Back to Journals » Journal of Blood Medicine » Volume 7
Update and new approaches in the treatment of Castleman disease
Authors Chan K, Lade S, Prince HM, Harrison S
Received 22 March 2016
Accepted for publication 5 May 2016
Published 3 August 2016 Volume 2016:7 Pages 145—158
DOI https://doi.org/10.2147/JBM.S60514
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin Bluth
Video abstract presented by Kah-Lok Chan.
Views: 375
Kah-Lok Chan,1 Stephen Lade,2 H Miles Prince,1,3 Simon J Harrison1,3
1Department of Haematology, 2Department of Anatomical Pathology, Peter MacCallum Cancer Centre, 3Sir Peter MacCallum Department of Oncology, The University of Melbourne, Melbourne, VIC, Australia
Abstract: First described 60 years ago, Castleman disease comprises a rare and heterogeneous cluster of disorders, characterized by lymphadenopathy with unique histological features and associated with cytokine-driven constitutional symptoms and biochemical disturbances. Although unicentric Castleman disease is curable with complete surgical excision, its multicentric counterpart is a considerable therapeutic challenge. The recent development of biological agents, particularly monoclonal antibodies to interleukin-6 and its receptor, allow for more targeted disease-specific intervention that promises improved response rates and more durable disease control; however, further work is required to fill knowledge gaps in terms of underlying pathophysiology and to facilitate alternative treatment options for refractory cases.
Keywords: Castleman disease, angiofollicular lymph node hyperplasia, biologics, siltuximab, tocilizumab, rituximab
Introduction
Castleman disease (CD), otherwise known as angiofollicular lymph node hyperplasia, was first reported in 1954 by Benjamin Castleman, who described a 40-year-old male with a mediastinal mass characterized histologically by lymph node hyperplasia and follicles with small, hyalinized foci.1 Subsequent contributions to the literature have delineated CD as a heterogeneous cluster of disorders, with distinct unicentric CD (UCD) and multicentric CD (MCD) subtypes, and identified the fundamental roles of human herpesvirus-8 (HHV-8) and interleukin-6 (IL-6) in a significant proportion of cases. These advances have paved the way for the introduction of novel targeted therapies into the CD armamentarium, particularly monoclonal antibodies directed against IL-6 and its receptor. In this paper, we review the current understanding of CD classification and pathophysiology, summarize current and emerging therapeutic strategies, and outline potential avenues for future research.
Epidemiology
The epidemiology of CD is difficult to characterize accurately due to its rarity and clinical heterogeneity. Based on analysis of insurance claims from two American databases, the incidence of CD is estimated at 21–25 cases per million person-years, with 23% of those cases potentially representing MCD.2 The incidence of human immunodeficiency virus (HIV)/HHV-8-associated MCD also appears to be increasing, from 2.3/10,000 patient-years prior to highly active antiretroviral therapy (1983–1996) to 8.3/10,000 patient-years after widespread implementation of highly active antiretroviral therapy (2002–2007).3
Extrapolating from larger case series,4–11 the median age at presentation for UCD is much lower (30–34 years) than for HIV-negative MCD (49–66 years), with HIV-positive MCD falling in between (36–40 years). The sex distribution is approximately equal, though some series have reported a male predominance, generally in the HIV-positive population. Limited data exist regarding geographic variations, though there is a predominance of HIV- and HHV-8-negative MCD in the Polynesian population, with more favorable disease outcome.12 Although other geographic variations likely exist, it is unclear whether these relate mainly to epidemiological characteristics or if patterns of clinical presentation also differ according to location.
Etiology and pathogenesis
Despite notable advances over the past 20 years, the overall understanding of the etiology and pathogenesis of CD remains limited. MCD may be subdivided by HHV-8 status, because all cases of HHV-8-related MCD appear to be a unified clinicopathological entity with consistent clinical features and outcomes, irrespective of HIV status.10 As all HIV-positive MCD cases are strongly associated with HHV-8 infection,6,11,13 the distinct phenomenon of HIV- and HHV-8-negative MCD has been termed “idiopathic MCD”, reflecting the paucity of knowledge regarding its pathophysiology.14 UCD is not typically associated with HIV or HHV-8 infection. Nevertheless, the discovery of the link to HHV-8 and the causal role of IL-6 have provided valuable insights into CD pathophysiology and formed a foundation for development of new treatment strategies and ongoing research.
