")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 11
Update on therapeutic management of idiopathic pulmonary fibrosis
Authors Tzouvelekis A, Bonella F, Spagnolo P
Received 9 January 2015
Accepted for publication 2 February 2015
Published 3 March 2015 Volume 2015:11 Pages 359—370
DOI https://doi.org/10.2147/TCRM.S69716
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Argyris Tzouvelekis,1 Francesco Bonella,2 Paolo Spagnolo3
1Department of Internal Medicine, Section of Pulmonary, Critical Care and Sleep Medicine, Yale School of Medicine, New Haven, CT, USA; 2Interstitial and Rare Lung Disease Unit, Ruhrlandklinik, University Hospital, University of Duisburg-Essen, Germany; 3Medical University Clinic, Canton Hospital Baselland and University of Basel, Liestal, Switzerland
Abstract: Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive diffuse parenchymal lung disease of unknown origin, with a mortality rate exceeding that of many cancers. The diagnostic process is complex and relies on the clinician integrating clinical, laboratory, radiological, and histological data. In the last decade, major advances in our understanding of the pathogenesis of IPF have shifted the paradigm from a primarily inflammatory process evolving to fibrosis to a condition driven by aberrant wound healing following alveolar epithelial cell injury that results in scarring of the lung, architectural distortion, and irreversible loss of function. Improved understanding of disease pathogenesis has led to the identification of several therapeutic targets and the design of high-quality clinical trials evaluating novel compounds. However, the results of these studies have been mostly disappointing, probably due to the plethora of mediators, growth factors, and signaling pathways involved in the fibrotic process. Most recently, pirfenidone and nintedanib, two compounds with pleiotropic anti-fibrotic properties, have been proven effective in reducing functional decline and disease progression in IPF. This is a major breakthrough. Nevertheless, we still have a long way to go. In fact, neither pirfenidone nor nintedanib is a cure for IPF, and most patients continue to progress despite treatment. As such, comprehensive care of patients with IPF, including management of concomitant conditions and physical debility, as well as timely referral for lung transplantation, remains essential. Several agents with a high potential are currently being tested, and many more are ready for clinical trials. Their completion is critical for achieving the ultimate goal of curing patients with IPF.
Keywords: pulmonary fibrosis, therapy, nintedanib, pirfenidone, treatment
Disease overview
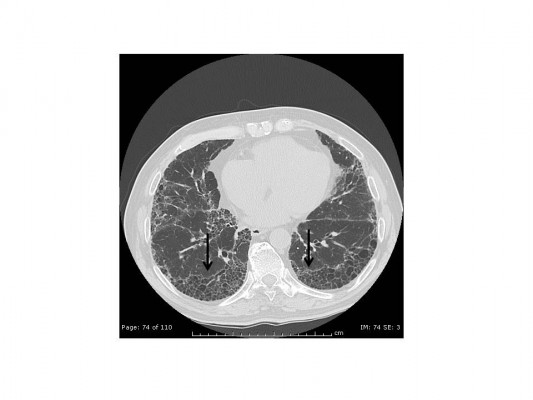
Idiopathic pulmonary fibrosis (IPF), the most common and severe form of the idiopathic interstitial pneumonias, is a chronic, progressive, and irreversible condition of unknown origin with a mean survival of approximately 3–5 years from the time of diagnosis.1–5 The disease, which affects primarily middle-aged and elderly adults (mainly male ex-smokers), is limited to the lungs and, contrary to the other idiopathic interstitial pneumonias, is associated with a radiological or histopathological pattern of usual interstitial pneumonia (UIP).1,6 Radiologically, UIP pattern consists of reticular opacities often associated with traction bronchiectasis and honeycombing. Architectural distortion, which reflects lung fibrosis, is frequently prominent. The distribution of UIP on high-resolution computed tomography (HRCT) images is characteristically basal and peripheral, although often patchy7 (Figure 1). Histologically, UIP is defined by a combination of: patchy interstitial fibrosis with alternating areas of normal lung; temporal heterogeneity of fibrosis characterized by scattered clusters of actively proliferating fibroblasts/myofibroblasts embedded into a myxoid stroma (“fibroblastic foci”); and architectural distortion due to chronic scarring or honeycomb change1 (Figure 2). The term UIP is often used interchangeably with IPF. However, the UIP pattern can be found in several clinical settings, including collagen vascular disease, drug toxicity, chronic hypersensitivity pneumonitis, asbestosis, familial IPF, and Hermansky-Pudlak syndrome.1 Therefore, the diagnosis of IPF requires exclusion of all known causes of fibrotic interstitial pneumonia.
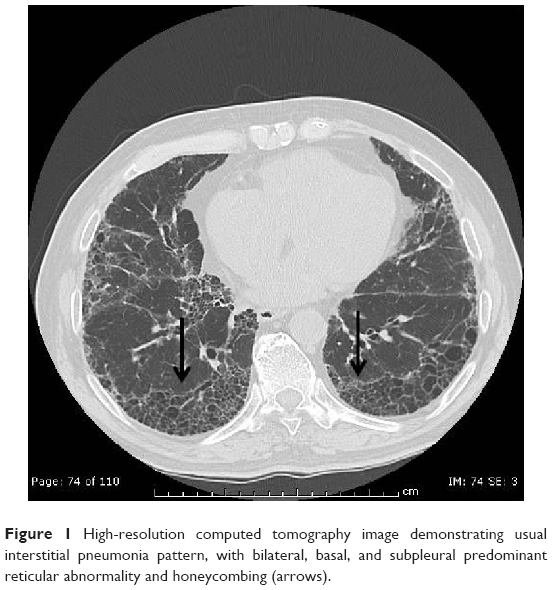 | Figure 1 High-resolution computed tomography image demonstrating usual interstitial pneumonia pattern, with bilateral, basal, and subpleural predominant reticular abnormality and honeycombing (arrows). |
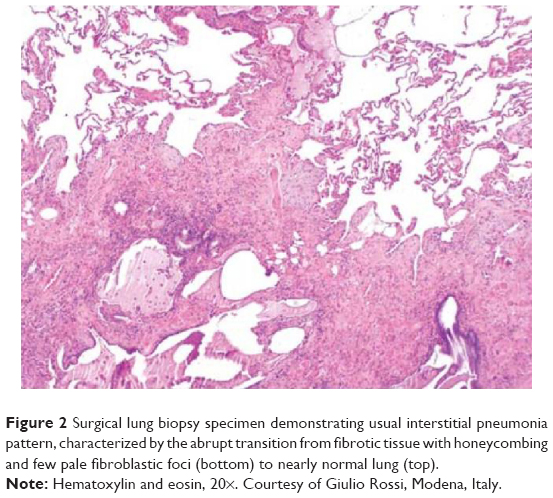 | Figure 2 Surgical lung biopsy specimen demonstrating usual interstitial pneumonia pattern, characterized by the abrupt transition from fibrotic tissue with honeycombing and few pale fibroblastic foci (bottom) to nearly normal lung (top). |
Patients with IPF usually seek medical attention because they suffer slowly progressive shortness of breath on exertion and non-productive cough. Other common clinical findings include bibasilar end-inspiratory (“velcro-like”) crackles heard on chest auscultation and digital clubbing. The natural history of IPF has been characterized as a slowly progressive lung disorder, and most patients follow this pattern. However, IPF is a highly heterogeneous and largely unpredictable disease in which periods of relative stability may be punctuated by episodes of accelerated decline, often resulting in respiratory failure and death.8,9
Consistent with the “inflammatory hypothesis” (ie, IPF was originally considered a predominantly inflammatory disorder), the 2,000 guidelines recommended corticosteroids in addition to cytotoxic agents (either azathioprine or cyclophosphamide) as the “standard treatment” for IPF, despite the very low level of evidence supporting this recommendation.10
In the past decade, our understanding of the pathobiology of IPF has undergone dramatic change. Current concepts suggest that the disease results from an aberrant reparative response to alveolar epithelial cell injury characterized by migration, proliferation, and activation of fibroblasts, and secretion of excessive amounts of extracellular matrix components, leading to scarring of the lung, architectural distortion, and irreversible loss of function. A complex interplay between environmental and host factors is thought to contribute to the development of IPF, although the inciting factors remain elusive and the disease pathogenesis is incompletely understood.11–13 The most recent clinical trials have therefore tested the efficacy of compounds that target the wound healing cascade and fibrogenesis. However, the results of these studies have mostly been disappointing, probably due to the multitude of mediators, growth factors, and signaling pathways involved in the fibrotic process.14 As such, the 2011 guidelines, which adopted the GRADE (Grades of Recommendation, Assessment, Development, and Evaluation) methodology to assess the quality of available data, did not strongly recommend any pharmacological treatments for patients with IPF.1 Importantly, however, the historical therapeutic strategy based on steroids and immunomodulators was no longer recommended, whereas some treatment regimens (combination of N-acetylcysteine [NAC]/prednisone/azathioprine, NAC monotherapy, warfarin, and pirfenidone) were considered unproven but reasonable therapeutic choices for a minority of IPF patients, based on their individual values and preferences.
