicate the normal flow of aqueous humor in open-angle which is blocked in angle-closure. |
The initial linkage mapping of nanophthalmos 1 (NNO1) gene on chromosome 11p in a large family with traits of autosomal dominant nanophthalmos, hyperopia, and a severe late-stage phenotype of angle-closure provided the first strong evidence for a causal role of genetic components in the pathogenesis of PACG.13 Since then, several studies have investigated the association of genes and genetic polymorphisms in PACG using the candidate-gene or genome-wide approaches.
With advances in genomic technologies, genome-wide association studies (GWAS) in the recent past have led to the successful identification of several genes and genetic variants associated with PACG in investigations across different ethnicities.14,15 While the precise role of these genes and genetic variants in the progression and/or development of PACG is still not completely understood, these studies have opened new perspectives in understanding the emerging cellular processes and biological pathways that might provide greater insight into the genetic etiology of the disease and are the focus of this review with an aim to provide an update on PACG genetic analysis research.
Genome-Wide Association Studies in PACG
GWAS method has been successfully applied to identify genetic loci for POAG16,17 and exfoliation glaucoma18 in the past. In regards to the genetics of PACG, two principal GWAS analyses in large populations of multi-ethnicities have lead to the identification of eight susceptibility genetic loci in PLEKHA7 (pleckstrin homology domain containing A7), COL11A1 (collagen type XI alpha 1 chain), PCMTD1 (protein-L-isoaspartate (D-aspartate) O-methyltransferase domain containing 1)-ST18 (ST18 C2H2C-type zinc finger transcription factor), EPDR1 (ependymin related 1), GLIS3 (GLIS family zinc finger 3), DPM2-FAM102A (dolichyl-phosphate mannosyltransferase subunit 2, regulatory and family sequence similarity 102 member A, respectively), CHAT (choline O-acetyltransferase, also designated as C10orf53), FERMT2 (fermitin family member 2, also known as PLEKHC1), all proven to be associated with PACG (Table 1).19,20
 | 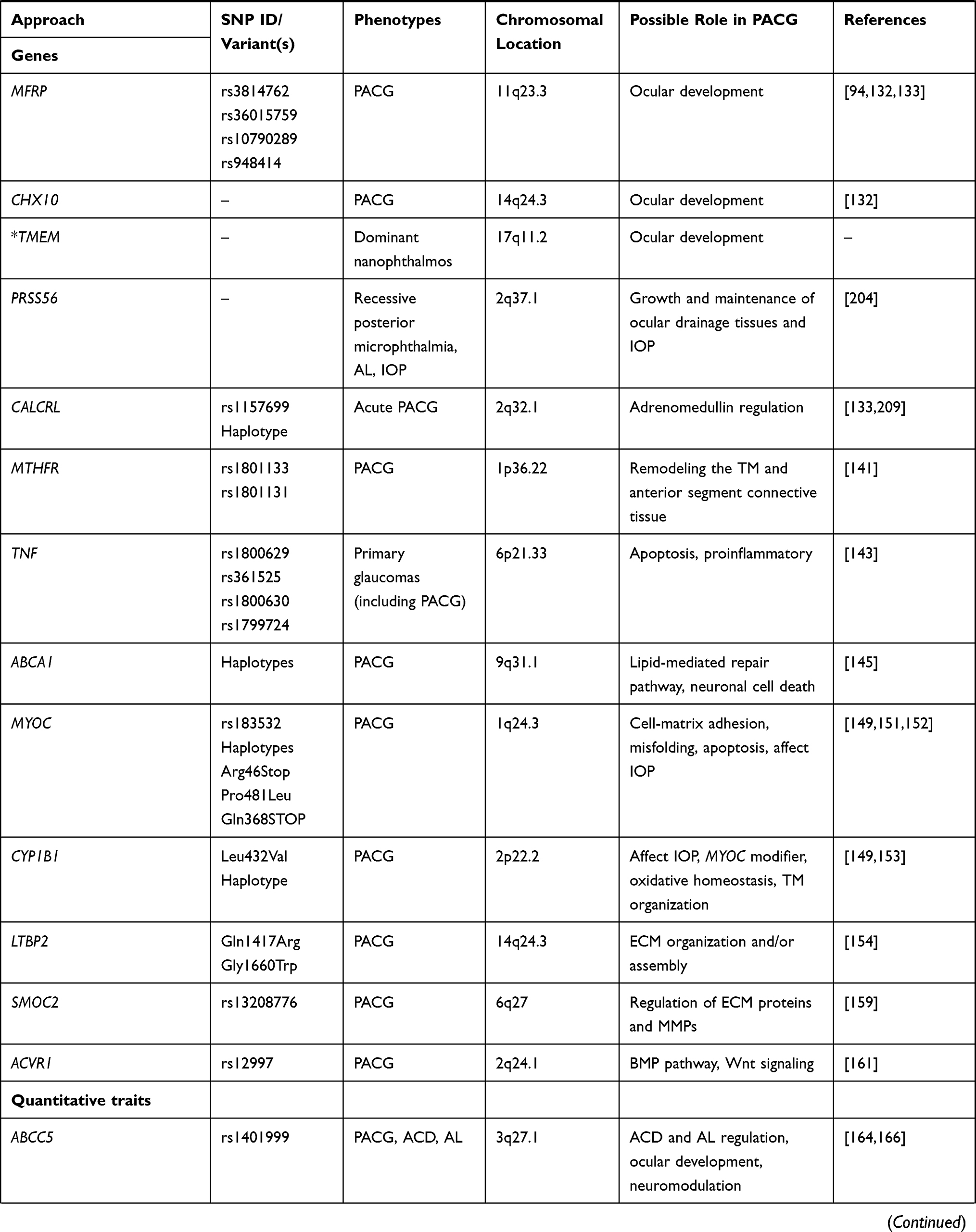 | 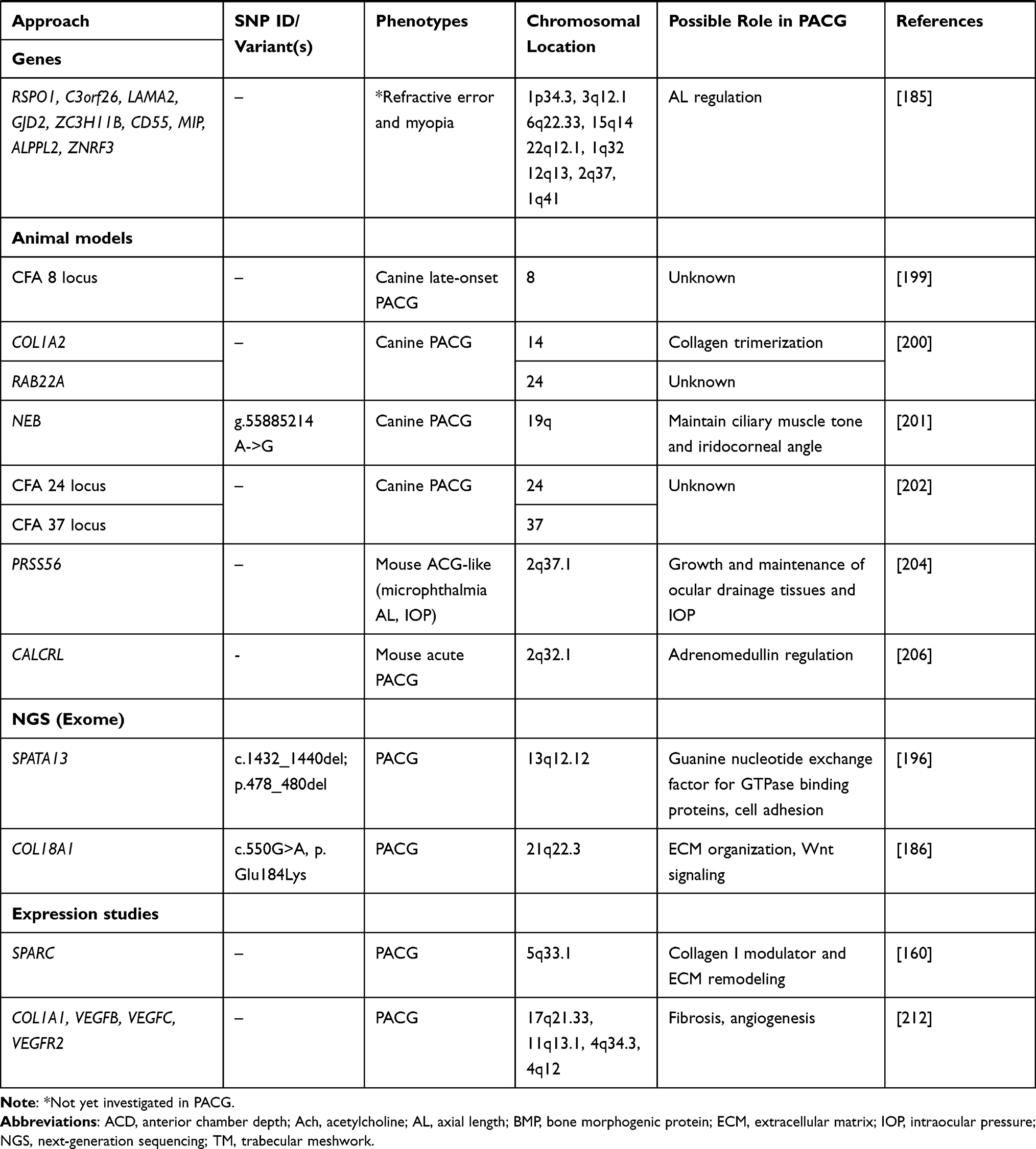 |
Table 1 List of Genes Associated with PACG and Related Phenotypes |
The first GWAS was conducted by Vithana et al on 1,854 PACG cases and 9,608 control participants from 5 Asian countries (Singapore, Hong Kong, India, Malaysia and Vietnam), with a second-stage replication in additional 1,917 PACG cases and 8,943 controls across six countries, including China, Singapore, India, Saudi Arabia and the United Kingdom.19 The study reported three novel loci: rs11024102 in PLEKHA7 (per-allele odds ratio (OR) = 1.22; P = 5.33 × 10−12), rs3753841 in COL11A1 (per-allele OR = 1.20; P = 9.22 × 10−10) and rs1015213 located between PCMTD1 and ST18 locus on chromosome 8q (per-allele OR = 1.50; P = 3.29 × 10−9) to be associated with PACG.19
PLEKHA7 on chromosome 11p15 encodes pleckstrin homology domain-containing protein 7, an adherens junction protein that plays a significant role in maintaining the stability of adherens junctions to regulate paracellular permeability and signaling pathways important for biological processes.21 In the eye, the tight junctions and adherens junctions play a significant role in cell-cell adhesion and paracellular permeability, thus maintaining the stability of structures such as the ciliary body, iris, aqueous flow system, and choroid, which are particularly relevant to glaucoma.19,22 The specific association of PLEKHA7 with apical junctional complexes (AJCs) and its particular localization to PACG-related anterior segment structures (iris, ciliary body, trabecular meshwork (TM)) and blood-aqueous barrier (BAB) components, including the vascular endothelium within the iris and ciliary microvasculature, indicates that PLEKHA7 might have a potential role for in PACG via fluidic regulation.23 Changes in the iris volume during pupillary dilation and choroidal effusion have been suggested to be involved in the pathogenesis of PACG.24 An aberrant fluidic movement in the iris microvasculature and pigmented iris epithelium due to a dynamic increase in iris volume (or lesser reduction in volume) has been observed in angle-closure eyes during dilation.25 In agreement, Lee et al reported down-regulation of PLEKHA7 expression in lens and iris of PACG patients, which also correlated with the carriers of the rs11024102 risk allele.22
On the other hand, disruption of BAB and leakage of inflammatory proteins and cells into the anterior chamber of the eye was found in both acute and chronic angle-closure glaucoma and suggested contributing to an increase in IOP be another mechanism in angle-closure development.26 There is evidence to support the role of PLEKHA7 in BAB maintenance. Silencing of PLEKHA7 in human non-pigmented epithelial and primary TM cells was shown to affect actin cytoskeleton organization, thereby compromising BAB integrity and aqueous outflow via Rac1/Cdc42 GAP activity of PLEKHA7.22 Thus, it has been hypothesized that PLEKHA7 variant may result in reduced expression of PLEKHA7, leading to “leaky” BAB, due to reduced tight junction or adherens junction proteins and altered regulation of fluid dynamics across the Schlemm’s canal, resulting in the clinical manifestations of PACG. However, these hypotheses need further validation by in vivo functional studies and animal studies.19,22,27
COL11A1 on 1p21.1 encodes one of the two α-chains of type XI collagen, a relatively minor fibrillar collagen. Diseases associated with mutations in this gene include type II Stickler syndrome and Marshall syndrome. These congenital syndromes are associated with manifestations including high myopia and blindness from retinal detachment.28–30 Collagen contributes to the tissues’ structural and mechanical properties, including the TM, sclera, and lamina cribrosa in the optic nerve head.31 Many studies have highlighted collagen’s significant role in high myopia and glaucoma.32,33 Alterations in the biomechanical features of the extracellular matrix (ECM) due to dysfunctional or structural changes in collagen might affect the TM function, resulting in decreased outflow and elevated IOP.33,34 Variations of collagen levels may lead to inter-individual differences in scleral and lamina cribrosa biomechanical properties.35,36 These alterations may modify the microenvironment of the optic nerve and possibly increase the susceptibility to axonal injury in glaucomatous eyes.37,38 Eyes predisposed to PACG are generally small and hyperopic. The COL11A1 causal variant associated with PACG is hypothesized to alter its function to persuade refractive error development, resulting in smaller hyperopic eyes and thereby predispose to PACG.19 Besides, abnormality of collagen in scleral and lamina cribrosa may increase susceptibility to AL changes and predispose to PACG.39 Considering the presence of COL11A1 in human TM cells,40 it is also possible that differential expression of COL11A1 in the TM may have a critical role in regulating the aqueous outflow pathway to influence the disease risk.19
In the PCMTD1 and ST18 locus, PCMTD1 encodes protein-l-isoaspartate O-methyltransferase domain-containing protein 1 of unknown function. The gene ST18 encodes a protein with zinc finger DNA binding transcription factor activity that functions as a tumor suppressor in breast cancer41 and a regulator of proapoptotic and proinflammatory genes in fibroblasts.42 Based on the linkage disequilibrium (LD) analysis and expression levels in the TM, PCMTD1 was suggested to be a more likely candidate for PACG susceptibility than ST18.19 PCMTD1-ST18 has been reported to be associated with the primary angle-closure suspect (PACS), providing further support for the role of this locus in angle-closure. PACS is the earliest stage of PACG, suggesting that this locus might increase the risk of narrow angle configurations.43,44
The original GWAS investigation19 was later significantly expanded to over 40,000 participants including 10,503 PACG cases from 24 countries across Asia, Australia, Europe, North America, and South America and is one of the largest GWAS performed so far for any glaucoma types.20 The study reported evidence of disease association at five new genetic loci upon meta-analysis of all patient collections. These loci included: FERMT2 rs7494379 (OR = 1.14, P = 3.43 × 10−11) on chromosome 14q22.1, EPDR1 rs3816415 (OR = 1.24, P = 5.94 × 10−15) on chromosome 7p14.1, GLIS3 rs736893 (OR = 1.18, P = 1.43 × 10−14) on chromosome 9p24.2, rs3739821 (OR = 1.15, P = 8.32 × 10−12) mapped in between DPM2–FAM102A on chromosome 9q34.11, and CHAT rs1258267 (OR = 1.22, P = 2.85 × 10−16) on chromosome 10q11.23 (Table 1).