HHV-8 in MCD and other associated disorders
HHV-8, or Kaposi’s sarcoma-associated herpesvirus, is a gamma herpesvirus that was first identified in 1994 from biopsy samples of cutaneous Kaposi’s sarcoma.15 Since that time, it has also been strongly implicated in the pathogenesis of acquired immunodeficiency syndrome-related primary effusion lymphoma,16,17 as well as a subset of MCD cases. HHV-8 has the capacity to infect many different cell types, including B-lymphocytes, macrophages, and endothelial cells;18 given that Kaposi’s sarcoma is of endothelial origin19 while primary effusion lymphoma and MCD are B-cell lymphoproliferative disorders, it is probable that the type of HHV-8-infected cell (as well as the presence or absence of other potential contributory factors, such as HIV coinfection) determines the spectrum of disease that manifests. In MCD, it appears that HHV-8 appears to preferentially infect IgM-positive memory B-cells, inducing their proliferation and differentiation into the characteristic plasmablast phenotype observed.20 To date, it remains uncertain what exactly influences this process in MCD compared to other HHV-8-associated B-cell lymphoproliferative disorders, where varying extents of lymphoplasmacytic differentiation are seen.
Akin to other gamma herpesviruses, HHV-8 undergoes latent and lytic phases in its replication cycle – in the latent phase, gene expression is highly restricted, whereas in the lytic phase, gene expression is upregulated and viral replication is enabled.21 In both latent and lytic phases, a recurring theme is the distinctive ability of HHV-8 to produce viral homologs of endogenous human regulatory proteins, facilitating a variety of functions, including viral persistence, inhibition of apoptosis, and cytokine generation.18
The pathogenic contribution of IL-6
One of the earliest links between IL-6 and CD was the demonstration that the site of excess IL-6 production in CD patients was within the germinal centers of involved lymph nodes.22 Following lymph node resection in a patient with UCD, there was a correlation between the reduction in patient symptoms and the fall in serum IL-6, acute phase reactants such as C-reactive protein (CRP) and hypergammaglobulinemia.
Further data supporting this association came from separate experiments involving transgenic mice constitutively expressing murine IL-623 and viral IL-6 (vIL-6),24 both of which developed an MCD-like syndrome comprising splenomegaly, multifocal lymphadenopathy, hypergammaglobulinemia, and plasmacytosis. However, when the experiment involving vIL-6 was replicated in a murine IL-6 knockout model, the features of MCD did not develop, implying that endogenous IL-6 plays a vital role in the pathogenesis of HHV-8-associated MCD.
The need for endogenous IL-6 may be explained by the greater relative potency of human IL-6 (hIL-6) compared to vIL-6.25 Overproduction of hIL-6 may occur via activation of the nuclear factor kappa B pathway,26 as well as HHV-8 latency-associated nuclear antigen (LANA)-mediated upregulation of IL-6 transcription.27 vIL-6 has also been shown to induce hIL-6 production in a variety of cell lines, including those from MCD patients,28 and promotes the secretion of vascular endothelial growth factor (VEGF), resulting in angiogenesis.29 This may contribute to the prominent capillary proliferation that is a histological characteristic of the hyaline vascular subtype of CD. Nevertheless, despite the greater potency of hIL-6, Polizzotto et al30 demonstrated that hIL-6 and vIL-6 can independently or jointly lead to clinical flares of HHV-8-associated MCD. Furthermore, flares were more severe when levels of hIL-6 and vIL-6 were concurrently elevated, suggesting that both human and viral cytokines contribute to disease severity.
The anemia that is commonly seen in MCD appears to be mediated by IL-6-driven hepcidin overproduction, based on a report of anti-IL-6 monoclonal antibody administration to two CD patients with anemia and elevated serum hepcidin levels, which resulted in a rapid decline in hepcidin levels and correction of anemia.31 Anti-IL-6 monoclonal antibody therapy also effectively abolishes CRP production in vivo, establishing it as a convenient marker of IL-6 bioactivity and facilitating monitoring of pharmacodynamic effects.32
Clinical manifestations
The majority of patients with UCD present with isolated lymphadenopathy and may therefore be asymptomatic or have symptoms relating to mass effects on surrounding structures. From a systematic case review including 278 patients with UCD, the mean size of involved lymph nodes at baseline was 5.5 cm, compared to 3.8 cm for MCD cases.33 In this series, the main sites of disease were the chest (29%), neck (23%), abdomen (21%), and retroperitoneum (17%), though other lymph node groups (axillary, inguinal) and the pelvis were also potential sites of involvement.