Four years is a long time in IPF. In fact, since the publication of the most recent IPF guidelines, two compounds, ie, pirfenidone and nintedanib, have consistently proven effective in reducing functional decline and disease progression,15,16 and are likely to become the standard of care worldwide in the near future. In addition, several agents with a high potential are currently being tested, and many more are ready for clinical trials. Here, we review and place in context the recent advances in and insights into pharmacological treatment of IPF.
Disease management
IPF requires a methodological therapeutic approach integrating both pharmacological and non-pharmacological management strategies.1
Pharmacological therapy
Following innumerable therapeutic efforts with disappointing results, the year 2014 has been highlighted by the emergence of two promising drugs, namely pirfenidone and nintedanib, which have finally shifted the dial in the therapeutic field of IPF (Table 1). The results of the most recent randomized controlled trials (RCTs) are summarized and briefly discussed below.
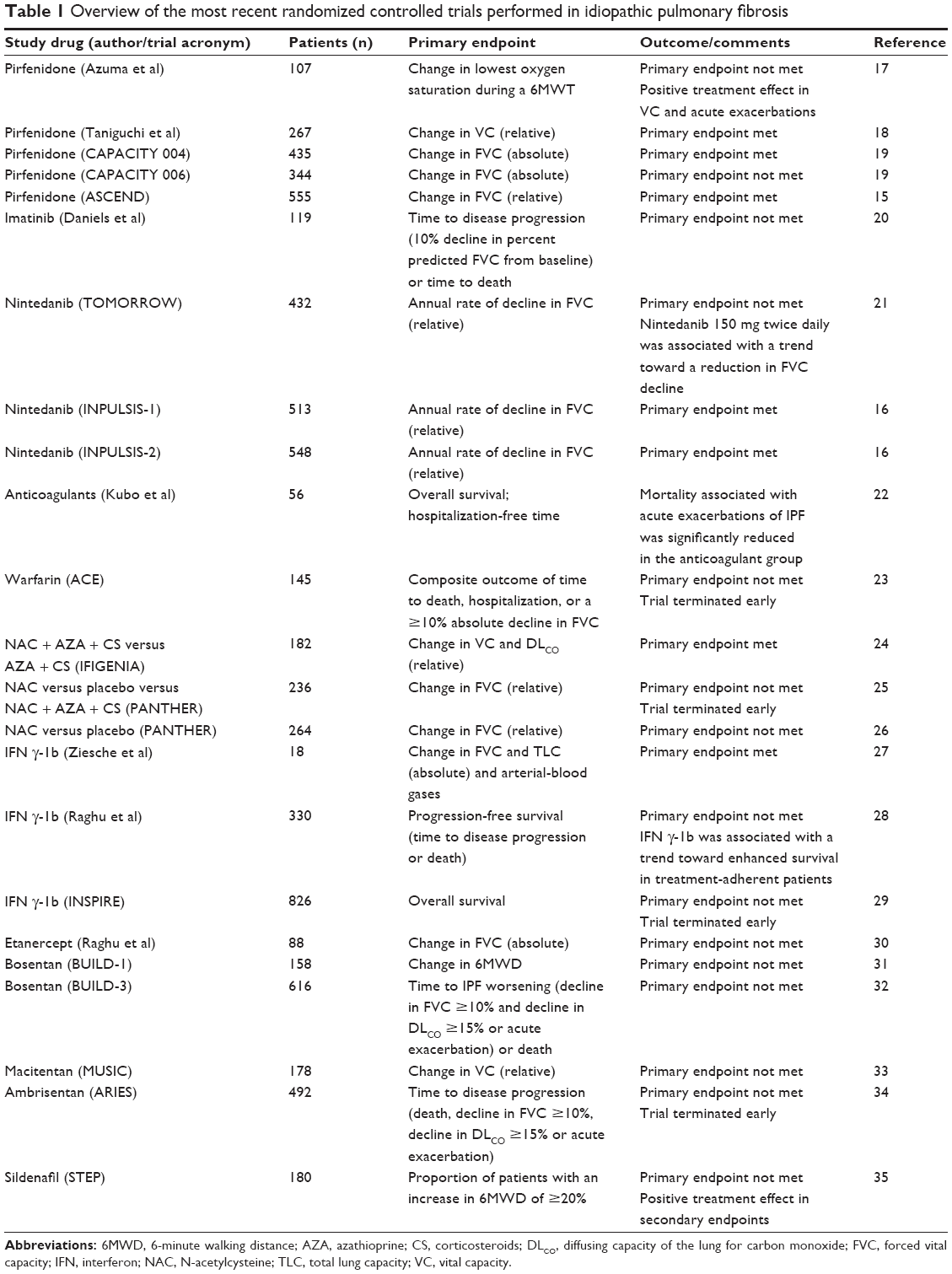 | Table 1 Overview of the most recent randomized controlled trials performed in idiopathic pulmonary fibrosis |
Compounds with Phase III RCT evidence of efficacy
Pirfenidone
Pirfenidone, an orally available compound with versatile anti-fibrotic and anti-inflammatory properties,36 has been clinically evaluated for its safety and efficacy in IPF in five RCTs comprising overall 1,710 patients.15,17–19 Original observation regarding its therapeutic potential17 fueled the undertaking of three RCTs, one in Japan and two multinational, involving centers in the USA, Europe, and Australia. A meta-analysis of all three studies showed a reduction in the risk of disease progression of approximately 30% compared with placebo.37 However, while pirfenidone reduced the rate of decline in vital capacity in the Japanese trial,18 it produced a similar beneficial effect in the rate of decline of the percentage of forced vital capacity (FVC) in only one of two CAPACITY studies (ie, Study 004).19 The drug was approved for the treatment of IPF in patients with mild to moderate functional impairment in Europe and Japan; conversely, these controversial results prompted the US Food and Drug Administration to request an additional trial to support the approval of pirfenidone. In the recent ASCEND trial, 555 patients with IPF were randomized to receive either the maximum oral dose of pirfenidone (2,403 mg/day) or placebo.15 Pirfenidone significantly reduced disease progression, as measured by changes in the mean decline of absolute (193 mL) or percent predicted (45%) FVC and changes in the 6-minute walking test (6MWT). Moreover, pirfenidone reduced disease progression or death by 43% compared with placebo. Although there was no statistically significant difference in rates of all-cause mortality, by pooling data from all three studies (ASCEND and Studies 004 and 006), the investigators demonstrated a reduction of all-cause and IPF-related mortality of 48% and 68%, respectively.