FERMT2 encodes a protein known as pleckstrin homology domain-containing, family C member 1 (PLEKHC1) that belongs to the same pleckstrin family of proteins similar to PLEKHA7, which was associated with PACG in the previous GWAS.19 It is also known as Kindlin-2 (KIND2) and mitogen inducible gene 2 (MIG2).45,46 The protein is a component of the ECM with a role in integrin activation and cell-ECM adhesion.47 FERMT2 (MIG2), together with migfilin and filamin, has been shown to link cell-matrix adhesions and orchestrate the actin cytoskeleton assembly and cell shape modulation.48 Besides, FERMT2 (KIND2) has also been demonstrated to interact with ß-catenin and T-cell factor 4 (TCF4) to enhance wingless/integrated (Wnt) signaling.49 Actin cytoskeleton assembly, cell-to-cell adhesion processes, and Wnt signaling are known to play a significant role in the development and progression of glaucoma, thereby implicating the role of FERMT2 in PACG pathogenesis.50–52
EPDR1 is a lysosomal protein of unknown function. It encodes a glycosylated type II transmembrane protein related to ependymins families of cell adhesion molecules and possibly may have a similar role.53,54 EPDR1 has been associated with Dupuytren’s disease of the connective tissues.55 Besides, considering the observed associations between EDPR1, FERMT2, and PLEKHA7 and PACG, the studies suggest a significant role of cell-to-cell adhesion processes in PACG pathogenesis.19,20 A recent crystal structure study revealed that EPDR1 might function as a lysosomal activator protein or a lipid transporter.54 Several POAG-linked genetic variants have identified proteins (eg, MYOC, CAV1/2, ABCA1) that play a role in removing or repairing damaged lipids influencing membrane surface tension.56 EDPR1 may have a similar role in PACG and merits further investigations.
GLIS3 is a transcription factor belonging to the family of Kruppel-like zinc finger proteins. It functions as an activator and repressor of transcription in several cellular processes, including proliferation, apoptosis, differentiation, and development.57–59 Mutations in GLIS3 have been associated with diabetes, renal disease, congenital hypothyroidism, and cancers.60–62 Calderari et al have provided experimental evidence for a pleiotropic role of GLIS3 in diabetes and neurological disorder and its effect on gene transcription in β-cells and neuron function through regulation of genes involved in autophagy.63 There is evidence to suggest that the proper functioning of GLIS3 is required for appropriate ß-cell survival.64 Diabetes is a risk factor for glaucoma, well supported by epidemiological and experimental studies.65,66 Also, GLIS3 has been demonstrated to function as an upstream transcriptional activator of Wnt signaling genes and induce posterior specification of neural progenitor cells.59 Although the exact molecular mechanism by which a gene involved in a metabolic pathway may contribute to PACG pathogenesis is unknown, the identification of GLIS3 variants associated with diabetes and PACG suggests a high relevance of GLIS3 in cross-phenotype association and overlapping etiology of complex diseases such as PACG. Besides, it is also possible that GLIS expression might affect the development or cell survival of the anterior segment tissue structures and influence PACG risk.
DPM2 and FAM102A are uncharacterized genes mapped between intergenic loci rs3739821on 9q34.11 found to be associated with PACG.20 DPM2 encodes a protein involved in glycosylation. It is associated with congenital disorders of glycosylation that cause severe pathological phenotypes related to the nervous system.67 Altered glycosylation has been reported in glaucomatous TM.68 These alterations may elicit biological and biochemical changes in the ECM that may contribute to glaucoma pathophysiology. FAM102A was identified as an early estrogen-induced gene 1 (also known as EEIG1) in response to 17β-estradiol, suggesting a role in estrogen metabolism.69 Accordingly, gender and/or hormonal changes have been observed to influence glaucoma outcomes.9 Besides, a recent meta-analysis study reported a significant association of the estrogen signaling pathway in open-angle glaucoma with a protective effect of hormone replacement therapy in lowering IOP.70 Furthermore, EEIG1 (FAM102A) was demonstrated to serve as a novel receptor activator of NF-κB (RANK) signaling component with a role in osteoclast formation,71 suggesting its role in tissue maintenance, repair, and remodeling. Besides, PIP5KL1 (Phosphatidylinositol-4-phosphate 5-kinase-like 1) was also found to be the nearby gene on this loci20 that has been reported to inhibit cell proliferation and migration and may have a role in tumorigenesis.72
CHAT encodes an enzyme choline O-acetyltransferase (ChAT), responsible for synthesizing the neurotransmitter acetylcholine (ACh), which has a role in pupillary constriction.73,74 Studies have demonstrated ChAT presence in amacrine cells but not RGCs.75,76 However, an alternatively spliced form of ChAT was identified by Tooyama and Kimura77 and confirmed in rat retina and optic nerve78 to show that the RGCs also possess a viable ChAT system which may help regulate ACh synthesis and function. Studies suggest the presence of FOS (Fos proto-oncogene, AP-1 transcription factor subunit) gene, a transcription factor for Ach that mediates regulation and expression of ChAT and ACh function in the RGCs.79 ACh functions as a neurotransmitter in both the pre-ganglionic sympathetic and parasympathetic neurons in the autonomic nervous system.80 Autonomic innervations of the eye control many ocular functions. The autonomic regulation of ocular blood flow, aqueous humor production, and IOP, among others (as reviewed elsewhere81,82), can be significant determinants of glaucoma. Interestingly, anticholinergic agents have been found to mediate pupillary block and increase acute PACG risk.73 Therefore, considering the potential role of the cholinergic system of the eye, it is plausible that natural genetic variation in CHAT could alter the risk for PACG by regulating ACh metabolism and influence the autonomic regulation of PACG-related structures.83
Numerous replication studies have been performed to validate the GWAS findings19,20 in other ethnicities with mixed outcomes. Studies have examined these loci for their association with the early-stage angle-closure disease.43,44,84 Even though PLEKHA7 (rs11024102), COL11A1 (rs3753841), and PCMTD1-ST18 (rs1015213) were strongly associated with PACG among the Asian, these loci were not associated with PAC in a Han Chinese population.84 A study in the South Indian population consisting of PACS and PAC/PACG patients examined the three variants reported in PLEKHA7 (rs11024102), COL11A1 (rs3753841), and PCMTD1-ST18 (rs1015213) genes.43 The study failed to replicate the findings of PLEKHA7 and COL11A1 but confirmed the earlier reported association between PCMTD1-ST18 variant and PAC/PACG. Also, none of these 3 variants were found to be associated with PACS. However, in a study by Nongpiur et al in 1397 PACS patients of Chinese ethnicity from Singapore and 604 PACS patients of Indian origin,44 rs1015213 [A] in PCMTD1-ST18, rs3816415 [A] in EPDR1, and rs3739821 [G] in DPM2-FAM102A showed evidence of significant association in the Chinese cohort. But, only PCMTD1-ST18 was replicated modestly in the Indian PACS patients. Besides, a meta-analysis showed a significant association of PCMTD1-ST18 and DPM2-FAM102A variants with PACS status,85 suggesting a possible link of these loci with narrow angle configuration.44 A study from China consisting of 51 PACG cases and an equal number of controls reported a significant association of COL11A1 variants, including rs3753841, rs1676486 and rs12138977 with PACG.86 Also, variant rs3753841 in COL11A1 was found to be significantly associated with PACG in the Australian cohort (p = 0.017; OR = 1.34); whereas the PLEKHA7 variant rs11024102 (p = 0.039; OR 1.43) and PCMTD1-ST18 variant rs1015213 (p = 0.014; OR 2.35) were found to be significantly associated with the disease development in the Nepalese cohort in the same study.87 However, none of these variants survived multiple testing corrections, indicating a need to investigate large population-based cohorts, nonetheless supporting the role of these GWAS variants in PACG to a large extent. The meta-analysis studies have shown that PLEKHA7 rs11024102 is strongly associated with PACG in the Asian population but not among the white population,85,88 whereas COL11A1 rs3753841 was significantly associated with PACG both in Caucasian and Asian populations.85,88 Furthermore, the PCMTD1-ST18 locus was associated with combined PACS and PAC.85 In another recent study, the five variants identified by Khor et al20 were examined in the northeast Iranian PACG patients.89 Except for the variant rs3739821 in the DPM2-FAM102A locus, the study reported a significant association of all other variants in GLIS3 (rs736893), EPDR1 (rs3816415), FERMT2 (rs7494379), and CHAT (rs1258267) genes with PACG susceptibility.89
Thus far, the GWAS investigations have provided strong evidence for the role of multiple genetic factors in PACG and highlighted the complex and polygenic nature of PACG. Although the findings of the replication studies in other ethnicities are variable, they largely support the role of these genes in PACG.85,88 The variability in the clinical presentation of PAC/PACG patients, race and sample size are some factors that could contribute to such discrepancies. Besides, the expression of all these genes in the cornea, lens, retina, choroid, and optic nerve tissues lend further support to a significant role of these genes in these structures and plausibly in PACG pathogenesis.19,20 Nevertheless, the precise pathological function of these genes in PACG is still not completely understood. Future functional studies of these gene products in the ocular system may be able to explain their causal role in the development or progression of PACG.
Candidate Gene Studies in PACG
The candidate gene approach has also been used to identify genes and genetic variants contributing to the pathogenesis of PACG and to replicate the GWAS findings as discussed above. Unlike GWAS, which represents an unbiased genome-wide approach, this method involves investigating genetic associations of one or more allelic variants within a specific target gene or genes hypothesized to have a role in certain traits or phenotype of the disease. Several studies have examined the association between genetic variants in several genes and PACG (Table 1). A meta-analysis by Rong et al revealed polymorphisms in five candidate genes that may contribute to the risk of PACG.85 These include rs3918249 in matrix metalloproteinases 9 (MMP9), rs17427817 and rs5745718 in hepatocyte-growth factor (HGF), rs2510143 and rs3814762 in membrane-type frizzled-related protein (MFRP), rs7830 in nitric oxide synthase 3 (NOS3), and rs1043618 in heat shock protein 70 (HSP70).
Considering the significant role of MMPs in ECM turnover in the TM and regulation of aqueous outflow dynamics, MMPs are important candidates in glaucoma and many investigators have examined the association between MMP9 gene variants and PACG.90,91 MMP9 is located on chromosome 20q11.2 and encodes a 92-kDa multidomain enzyme known as gelatinase or type V collagenase, which is actively involved in scleral ECM remodeling.90,92 Besides the functional evidence in Mmp9 null mice,91 molecular genetics analyses across different ethnic groups have reported a variable link between the MMP9 gene polymorphisms and PACG in Taiwanese,93 Chinese,94 Singaporean,95 Caucasians,96 Indian97 and Pakistani98 populations. Apart from rs3918249 revealed by Rong et al85 another meta-analysis of six tagged SNPs in MMP9 by Chen et al showed a significant association between rs17576 (or rs2664538, now merged with rs17576) and non-Chinese PACG patients.99 The association of this variant was first reported in Taiwanese PACG patients by Wang et al.93 Variant rs17576 was reported to be in high linkage disequilibrium with rs3918249 (r2=0.98) and associated with PACG in the Australian and Pakistani cohorts.96,98 However, the associations were not replicated in Singaporean95 and Han Chinese94 ethnicities. However, another study suggested rs3918254 in MMP9 to be a susceptible locus to PACG in Han Chinese.100 Likewise, rs2250889 was found to be significant in Southern Chinese PACG patients.101 Also, a functional promoter variant rs3918242 (−1562C/T), which affected the transcription of the gene and was not a part of the meta-analysis studies, was reported to be associated with PACG patients from North India,97 but not in PACG patients of Pakistani origin.98 Variants in MPP9 may alter MMP9 function and affect ECM restoration during ocular development and may shorten the AL, which is a significant determinant of PACG.102
NOS3 is the enzyme responsible for generating nitric oxide, an endogenous signaling molecule, and an emerging therapeutic target for lowering IOP.103 In the eye, NO synthesis is predominantly localized to the Schelmm’s canal cells, and the TM is considered a major site of action.104 NO is involved in a myriad of physiological processes contributing to vasodilatation, increase local blood flow, and decrease vascular outflow resistance in ocular circulation by activating downstream signaling via soluble guanylate cyclase and cyclic guanosine monophosphate.105 Also, NO plays a protective role in oxidative stress-induced tissue injury and apoptosis.106 However, unlike in POAG, the relationship between vascular dysregulation and PACG is not clearly defined. Furthermore, excessive stimulation of N-methyl-d-aspartate (NMDA) receptor (NMDAR) has been shown to induce RGC apoptosis via neuronal nitric oxide synthase (nNOS).107 This effect of NO on retinal cells was demonstrated to be mediated in part by MMP9 activation through S-nitrosylation, corroborated by highly reduced activity of MMP9 in nNOS−/- mice. The study suggested a role of NO-activated MMP9 in retinal excitotoxicity.107 NOS3 variants have been associated with PACG in the Pakistani (VNTR),108 Australian (rs3793342, rs3918188, rs7830),109 but not with PAC/PACG in the Han Chinese population (rs3793342 and rs11771443).110,111 NOS3 may have a role in PACG pathogenesis by affecting the anterior chamber depth or activation of the MMP9-related pathway.