Systemic symptoms are a common feature of MCD, in keeping with the elevated cytokine levels that are a key driver in its pathogenesis. These include traditional “B” symptoms such as fever, night sweats, and weight loss, as well as generalized lymphadenopathy and hepatosplenomegaly. More severe disease phenotypes include a severe, inflammatory vascular leak syndrome where patients may develop ascites, pericardial effusions, pleural effusions, and/or peripheral edema.5 Hematological associations include anemia driven by IL-6 or secondary to autoimmune hemolysis, immune thrombocytopenia,34 and acquired factor VIII deficiency.35 Bronchiolitis obliterans,5 glomerulonephritis,9 and pemphigus have also been reported, with the presence of pemphigus associated with an unfavorable prognosis.9
MCD may be seen in association with the POEMS syndrome, which comprises polyneuropathy, organomegaly, endocrinopathy, monoclonal protein, and skin changes.36 A clinicopathological variant of CD has also recently been reported in Japan, characterized by low-volume lymphadenopathy with CD histology (usually mixed-type or, less commonly, the hyaline vascular variant) in association with thrombocytopenia, ascites, myelofibrosis, renal dysfunction, and organomegaly, termed the TAFRO syndrome.37
Diagnosis
Given the rarity of CD and the typically nonspecific nature of symptoms, a high index of suspicion is required in patients presenting with a compatible clinical picture, such as unifocal or generalized lymphadenopathy, splenomegaly, and/or unexplained constitutional symptoms. Common laboratory findings include anemia, thrombocytopenia, hypoalbuminemia, polyclonal hypergammaglobulinemia, and elevation of acute phase reactants, such as CRP. Elevations in circulating cytokines such as IL-6 and VEGF may also be detected – although not essential for diagnosis, plasma VEGF levels can be useful in distinguishing and monitoring cases of CD associated with POEMS syndrome.38
Imaging findings are nonspecific and may demonstrate lymphadenopathy and organomegaly, as well as other features of disease, such as sclerotic bony lesions or pulmonary infiltrates. 18F-fluorodeoxyglucose positron emission tomography scanning may have clinical utility in terms of identifying suitable biopsy targets, tracking disease activity, and monitoring response to treatment.39–41
The diagnosis of CD is made on analysis of involved lymph node tissue, showing classic histological features as described. Excisional lymph node biopsy is preferred for diagnosis, particularly if the patient appears to have UCD. Immunohistochemical staining for LANA or polymerase chain reaction testing can be used to detect the presence of HHV-8 within lymph node sections. Detection of plasma HHV-8 viral DNA is not helpful because the test is not specific18 and will be negative in idiopathic cases. Histological confirmation therefore remains essential.
Pathological subtypes
CD is principally distinguished by the presence of localized or generalized lymphadenopathy, designated UCD and MCD, respectively. CD may also be classified according to specific histological features, as well as the presence or absence of viral coinfection.
Histological variants
- The hyaline vascular variant, which was described in Castleman’s original reports,1,42 usually presents as a unicentric lesion, often thoracic, involving a single lymph node or group of nodes. The histological pattern is usually diagnostic. Typically, there are multiple tight aggregates of follicular dendritic cells (FDCs) or atrophic follicles, often with radially penetrating vessels, contained within expanded circumscribed clusters of mantle zone lymphocytes. The lymphocytes often form concentric “onion skin” layers around the dendritic cells. The vascular pattern between the small lymphoid nodules is also typically prominent, and perivascular hyalinization may be a feature (Figure 1). FDC atypia or “dysplasia” is not uncommon. There is an association between this form of the disease, various vascular tumors, and dendritic cell proliferations, such as dendritic cell sarcomas.