Adverse events
Gastrointestinal adverse events, including nausea, dyspepsia, anorexia, and gastroesophageal reflux, were more common in the pirfenidone group than in the placebo group (36.0% versus 13.4%, 17.6% versus 6.1%, 15.8% versus 6.5%, and 11.9% versus 6.5%, respectively). Skin-related adverse events (eg, photosensitivity rash) were also more common in the pirfenidone group than in the placebo group (28.1% versus 8.7%). Finally, elevations in levels of alanine or aspartate aminotransferase that were three or more times the upper limit of the normal range occurred more frequently in the pirfenidone group (2.9% versus 0.7%), but were reversible and without clinically significant sequelae. Overall, pirfenidone was safe and well tolerated with only 3.6% more patients in the intervention arm discontinuing the drug due to an adverse event.
Nintedanib
Nintedanib is an intracellular multiple tyrosine kinase inhibitor that targets receptors of vascular endothelial growth factor, fibroblast growth factor, and platelet-derived growth factor, all thought to be involved in the pathogenesis of IPF.38 An earlier Phase IIb study (TOMORROW) of 432 patients with mild to moderate disease (FVC >50%) reported that treatment with 150 mg of nintedanib twice daily was effective in reducing FVC decline and preventing acute exacerbations, while preserving quality of life.21 Encouraged by these findings, the investigators conducted two replicate studies (INPULSIS-1 and INPULSIS-2) enrolling 1,061 IPF patients who received either nintedanib (150 mg twice daily) or placebo.16 Both trials met their primary endpoint (ie, the difference in adjusted annual rate of change in FVC was approximately 115 mL in both trials, favoring nintedanib). Nintedanib was also associated with a significant delay in time to first exacerbation in INPULSIS-2 but not in INPULSIS-1. No survival benefit was observed in either trial, although these studies were not powered to detect statistically significant differences in mortality.
Adverse events
In both trials, the most frequent adverse event in the nintedanib arms was diarrhea (61.5% versus 18.6% in the placebo arm in INPULSIS-1 and 63.2% versus 18.3% in INPULSIS-2, respectively). Diarrhea led to premature discontinuation of the study drug in 4.5% of patients receiving nintedanib in INPULSIS-1 (versus none of the patients receiving placebo) and 4.3% of patients in INPULSIS-2 (versus 0.5% of patients receiving placebo). Other adverse events more frequent in the nintedanib arms than in the placebo arms in both trials were nausea (22.7% versus 5.9% in INPULSIS-1 and 26.1% versus 7.3% in INPULSIS-2, respectively) and vomiting (12.9% versus 2.0% in INPULSIS-1 and 10.3% versus 3.2% in INPULSIS-2). Levels of alanine aminotransferase, aspartate aminotransferase, or both that were three or more times the upper limit of the normal range occurred more frequently in the nintedanib groups than in the placebo groups (4.9% versus 0.5% in INPULSIS-1 and 5.2% versus 0.9% in INPULSIS-2, respectively), but were reversible and without clinically significant sequelae. Moreover, in both trials, the proportion of patients reporting serious adverse events was similar in the nintedanib and placebo groups (31.1% versus 27.0% in INPULSIS-1 and 29.8% versus 32.9% in INPULSIS-2, respectively) and most patients continued to receive nintedanib for the duration of the study.
Pirfenidone and nintedanib have been approved by both the European Medicines Agency and the US Food and Drug Administration, although with slightly different indications. In the USA, both drugs are approved for the treatment of IPF patients regardless of disease severity; in Europe, nintedanib has the same indication as in the USA, whereas pirfenidone is approved for the treatment of patients with mild to moderate disease (FVC ≥50% and ≤90% and diffusing capacity of the lung for carbon monoxide [DLCO] ≥35% and ≤90%). Both drugs are expensive (the expected cost of pirfenidone and nintedanib will be approximately $100,000 per patient per year in the USA), and their cost-effectiveness is difficult to estimate. Nevertheless, we can expect these two drugs to be widely used due to the high unmet medical need in IPF.
Other compounds/treatment regimes
Anticoagulants
Anticoagulants have been evaluated in IPF based on their efficacy in experimentally-induced lung fibrosis when given either prophylactically or therapeutically.39 Kubo et al originally reported a one-year survival benefit of anticoagulant (warfarin or heparin) therapy in patients with IPF (n=56) experiencing acute exacerbations.22 However, this trial was underpowered (as well as having an open-label design), so there was still uncertainty regarding the benefits and harms of anticoagulation in IPF. Therefore, Noth et al conducted an RCT (ACE-IPF) to test the hypothesis that treatment with warfarin at recommended doses would reduce the composite endpoint of mortality, hospitalization, and a 10% annual rate of FVC decline.23 An increased number of deaths associated with anticoagulation (14 versus three in the placebo limb) led to early discontinuation of the trial, thus arguing against the use of this therapeutic strategy in IPF, although the study exhibited substantial methodological caveats.40
N-acetylcysteine
NAC, a precursor of the endogenous antioxidant glutathione, has been used in IPF based on the assumption that an oxidant-antioxidant imbalance plays a role in the pathogenesis of the disease.41 IFIGENIA, a double-blind, randomized, placebo-controlled multicenter study, assessed the efficacy over one year of a high oral dose of NAC (600 mg three times daily) in combination with prednisone and azathioprine.24 While this so-called “triple therapy” significantly slowed the decline of both vital capacity and DLCO (the primary endpoints) compared with prednisone plus azathioprine (the “placebo” arm), this study had several drawbacks, mainly related to the lack of a true placebo arm (ie, patients not taking any potentially effective drug) and the high rate of patients lost to follow-up at 12 months due to death or withdrawal (about 30%). Therefore, the IPFnet Consortium designed PANTHER-IPF, a placebo-controlled, randomized, three-arm trial, to verify the efficacy of NAC in IPF.25 Patients were assigned in a 1:1:1 ratio to prednisone, azathioprine, and NAC (combination therapy), NAC alone, or placebo. Unexpectedly, a prespecified interim analysis of efficacy and safety planned at approximately 50% of data collection showed that the combination therapy, as compared with placebo, was associated with a statistically significant increase in all-cause mortality, all-cause hospitalizations, and treatment-related severe adverse events. Therefore, the three-drug regimen limb was stopped and PANTHER was continued as a double-blind, two-group study (ie, NAC versus placebo). While it is difficult to assess which components of the three-drug regimen might have been responsible for the observed negative outcomes, one possibility relates to the excessive dose of corticosteroids administered (prednisolone 40 mg/day) that does not reflect the low-dose regimen (ie, 10–15 mg/day) applied in most referral centers. Similarly, the modified PANTHER-IPF study did not demonstrate any effect of NAC monotherapy compared with placebo on the rate of change in FVC over a 60-week period.26 Nevertheless, a pre-interim analysis treatment paradox was observed (ie, improvement in FVC, DLCO, and 6MWT in the NAC group compared with placebo before the interim analysis). Although investigators were unable to provide a reasonable explanation for this paradox, it is likely that this event simply represents a regression to the mean for all these indicators, which randomly occurred after the interim analysis.