HSP70, also known as HSPA1A (heat shock protein family A (Hsp70) Member 1A), is an intron-less gene encoding a 70kDa heat shock protein. HSP70 is a stress response protein that functions as a molecular chaperon to regulate protein folding, translocation, misfolding, and degradation to maintain protein homeostasis and cell survival. Hsp70 has been implicated in several neurodegenerative diseases and RGC survival.112 Variant rs1043618, located at the 5ʹ-untranslated region of HSPA1A, down-regulates the expression of HSPA1A113 and has been strongly associated with PACG in the Pakistani108 and modestly in the Han Chinese94 populations. Similar to NOS3, HSPA1A has also been shown to induce MMP9 transcription through activation of nuclear factor kappa B (NF-κB) and activating protein-1 (AP-1) and may thus have an indirect role in PACG pathogenesis.114
HGF gene located on chromosome 7q21.11 encodes a hepatocyte growth factor protein, which belongs to a family of soluble cytokines and plasminogen-related growth factors. Activated HGF binds to c-Met (mesenchymal-epithelial transition factor) to induce HGF/c-Met signaling and stimulate cell growth, migration, morphogenesis and angiogenesis in numerous cell and tissue types.115 Over-activation of HGF/c-Met signaling can promote cancer development.115 HGF/c-Met signaling can stimulate various downstream signaling pathways, including Wnt/β-catenin signaling,115,116 which is known to have a significant role in glaucoma.117 In accordance, HGF receptors are expressed in multiple eye tissues, including the TM,118 and HGF is over-expressed in glaucomatous eyes.119 HGF has also been shown to regulate the barrier function of retinal pigment epithelium (RPE) cells. Over-expression of HGF in the RPE cells of the rabbit was found to induce retinal detachment.120 In vitro and in vivo studies have shown that HGF can confer protection to RGCs by increasing neuronal survival and promoting axonal regeneration.121 HGF was found to confer distinct advantages in sustaining long-term ganglion cell survival and axonal regeneration to respond to favorable stimuli than the two well-established trophic factors, ciliary neurotrophic factor (CNTF) and brain-derived neurotrophic factor (BDNF).122 Many variants in HGF were first reported to be associated with hyperopia and suggested to regulate human ocular development (emmetropization).123 Since angle-closure glaucoma and hyperopia share the same feature of a short AL, the HGF gene has been considered a risk factor of PACG. Studies have shown that variants in HGF (rs5745718 and rs17427817) were associated with susceptibility to PACG in the Nepalese124 and Han Chinese125 populations. The findings were also corroborated in the meta-analysis by Rong et al.85 Although the exact role of HGF in PACG pathogenesis is unknown, given its role in ocular tissues, it can be speculated that these variants may affect HGF expression and over-activate downstream HGF/c-Met/Wnt signaling pathway to induce morphological and physiological changes of the anterior segment structure, disrupting normal aqueous regulation and increase the risk of PACG.
MFRP on chromosome 11q23.3 has been previously associated with microphthalmia, isolated 5,126 nanophthalmos2127 and high hyperopia.127,128 These disorders are characterized by very small, hyperopic eyes exhibiting an unusually short axial length.129 In humans, MFRP is essential for prenatal ocular growth and postnatal emmetropization, a complicated process involving the regulation of axial growth of the eye in MFRP−/- null homozygotes.130 The strong association of ocular size (small eye) and angle-closure disease makes the genes involved in eye development potential candidates for PACG. Some studies have investigated the association between MFRP variants and PACG with inconsistent findings. MFRP variants rs3814762, rs36015759, and rs2510143 were not associated with the development of acute angle-closure glaucoma in Taiwanese subjects.131 No association of MFRP was reported by Aung et al in Singaporean Chinese PACG patients.132 Two variants, rs948414 and rs36015759, and variant rs10790289 in MFRP showed nominal association in Australian and Nepalese cohorts, respectively.133 Likewise, rs3814762 showed modest association in Han Chinese PAC patients.94 The meta-analysis by Rong et al confirmed the association of rs3814762 in PACG.85 MFRP is specifically expressed in the RPE and ciliary body.134 The gene encodes a glycosylated transmembrane protein with an extracellular frizzled-related cysteine-rich domain and hence speculated to be a regulator of Wnt signaling.135 Besides, analysis of transgenic mice (Mfrprd6) deficient in Mfrp demonstrated that disruption of Mfrp leads to increased expression of Prss56 during postnatal development of the Mfrprd6 eye.136 Given the relation between Mfrp deficiency and the expression of Prss56 (serine protease 56), combined with the genetic link of MFRP and PRSS56 variants and ocular size in humans, the authors suggested a likelihood of these genes being part of a regulatory network that influences postnatal posterior eye maintenance and development.136 Considering the alleged role of genes involved in ocular development in PACG, Aung et al also explored the role of VSX2 (visual system homeobox 2; also known as CHX10) on chromosome 14q24.3 in PACG patients.132 Mutations in VSX2 have been associated with microphthalmia, isolated 2.137,138 The study identified a possibly disease-causing variant c.728G>A resulting in Gly243Asp substitution in one PACG patient, which was absent in 215 normal controls. However, the overall results did not support a significant role of this variant in PACG.132 Another gene involved in ocular development that can contribute to PACG and would be worth investigating is TMEM98 (transmembrane protein 98). Mutations in the TMEM98 gene have been associated with autosomal dominant nanophthalmos and is expressed in tissues of the outflow pathway.139,140 So far, this gene has not been investigated in PACG.
Other Candidate Genes in PACG
Besides the genes and genetic variants described above, several other genes/variants have also been investigated in PACG (Table 1). The C677T (rs1801133) and A1298C (rs1801131) polymorphisms of the MTHFR (methylenetetrahydrofolate reductase) gene involved in homocysteine metabolism141 were associated in Pakistani PACG but not in North Indian142 and Australian and Nepalese cohorts.133 Promoter variants and haplotypes in TNF (tumor necrosis factor), a pluripotent proinflammatory cytokine, were significantly associated with primary glaucoma (including PACG) in the North Indian cohort.143 Haplotypes in ABCA1 (ATP binding cassette subfamily A member 1), a gene involved in lipid metabolism and associated with neuroinflammation and neuronal death,144 were associated in Han Chinese PACG145 but showed no association in Jordanian Arabs146 and cohorts of Northern China.147 MYOC (myocilin), OPTN (optineurin), WDR36 (WD repeat domain 36), and CYP1B1 (cytochrome P450 family 1 subfamily B member 1) genes previously associated with POAG were not associated with the middle-eastern Saudi PACG patients.148 But a mutation in MYOC (Arg46Stop) and CYP1B1 (Leu432Val) was identified in a Chinese PACG family.149 In contrast, MYOC was found to confer no risk in Chinese PACG cohorts,150 despite a positive association in the PACG cohort from Quebec.151 Furthermore, a polymorphism rs183532 and haplotypes in MYOC were also associated with PACG in the Han Chinese.152 On the other hand, CYP1B1 haplotype (C-C-G-G-T-A) was reported to confer modest risk in PACG patients of Indian origin.153 Similarly, LTBP2, commonly associated with pseudoexfoliation, was observed to have mutations (p.Gln1417Arg and p.Gly1660Trp) that were suggested to contribute to Iranian PACG patients.154 The studies of genes related to oxidative stress pathways in Saudi PACG patients such as rs4880 in SOD2 (superoxide dismutase 2),155 rs1001179 in CAT (catalase 2),156 T1/M0 genotypes in GST (glutathione S-transferase),157 and mitochondrial DNA haplogroups158 support the role of oxidative stress in PACG. Furthermore, rs13208776 variant in SMOC2 (secreted protein acidic and rich in cysteine (SPARC)-related modular calcium-binding protein 2) encoding a matricellular glycoprotein and known to regulate the expression of ECM proteins and MMPs has been recently reported to be associated with PACG in the Saudi population.159,160 Also, rs12997 in ACVR1 (activin A receptor type I), a critical regulator of the bone morphogenetic protein (BMP)/Wnt signaling, was recently reported to be associated in Saudi PACG patients suggesting a significant role of BMP signaling in PACG.161
It is important to note that all of these studies either lack sufficient replication or were reported to have negative associations with PACG and thus need further validations to confirm their role in PACG. Interestingly, none of these candidate genes/variants discussed so far emerged in the GWAS investigations19,20 conducted in PACG. These discrepancies may primarily result from false-positive signals arising because of the number of samples investigated in these candidate gene studies. Second, it is evident from the GWAS and candidate gene investigation findings that PACG is a genetically complex and multifactorial disease with no clear inheritance pattern. Thus, it is highly likely that variations/genes involved in PACG development and/or progression would be ethnic-specific, as observed in other complex human diseases with no clear Mendelian inheritance. In contrast, the GWAS investigations included samples consisting of an admixture of different ethnicities (Singaporean Chinese, Hong Kong Chinese, Malaysians, Vietnamese, and Indian) in their discovery stage. As a result, it is possible that population-specific disease association signals could have probably been lost or remains to be verified. More importantly, it is possible that exploring gene-gene interactions between genetic variants to mimic the complex nature of PACG may reveal, at least in parts, the missing heritability in well designed and powered association studies as demonstrated in POAG.162 Finally, clinical disparities of stages or severity of PACG disease would have confounded the candidate gene and/or GWAS investigation findings.
Quantitative Trait Loci in PACG
Identifying quantitative trait loci (QTL), a genetic link between heritable quantitative traits or endophenotypes in complex multifactorial diseases, is a useful genetic tool to understand the role of a specific trait in the disease pathogenesis and gain further insights into disease mechanisms. Data from genomic studies have identified multiple genetic factors that have the potential to predispose individuals to a high risk of developing PACG. Studies in the past have demonstrated that ocular biometric parameters such as a shallow ACD and short AL are strong anatomical risk factors for PACG.8,10,163 Associations of the genetic factors with these ocular biometric parameters in PACG have been investigated.
A genome-wide based QTL analysis identified a common genetic variant tagged by marker rs1401999 mapped to ABCC5 (ATP-binding cassette subfamily C member 5) on chromosome 3 to be associated with ACD (per-allele effect size = −0.045 mm, P = 8.17×10−9) in a case-control dataset from multiple ethnicities across Asia consisting 4276 PACG cases and 18,801 controls.164 This association was further strengthened when a subgroup of open-angle glaucoma controls was included in the analysis, suggesting that the risk of PACG might, at least, partly be influenced by genetic variants affecting ACD. This variant was also reported to be associated with PACG (OR = 1.13; 95% CI: 1.06–1.22; P = 0.00046).164 The ABCC5 locus has been further replicated with a moderate association in a group of Chinese PACG patients that investigated the PARL-ABCC5-HTR3D-HTR3C region, which is in strong LD at the locus on chromosome 3q27165 and also recently reported to be nominally associated with AL.166
The ABC proteins, a large family of ATPases found in the cell membrane, are involved in the efflux of endogenous metabolites like cyclic nucleotides, folic acid, and other molecules across the cell and participate in tissue defense and cellular signal transduction processes.167,168 ABCC5 is also known as multidrug resistance protein isoform MRP5, best known for its roles in multidrug resistance observed in chemotherapy‐resistant tumors.169 Studies in Abcc5−/− mice showed that ABCC5 is a general glutamate conjugate and analog transporter that affects the disposition of endogenous metabolites, toxins, and drugs.170 ABCC5 is ubiquitously expressed, including the structures of the anterior segment of the eye such as iris, ciliary body, lens, and cornea.164,171 The exact mechanism(s) by which ABCC5 has a role in angle-closure is not known. The inhibition of endogenous ABCC5 activity in zebrafish showed a significant reduction of body length and ocular size,172 suggestive of a role of ABCC5 in eye growth and development via the regulation of cGMP signaling, which can potentially influence ACD.173
A GWA study predicted that ABCC5 might be the new susceptibility gene for type 2 diabetes in humans174 through regulation of glucagon-like peptide-1 (GLP-1) secretion as demonstrated in Abcc5-/- mice.175 Studies in the past have reported a link between type 2 diabetes and glaucomatous optic nerve damage.65,66 Besides, GLP-1 is expressed in the human retina, and GLP-1 receptor activation was found to prevent retinal neurodegeneration by reducing glutamate excitotoxicity and upregulation of prosurvival pathways indicating a plausible neuromodulatory role of ABCC5 in PACG through GLP-1 regulation.176,177
Among the other common variants identified in PACG through GWAS,19,20 PLEKHA7 and COL11A1 variants showed no association with AL or ACD in the European Prospective Investigation of Cancer-Norfolk eye study,178 four population-based studies that included three from Singapore Eye Study and one Beijing Eye Study,179 the Jiangsu eye Study in Han Chinese84 and in another study from Shanghai.86 However, the latter study did report a significant association of COL11A1 variants, including rs3753841, rs1676486, and rs12138977 with moderate-to-severe mean deviation-based glaucoma severity.86 Likewise, studies suggest a role of PLEKHA7 in modifying the disease risk via the IOP-related pathway.180,181 In contrast to these variants, the intergenic PACG susceptibility locus between PCMTD1 and ST18 (rs1015213) demonstrated consistent association with shallower ACD but not with AL in the above mentioned European and Asian population-based studies.178,179 Also, Wei et al reported no association between these three loci and disease severity or progression in Singaporean Chinese patients.182
In another study, the association between all the 8 PACG loci identified by GWAS19,20 and PACG endophenotypes such as AL and ACD were investigated in the Han Chinese population.183 The study reported nominal association of COL11A1 (rs3753841), CHAT (rs1258267), and GLIS3 (rs736893) with ACD (p = 0.023, 0.016, 0.01, respectively). However, these associations did not survive false discovery rate correction for multiple testing. Also, the analysis of multiple variants in MYOC and ABCA1 showed no association with ACD and AL.147 Likewise, variants in MFRP and HGF have shown no association with ACD and AL phenotypes. However, variant rs7290117 in ZNRF3 was suggested to be involved in the regulation of AL.183 In another recent study, Liu et al investigated the association of the eight susceptibility loci identified in PACG with the disease severity based on the visual field mean deviation.184 The study examined 436 mild-to-moderate PACG and 206 severe PACG patients. Only variant rs3816415 in EPDR1 was significantly associated with severe PACG (OR, 2.03; 95% CI, 1.49–2.78; P = 1 × 10−5), suggesting that the EPDR1 variant may predispose individuals to an aggressive form of PACG. The study also reported that PACG patients with a genetic risk score in the highest quartile have more than a 3-fold risk of developing severe PACG.184 Similarly, COL11A1 rs1676484 and rs12138977 polymorphisms have also been associated with disease severity in Chinese PACG patients.86 Shi et al reported that NOS3 variant rs11771443 was associated with deeper ACD but not with primary angle-closure (PAC), AL and diopter of spherical power in Han Chinese.110
In a genomic QTL meta-analysis of ocular AL conducted in 12,531 Europeans and 8,216 Asians of the Consortium for Refractive Error and Myopia cohorts, Cheng et al identified nine loci significantly related to AL (RSPO1 [R-spondin 1], C3orf26 [chromosome 3 open reading frame 26], LAMA2 [laminin subunit alpha 2), GJD2 [gap junction protein delta 2], ZC3H11B 9 [zinc finger CCCH-type containing 11B], ZNRF3 [zinc and ring finger 3], CD55 [CD55 molecule (Cromer blood group)], MIP [major intrinsic protein of lens fiber], and ALPPL2 [alkaline phosphatase, placental like 2]).185 It is interesting to note that although AL is an important clinical determinant of PACG, none of the loci identified by Cheng et al were reported in PACG GWA studies.19,20 Likewise, ABCC5 associated with ACD and PACG164 also did not emerge in these GWAS investigations.19,20 Taken together, these findings suggest that the contribution of ACD and AL to PACG pathogenesis are more complex events that are still not completely understood, and the involvement of other clinical attributes in PACG pathogenesis remains to be investigated.