- The plasma cell variants of CD (PCCD) are less well-defined histologically, and often the diagnosis is one of exclusion. It is more commonly multicentric than unicentric and is often associated with constitutional symptoms, cytopenias, hypergammaglobulinemia, splenomegaly, and increased IL-6 levels. The lymph nodes usually show preserved architecture, variable follicular hyperplasia, and marked paracortical plasmacytosis43 (Figure 2). The FDC meshworks are usually preserved, and FDC atypia is not a feature. The vascular pattern is often unremarkable, although a subset of patients may show some hyaline vascular changes (the so-called “mixed” pattern). Unfortunately, the histological features of PCCD may be replicated in a variety of reactive, inflammatory, and infective conditions, including HIV-associated lymphadenopathy and other immunodeficiencies, autoimmune diseases (in particular, rheumatoid arthritis), and IgG4-related lymphadenopathy. Some B-cell lymphomas and plasmacytic tumors may also cause confusion.
- A proportion of the multicentric PCCD cases are associated with HIV infection, and virtually all of these are HHV-8-positive. HHV-8 has also been demonstrated in a significant number of the HIV-negative multicentric PCCD cases. The latter group of patients are often from areas with endemic HHV-8 infection. The term “plasmablastic variant” has been used for these HHV-8-associated cases, identifying the distinctive IgM-positive “plasmablasts” present in the expanded mantle zones in involved lymph nodes. However, the immunophenotype of these cells is not truly plasmablastic, as they may be positive for CD20, and CD138 is often negative. They are usually lambda light chain restricted and positive for HHV-8 antigen, but polyclonal on molecular testing. There is a significant risk of progression of these HHV-8-associated cases to an unusual form of diffuse large B-cell lymphoma (WHO designation – large B-cell lymphoma arising in HHV-8-associated MCD). These tumors are also IgM and lambda light chain positive.44,45
 | Figure 1 The hyaline vascular variant of CD. Notes: Hematoxylin and eosin–stained sections (panel A, 200× magnification) demonstrate a vascular, atrophic germinal center with surrounding concentric “onion skin” layers of lymphocytes. Immunohistochemical staining for CD23 (panel B, 200× magnification) highlights several follicular dendritic cell clusters within an expanded mantle zone of CD23-positive cells. CD34 (panel C, 200× magnification) identifies a positively staining blood vessel within an atrophic follicle. Abbreviation: CD, Castleman disease. |
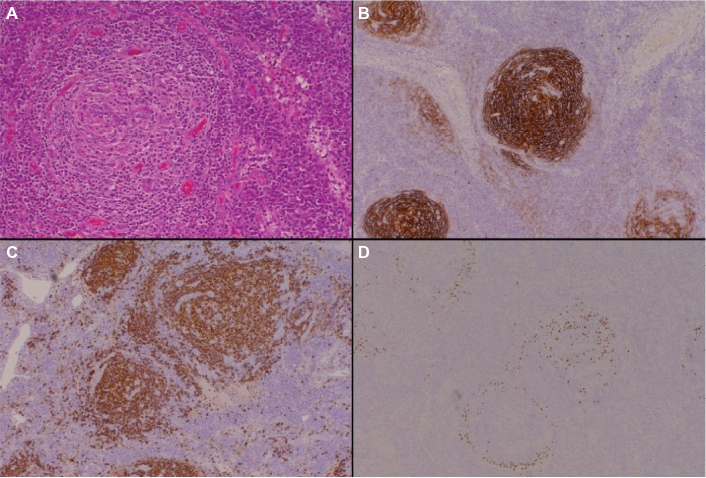 | Figure 2 The plasma cell variant of CD. Notes: Hematoxylin and eosin–stained sections (panel A, 200× magnification) demonstrate preservation of the lymph node germinal center, with associated paracortical plasmacytosis. Immunohistochemical staining for CD21 (panel B, 100× magnification) highlights the follicular dendritic cells within the germinal centers, while CD20 (panel C, 100× magnification) identifies the normal background B-cell population. Immunoperoxidase staining for HHV-8 (panel D, 100× magnification) reveals HHV-8-positive plasma cells within the expanded mantle zones, which play a key role in the pathogenesis of this CD variant. Abbreviations: CD, Castleman disease; HHV-8, human herpesvirus-8. |
Due to the inherent complexities, diagnosing CD (and excluding potential alternative processes) requires careful assessment of clinical, biochemical, radiographic, and histological features. Table 1 summarizes the different subtypes of CD discussed earlier, as well as notable disease associations and important differential diagnoses that need to be considered, particularly if patients do not respond as anticipated to treatment.