Interferon gamma-1b
Interferon gamma-1b (IFN γ-1b) is a compound with anti-fibrotic and immunomodulatory properties. The seminal report from Ziesche et al in 18 patients with IPF demonstrated functional benefit of subcutaneous IFN-γ-1b plus prednisolone compared with prednisolone monotherapy.27 Following this observation, an RCT of IFN γ-1b versus placebo did not show any significant benefits in terms of progression-free survival, pulmonary function parameters, or quality of life in IPF.28 However, subgroup analyses of patients with close treatment adherence revealed a trend towards increased survival. Based on these observations, a larger RCT (INSPIRE) was specifically designed to assess the effect of IFN γ-1b on survival time in IPF patients with mild to moderate impairment of baseline pulmonary function.29 A protocol-defined interim analysis revealed that the hazard ratio for mortality among patients treated with IFN γ-1b crossed the predefined stopping boundary for lack of minimal benefit and the study was terminated early.
Anti-acids
Abnormal acid gastroesophageal reflux has been reported in as many as 88% of patients with IPF and chronic microaspiration has been hypothesized to trigger development of the disease.42 Moreover, anti-reflux treatment has been associated with improved survival and significant functional benefits in patients with IPF. A retrospective study by Lee et al reported a statistically significant difference in the decline of FVC and fewer exacerbations in a large cohort of patients with IPF (n=242) under treatment with anti-acid agents (proton pump inhibitors or histamine 2-receptor antagonists).43 A more recent study analyzed change in FVC in patients assigned to the placebo arms of three large RCTs. After adjustment for sex, baseline FVC, and baseline DLCO, patients taking anti-acid treatment at baseline (proton pump inhibitors or histamine 2-receptor antagonists) were shown to have a smaller decrease in FVC at 30 weeks compared with those not taking anti-acid treatment.44 While no definitive conclusions can be drawn from these studies given their retrospective design, they provide a compelling rationale for future trials of anti-reflux therapies in IPF. According to current guidelines, asymptomatic gastroesophageal reflux should be medically treated in the majority of patients (weak recommendation, very low-quality evidence).1 However, the optimal dose of anti-acids and whether their administration may affect plasma concentrations of anti-fibrotic agents (eg, pirfenidone) need to be explored further.45
Anti-inflammatory and vasodilator agents
The therapeutic potential of several anti-inflammatory and vasodilator agents has been tested in patients with IPF, with results being rather disappointing. While the 2,000 guidelines on IPF recommended a combination of corticosteroids and azathioprine (or cyclophosphamide) as the standard of care,10 the recent PANTHER trial provided robust evidence of a poor safety and efficacy profile for this drug regimen.25 Etanercept, a tumor necrosis factor-α inhibitor, was evaluated in a randomized, prospective, double-blind, placebo-controlled Phase II trial.30 However, after 48 weeks of treatment, no significant effect on any of the efficacy outcomes (change in percentage of predicted FVC or DLCO, and change in the alveolar to arterial oxygen pressure difference [P{A-a}O2] at rest from baseline) was observed.
Data from basic science, animal, and translational studies suggest that the endothelin system, and endothelin (ET)-1 in particular, contributes significantly to the pathobiology of several fibrotic disorders, including IPF, by promoting fibroblast proliferation, myofibroblast differentiation, and synthesis of collagen.46 However, vasodilators such as bosentan (an ET-1 receptor antagonist),31,32 macitentan (an ET receptor antagonist),33 and ambrisentan (a selective ET-A receptor antagonist),34 have not only been proven ineffective in changing the natural history of the disease, but have also been associated with increased mortality rates (ambrisentan). In fact, an RCT evaluating the safety and efficacy of ambrisentan in patients with IPF (ARIES) was terminated prematurely due to lack of efficacy and a drug-related increase in disease progression compared with placebo.34 A possible explanation for this negative outcome relates to aberrant vasodilation, increased ventilation/perfusion inequalities, and worsening hypoxemia. So far, the only vasodilator that has demonstrated a favorable effect on functional parameters (DLCO) and indicators of dyspnea and quality of life is sildenafil.35 However, the trial investigating this drug (STEP), in which 180 subjects were randomized to sildenafil (20 mg three times daily) or placebo for 12 weeks, did not meet its primary endpoint, as only 10% of patients in the sildenafil arm and 7% in the placebo arm showed an improvement of ≥20 m in the 6MWT.
Novel therapeutic agents
A number of novel compounds are currently in the therapeutic pipeline for IPF (summarized in Table 2). The application status of stem cell therapy in patients with IPF is still exploratory and in its infancy.47 At present, there is only one safety clinical trial showing promising results from endobronchial infusion of autologous adipose-derived stem cells in a small cohort of patients with IPF (n=14). This study met its primary endpoint of absence of serious adverse events, including all-cause mortality. However, its results should be interpreted cautiously before rigid conclusions can be drawn.48 Significant limitations severely hampering the widespread implementation of stem cell therapy in IPF relate mainly to our limited knowledge of the fate of these cells within the profibrotic microenvironment given their mesenchymal origin and their potential to differentiate into myofibroblasts, thus causing disease progression. In addition, there are many unanswered questions including the time (early or advanced disease) and optimal route of administration (intravenous or endobronchial), source of mesenchymal stem cells (eg, adipose tissue, bone marrow, or umbilical cord), frequency of infusions, and choice of the appropriate primary endpoints to show benefit.49 An RCT approved by the US Food and Drug Administration is currently recruiting patients for a study investigating the safety and efficacy of a single intravenous administration of allogeneic bone marrow-derived mesenchymal stem cells, and its results are eagerly awaited.