Next-Generation Sequencing Studies in PACG
With advances in next-generation sequencing (NGS), a combination of linkage analysis and whole genome/exome sequencing has been utilized to identify causal genes in families with PACG. Using a linkage and whole-exome sequencing approach, Suri et al identified the genetic cause of iridocorneal angle-closure in three unrelated Iranian families with at least ten individuals diagnosed with PACS, PAC, or PACG.186 A mutation (c.550G>A, p.Glu184Lys) identified in COL18A1 (collagen type XVIII alpha 1 chain) at 21q22.3 encoding collagen type XVIII was found to be the cause of angle-closure in the pedigree. The inheritance pattern of angle-closure causing mutations in COL18A1 was autosomal dominant. Other COL18A1 mutations were also identified in two unrelated PACS families, lending further support for a causative role of COL18A1 in angle-closure. However, it is important to note that the individuals of these two unrelated PACS families were parents or grandparents of Knobloch syndrome (KS) patients. KS is a rare autosomal recessive disorder with considerable clinical variability and is classically characterized by severe ocular abnormalities, including high myopia, retinal detachment, and occipital encephalocele that often leads to bilateral blindness.187 COL18A1 is a significant gene associated with KS type 1.187–189 Interestingly, glaucoma has also been observed in patients with KS.190–192
COL18A1 is expressed in human eye tissues, including the TM, cornea and ciliary body.186 The potential significance of COL18A1 in causing PACG emphasizes the importance of collagen and the ECM structure in glaucoma pathophysiology.33,193 Type XVIII collagen is a component of the ECM proteins that contain multiple triple-helix domains (collagenous domains) interrupted by non-collagenous domains.194 The N-terminal of the long isoform of the protein consists of a cysteine-rich frizzled domain that is homologous to the extracellular part of frizzled receptors involved in Wnt signaling. The exact effect of the p.Glu184Lys mutation on COLA18A1 function is still unknown but may probably interfere with the regulation of the Wnt signaling pathway. Wnt signaling is involved in the regulation of eyeball size, a trait commonly associated with PACG.195 And interestingly, MFRP that has also been implicated in the etiology of PACG, also contains a frizzled-like domain,135 provides further support to their role in PACG development.
In another recent study using a combination of linkage and whole-genome sequencing, Waseem et al identified a causal genetic variant in SPATA13 (spermatogenesis associated protein 13) in a seven-generation PACG family of British origin.196 A single 9bp in-frame deletion variant (c.1432_1440del; p.478_480del) in SPATA13 on chromosome 13q12.12 was found to segregate in all the affected individuals of the family with variable expression and decreased penetrance. Additional rare variants in SPATA13 were also observed in unrelated PACS, PAC or PACG cohorts, including the 9bp deletion, supporting the causal evidence of this variant.196
SPATA13, also known as ASEF2 (adenomatous polyposis coli-stimulated guanine nucleotide exchange factor 2), acts as guanine nucleotide exchange factor (GEF) for RhoA, Rac1, and Cdc42 GTPases.197,198 SPATA13 transcripts encode a 652 amino acid (SP-652) and 1277 amino acid protein (SP-1277) due to alternate N-terminal splicing, which shows nuclear and cytoplasmic localization with partial co-localization.196 The transcripts show ubiquitous expression and are highly expressed in the iris, cornea, ciliary body, and retina, the tissues most affected by PACG and suggested to regulate tissue homeostasis.196 In RPE-cells, SP-1277 showed dramatic redistribution during various cell division stages, suggesting a role in mitosis.196 Isoform SP-652 has been shown to regulate actin cytoskeletal reorganization198 and is involved in angiogenesis.198 The 9bp deletion reported in this study was found to increases the Rac1-dependent GEF activity, an effect that was consistent with three other variants reported in this study.196 Taken together, SPATA13 was suggested to have a regulatory role in cell division and cell adhesion in the anterior segment of the eye, affecting tissue homeostasis and influencing PACG pathogenesis.196 Similar to SPATA13, PLEKHA7, another protein implicated in PACG,19,23 encodes a Rac1/Cdc42 GAP activity,22 implicating the role of the Rho-GTPase pathway in PACG.
Animal-Model Studies in PACG
Further insights into the genetic predisposition towards PACG have also been gained through genetic analysis of animal models. An initial genome-wide study in a Dandie Dinmont Terrier cohort representing a late-onset form of PACG led to identifying a novel 9.5Mb susceptibility locus on canine chromosome 8.199 Newer susceptibility genes have also been identified as possible contributors to acute PACG in American Basset Hounds. These include: COL1A2 (collagen type I alpha 2 chain) on chromosome 14, RAB22A (RAB22A, member RAS oncogene family) on chromosome 24200 and NEB (nebulin) mapped to chromosome 19q.201 NEB variant (g.55885214 A->G) identified by exome sequencing was also associated with PACG in the second cohort of unrelated Basset Hounds.201 Given the significant role of collagen in glaucoma33 and the identification of a collagen gene (COL11A1) variant in GWAS19 implies that COL1A2 might also represent a highly potential candidate in PACG pathogenesis. RAB22A, are members of the Ras-related family of GTPases and their role in glaucoma is unknown.200 NEB is a large protein that promotes the contractile function of sarcomeres and is prominently expressed in the ciliary eye muscles, indicating its role in the maintenance of ciliary muscle tone and iridocorneal angle.201
In a recent genome-wide and RNA sequencing study in European Basset Hounds, Oliver et al identified two novel loci of 1.4 and 0.2 Mb regions, on chromosomes 24 and 37, respectively, that were significantly associated with PACG and also revealed differential expression of eight genes within these two loci.202 The locus on chromosome 24, consisting of RNF24/PANK2 (ring finger protein 24 and pantothenate kinase 2, respectively), the nearest upstream genes, has previously been associated with glaucoma traits in humans.203 However, this study failed to replicate the previous GWAS findings of NEB (including the variant), COL1A2, and RAB22A associations observed in American Basset Hounds,200,201 probably due to differences in the frequency distribution of the risk loci in the American and European Basset Hounds.202
Animal-based studies have also been utilized to investigate genetic links in AL regulation.204 The genetic alterations in a novel serine protease-encoding gene (Prss56) were found to be associated with decreased AL in a mouse model resembling ACG-like phenotype.204 Besides, mutations in human PRSS56 in six families with autosomal recessive posterior microphthalmia were also found to cause a significant reduction in the ocular AL in the same study.204 Prss56 is expressed in the retina.204 Furthermore, Prss56-/- mice have been recently shown to exhibit ocular angle defects and increased risk of high IOP.205 The study suggested a critical role for PRSS56 in the development and maintenance of ocular drainage tissues and IOP homeostasis.205
In a transgenic mouse model of acute PAC, overexpression of calcitonin receptor-like receptor (CALCRL) in the pupillary sphincter muscle exhibited pupillary palsy due to relaxant effect of adrenomedullin leading to obstruction of aqueous outflow and acutely and transiently elevated IOP, resembling the phenotypic characteristics of acute PACG in humans.206,207 It was hypothesized that defective adrenomedullin regulation in the pupillary sphincter muscle might result in the development of an acute attack of angle-closure in humans. CALCRL belongs to a family of G-protein-coupled receptors. The transport of CALCRL to the plasma membrane is facilitated by receptor-activity-modifying proteins (RAMPs). Depending on which RAMP members are expressed, CALCRL can function as either a calcitonin-gene-related peptide (CGRP) receptor via RAMP1 or an adrenomedullin receptor via RAMP2.208 CALCRL and RAMP2 (receptor-activity-modifying protein 2) heterodimers have been identified in the pupillary sphincter muscle.206 In agreement with the animal studies data, a common variant rs1157699 was nominally associated with acute but not chronic PACG in the Southern Chinese population.209 However, a rare haplotype AATACAGAT in the CALCRL gene was found to exhibit a significant protective effect in the Australian Caucasian cohort (corrected p-value=0.024).133 The implications of this association are still unclear and were absent in the Nepalese PACG cohort in the same study, indicating a need for replication in other ethnicities and further validation for the role of this gene in human PACG.133
Expression Studies in PACG
There are no reports of blood-based or tissue-based mRNA expression profiling studies in PACG. However, few studies have been conducted to investigate altered gene expression of the iris in glaucoma. Among several ocular biometric risk factors associated with PACG, anterior segment optical coherence tomography studies have reported the association between angle-closure and increased quantitative iris parameters such as iris curvature, iris area, and iris thickness.210,211 These studies suggest biomechanical and structural changes of the iris as significant clinical determinants of PACG. Based on these observations, it was hypothesized that a differential iris response to pupil dilation and choroidal effusion occurrence might have a role in PACG pathogenesis.212 Increased expression of SPARC (secreted protein, acidic and rich in cysteine) and collagen I transcripts were reported in PACG iris than POAG.160 This finding was in agreement with the previously observed increased deposition of mature collagen I in PACG iris.213 The study reinforced the established role of SPARC as a modulator of collagen I production and suggested increased levels of iris collagen to be a discrete biological signature for PACG.160 SPARC, a collagen-binding matricellular glycoprotein, is involved in regulating collagen deposition in the ECM and was proposed to play a role in PACG by influencing biomechanical properties of the iris through ECM reorganization.160,214 In another study, Seet et al examined the expression of known genes implicated in glaucoma, such as COL1A1 (collagen type 1 alpha 1 chain), VEGFA, VEGFB, VEGFC (vascular endothelial growth factors A, B and C, respectively) and members of the VEGF receptors 1 (VEGFR1) and 2 (VEGFR2).212 The study reported distinct increased mRNA expression of COL1A1, VEGFB, VEGFC, and VEGFR2 in PACG iris than POAG. Furthermore, a combination of gene expression levels and biometric features such as lens vault and anterior chamber volume augmented cross-validated differentiation between PACG and POAG more effectively with the highest accuracy. The study highlighted distinct molecular disparities between glaucoma types and the importance of combining molecular profiles with known biometric ocular features to gain a better understanding of the disease etiology and their subtypes.212 A functional and pathway enrichment analysis study of differently expressed genes from two different datasets215 revealed three important differentially expressed genes, including HGF, AKR1B10 (aldo-ketoreductase family 1, member B10), and AKR1C3 (aldo-ketoreductase family 1, member C3) that were suggested to serve as important biomarkers and targets for glaucoma diagnosis and treatment.216 A more detailed and systematic gene expression studies are needed to identify differentially expressed functional targets in PACG. These transcripts may allow the identification of their regulatory counter-parts such as microRNAs (miRNAs) using in-silico bioinformatics tools. The functional characterization of mRNA-miRNA co-expression patterns can shed light on the pathological mechanisms of the disease, and their molecular signatures might possibly serve as diagnostic markers.
Potential Pathways Involved in PACG
Genetic studies performed thus far have yielded significant genes contributing to PACG. The proteins encoded by the current set of PACG genes are involved in a broad range of cellular processes and biological functions. Based on these functions, a simple pathway-based enrichment analysis using Enrichr online tool (https://maayanlab.cloud/Enrichr/) predicted overrepresentation of the syndecan-1, ECM organization and integrin-1 pathways to be the top 3 ranking pathways among others that may contribute to PACG pathogenesis (Figure 2).
 |
Figure 2 Bar chart showing an overview of pathways significantly overrepresented by a set of genes identified in PACG using Enrichr online tool (https://maayanlab.cloud/Enrichr/). An asterisk (*) next to a p-value indicates the term also has a significant adjusted p-value (<0.05). |
The ECM of the TM comprises of collagens, elastin-containing microfibrils, matricellular and structural organizing proteins, glycosaminoglycans (GAGs), and proteoglycans that are essential for maintaining the integrity of the TM.217 ECM is believed to be a critical component to outflow resistance and IOP regulation in normal and glaucomatous eyes.218 Matrix remodeling constitutes an essential aspect of mechanically regulated pathways.219 Forces created by high IOP probably induce mechanical stretching or distortion of the ECM that may lead to clinical manifestations of the disease.219 Although no genetic variants significantly associated with PACG were directly linked to the ECM, several genetic loci were found to be associated with this process, including COL11A1, COL18A1, SPARC, COL1A2, MMP9, LAMA2 and LTBP2 as discussed earlier.
Cell adhesion proteins, cell surface ECM receptors, and related binding proteins are also present in the TM beams.217 Many biological activities of ECM are mediated via integrin-ECM interactions.220 Integrins are membrane-spanning receptors that mediate cell adhesion to ECM proteins and provide critical connections between actin-mediated cell processes and the ECM (eg, PLEKHA7, FERMT2, EPDR1).221 Syndecans are a family of cell surface ECM receptors expressed in the TM.222 Syndecan-1 (CD138) is a heparan sulfate proteoglycan that binds to bind to integrins, collagens, associates with actin cytoskeletal structures, growth factors (such as HGF) and participates in diverse biological responses including cell-cell and cell-ECM adhesions, apoptosis, growth factor regulation and angiogenesis.223 Syndecan-1 has been shown to modulate matrix assembly in several models of inflammatory diseases.224 Deficiency of Sdc1-/- was associated with the assembly of a disorganized matrix and impaired collagen cross-linking in the myocardial infarction mice model.225 It is tempting to speculate that these proteoglycans may have a similar effect in PACG eyes.213 More investigations are needed to verify this process.