 | Table 1 Summary of CD subtypes and important differential diagnoses Note: “–” there are no relevant differential diagnoses for these categories. Abbreviations: CD, Castleman disease; HIV, human immunodeficiency virus; HHV-8, human herpesvirus-8. |
Overview of current therapeutic strategies and outcomes
Unicentric Castleman disease
Complete surgical resection is curative for UCD, leading to excellent long-term outcomes with 10-year overall survival rates in excess of 95%.33 If this is not feasible, the optimal treatment strategy is not well-defined. Debulking surgery should be considered, particularly if there are local symptoms at presentation or if there is a risk of compressing nearby critical structures. Systemic options (as used for MCD, detailed in the “Chemotherapy” section) may also be utilized, potentially facilitating a reduction in lymph node size to an extent, where subsequent resection is possible.46
Although the available literature is confined to a small number of cases,47–58 as summarized in Table 2, radiotherapy appears to be a reasonable alternative treatment option in unresectable cases of UCD. Of the 17 identified patients that were treated with radiotherapy alone, the majority received 40–45 Gy (range 27–60), with six patients achieving complete response (CR, 35%) and seven achieving partial response (PR, 41%). After median follow-up of 20 months (range 5–175), three patients (18%) had died, though only one death was disease-related.
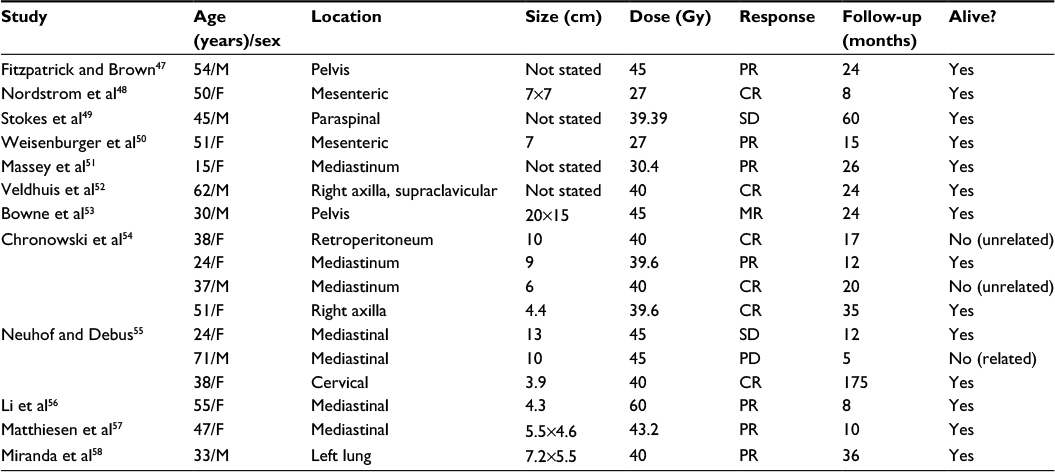 | Table 2 Summary of radiotherapy outcomes in unresectable UCD Abbreviations: UCD, unicentric Castleman disease; cm, centimeters; Gy, Gray; M, male; F, female; SD, stable disease; MR, minor response; PR, partial response; CR, complete response; PD, progressive disease. |
Multicentric Castleman disease
A range of systemic therapies have been utilized in MCD, including cytotoxic chemotherapy, antibodies directed against CD20 as well as IL-6 and its receptor, immunomodulators, bortezomib, and antiviral agents. While they all appear to have disease activity, the literature documenting their use is mainly confined to case reports or small series of patients, limiting overall assessment of efficacy and direct comparisons between regimens.
Chemotherapy
Chemotherapeutic options include the following:
- Low-dose single-agent chemotherapy, such as daily oral etoposide59 or intermittent etoposide or vinblastine13 – these may facilitate symptom relief, but disease control tends to be lost rapidly after treatment cessation.