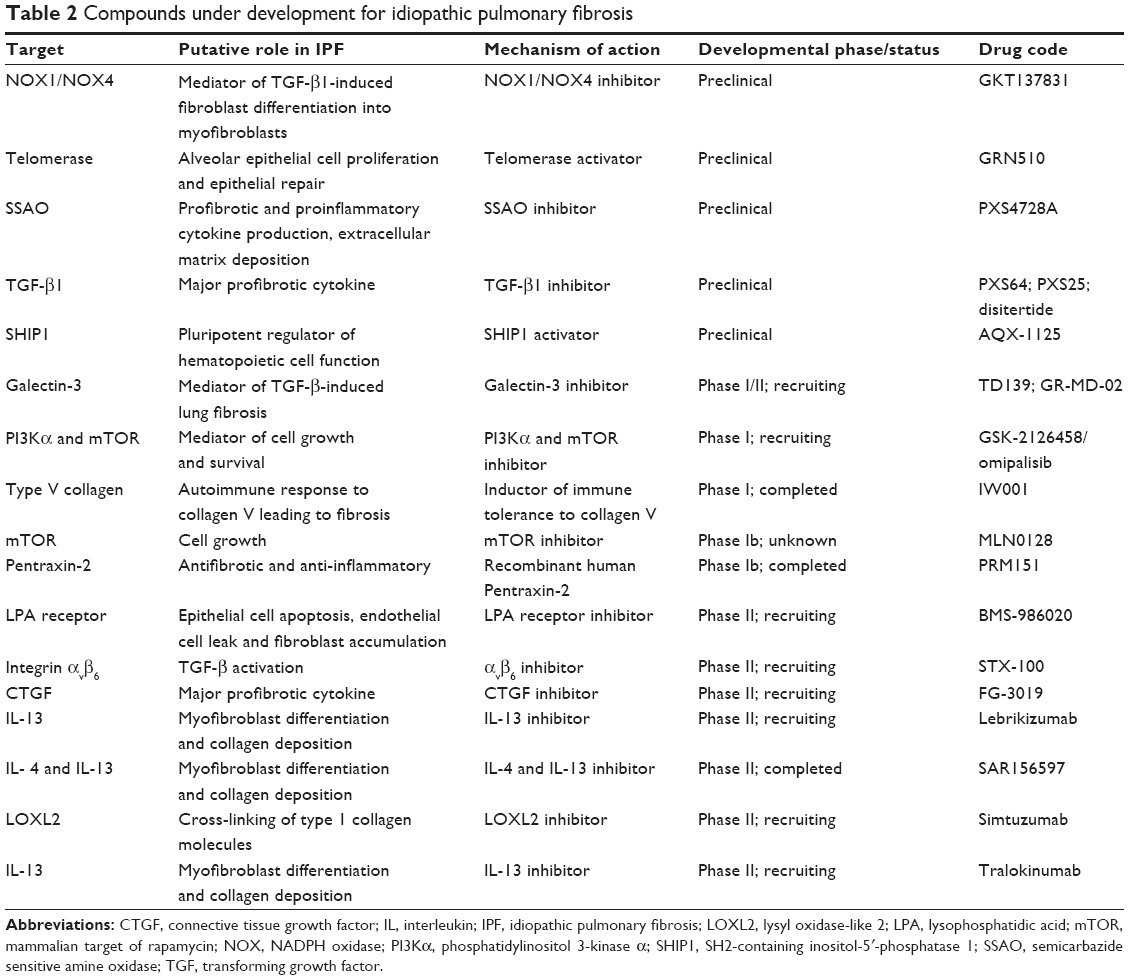 | Table 2 Compounds under development for idiopathic pulmonary fibrosis |
Non-pharmacological management
Lung transplantation
The most recent guidelines advocate lung transplantation as the most effective and reliable treatment for patients with IPF.1 Five-year survival rates range from 50% to 56%, whereas 10-year survival rates drop to 30%.1 A reduction in the risk of death by 75% after lung transplantation has been demonstrated in a series of 46 patients with IPF.50 Further data reveal that patients with IPF undergoing lung transplantation have more favorable long-term survival compared with patients transplanted for other indications.51 Whether single or double lung transplantation represents the most beneficial approach is a matter of ongoing controversy. Current evidence suggests that although bilateral procedures are associated with increased early mortality, they also promote long-term survival. On the other hand, patients put on a waiting list for bilateral lung transplantation have to wait longer, which may outweigh any improved survival benefits.51
Due to the unpredictable nature of the disease and the high waiting list mortality rates, the scientific community strongly recommends that patients with IPF be referred early for lung transplantation. The implementation of a lung allocation score, which prioritizes patients for lung transplantation based on disease severity, has led to a reduction in waiting list mortality from 47% to 11%.52 Nevertheless, mortality is still higher than for other diseases, such as chronic obstructive pulmonary disease (COPD). In addition, preoperative quality of life in patients with IPF has been demonstrated to be much poorer than that of patients with COPD, whereas in IPF patients lung function deteriorates much more rapidly. Following the introduction of the lung allocation score, IPF has become the most common indication for lung transplantation.51,52 Yet, preoperative and postoperative mortality rates are still unacceptably high, necessitating further refinement of the lung allocation score (eg, by incorporating genomic and proteomic prognosticators). In this regard, a recent study by Silhan et al demonstrated that telomerase mutation carriers undergoing lung transplantation may need specific attention, including adjustment of immunosuppressive agents and platelet transfusion support.53
Pulmonary rehabilitation
Pulmonary rehabilitation (PR) has been proven to be the standard of care for chronic lung diseases because of its efficacy in alleviating symptoms, and improving exercise tolerance, functional capacity, and dyspnea scores.54–56 Moreover, rehabilitation has been associated with improvement in symptoms such as anxiety and depression.57 Reductions in exercise lactic acidosis and ventilation-efficient exercise breathing pattern along with avoidance of infection are some of the pathophysiological mechanisms proposed for the beneficial effect of PR. In patients with IPF, PR has been associated with a significant improvement in 6MWT, dyspnea, and quality of life.1,58,59 While PR was originally indicated for end-stage patients with limited daily activity, current concepts support referral of IPF patients at the time of diagnosis. Moreover, the benefits of PR expand to preoperative and postoperative lung transplantation procedures, helping clinicians to select appropriate candidates and at the same time psychologically prepare patients for the operation.57 Successful PR includes behavioral changes, such as weight loss, pacing, and energy conservation strategies and adaptation of specific breathing and exercise patterns.57 Future studies focusing not only on improving exercise tolerance but also on patient education and psychological support should be aggressively pursued.