It is not surprising that ECM remodeling is emerging as a critical mechanism for PACG.217,218 The ECM is a highly dynamic structure, continually undergoing a remodeling process in response to the microenvironment changes regulating cell behaviors. It is essential to understand the mechanisms of ECM remodeling and its regulation; and how the ECM’s biomechanical properties influence cell behaviors during normal development and in the disease stage. A better understanding of the modulation of ECM dynamics and its components may help develop effective strategies to regulate cell behaviors and maintain tissue integrity and function. These observations also support the role of epistatic gene interactions in PACG.162
Concluding Remarks
PACG has long been believed to have a significant genetic premise exhibiting geographical and racial differences in the incidence of PACG. Although the common disease-causing variants/mutations have not been identified yet, the genetics studies have yielded important loci/genes that may contribute towards the pathogenesis of PACG. GWAS investigations have revealed eight genetic loci in PLEKHA7, COL11A1, PCMTD1-ST18, FERMT2, EPDR1, GLIS3, DPM2-FAM102A, and CHAT that may have a significant role in PACG development or progression. Moreover, additional candidate genes: HGF, MMP9, MFRP, NOS3, and HSPA1A (HSP70) have also been suggested to influence the risk of PACG by meta-analysis. Furthermore, GWAS of quantitative traits has shown that ABCC5 could be an important marker of PACG phenotype, ACD. Besides, the findings of certain candidate genes (eg, SMOC2, ACVR1), animal studies, and expression studies are promising. Genes involved in ECM organization and/or remodeling pathways seem to represent critical targets in PACG pathogenesis. These findings provide a newer perspective in understanding the genetic mechanisms that may plausibly contribute to PAC/PACG and highlight the complex genetic etiology of PACG. They also offer new avenues for future research of proteins encoded by these genes, which participate in a broad range of cellular processes and biological pathways. However, in the absence of any direct functional evidence, the mere presence of these genes in ocular tissues does not confirm their role in PACG etiology. Functional, molecular and animal model studies are further needed to elucidate their causal role in PACG pathogenesis. Also, as is commonly observed in genetic association studies of complex human diseases, most of these susceptibility loci account for a relatively small fraction of PACG cases. They exhibit a moderate effect with an odds of 1.0–1.5 risk per allele, indicating the presence of additional genes/genetic variant(s) that may have been missed or not covered (eg, insertions, deletions, copy number variants) and the role of gene-gene interactions that remains to be explored. The functions of non-coding RNAs, miRNAs and epigenetic regulators in PACG also remain to be investigated. NGS may represent a useful tool in PACG with strong familial links. The use of integrated genome analysis consisting of whole genome/exome sequencing, transcriptome analysis, and epigenetic regulations might highlight the underlying genetic and environmental abnormalities of clinical relevance. Until then, the importance of accurate clinical phenotyping cannot be overemphasized, and the diagnosis of angle-closure between iris and trabecular meshwork remains the hallmark of PACG.
Acknowledgments
The author would like to thank the Vice Deanship of Scientific Research Chair, Glaucoma Research Chair in Ophthalmology at the King Saud University for their support; Mr. Taif A. Azad and Dr. Tahira Sultan for their help with figures and references.
Disclosure
The author reports no conflicts of interest in this work.
References
1. Weinreb RN, Aung T, Medeiros FA. The pathophysiology and treatment of glaucoma: a review. JAMA. 2014;311(18):1901–1911. doi:10.1001/jama.2014.3192
2. Tham YC, Li X, Wong TY, Quigley HA, Aung T, Cheng CY. Global prevalence of glaucoma and projections of glaucoma burden through 2040: a systematic review and meta-analysis. Ophthalmology. 2014;121(11):2081–2090. doi:10.1016/j.ophtha.2014.05.013
3. Chan EW, Li X, Tham YC, et al. Glaucoma in Asia: regional prevalence variations and future projections. Br J Ophthalmol. 2016;100(1):78–85. doi:10.1136/bjophthalmol-2014-306102
4. Cheng JW, Zong Y, Zeng YY, Wei RL. The prevalence of primary angle closure glaucoma in adult Asians: a systematic review and meta-analysis. PLoS One. 2014;9(7):e103222. doi:10.1371/journal.pone.0103222
5. Foster PJ. The epidemiology of primary angle closure and associated glaucomatous optic neuropathy. Semin Ophthalmol. 2002;17(2):50–58. doi:10.1076/soph.17.2.50.14718
6. Foster PJ, Buhrmann R, Quigley HA, Johnson G. The definition andclassification of glaucoma in prevalence surveys. Br J Ophthalmol. 2002;86:238–242. doi:10.1136/bjo.86.2.238
7. Wang N, Ouyang J, Zhou W, et al. [Multiple patterns of angle closure mechanisms in primary angle closure glaucoma in Chinese]. Zhonghua Yan Ke Za Zhi. 2000;36(1):46–51, 45, 46. Chinese.
8. Sun X, Dai Y, Chen Y, et al. Primary angle closure glaucoma: what we know and what we don’t know. Prog Retin Eye Res. 2017;57:26–45. doi:10.1016/j.preteyeres.2016.12.003
9. Vajaranant TS, Nayak S, Wilensky JT, Joslin CE. Gender and glaucoma: what we know and what we need to know. Curr Opin Ophthalmol. 2010;21(2):91–99. doi:10.1097/ICU.0b013e3283360b7e
10. Amerasinghe N, Aung T. Angle-closure: risk factors, diagnosis and treatment. Prog Brain Res. 2008;173:31–45.
11. Kavitha S, Zebardast N, Palaniswamy K, et al. Family history is a strong risk factor for prevalent angle closure in a South Indian population. Ophthalmology. 2014;121(11):2091–2097. doi:10.1016/j.ophtha.2014.05.001
12. Subak-Sharpe I, Low S, Nolan W, Foster PJ. Pharmacological and environmental factors in primary angle-closure glaucoma. Br Med Bull. 2010;93(1):125–143. doi:10.1093/bmb/ldp042
13. Othman MI, Sullivan SA, Skuta GL, et al. Autosomal dominant nanophthalmos (NNO1) with high hyperopia and angle-closure glaucoma maps to chromosome 11. Am J Hum Genet. 1998;63(5):1411–1418. doi:10.1086/302113
14. Chandra A, Mitry D, Wright A, Campbell H, Charteris DG. Genome-wide association studies: applications and insights gained in Ophthalmology. Eye (Lond). 2014;28(9):1066–1079. doi:10.1038/eye.2014.145
15. Aung T, Khor CC. Glaucoma genetics: recent advances and future directions. Asia Pac J Ophthalmol (Phila). 2016;5(4):256–259. doi:10.1097/APO.0000000000000229
16. Thorleifsson G, Walters GB, Hewitt AW, et al. Common variants near CAV1 and CAV2 are associated with primary open-angle glaucoma. Nat Genet. 2010;42(10):906–909. doi:10.1038/ng.661
17. Burdon KP, Macgregor S, Hewitt AW, et al. Genome-wide association study identifies susceptibility loci for open angle glaucoma at TMCO1 and CDKN2B-AS1. Nat Genet. 2011;43(6):574–578. doi:10.1038/ng.824
18. Thorleifsson G, Magnusson KP, Sulem P, et al. Common sequence variants in the LOXL1 gene confer susceptibility to exfoliation glaucoma. Science. 2007;317(5843):1397–1400. doi:10.1126/science.1146554
19. Vithana EN, Khor CC, Qiao C, et al. Genome-wide association analyses identify three new susceptibility loci for primary angle closure glaucoma. Nat Genet. 2012;44(10):1142–1146. doi:10.1038/ng.2390
20. Khor CC, Do T, Jia H, et al. Genome-wide association study identifies five new susceptibility loci for primary angle closure glaucoma. Nat Genet. 2016;48(5):556–562. doi:10.1038/ng.3540
21. Pulimeno P, Paschoud S, Citi S. A role for ZO-1 and PLEKHA7 in recruiting paracingulin to tight and adherens junctions of epithelial cells. J Biol Chem. 2011;286(19):16743–16750. doi:10.1074/jbc.M111.230862
22. Lee MC, Shei W, Chan AS, et al. Primary angle closure glaucoma (PACG) susceptibility gene PLEKHA7 encodes a novel Rac1/Cdc42 GAP that modulates cell migration and blood-aqueous barrier function. Hum Mol Genet. 2017;26(20):4011–4027. doi:10.1093/hmg/ddx292
23. Lee MC, Chan AS, Goh SR, et al. Expression of the primary angle closure glaucoma (PACG) susceptibility gene PLEKHA7 in endothelial and epithelial cell junctions in the eye. Invest Ophthalmol Vis Sci. 2014;55(6):3833–3841. doi:10.1167/iovs.14-14145
24. Nongpiur ME, Ku JY, Aung T. Angle closure glaucoma: a mechanistic review. Curr Opin Ophthalmol. 2011;22(2):96–101. doi:10.1097/ICU.0b013e32834372b9
25. Quigley HA, Silver DM, Friedman DS, et al. Iris cross-sectional area decreases with pupil dilation and its dynamic behavior is a risk factor in angle closure. J Glaucoma. 2009;18(3):173–179. doi:10.1097/IJG.0b013e31818624ce
26. Kong X, Liu X, Huang X, Mao Z, Zhong Y, Chi W. Damage to the blood-aqueous barrier in eyes with primary angle closure glaucoma. Mol Vis. 2010;16:2026–2032.
27. Heimark RL, Kaochar S, Stamer WD. Human Schlemm’s canal cells express the endothelial adherens proteins, VE-cadherin and PECAM-1. Curr Eye Res. 2002;25(5):299–308. doi:10.1076/ceyr.25.5.299.13495
28. Acke FR, Malfait F, Vanakker OM, et al. Novel pathogenic COL11A1/COL11A2 variants in Stickler syndrome detected by targeted NGS and exome sequencing. Mol Genet Metab. 2014;113(3):230–235. doi:10.1016/j.ymgme.2014.09.001
29. Richards AJ, McNinch A, Martin H, et al. Stickler syndrome and the vitreous phenotype: mutations in COL2A1 and COL11A1. Hum Mutat. 2010;31(6):E1461–1471. doi:10.1002/humu.21257
30. Khalifa O, Imtiaz F, Allam R, et al. A recessive form of Marshall syndrome is caused by a mutation in the COL11A1 gene. J Med Genet. 2012;49(4):246–248. doi:10.1136/jmedgenet-2012-100783
31. Tengroth B, Rehnberg M, Amitzboll T. A comparative analysis of the collagen type and distribution in the trabecular meshwork, sclera, lamina cribrosa and the optic nerve in the human eye. Acta Ophthalmol Suppl. 1985;173:91–93. doi:10.1111/j.1755-3768.1985.tb06856.x
32. McBrien NA, Metlapally R, Jobling AI, Gentle A. Expression of collagen-binding integrin receptors in the mammalian sclera and their regulation during the development of myopia. Invest Ophthalmol Vis Sci. 2006;47(11):4674–4682. doi:10.1167/iovs.05-1150
33. Huang W, Fan Q, Wang W, Zhou M, Laties AM, Zhang X. Collagen: a potential factor involved in the pathogenesis of glaucoma. Med Sci Monit Basic Res. 2013;19:237–240. doi:10.12659/MSMBR.889061
34. Last JA, Pan T, Ding Y, et al. Elastic modulus determination of normal and glaucomatous human trabecular meshwork. Invest Ophthalmol Vis Sci. 2011;52(5):2147–2152. doi:10.1167/iovs.10-6342
35. Wang B, Hua Y, Brazile BL, Yang B, Sigal IA. Collagen fiber interweaving is central to sclera stiffness. Acta Biomater. 2020;113:429–437. doi:10.1016/j.actbio.2020.06.026
36. Behkam R, Kollech HG, Jana A, et al. Racioethnic differences in the biomechanical response of the lamina cribrosa. Acta Biomater. 2019;88:131–140. doi:10.1016/j.actbio.2019.02.028
37. Norman RE, Flanagan JG, Sigal IA, Rausch SM, Tertinegg I, Ethier CR. Finite element modeling of the human sclera: influence on optic nerve head biomechanics and connections with glaucoma. Exp Eye Res. 2011;93(1):4–12. doi:10.1016/j.exer.2010.09.014
38. Wallace DM, O’Brien CJ. The role of lamina cribrosa cells in optic nerve head fibrosis in glaucoma. Exp Eye Res. 2016;142:102–109. doi:10.1016/j.exer.2014.12.006
39. Ren R, Wang N, Li B, et al. Lamina cribrosa and peripapillary sclera histomorphometry in normal and advanced glaucomatous Chinese eyes with various axial length. Invest Ophthalmol Vis Sci. 2009;50(5):2175–2184. doi:10.1167/iovs.07-1429
40. Michael I, Shmoish M, Walton DS, Levenberg S. Interactions between trabecular meshwork cells and lens epithelial cells: a possible mechanism in infantile aphakic glaucoma. Invest Ophthalmol Vis Sci. 2008;49(9):3981–3987. doi:10.1167/iovs.08-1674
41. Jandrig B, Seitz S, Hinzmann B, et al. ST18 is a breast cancer tumor suppressor gene at human chromosome 8q11.2. Oncogene. 2004;23(57):9295–9302. doi:10.1038/sj.onc.1208131
42. Yang J, Siqueira MF, Behl Y, Alikhani M, Graves DT. The transcription factor ST18 regulates proapoptotic and proinflammatory gene expression in fibroblasts. FASEB J. 2008;22(11):3956–3967. doi:10.1096/fj.08-111013
43. Duvesh R, Verma A, Venkatesh R, et al. Association study in a South Indian population supports rs1015213 as a risk factor for primary angle closure. Invest Ophthalmol Vis Sci. 2013;54(8):5624–5628. doi:10.1167/iovs.13-12186
44. Nongpiur ME, Cheng CY, Duvesh R, et al. Evaluation of primary angle-closure glaucoma susceptibility loci in patients with early stages of angle-closure disease. Ophthalmology. 2018;125(5):664–670. doi:10.1016/j.ophtha.2017.11.016
45. Ma YQ, Qin J, Wu C, Plow EF. Kindlin-2 (Mig-2): a co-activator of beta3 integrins. J Cell Biol. 2008;181(3):439–446. doi:10.1083/jcb.200710196
46. Liu J, Fukuda K, Xu Z, et al. Structural basis of phosphoinositide binding to kindlin-2 protein pleckstrin homology domain in regulating integrin activation. J Biol Chem. 2011;286(50):43334–43342. doi:10.1074/jbc.M111.295352
47. Shen Z, Ye Y, Kauttu T, et al. Novel focal adhesion protein kindlin-2 promotes the invasion of gastric cancer cells through phosphorylation of integrin beta1 and beta3. J Surg Oncol. 2013;108(2):106–112. doi:10.1002/jso.23353
48. Tu Y, Wu S, Shi X, Chen K, Wu C. Migfilin and Mig-2 link focal adhesions to filamin and the actin cytoskeleton and function in cell shape modulation. Cell. 2003;113(1):37–47. doi:10.1016/S0092-8674(03)00163-6
49. Yu Y, Wu J, Wang Y, et al. Kindlin 2 forms a transcriptional complex with beta-catenin and TCF4 to enhance Wnt signalling. EMBO Rep. 2012;13(8):750–758. doi:10.1038/embor.2012.88
50. Tian B, Geiger B, Epstein DL, Kaufman PL. Cytoskeletal involvement in the regulation of aqueous humor outflow. Invest Ophthalmol Vis Sci. 2000;41(3):619–623.
51. Bermudez JY, Montecchi-Palmer M, Mao W, Clark AF. Cross-linked actin networks (CLANs) in glaucoma. Exp Eye Res. 2017;159:16–22. doi:10.1016/j.exer.2017.02.010
52. Dhamodaran K, Baidouri H, Sandoval L, Raghunathan V. Wnt activation after inhibition restores trabecular meshwork cells toward a normal phenotype. Invest Ophthalmol Vis Sci. 2020;61(6):30. doi:10.1167/iovs.61.6.30
53. Apostolopoulos J, Sparrow RL, McLeod JL, et al. Identification and characterization of a novel family of mammalian ependymin-related proteins (MERPs) in hematopoietic, nonhematopoietic, and malignant tissues. DNA Cell Biol. 2001;20(10):625–635. doi:10.1089/104454901753340613
54. Wei Y, Xiong ZJ, Li J, et al. Crystal structures of human lysosomal EPDR1 reveal homology with the superfamily of bacterial lipoprotein transporters. Commun Biol. 2019;2:52. doi:10.1038/s42003-018-0262-9
55. Ng M, Thakkar D, Southam L, et al. A genome-wide association study of dupuytren disease reveals 17 additional variants implicated in fibrosis. Am J Hum Genet. 2017;101(3):417–427. doi:10.1016/j.ajhg.2017.08.006
56. Petty HR. Frontiers of complex disease mechanisms: membrane surface tension may link genotype to phenotype in glaucoma. Front Cell Dev Biol. 2018;6:32. doi:10.3389/fcell.2018.00032
57. Kim YS, Nakanishi G, Lewandoski M, Jetten AM. GLIS3, a novel member of the GLIS subfamily of Kruppel-like zinc finger proteins with repressor and activation functions. Nucleic Acids Res. 2003;31(19):5513–5525. doi:10.1093/nar/gkg776
58. Lichti-Kaiser K, ZeRuth G, Jetten AM. Transcription factor Gli-Similar 3 (Glis3): implications for the development of congenital hypothyroidism. J Endocrinol Diabetes Obes. 2014;2(2):1024.
59. Jeon K, Kumar D, Conway AE, Park K, Jothi R, Jetten AM. GLIS3 transcriptionally activates WNT genes to promote differentiation of human embryonic stem cells into posterior neural progenitors. Stem Cells. 2019;37(2):202–215. doi:10.1002/stem.2941
60. Wen X, Yang Y. Emerging roles of GLIS3 in neonatal diabetes, type 1 and type 2 diabetes. J Mol Endocrinol. 2017;58(2):R73–R85. doi:10.1530/JME-16-0232
61. Rurale G, Persani L, Marelli F. GLIS3 and thyroid: a pleiotropic candidate gene for congenital hypothyroidism. Front Endocrinol (Lausanne). 2018;9:730. doi:10.3389/fendo.2018.00730
62. Chou CK, Tang CJ, Chou HL, et al. The potential role of kruppel-like zinc-finger protein Glis3 in genetic diseases and cancers. Arch Immunol Ther Exp (Warsz). 2017;65(5):381–389. doi:10.1007/s00005-017-0470-x
63. Calderari S, Ria M, Gerard C, et al. Molecular genetics of the transcription factor GLIS3 identifies its dual function in beta cells and neurons. Genomics. 2018;110(2):98–111. doi:10.1016/j.ygeno.2017.09.001
64. Nogueira TC, Paula FM, Villate O, et al. GLIS3, a susceptibility gene for type 1 and type 2 diabetes, modulates pancreatic beta cell apoptosis via regulation of a splice variant of the BH3-only protein Bim. PLoS Genet. 2013;9(5):e1003532. doi:10.1371/journal.pgen.1003532
65. Nakamura M, Kanamori A, Negi A. Diabetes mellitus as a risk factor for glaucomatous optic neuropathy. Ophthalmologica. 2005;219(1):1–10. doi:10.1159/000081775
66. Zhao D, Cho J, Kim MH, Friedman DS, Guallar E. Diabetes, fasting glucose, and the risk of glaucoma: a meta-analysis. Ophthalmology. 2015;122(1):72–78. doi:10.1016/j.ophtha.2014.07.051
67. Barone R, Aiello C, Race V, et al. DPM2-CDG: a muscular dystrophy-dystroglycanopathy syndrome with severe epilepsy. Ann Neurol. 2012;72(4):550–558. doi:10.1002/ana.23632
68. Sienkiewicz AE, Rosenberg BN, Edwards G, Carreon TA, Bhattacharya SK. Aberrant glycosylation in the human trabecular meshwork. Proteomics Clin Appl. 2014;8(3–4):130–142. doi:10.1002/prca.201300031
69. Wang DY, Fulthorpe R, Liss SN, Edwards EA. Identification of estrogen-responsive genes by complementary deoxyribonucleic acid microarray and characterization of a novel early estrogen-induced gene: EEIG1. Mol Endocrinol. 2004;18(2):402–411. doi:10.1210/me.2003-0202
70. Ulhaq ZS. The association of estrogen-signaling pathways and susceptibility to open-angle glaucoma. Beni-Suef Univ J Basic Appl Sci. 2020;9(1):7. doi:10.1186/s43088-020-0034-8
71. Choi HK, Kang HR, Jung E, Kim TE, Lin JJ, Lee SY. Early estrogen-induced gene 1, a novel RANK signaling component, is essential for osteoclastogenesis. Cell Res. 2013;23(4):524–536. doi:10.1038/cr.2013.33
72. Shi L, Zhao M, Luo Q, et al. Overexpression of PIP5KL1 suppresses cell proliferation and migration in human gastric cancer cells. Mol Biol Rep. 2010;37(5):2189–2198. doi:10.1007/s11033-009-9701-5
73. Lachkar Y, Bouassida W. Drug-induced acute angle closure glaucoma. Curr Opin Ophthalmol. 2007;18(2):129–133. doi:10.1097/ICU.0b013e32808738d5
74. Mandak JS, Minerva P, Wilson TW, Smith EK. Angle closure glaucoma complicating systemic atropine use in the cardiac catheterization laboratory. Cathet Cardiovasc Diagn. 1996;39(3):262–264. doi:10.1002/(SICI)1097-0304(199611)39:3<262::AID-CCD11>3.0.CO;2-H
75. Eckenstein F, Thoenen H. Production of specific antisera and monoclonal antibodies to choline acetyltransferase: characterization and use for identification of cholinergic neurons. EMBO J. 1982;1(3):363–368. doi:10.1002/j.1460-2075.1982.tb01175.x
76. Voigt T. Cholinergic amacrine cells in the rat retina. J Comp Neurol. 1986;248(1):19–35. doi:10.1002/cne.902480103
77. Tooyama I, Kimura H. A protein encoded by an alternative splice variant of choline acetyltransferase mRNA is localized preferentially in peripheral nerve cells and fibers. J Chem Neuroanat. 2000;17(4):217–226. doi:10.1016/S0891-0618(99)00043-5
78. Yasuhara O, Tooyama I, Aimi Y, et al. Demonstration of cholinergic ganglion cells in rat retina: expression of an alternative splice variant of choline acetyltransferase. J Neurosci. 2003;23(7):2872–2881. doi:10.1523/JNEUROSCI.23-07-02872.2003
79. Koistinaho J, Sagar SM. Light-induced c-fos expression in amacrine cells in the rabbit retina. Brain Res Mol Brain Res. 1995;29(1):53–63. doi:10.1016/0169-328X(94)00218-4
80. McCorry LK. Physiology of the autonomic nervous system. Am J Pharm Educ. 2007;71(4):78. doi:10.5688/aj710478
81. McDougal DH, Gamlin PD. Autonomic control of the eye. Compr Physiol. 2015;5(1):439–473.
82. Neuhuber W, Schrodl F. Autonomic control of the eye and the iris. Auton Neurosci. 2011;165(1):67–79.
83. Faiq MA, Wollstein G, Schuman JS, Chan KC. Cholinergic nervous system and glaucoma: from basic science to clinical applications. Prog Retin Eye Res. 2019;72:100767.
84. Shi H, Zhu R, Hu N, et al. An extensive replication study on three new susceptibility Loci of primary angle closure glaucoma in han chinese: Jiangsu eye study. J Ophthalmol. 2013;2013:641596. doi:10.1155/2013/641596
85. Rong SS, Tang FY, Chu WK, et al. Genetic associations of primary angle-closure disease: a systematic review and meta-analysis. Ophthalmology. 2016;123(6):1211–1221. doi:10.1016/j.ophtha.2015.12.027
86. Wan Y, Li S, Gao Y, Tang L, Cao W, Sun X. COL11A1 polymorphisms are associated with primary angle-closure glaucoma severity. J Ophthalmol. 2019;2019:2604386. doi:10.1155/2019/2604386
87. Awadalla MS, Thapa SS, Hewitt AW, Burdon KP, Craig JE. Association of genetic variants with primary angle closure glaucoma in two different populations. PLoS One. 2013;8(6):e67903. doi:10.1371/journal.pone.0067903
88. Shuai P, Yu M, Li X, et al. Genetic associations in PLEKHA7 and COL11A1 with primary angle closure glaucoma: a meta-analysis. Clin Exp Ophthalmol. 2015;43(6):523–530. doi:10.1111/ceo.12516
89. Yousefian A, Shokoohi-Rad S, Abbaszadegan MR, et al. Primary angle closure glaucoma-associated genetic polymorphisms in Northeast Iran. J Ophthalmic Vis Res. 2020;15(1):45–52. doi:10.18502/jovr.v15i1.5942
90. Wong TT, Sethi C, Daniels JT, Limb GA, Murphy G, Khaw PT. Matrix metalloproteinases in disease and repair processes in the anterior segment. Surv Ophthalmol. 2002;47(3):239–256.
91. De Groef L, Andries L, Siwakoti A, et al. Aberrant collagen composition of the trabecular meshwork results in reduced aqueous humor drainage and elevated IOP in MMP-9 null mice. Invest Ophthalmol Vis Sci. 2016;57(14):5984–5995. doi:10.1167/iovs.16-19734
92. St Jean PL, Zhang XC, Hart BK, et al. Characterization of a dinucleotide repeat in the 92 kDa type IV collagenase gene (CLG4B), localization of CLG4B to chromosome 20 and the role of CLG4B in aortic aneurysmal disease. Ann Hum Genet. 1995;59(1):17–24. doi:10.1111/j.1469-1809.1995.tb01602.x
93. Wang IJ, Chiang TH, Shih YF, et al. The association of single nucleotide polymorphisms in the MMP-9 genes with susceptibility to acute primary angle closure glaucoma in Taiwanese patients. Mol Vis. 2006;12:1223–1232.
94. Shi H, Zhu R, Hu N, et al. Association of frizzled-related protein (MFRP) and heat shock protein 70 (HSP70) single nucleotide polymorphisms with primary angle closure in a Han Chinese population: Jiangsu eye study. Mol Vis. 2013;19:128–134.
95. Aung T, Yong VH, Lim MC, et al. Lack of association between the rs2664538 polymorphism in the MMP-9 gene and primary angle closure glaucoma in Singaporean subjects. J Glaucoma. 2008;17(4):257–258. doi:10.1097/IJG.0b013e31815c3aa5
96. Awadalla MS, Burdon KP, Kuot A, Hewitt AW, Craig JE. Matrix metalloproteinase-9 genetic variation and primary angle closure glaucoma in a Caucasian population. Mol Vis. 2011;17:1420–1424.
97. Thakur N, Kupani M, Pandey RK, Mannan R, Pruthi A, Mehrotra S. Genetic association of −1562C>T polymorphism in the MMP9 gene with primary glaucoma in a north Indian population. PLoS One. 2018;13(2):e0192636. doi:10.1371/journal.pone.0192636
98. Micheal S, Yousaf S, Khan MI, et al. Polymorphisms in matrix metalloproteinases MMP1 and MMP9 are associated with primary open-angle and angle closure glaucoma in a Pakistani population. Mol Vis. 2013;19:441–447.
99. Chen X, Chen Y, Wiggs JL, Pasquale LR, Sun X, Fan BJ. Association of matrix metalloproteinase-9 (MMP9) variants with primary angle closure and primary angle closure glaucoma. PLoS One. 2016;11(6):e0157093. doi:10.1371/journal.pone.0157093
100. Gao XJ, Hou SP, Li PH. The association between matrix metalloprotease-9 gene polymorphisms and primary angle-closure glaucoma in a Chinese Han population. Int J Ophthalmol. 2014;7(3):397–402. doi:10.3980/j.issn.2222-3959.2014.03.02
101. Cong Y, Guo X, Liu X, et al. Association of the single nucleotide polymorphisms in the extracellular matrix metalloprotease-9 gene with PACG in southern China. Mol Vis. 2009;15:1412–1417.
102. Harper AR, Summers JA. The dynamic sclera: extracellular matrix remodeling in normal ocular growth and myopia development. Exp Eye Res. 2015;133:100–111. doi:10.1016/j.exer.2014.07.015
103. Aliancy J, Stamer WD, Wirostko BA. Review of nitric oxide for the treatment of glaucomatous disease. Ophthalmol Ther. 2017;6(2):221–232. doi:10.1007/s40123-017-0094-6
104. Chang JY, Stamer WD, Bertrand J, et al. Role of nitric oxide in murine conventional outflow physiology. Am J Physiol Cell Physiol. 2015;309(4):C205–214. doi:10.1152/ajpcell.00347.2014
105. Toda N, Nakanishi-Toda M. Nitric oxide: ocular blood flow, glaucoma, and diabetic retinopathy. Prog Retin Eye Res. 2007;26(3):205–238. doi:10.1016/j.preteyeres.2007.01.004
106. Wink DA, Miranda KM, Espey MG, et al. Mechanisms of the antioxidant effects of nitric oxide. Antioxid Redox Signal. 2001;3(2):203–213. doi:10.1089/152308601300185179
107. Manabe S, Lipton SA. Divergent NMDA signals leading to proapoptotic and antiapoptotic pathways in the rat retina. Invest Ophthalmol Vis Sci. 2003;44(1):385–392. doi:10.1167/iovs.02-0187
108. Ayub H, Khan MI, Micheal S, et al. Association of eNOS and HSP70 gene polymorphisms with glaucoma in Pakistani cohorts. Mol Vis. 2010;16:18–25.
109. Awadalla MS, Thapa SS, Hewitt AW, Craig JE, Burdon KP. Association of eNOS polymorphisms with primary angle-closure glaucoma. Invest Ophthalmol Vis Sci. 2013;54(3):2108–2114. doi:10.1167/iovs.12-11391
110. Shi H, Zhu R, Hu N, et al. Association of eNOS polymorphisms with anterior chamber depth in han chinese: Jiangsu eye study. J Ophthalmol. 2014;2014:164104. doi:10.1155/2014/164104
111. Liao Q, Wang DH, Sun HJ. Association of genetic polymorphisms of eNOS with glaucoma. Mol Vis. 2011;17:153–158.
112. Piri N, Kwong JM, Gu L, Caprioli J. Heat shock proteins in the retina: focus on HSP70 and alpha crystallins in ganglion cell survival. Prog Retin Eye Res. 2016;52:22–46.