Oxygen therapy
Long-term oxygen treatment is recommended for all IPF patients with resting or nocturnal hypoxemia.1,57 There are no RCTs evaluating the efficacy of long-term oxygen treatment due to ethical concerns about withholding oxygen in patients with respiratory failure.1,57 A retrospective trial comparing oxygen therapy, colchicine, and corticosteroids showed no survival benefit in patients receiving oxygen compared with the other two groups.60 Studies support the notion that oxygen therapy may improve quality of life by impacting daytime social and physical functioning.57 Therefore, oxygen therapy represents a fundamental component of the management of patients with IPF.1
Prognostic indicators
Functional and physiological indices
IPF has a mean survival ranging from 3 to 5 years, with a rate of progression that is highly variable and heterogeneous. Clinically, three different forms of IPF can be identified, ie, slowly progressive, rapidly progressive, and relatively stable punctuated by rapid disease acceleration (known as acute exacerbation).61 There have been tremendous efforts to accurately identify these phenotypes using clinical, functional, and physiological indicators. Results have been rather conflicting and controversial. Pulmonary function tests and HRCT have provided us with scattered statements. Flaherty et al have demonstrated that short-term changes in pulmonary function tests are strong predictors of long-term survival, whereas serial changes in HRCT were of limited value.62 Conversely, baseline HRCT information may reliably distinguish a subset of patients in whom disease is clearly irreversible (UIP pattern) and a subgroup in which treatment response is likely (organizing pneumonia pattern). On the other hand, Latsi et al demonstrated that serial pulmonary function tests predict survival and longitudinal behavior more accurately than baseline values.63 An absolute decline of ≥10% in FVC and ≥15% in DLCO has been associated with worst outcome and represents an acceptable approach to assess disease progression and estimate the risk of mortality.1 As such, an absolute decline of FVC ≥10% affects management decisions (eg, prioritization for lung transplantation) and patient counseling.64 Furthermore, it has been shown that even changes in FVC <10% may predict outcome.65 Whether the absolute or the relative difference in FVC represents the most accurate method for predicting outcome is unclear. Richeldi et al evaluated both methods in a cohort of patients with IPF and showed that the relative method predicts almost twice as many events of disease progression when compared with the absolute method.66 Therefore, this approach may maximize chances of identifying clinically meaningful alterations with high prognostic accuracy. The absolute changes in FVC and DLCO over time have been used to create a weighted-point algorithm to predict outcome, ie, the GAP model, which also includes sex and age. The GAP model was evaluated in 558 patients with IPF and successfully stratified outcome.67
Besides GAP, which represents a true breakthrough in risk-stratification algorithms, other scoring systems have been proposed.68 Wells et al reconciled indices of functional severity (FVC, DLCO, and forced expiratory volume in one second) and extent of disease (HRCT) to create a prognostic index (the composite physiological index [CPI]) that quantifies functional abnormality attributable to fibrosis while excluding that due to emphysema.68 The CPI score was further validated as a component of another risk stratification score (ROSE), that combined data such as dyspnea, 6MWT, and CPI. However, ROSE showed very low specificity in predicting 3-year survival, and this is mainly attributed to the absence of reliable information that could predict acute exacerbations.69
The 6MWT has been evaluated by several studies as a surrogate marker of disease progression and as a primary endpoint in clinical trials.70 However, these studies were underpowered and yielded controversial results.70 du Bois et al published the largest study investigating the clinical usefulness of 6MWT as a marker of disease progression in a cohort of 826 patients with IPF.71 They demonstrated that a decline as low as 24 m (the minimal clinically important difference) conferred a significantly higher mortality risk. Further, a 24-week decline of >50 m was associated with a nearly three-fold increase in mortality risk.71 These results strongly support the validity of 6MWT as a prognostic marker, which may improve upon the sensitivity of progression-free survival or change in FVC endpoints used in most clinical trials.
Insights from translational research in management of IPF
Several molecules have been proposed as biomarkers of disease progression and treatment responsiveness. While some of these biomarkers are particularly promising, they are largely unavailable for routine clinical use. Krebs von den Lungen-6 (KL-6) is a high molecular weight glycoprotein produced by regenerating type II pneumocytes.72 Serum levels of KL-6 are elevated in patients with IPF, and these levels may correlate with increased risk of acute exacerbation and mortality.73,74 In Japan, KL-6 is measured routinely in patients with IPF, but further validation in non-Japanese cohorts is required before these data can be generalized. Serum levels of surfactant protein A and D are also elevated in patients with IPF and are predictive of survival.75–77 CCL18, a CC-chemokine produced by human myeloid cells and a marker of macrophage activation, is a reliable predictor of mortality in patients with IPF. Prasse et al showed that elevated serum levels of CCL18 were strong predictors of disease progression and inversely correlated with functional parameters of disease severity.78 Matrix metalloproteinase-7 is upregulated in lung tissue and bronchoalveolar lavage from patients with IPF.79 Matrix metalloproteinase-7 levels correlate with disease severity,80 and appear to predict mortality in patients with IPF.81,82
Conclusion and future perspectives
According to current guidelines, the standard of care for patients with IPF includes early referral for lung transplantation, palliative care, and enrollment into therapeutic clinical trials.1 However, owing to the availability of novel, more robust data derived from recently completed RCTs, the therapeutic guidelines are currently being revised. The “weak negative” recommendation for pirfenidone will likely give place to a “strong positive” recommendation and a similar recommendation is likely to be made for nintedanib. However, these drugs at best slow down functional decline and disease progression and do not cure the disease itself. An oncological approach using cocktails of drugs may prove more efficacious, given the plethora of profibrotic pathways and mediators that need to be targeted simultaneously.83 In addition, investigators should focus on optimizing treatment for more severe cases of IPF (including acute exacerbations) and concomitant conditions/complications, such as pulmonary hypertension and extensive emphysema. With regard to blood biomarkers, standardization of collection matrices and identification of uniform cut-off levels in the context of large, multicenter clinical trials are urgently needed. Finally, and perhaps most importantly, the scientific community should implement personalized medicine approaches based on genomic and proteomic information in order to identify subjects who would benefit from individually tailored therapeutic regimens while minimizing side effects.84 Discoveries of gene variants and changes in gene expression that reliably predict outcome have the potential to revolutionize our prognostic perspective and impact on therapeutic approaches. The advent of next-generation genome sequencing to collect epigenomic profiles may provide the way forward.
Disclosure
Paolo Spagnolo serves as consultant for Roche and has received consulting fees from Boehringer Ingelheim. AT and FB have no conflicts of interest to disclose.