113. He M, Guo H, Yang X, et al. Functional SNPs in HSPA1A gene predict risk of coronary heart disease. PLoS One. 2009;4(3):e4851. doi:10.1371/journal.pone.0004851
114. Lee KJ, Kim YM, Kim DY, et al. Release of heat shock protein 70 (Hsp70) and the effects of extracellular Hsp70 on matric metalloproteinase-9 expression in human monocytic U937 cells. Exp Mol Med. 2006;38(4):364–374. doi:10.1038/emm.2006.43
115. Zhang Y, Xia M, Jin K, et al. Function of the c-Met receptor tyrosine kinase in carcinogenesis and associated therapeutic opportunities. Mol Cancer. 2018;17(1):45. doi:10.1186/s12943-018-0796-y
116. Previdi S, Maroni P, Matteucci E, Broggini M, Bendinelli P, Desiderio MA. Interaction between human-breast cancer metastasis and bone microenvironment through activated hepatocyte growth factor/Met and beta-catenin/Wnt pathways. Eur J Cancer. 2010;46(9):1679–1691. doi:10.1016/j.ejca.2010.02.036
117. Webber HC, Bermudez JY, Millar JC, Mao W, Clark AF. The role of Wnt/beta-catenin signaling and K-cadherin in the regulation of intraocular pressure. Invest Ophthalmol Vis Sci. 2018;59(3):1454–1466. doi:10.1167/iovs.17-21964
118. Wordinger RJ, Clark AF, Agarwal R, et al. Cultured human trabecular meshwork cells express functional growth factor receptors. Invest Ophthalmol Vis Sci. 1998;39(9):1575–1589.
119. Hu DN, Ritch R. Hepatocyte growth factor is increased in the aqueous humor of glaucomatous eyes. J Glaucoma. 2001;10(3):152–157. doi:10.1097/00061198-200106000-00002
120. Jin M, Chen Y, He S, Ryan SJ, Hinton DR. Hepatocyte growth factor and its role in the pathogenesis of retinal detachment. Invest Ophthalmol Vis Sci. 2004;45(1):323–329. doi:10.1167/iovs.03-0355
121. Tonges L, Ostendorf T, Lamballe F, et al. Hepatocyte growth factor protects retinal ganglion cells by increasing neuronal survival and axonal regeneration in vitro and in vivo. J Neurochem. 2011;117(5):892–903. doi:10.1111/j.1471-4159.2011.07257.x
122. Wong WK, Cheung AW, Yu SW, Sha O, Cho EY. Hepatocyte growth factor promotes long-term survival and axonal regeneration of retinal ganglion cells after optic nerve injury: comparison with CNTF and BDNF. CNS Neurosci Ther. 2014;20(10):916–929. doi:10.1111/cns.12304
123. Veerappan S, Pertile KK, Islam AF, et al. Role of the hepatocyte growth factor gene in refractive error. Ophthalmology. 2010;117(2):
124. Awadalla MS, Thapa SS, Burdon KP, Hewitt AW, Craig JE. The association of hepatocyte growth factor (HGF) gene with primary angle closure glaucoma in the Nepalese population. Mol Vis. 2011;17:2248–2254.
125. Jiang Z, Liang K, Ding B, et al. Hepatocyte growth factor genetic variations and primary angle-closure glaucoma in the Han Chinese population. PLoS One. 2013;8(4):e60950. doi:10.1371/journal.pone.0060950
126. Ayala-Ramirez R, Graue-Wiechers F, Robredo V, Amato-Almanza M, Horta-Diez I, Zenteno JC. A new autosomal recessive syndrome consisting of posterior microphthalmos, retinitis pigmentosa, foveoschisis, and optic disc drusen is caused by a MFRP gene mutation. Mol Vis. 2006;12:1483–1489.
127. Sundin OH, Leppert GS, Silva ED, et al. Extreme hyperopia is the result of null mutations in MFRP, which encodes a Frizzled-related protein. Proc Natl Acad Sci U S A. 2005;102(27):9553–9558. doi:10.1073/pnas.0501451102
128. Wang P, Yang Z, Li S, Xiao X, Guo X, Zhang Q. Evaluation of MFRP as a candidate gene for high hyperopia. Mol Vis. 2009;15:181–186.
129. Wasmann RA, Wassink-Ruiter JS, Sundin OH, Morales E, Verheij JB, Pott JW. Novel membrane frizzled-related protein gene mutation as cause of posterior microphthalmia resulting in high hyperopia with macular folds. Acta Ophthalmol. 2014;92(3):276–281. doi:10.1111/aos.12105
130. Sundin OH, Dharmaraj S, Bhutto IA, et al. Developmental basis of nanophthalmos: MFRP Is required for both prenatal ocular growth and postnatal emmetropization. Ophthalmic Genet. 2008;29(1):1–9. doi:10.1080/13816810701651241
131. Wang IJ, Lin S, Chiang TH, et al. The association of membrane frizzled-related protein (MFRP) gene with acute angle-closure glaucoma–a pilot study. Mol Vis. 2008;14:1673–1679.
132. Aung T, Lim MC, Wong TT, et al. Molecular analysis of CHX10 and MFRP in Chinese subjects with primary angle closure glaucoma and short axial length eyes. Mol Vis. 2008;14:1313–1318.
133. Awadalla MS, Burdon KP, Thapa SS, Hewitt AW, Craig JE. A cross-ethnicity investigation of genes previously implicated in primary angle closure glaucoma. Mol Vis. 2012;18:2247–2254.
134. Mandal MN, Vasireddy V, Jablonski MM, et al. Spatial and temporal expression of MFRP and its interaction with CTRP5. Invest Ophthalmol Vis Sci. 2006;47(12):5514–5521. doi:10.1167/iovs.06-0449
135. Katoh M. Molecular cloning and characterization of MFRP, a novel gene encoding a membrane-type Frizzled-related protein. Biochem Biophys Res Commun. 2001;282(1):116–123. doi:10.1006/bbrc.2001.4551
136. Soundararajan R, Won J, Stearns TM, et al. Gene profiling of postnatal Mfrprd6 mutant eyes reveals differential accumulation of Prss56, visual cycle and phototransduction mRNAs. PLoS One. 2014;9(10):e110299. doi:10.1371/journal.pone.0110299
137. Iseri SU, Wyatt AW, Nurnberg G, et al. Use of genome-wide SNP homozygosity mapping in small pedigrees to identify new mutations in VSX2 causing recessive microphthalmia and a semidominant inner retinal dystrophy. Hum Genet. 2010;128(1):51–60. doi:10.1007/s00439-010-0823-6
138. Reis LM, Khan A, Kariminejad A, Ebadi F, Tyler RC, Semina EV. VSX2 mutations in autosomal recessive microphthalmia. Mol Vis. 2011;17:2527–2532.
139. Awadalla MS, Burdon KP, Souzeau E, et al. Mutation in TMEM98 in a large white kindred with autosomal dominant nanophthalmos linked to 17p12-q12. JAMA Ophthalmol. 2014;132(8):970–977. doi:10.1001/jamaophthalmol.2014.946
140. Wagner AH, Anand VN, Wang WH, et al. Exon-level expression profiling of ocular tissues. Exp Eye Res. 2013;111:105–111. doi:10.1016/j.exer.2013.03.004
141. Micheal S, Qamar R, Akhtar F, Khan MI, Khan WA, Ahmed A. MTHFR gene C677T and A1298C polymorphisms and homocysteine levels in primary open angle and primary closed angle glaucoma. Mol Vis. 2009;15:2268–2278.
142. Gupta S, Bhaskar PK, Bhardwaj R, et al. MTHFR C677T predisposes to POAG but not to PACG in a North Indian population: a case control study. PLoS One. 2014;9(7):e103063. doi:10.1371/journal.pone.0103063
143. Passan S, Goyal S, Bhat MA, Singh D, Vanita V. Association of TNF-alpha gene alterations (c.-238G>A, c.-308G>A, c.-857C>T, c.-863C>A) with primary glaucoma in north Indian cohort. Gene. 2019;709:25–35. doi:10.1016/j.gene.2019.05.035
144. Karasinska JM, de Haan W, Franciosi S, et al. ABCA1 influences neuroinflammation and neuronal death. Neurobiol Dis. 2013;54:445–455. doi:10.1016/j.nbd.2013.01.018
145. Luo H, Chen Y, Ye Z, et al. Evaluation of the association between common genetic variants near the ABCA1 gene and primary angle closure glaucoma in a Han Chinese population. Invest Ophthalmol Vis Sci. 2015;56(11):6248–6254. doi:10.1167/iovs.15-16741
146. Alkhatib R, Abudhaim N, Al-Eitan L, Abdo N, Alqudah A, Aman H. Genetic analysis of ABCA1 gene of primary glaucoma in Jordanian Arab population. Appl Clin Genet. 2019;12:181–189. doi:10.2147/TACG.S213818
147. Wang SL, Piao SY, Xu MY, et al. Evaluating correlation between the ocular biometry and genetic variants of MYOC and ABCA1 with primary angle-closure glaucoma in a cohort from northern China. Int J Ophthalmol. 2019;12(8):1317–1322. doi:10.18240/ijo.2019.08.13
148. Abu-Amero KK, Morales J, Osman MN, Bosley TM. Nuclear and mitochondrial analysis of patients with primary angle-closure glaucoma. Invest Ophthalmol Vis Sci. 2007;48(12):5591–5596. doi:10.1167/iovs.07-0780
149. Dai X, Nie S, Ke T, Liu J, Wang Q, Liu M. [Two variants in MYOC and CYP1B1 genes in a Chinese family with primary angle-closure glaucoma]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2008;25(5):493–496. Chinese.
150. Aung T, Yong VH, Chew PT, et al. Molecular analysis of the myocilin gene in Chinese subjects with chronic primary-angle closure glaucoma. Invest Ophthalmol Vis Sci. 2005;46(4):1303–1306. doi:10.1167/iovs.04-1163
151. Faucher M, Anctil JL, Rodrigue MA, et al. Founder TIGR/myocilin mutations for glaucoma in the Quebec population. Hum Mol Genet. 2002;11(18):2077–2090. doi:10.1093/hmg/11.18.2077
152. Jin X, Wang D-J, Qu L-H, Hou B-K, Gong Y, Xu -W-W. Haplotype analysis of association of the MYOC gene with primary angle-closure glaucoma in a Han Chinese population. Genet Test Mol Biomarkers. 2015;19(1):3–8. doi:10.1089/gtmb.2014.0130
153. Chakrabarti S, Devi KR, Komatireddy S, et al. Glaucoma-associated CYP1B1 mutations share similar haplotype backgrounds in POAG and PACG phenotypes. Invest Ophthalmol Vis Sci. 2007;48(12):5439–5444. doi:10.1167/iovs.07-0629
154. Yazdani S, Elahi E, Safari I, Akbarian S. A possible role for LTBP2 in the etiology of primary angle closure glaucoma. J Ophthalmic Vis Res. 2015;10(2):123–129. doi:10.4103/2008-322X.163783
155. Abu-Amero KK, Azad TA, Mousa A, Osman EA, Sultan T, Al-Obeidan SA. Association of SOD2 mutation (c.47T > C) with various primary angle closure glaucoma clinical indices. Ophthalmic Genet. 2015;36(2):180–183. doi:10.3109/13816810.2013.838276
156. Abu-Amero KK, Azad TA, Mousa A, Osman EA, Sultan T, Al-Obeidan SA. A catalase promoter variant rs1001179 is associated with visual acuity but not with primary angle closure glaucoma in Saudi patients. BMC Med Genet. 2013;14:84. doi:10.1186/1471-2350-14-84
157. Abu-Amero KK, Morales J, Mohamed GH, Osman MN, Bosley TM. Glutathione S-transferase M1 and T1 polymorphisms in Arab glaucoma patients. Mol Vis. 2008;14:425–430.
158. Abu-Amero KK, Gonzalez AM, Osman EA, Larruga JM, Cabrera VM, Al-Obeidan SA. Susceptibility to primary angle closure glaucoma in Saudi Arabia: the possible role of mitochondrial DNA ancestry informative haplogroups. Mol Vis. 2011;17:2171–2176.
159. Al-Dabbagh N, Al-Shahrani H, Al-Dohayan N, Mustafa M, Arfin M, Al-Asmari AK. The SPARC-related modular calcium binding protein 2 (SMOC2) gene polymorphism in primary glaucoma: a case-control study. Clin Ophthalmol. 2017;11:549–555. doi:10.2147/OPTH.S126459
160. Chua J, Seet LF, Jiang Y, et al. Increased SPARC expression in primary angle closure glaucoma iris. Mol Vis. 2008;14:1886–1892.
161. Kondkar AA, Sultan T, Azad TA, Osman EA, Almobarak FA, Al-Obeidan SA. Association analysis of polymorphisms rs12997 in ACVR1 and rs1043784 in BMP6 genes involved in bone morphogenic protein signaling pathway in primary angle-closure and pseudoexfoliation glaucoma patients of Saudi origin. BMC Med Genet. 2020;21(1):145. doi:10.1186/s12881-020-01076-0
162. Verma SS, Cooke Bailey JN, Lucas A, et al. Epistatic gene-based interaction analyses for glaucoma in eMERGE and NEIGHBOR Consortium. PLoS Genet. 2016;12(9):e1006186. doi:10.1371/journal.pgen.1006186
163. Mansoori T, Balakrishna N. Anterior segment morphology in primary angle closure glaucoma using ultrasound biomicroscopy. J Curr Glaucoma Pract. 2017;11(3):86–91.
164. Nongpiur ME, Khor CC, Jia H, et al. ABCC5, a gene that influences the anterior chamber depth, is associated with primary angle closure glaucoma. PLoS Genet. 2014;10(3):e1004089. doi:10.1371/journal.pgen.1004089
165. Tang FY, Ma L, Tam POS, Pang CP, Tham CC, Chen LJ. Genetic association of the PARL-ABCC5-HTR3D-HTR3C locus with primary angle-closure glaucoma in Chinese. Invest Ophthalmol Vis Sci. 2017;58(10):4384–4389. doi:10.1167/iovs.17-22304
166. Wang S, Zhuang W, Zhang W, et al. The association of single nucleotide polymorphisms in ABCC5 gene with primary angle closure glaucoma and the ocular biometric parameters in a Northern Chinese Population. Ophthalmic Res. 2020. doi:10.1159/000511454
167. Jedlitschky G, Burchell B, Keppler D. The multidrug resistance protein 5 functions as an ATP-dependent export pump for cyclic nucleotides. J Biol Chem. 2000;275(39):30069–30074. doi:10.1074/jbc.M005463200
168. Wielinga P, Hooijberg JH, Gunnarsdottir S, et al. The human multidrug resistance protein MRP5 transports folates and can mediate cellular resistance against antifolates. Cancer Res. 2005;65(10):4425–4430. doi:10.1158/0008-5472.CAN-04-2810
169. Robey RW, Pluchino KM, Hall MD, Fojo AT, Bates SE, Gottesman MM. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat Rev Cancer. 2018;18(7):452–464. doi:10.1038/s41568-018-0005-8
170. Jansen RS, Mahakena S, de Haas M, Borst P, van de Wetering K. ATP-binding cassette subfamily C member 5 (ABCC5) functions as an efflux transporter of glutamate conjugates and analogs. J Biol Chem. 2015;290(51):30429–30440. doi:10.1074/jbc.M115.692103
171. Karla PK, Quinn TL, Herndon BL, Thomas P, Pal D, Mitra A. Expression of multidrug resistance associated protein 5 (MRP5) on cornea and its role in drug efflux. J Ocul Pharmacol Ther. 2009;25(2):121–132. doi:10.1089/jop.2008.0084
172. Long Y, Li Q, Li J, Cui Z. Molecular analysis, developmental function and heavy metal-induced expression of ABCC5 in zebrafish. Comp Biochem Physiol B Biochem Mol Biol. 2011;158(1):46–55. doi:10.1016/j.cbpb.2010.09.005
173. Fang F, Pan M, Yan T, et al. The role of cGMP in ocular growth and the development of form-deprivation myopia in guinea pigs. Invest Ophthalmol Vis Sci. 2013;54(13):7887–7902. doi:10.1167/iovs.13-11999
174. Direk K, Lau W, Small KS, Maniatis N, Andrew T. ABCC5 transporter is a novel type 2 diabetes susceptibility gene in European and African American populations. Ann Hum Genet. 2014;78(5):333–344. doi:10.1111/ahg.12072
175. Cyranka M, Veprik A, McKay EJ, et al. Abcc5 knockout mice have lower fat mass and increased levels of circulating GLP-1. Obesity (Silver Spring). 2019;27(8):1292–1304. doi:10.1002/oby.22521
176. Hernandez C, Bogdanov P, Corraliza L, et al. Topical administration of GLP-1 receptor agonists prevents retinal neurodegeneration in experimental diabetes. Diabetes. 2016;65(1):172–187. doi:10.2337/db15-0443
177. Zhang R, Zhang H, Xu L, Ma K, Wallrapp C, Jonas JB. Neuroprotective effect of intravitreal cell-based glucagon-like peptide-1 production in the optic nerve crush model. Acta Ophthalmol. 2011;89(4):e320–326. doi:10.1111/j.1755-3768.2010.02044.x
178. Day AC, Luben R, Khawaja AP, et al. Genotype-phenotype analysis of SNPs associated with primary angle closure glaucoma (rs1015213, rs3753841 and rs11024102) and ocular biometry in the EPIC-Norfolk Eye Study. Br J Ophthalmol. 2013;97(6):704–707. doi:10.1136/bjophthalmol-2012-302969
179. Nongpiur ME, Wei X, Xu L, et al. Lack of association between primary angle-closure glaucoma susceptibility loci and the ocular biometric parameters anterior chamber depth and axial length. Invest Ophthalmol Vis Sci. 2013;54(8):5824–5828. doi:10.1167/iovs.13-11901
180. Khawaja AP, Cooke Bailey JN, Wareham NJ, et al. Genome-wide analyses identify 68 new loci associated with intraocular pressure and improve risk prediction for primary open-angle glaucoma. Nat Genet. 2018;50(6):778–782. doi:10.1038/s41588-018-0126-8
181. MacGregor S, Ong JS, An J, et al. Genome-wide association study of intraocular pressure uncovers new pathways to glaucoma. Nat Genet. 2018;50(8):1067–1071. doi:10.1038/s41588-018-0176-y
182. Wei X, Nongpiur ME, de Leon MS, et al. Genotype-phenotype correlation analysis for three primary angle closure glaucoma-associated genetic polymorphisms. Invest Ophthalmol Vis Sci. 2014;55(2):1143–1148. doi:10.1167/iovs.13-13552
183. Zhuang W, Wang S, Hao J, et al. Genotype-ocular biometry correlation analysis of eight primary angle closure glaucoma susceptibility loci in a cohort from Northern China. PLoS One. 2018;13(11):e0206935. doi:10.1371/journal.pone.0206935
184. Liu C, Nongpiur ME, Cheng CY, et al. Evaluation of primary angle-closure glaucoma susceptibility loci for estimating angle closure disease severity. Ophthalmology. 2020.
185. Cheng CY, Schache M, Ikram MK, et al. Nine loci for ocular axial length identified through genome-wide association studies, including shared loci with refractive error. Am J Hum Genet. 2013;93(2):264–277. doi:10.1016/j.ajhg.2013.06.016
186. Suri F, Yazdani S, Chapi M, et al. COL18A1 is a candidate eye iridocorneal angle-closure gene in humans. Hum Mol Genet. 2018;27(21):3772–3786. doi:10.1093/hmg/ddy256
187. Sertie AL, Quimby M, Moreira ES, et al. A gene which causes severe ocular alterations and occipital encephalocele (Knobloch syndrome) is mapped to 21q22.3. Hum Mol Genet. 1996;5(6):843–847. doi:10.1093/hmg/5.6.843
188. Menzel O, Bekkeheien RC, Reymond A, et al. Knobloch syndrome: novel mutations in COL18A1, evidence for genetic heterogeneity, and a functionally impaired polymorphism in endostatin. Hum Mutat. 2004;23(1):77–84. doi:10.1002/humu.10284
189. Keren B, Suzuki OT, Gerard-Blanluet M, et al. CNS malformations in Knobloch syndrome with splice mutation in COL18A1 gene. Am J Med Genet A. 2007;143A(13):1514–1518. doi:10.1002/ajmg.a.31784
190. Paisan-Ruiz C, Scopes G, Lee P, Houlden H. Homozygosity mapping through whole genome analysis identifies a COL18A1 mutation in an Indian family presenting with an autosomal recessive neurological disorder. Am J Med Genet B Neuropsychiatr Genet. 2009;150B(7):993–997. doi:10.1002/ajmg.b.30929
191. Khaliq S, Abid A, White DR, et al. Mapping of a novel type III variant of Knobloch syndrome (KNO3) to chromosome 17q11.2. Am J Med Genet A. 2007;143A(23):2768–2774. doi:10.1002/ajmg.a.31739
192. Joyce S, Tee L, Abid A, Khaliq S, Mehdi SQ, Maher ER. Locus heterogeneity and Knobloch syndrome. Am J Med Genet A. 2010;152A(11):2880–2881. doi:10.1002/ajmg.a.33619
193. Vithana EN, Aung T, Khor CC, et al. Collagen-related genes influence the glaucoma risk factor, central corneal thickness. Hum Mol Genet. 2011;20(4):649–658. doi:10.1093/hmg/ddq511
194. Elamaa H, Snellman A, Rehn M, Autio-Harmainen H, Pihlajaniemi T. Characterization of the human type XVIII collagen gene and proteolytic processing and tissue location of the variant containing a frizzled motif. Matrix Biol. 2003;22(5):427–442. doi:10.1016/S0945-053X(03)00073-8
195. Fuhrmann S. Wnt signaling in eye organogenesis. Organogenesis. 2008;4(2):60–67. doi:10.4161/org.4.2.5850
196. Waseem NH, Low S, Shah AZ, et al. Mutations in SPATA13/ASEF2 cause primary angle closure glaucoma. PLoS Genet. 2020;16(4):e1008721. doi:10.1371/journal.pgen.1008721
197. Bristow JM, Sellers MH, Majumdar D, Anderson B, Hu L, Webb DJ. The Rho-family GEF Asef2 activates Rac to modulate adhesion and actin dynamics and thereby regulate cell migration. J Cell Sci. 2009;122(Pt 24):4535–4546. doi:10.1242/jcs.053728
198. Sagara M, Kawasaki Y, Iemura S-I, Natsume T, Takai Y, Akiyama AT. Asef2 and Neurabin2 cooperatively regulate actin cytoskeletal organization and are involved in HGF-induced cell migration. Oncogene. 2009;28(10):1357–1365. doi:10.1038/onc.2008.478
199. Ahonen SJ, Pietila E, Mellersh CS, et al. Genome-wide association study identifies a novel canine glaucoma locus. PLoS One. 2013;8(8):e70903. doi:10.1371/journal.pone.0070903
200. Ahram DF, Cook AC, Kecova H, Grozdanic SD, Kuehn MH. Identification of genetic loci associated with primary angle-closure glaucoma in the basset hound. Mol Vis. 2014;20:497–510.
201. Ahram DF, Grozdanic SD, Kecova H, Henkes A, Collin RW, Kuehn MH. Variants in nebulin (NEB) are linked to the development of familial primary angle closure glaucoma in Basset Hounds. PLoS One. 2015;10(5):e0126660. doi:10.1371/journal.pone.0126660
202. Oliver JAC, Ricketts SL, Kuehn MH, Mellersh CS. Primary closed angle glaucoma in the Basset Hound: genetic investigations using genome-wide association and RNA sequencing strategies. Mol Vis. 2019;25:93–105.
203. Axenovich T, Zorkoltseva I, Belonogova N, et al. Linkage and association analyses of glaucoma related traits in a large pedigree from a Dutch genetically isolated population. J Med Genet. 2011;48(12):802–809. doi:10.1136/jmedgenet-2011-100436
204. Nair KS, Hmani-Aifa M, Ali Z, et al. Alteration of the serine protease PRSS56 causes angle-closure glaucoma in mice and posterior microphthalmia in humans and mice. Nat Genet. 2011;43(6):579–584. doi:10.1038/ng.813
205. Labelle-Dumais C, Pyatla G, Paylakhi S, et al. Loss of PRSS56 function leads to ocular angle defects and increased susceptibility to high intraocular pressure. Dis Model Mech. 2020;13(5):dmm042853. doi:10.1242/dmm.042853
206. Ittner LM, Schwerdtfeger K, Kunz TH, et al. Transgenic mice with ocular overexpression of an adrenomedullin receptor reflect human acute angle-closure glaucoma. Clin Sci (Lond). 2008;114(1):49–58. doi:10.1042/CS20070163
207. Yousufzai SY, Ali N, Abdel-Latif AA. Effects of adrenomedullin on cyclic AMP formation and on relaxation in iris sphincter smooth muscle. Invest Ophthalmol Vis Sci. 1999;40(13):3245–3253.
208. McLatchie LM, Fraser NJ, Main MJ, et al. RAMPs regulate the transport and ligand specificity of the calcitonin-receptor-like receptor. Nature. 1998;393(6683):333–339. doi:10.1038/30666
209. Cao D, Liu X, Guo X, Cong Y, Huang J, Mao Z. Investigation of the association between CALCRL polymorphisms and primary angle closure glaucoma. Mol Vis. 2009;15:2202–2208.
210. Wang B, Sakata LM, Friedman DS, et al. Quantitative iris parameters and association with narrow angles. Ophthalmology. 2010;117(1):11–17. doi:10.1016/j.ophtha.2009.06.017
211. Wang BS, Narayanaswamy A, Amerasinghe N, et al. Increased iris thickness and association with primary angle closure glaucoma. Br J Ophthalmol. 2011;95(1):46–50. doi:10.1136/bjo.2009.178129
212. Seet LF, Narayanaswamy A, Finger SN, et al. Distinct iris gene expression profiles of primary angle closure glaucoma and primary open angle glaucoma and their interaction with ocular biometric parameters. Clin Exp Ophthalmol. 2016;44(8):684–692. doi:10.1111/ceo.12743
213. He M, Lu Y, Liu X, Ye T, Foster PJ. Histologic changes of the iris in the development of angle closure in Chinese eyes. J Glaucoma. 2008;17(5):386–392. doi:10.1097/IJG.0b013e31815c5f69
214. Bradshaw AD. The role of SPARC in extracellular matrix assembly. J Cell Commun Signal. 2009;3(3–4):239–246. doi:10.1007/s12079-009-0062-6
215. Kennedy KD, AnithaChristy SA, Buie LK, Borras T. Cystatin a, a potential common link for mutant myocilin causative glaucoma. PLoS One. 2012;7(5):e36301. doi:10.1371/journal.pone.0036301
216. Nie Q, Zhang X. Transcriptional profiling analysis predicts potential biomarkers for glaucoma: HGF, AKR1B10 and AKR1C3. Exp Ther Med. 2018;16(6):5103–5111. doi:10.3892/etm.2018.6875
217. Acott TS, Kelley MJ. Extracellular matrix in the trabecular meshwork. Exp Eye Res. 2008;86(4):543–561.
218. Vranka JA, Kelley MJ, Acott TS, Keller KE. Extracellular matrix in the trabecular meshwork: intraocular pressure regulation and dysregulation in glaucoma. Exp Eye Res. 2015;133:112–125. doi:10.1016/j.exer.2014.07.014
219. Vittal V, Rose A, Gregory KE, Kelley MJ, Acott TS. Changes in gene expression by trabecular meshwork cells in response to mechanical stretching. Invest Ophthalmol Vis Sci. 2005;46(8):2857–2868. doi:10.1167/iovs.05-0075
220. Faralli JA, Schwinn MK, Gonzalez JM, Filla MS, Peters DM. Functional properties of fibronectin in the trabecular meshwork. Exp Eye Res. 2009;88(4):689–693. doi:10.1016/j.exer.2008.08.019
221. Filla MS, Faralli JA, Peotter JL, Peters DM. The role of integrins in glaucoma. Exp Eye Res. 2017;158:124–136. doi:10.1016/j.exer.2016.05.011
222. Filla MS, David G, Weinreb RN, Kaufman PL, Peters DM. Distribution of syndecans 1–4 within the anterior segment of the human eye: expression of a variant syndecan-3 and matrix-associated syndecan-2. Exp Eye Res. 2004;79(1):61–74. doi:10.1016/j.exer.2004.02.010
223. Stringer SE, Gallagher JT. Heparan sulphate. Int J Biochem Cell Biol. 1997;29(5):709–714. doi:10.1016/S1357-2725(96)00170-7
224. Teng YH, Aquino RS, Park PW. Molecular functions of syndecan-1 in disease. Matrix Biol. 2012;31(1):3–16. doi:10.1016/j.matbio.2011.10.001
225. Vanhoutte D, Schellings MW, Gotte M, et al. Increased expression of syndecan-1 protects against cardiac dilatation and dysfunction after myocardial infarction. Circulation. 2007;115(4):475–482. doi:10.1161/CIRCULATIONAHA.106.644609
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.