References
Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183(6):788–824. | ||
Bjoraker JA, Ryu JH, Edwin MK, et al. Prognostic significance of histopathologic subsets in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1998;157(1):199–203. | ||
Flaherty KR, Toews GB, Travis WD, et al. Clinical significance of histological classification of idiopathic interstitial pneumonia. Eur Respir J. 2002;19(2):275–283. | ||
Nicholson AG, Colby TV, du Bois RM, Hansell DM, Wells AU. The prognostic significance of the histologic pattern of interstitial pneumonia in patients presenting with the clinical entity of cryptogenic fibrosing alveolitis. Am J Respir Crit Care Med. 2000;162(6):2213–2217. | ||
King TE Jr, Schwarz MI, Brown K, et al. Idiopathic pulmonary fibrosis: relationship between histopathologic features and mortality. Am J Respir Crit Care Med. 2001;164(6):1025–1032. | ||
Travis WD, Costabel U, Hansell DM, et al. An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188(6):733–748. | ||
Lynch DA, Travis WD, Müller NL, et al. Idiopathic interstitial pneumonias: CT features. Radiology. 2005;236(1):10–21. | ||
King TE Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet. 2011;378(9807):1949–1961. | ||
Ley B, Collard HR, King TE Jr. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;183(4):431–440. | ||
American Thoracic Society. Idiopathic pulmonary fibrosis: diagnosis and treatment. International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS). Am J Respir Crit Care Med. 2000;161(2 Pt 1):646–664. | ||
Spagnolo P, Grunewald J, du Bois RM. Genetic determinants of pulmonary fibrosis: evolving concepts. Lancet Respir Med. 2014;2(5):416–428. | ||
Wolters PJ, Collard HR, Jones KD. Pathogenesis of idiopathic pulmonary fibrosis. Annu Rev Pathol. 2014;9:157–179. | ||
Spagnolo P, Rossi G, Cavazza A. Pathogenesis of idiopathic pulmonary fibrosis and its clinical implications. Expert Rev Clin Immunol. 2014;10(8):1005–1017. | ||
Maher TM. Idiopathic pulmonary fibrosis: pathobiology and novel approaches to treatment. Clin Chest Med. 2012;33(1):69–83. | ||
King TE Jr, Bradford WZ, Castro-Bernardini S, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2083–2092. | ||
Richeldi L, du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2071–2082. | ||
Azuma A, Nukiwa T, Tsuboi E, et al. Double-blind, placebo-controlled trial of pirfenidone in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2005;171(9):1040–1047. | ||
Taniguchi H, Ebina M, Kondoh Y, et al. Pirfenidone in idiopathic pulmonary fibrosis. Eur Respir J. 2010;35(4):821–829. | ||
Noble PW, Albera C, Bradford WZ, et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet. 2011;377(9779):1760–1769. | ||
Daniels CE, Lasky JA, Limper AH, et al. Imatinib treatment for IPF: randomised placebo controlled trial results. Am J Respir Crit Care Med. 2010;181(6):604–610. | ||
Richeldi L, Costabel U, Selman M, et al. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N Engl J Med. 2011;365(12):1079–1087. | ||
Kubo H, Nakayama K, Yanai M, et al. Anticoagulant therapy for idiopathic pulmonary fibrosis. Chest. 2005;128(3):1475–1482. | ||
Noth I, Anstrom KJ, Calvert SB, et al. A placebo-controlled randomized trial of warfarin in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2012;186(1):88–95. | ||
Demedts M, Behr J, Buhl R, et al. High-dose acetylcysteine in idiopathic pulmonary fibrosis. N Engl J Med. 2005;353(21):2229–2242. | ||
Idiopathic Pulmonary Fibrosis Clinical Research Network, Martinez FJ, de Andrade JA, Anstrom KJ, King TE Jr, Raghu G. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med. 2012;366(21):1968–1977. | ||
Idiopathic Pulmonary Fibrosis Clinical Research Network, Martinez FJ, de Andrade JA, Anstrom KJ, King TE Jr, Raghu G. Randomized trial of acetylcysteine in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2093–3101. | ||
Ziesche R, Hofbauer E, Wittmann K, et al. A preliminary study of long-term treatment with interferon gamma-1b and low-dose prednisolone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 1999;341(17):1264–1269. | ||
Raghu G, Brown KK, Bradford WZ, et al. A placebo-controlled trial of interferon gamma-1b in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2004;350(2):125–133. | ||
King TE Jr, Albera C, Bradford WZ, et al. Effect of interferon gamma-1b on survival in patients with idiopathic pulmonary fibrosis (INSPIRE): a multicentre, randomised, placebo-controlled trial. Lancet. 2009;374(9685):222–228. | ||
Raghu G, Brown KK, Costabel U, et al. Treatment of idiopathic pulmonary fibrosis with etanercept: an exploratory, placebo-controlled trial. Am J Respir Crit Care Med. 2008;178(9):948–955. | ||
King TE Jr, Behr J, Brown KK, et al. BUILD-1: a randomized placebo-controlled trial of bosentan in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2008;177(1):75–81. | ||
King TE Jr, Brown KK, Raghu G, et al. BUILD-3: a randomized, controlled trial of bosentan in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;184(1):92–99. | ||
Raghu G, Million-Rousseau R, Morganti A, et al. Macitentan for the treatment of idiopathic pulmonary fibrosis: the randomized controlled MUSIC trial. Eur Respir J. 2013;42(6):1622–1632. | ||
Raghu G, Behr J, Brown KK, et al. Treatment of idiopathic pulmonary fibrosis with ambrisentan: a parallel, randomized trial. Ann Intern Med. 2013;158(9):641–649. | ||
Idiopathic Pulmonary Fibrosis Clinical Research Network, Zisman DA, Schwarz M, Anstrom KJ, Collard HR, Flaherty KR, Hunninghake GW. A controlled trial of sildenafil in advanced idiopathic pulmonary fibrosis. N Engl J Med. 2010;363(7):620–628. | ||
Iyer SN, Gurujeyalakshmi G, Giri SN. Effects of pirfenidone on transforming growth factor-b gene expression at the transcriptional level in bleomycin hamster model of lung fibrosis. J Pharmacol Exp Ther. 1999;291(1):367–373. | ||
Spagnolo P, Del Giovane C, Luppi F, et al. Non-steroid agents for idiopathic pulmonary fibrosis. Cochrane Database Syst Rev. 2010;9:CD003134. | ||
Chaudhary NI, Roth GJ, Hilberg F, et al. Inhibition of PDGF, VEGF and FGF signalling attenuates fibrosis. Eur Respir J. 2007;29(5):976–985. | ||
Scotton CJ, Krupiczojc MA, Königshoff M, et al. Increased local expression of coagulation factor X contributes to the fibrotic response in human and murine lung injury. J Clin Invest. 2009;119(9):2550–2563. | ||
Tzouvelekis A, Margaritopoulos G, Loukides S, Bouros D. Warfarin in idiopathic pulmonary fibrosis: friend or foe, is it a matter of genes and heparin? Am J Respir Crit Care Med. 2013;187(2):213–214. | ||
Cantin AM, Hubbard RC, Crystal RG. Glutathione deficiency in the epithelial lining fluid of the lower respiratory tract in idiopathic pulmonary fibrosis. Am Rev Respir Dis. 1989;139(2):370–372. | ||
Raghu G, Freudenberger TD, Yang S, et al. High prevalence of abnormal acid gastro-oesophageal reflux in idiopathic pulmonary fibrosis. Eur Respir J. 2006;27(1):136–142. | ||
Lee JS, Ryu JH, Elicker BM, et al. Gastroesophageal reflux therapy is associated with longer survival in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;184(12):1390–1394. | ||
Lee JS, Collard HR, Anstrom KJ, et al. Anti-acid treatment and disease progression in idiopathic pulmonary fibrosis: an analysis of data from three randomised controlled trials. Lancet Respir Med. 2013;1(5):369–376. | ||
Tzouvelekis A, Bouros D. Anti-acid treatment for idiopathic pulmonary fibrosis. Lancet Respir Med. 2013;1(5):348–349. | ||
Swigris JJ, Brown KK. The role of endothelin-1 in the pathogenesis of idiopathic pulmonary fibrosis. Bio Drugs. 2010;24(1):49–54. | ||
Tzouvelekis A, Antoniadis A, Bouros D. Stem cell therapy in pulmonary fibrosis. Curr Opin Pulm Med. 2011;17(5):368–373. | ||
Tzouvelekis A, Paspaliaris V, Koliakos G, et al. A prospective, non-randomized, no placebo-controlled, phase Ib clinical trial to study the safety of the adipose derived stromal cells-stromal vascular fraction in idiopathic pulmonary fibrosis. J Transl Med. 2013;11:171. | ||
Tzouvelekis A, Bouros D. Steep barriers to overcome for successful application of stem cell treatment in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2013;188(2):251–252. | ||
Thabut G, Mal H, Castier Y, et al. Survival benefit of lung transplantation for patients with idiopathic pulmonary fibrosis. J Thorac Cardiovasc Surg. 2003;126(2):469–475. | ||
George TJ, Arnaoutakis GJ, Shah AS. Lung transplant in idiopathic pulmonary fibrosis. Arch Surg. 2011;146(10):1204–1209. | ||
Christie JD, Edwards LB, Kucheryavaya AY, et al. The Registry of the International Society for Heart and Lung Transplantation: Twenty-Eighth Adult Lung and Heart-Lung Transplant Report – 2011. J Heart Lung Transplant. 2011;30(10):1104–1122. | ||
Silhan LL, Shah PD, Chambers DC, et al. Lung transplantation in telomerase mutation carriers with pulmonary fibrosis. Eur Respir J. 2014;44(1):178–187. | ||
Arizono S, Taniguchi H, Sakamoto K, et al. Endurance time is the most responsive exercise measurement in idiopathic pulmonary fibrosis. Respir Care. 2014;59(7):1108–1115. | ||
Nishiyama O, Kondoh Y, Kimura T, et al. Effects of pulmonary rehabilitation in patients with idiopathic pulmonary fibrosis. Respirology. 2008;13(3):394–399. | ||
Nishiyama O, Taniguchi H, Kondoh Y, et al. Quadriceps weakness is related to exercise capacity in idiopathic pulmonary fibrosis. Chest. 2005;127(6):2028–2033. | ||
Egan JJ. Follow-up and nonpharmacological management of the idiopathic pulmonary fibrosis patient. Eur Respir Rev. 2011;20(120):114–117. | ||
Belkin A, Swigris JJ. Health-related quality of life in idiopathic pulmonary fibrosis: where are we now? Curr Opin Pulm Med. 2013;19(5):474–479. | ||
Swigris JJ, Fairclough DL, Morrison M, et al. Benefits of pulmonary rehabilitation in idiopathic pulmonary fibrosis. Respir Care. 2011;56(6):783–789. | ||
Douglas WW, Ryu JH, Schroeder DR. Idiopathic pulmonary fibrosis: impact of oxygen and colchicine, prednisone, or no therapy on survival. Am J Respir Crit Care Med. 2000;161(4 Pt 1):1172–1178. | ||
Selman M, Carrillo G, Estrada A, et al. Accelerated variant of idiopathic pulmonary fibrosis: clinical behavior and gene expression pattern. PLoS One. 2007;2(5):e482. | ||
Flaherty KR, Mumford JA, Murray S, et al. Prognostic implications of physiologic and radiographic changes in idiopathic interstitial pneumonia. Am J Respir Crit Care Med. 2003;168(5):543–548. | ||
Latsi PI, du Bois RM, Nicholson AG, et al. Fibrotic idiopathic interstitial pneumonia: the prognostic value of longitudinal functional trends. Am J Respir Crit Care Med. 2003;168(5):531–537. | ||
du Bois RM, Weycker D, Albera C, et al. Forced vital capacity in patients with idiopathic pulmonary fibrosis: test properties and minimal clinically important difference. Am J Respir Crit Care Med. 2011;184(12):1382–1389. | ||
Zappala CJ, Latsi PI, Nicholson AG, et al. Marginal decline in forced vital capacity is associated with a poor outcome in idiopathic pulmonary fibrosis. Eur Respir J. 2010;35(4):830–836. | ||
Richeldi L, Ryerson CJ, Lee JS, et al. Relative versus absolute change in forced vital capacity in idiopathic pulmonary fibrosis. Thorax. 2012;67(5):407–411. | ||
Ley B, Ryerson CJ, Vittinghoff E, et al. A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med. 2012;156(10):684–691. | ||
Wells AU, Desai SR, Rubens MB, et al. Idiopathic pulmonary fibrosis: a composite physiologic index derived from disease extent observed by computed tomography. Am J Respir Crit Care Med. 2003;167(7):962–969. | ||
Mura M, Porretta MA, Bargagli E, et al. Predicting survival in newly diagnosed idiopathic pulmonary fibrosis: a 3-year prospective study. Eur Respir J. 2012;40(1):101–109. | ||
Swigris JJ, Wamboldt FS, Behr J, et al. The 6 minute walk in idiopathic pulmonary fibrosis: longitudinal changes and minimum important difference. Thorax. 2010;65(2):173–177. | ||
du Bois RM, Weycker D, Albera C, et al. Six-minute-walk test in idiopathic pulmonary fibrosis: test validation and minimal clinically important difference. Am J Respir Crit Care Med. 2011;183(9):1231–1237. | ||
Stahel RA, Gilks WR, Lehmann HP, Schenker T. Third International Workshop on Lung Tumor and Differentiation Antigens: overview of the results of the central data analysis. Int J Cancer Suppl. 1994;8:6–26. | ||
Kohno N, Kyoizumi S, Awaya Y, Fukuhara H, Yamakido M, Akiyama M. New serum indicator of interstitial pneumonitis activity: sialylated carbohydrate antigen KL-6. Chest. 1989;96(1):68–73. | ||
Ohshimo S, Ishikawa N, Horimasu Y, et al. Baseline KL-6 predicts increased risk for acute exacerbation of idiopathic pulmonary fibrosis. Respir Med. 2014;108(7):1031–1039. | ||
Greene KE, King TE Jr, Kuroki Y, et al. Serum surfactant proteins-A and -D as biomarkers in idiopathic pulmonary fibrosis. Eur Respir J. 2002;19(3):439–446. | ||
Kinder BW, Brown KK, McCormack FX, et al. Serum surfactant protein-A is a strong predictor of early mortality in idiopathic pulmonary fibrosis. Chest. 2009;135(6):1557–1563. | ||
Takahashi H, Fujishima T, Koba H, et al. Serum surfactant proteins A and D as prognostic factors in idiopathic pulmonary fibrosis and their relationship to disease extent. Am J Respir Crit Care Med. 2000;162(3 Pt 1):1109–1114. | ||
Prasse A, Probst C, Bargagli E, et al. Serum CC-chemokine ligand 18 concentration predicts outcome in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2009;179(8):717–723. | ||
Zuo F, Kaminski N, Eugui E, et al. Gene expression analysis reveals matrilysin as a key regulator of pulmonary fibrosis in mice and humans. Proc Natl Acad Sci U S A. 2002;99(9):6292–6297. | ||
Rosas IO, Richards TJ, Konishi K, et al. MMP1 and MMP7 as potential peripheral blood biomarkers in idiopathic pulmonary fibrosis. PLoS Med. 2008;5:e93. | ||
Herazo-Maya JD, Noth I, Duncan SR, et al. Peripheral blood mononuclear cell gene expression profiles predict poor outcome in idiopathic pulmonary fibrosis. Sci Transl Med. 2013;5(205):205ra136. | ||
Song JW, Do KH, Jang SJ, Colby TV, Han S, Kim DS. Blood biomarkers MMP-7 and SP-A: predictors of outcome in idiopathic pulmonary fibrosis. Chest. 2013;143(5):1422–1429. | ||
Spagnolo P, Sverzellati N, Rossi G, et al. Idiopathic pulmonary fibrosis: an update. Ann Med. January 22, 2015. [Epub ahead of print]. | ||
Bouros D, Tzouvelekis A. Idiopathic pulmonary fibrosis: on the move. Lancet Respir Med. 2014;2(1):17–19. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.