- Single-agent cladribine is efficacious60,61 and, from a report of two cases (one idiopathic, one HHV-8-related), can result in complete remission lasting up to 24 months. However, both patients in this report subsequently developed non-Hodgkin lymphoma, which was refractory to salvage chemotherapy.61
- Combination chemotherapy, such as CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisolone) or equivalent,54,62,63 as summarized in Table 3. From limited patient numbers (22), these regimens have shown a reasonable rate of CR (eight patients, 36%), with the majority of patients alive (19, 86%) at an overall median follow-up of 38 months. Of note, in the study by Zhu et al,63 all patients were HIV-negative, whereas the other two studies did not report HIV or HHV-8 status.
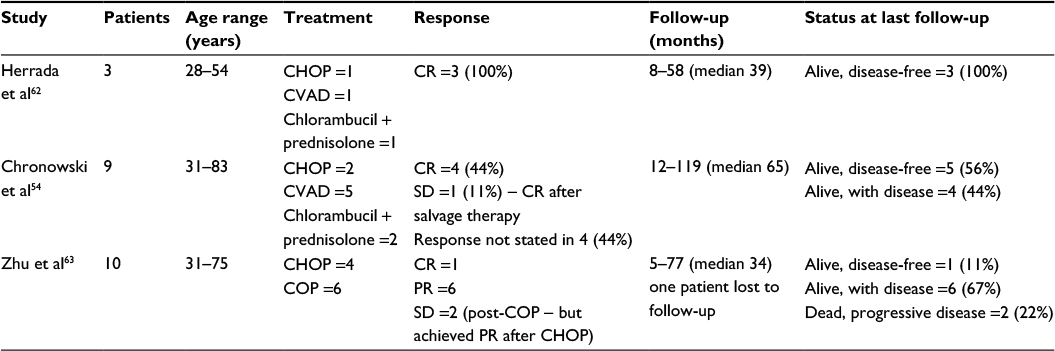 | Table 3 Summary of combination chemotherapy in MCD Abbreviations: MCD, multicentric Castleman disease; CHOP, cyclophosphamide, doxorubicin, vincristine, prednisolone; CVAD, cyclophosphamide, vincristine, doxorubicin, dexamethasone; COP, cyclophosphamide, vincristine, prednisolone; SD, stable disease; PR, partial response; CR, complete response. |
Rituximab
Rituximab, a humanized monoclonal antibody to CD20 (which is typically found on CD plasmablasts), has shown significant activity in HIV-positive and idiopathic MCD patients, when used either as monotherapy or in combination with chemotherapy. The strongest evidence to support its use is in the HIV-positive population, with single-agent rituximab achieving sustained remissions in the majority of patients (treated with four infusions of 375 mg/m2 at weekly intervals) from two Phase II studies. In the first study of 24 patients by Gérard et al,64 the overall survival rate was 92% at 1 year, with 71% in ongoing remission. In the second study of 21 patients by Bower et al,65 the overall survival rate was 95% at 2 years, with 79% in ongoing remission. Toxicity was mild in both groups, with reactivation of Kaposi’s sarcoma being the major adverse effect. A small study of three patients also demonstrated the efficacy of rituximab monotherapy in the idiopathic MCD cohort, with two patients achieving durable PR (with 16 and 40 months of follow-up) and the last patient having a significant decrease in inflammatory markers, maintained for over 10 months with subsequent chemotherapy.66
Rituximab has also been combined with chemotherapy in the HIV-positive population, with the largest patient numbers receiving rituximab with intravenous etoposide or liposomal doxorubicin. In the first study of 14 patients by Bower et al6 (rituximab and etoposide), the overall survival rate was 86% at 2 years, compared to 97% in their rituximab monotherapy cohort. In the second study of 17 patients by Uldrick et al67 (rituximab and liposomal doxorubicin), the overall survival rate was 81% at 3 years, with 69% in ongoing remission.
From retrospective analyses, patients who received rituximab-containing regimens achieved superior CR rates and overall survival compared to patients who received non-rituximab-containing regimens,68,69 as well as a reduced risk of developing subsequent non-Hodgkin lymphoma.69 Rituximab also had significant efficacy in the retreatment of relapsed HIV-associated MCD,68 though the patient numbers in this subgroup were small. These data support the use of rituximab in MCD cases where anti-IL-6 therapy is not available; however, larger patient numbers and more mature follow-up data are required to ascertain if addition of chemotherapy improves outcomes compared to rituximab monotherapy.
Antiviral agents
Limited trials of antiviral agents in HIV- and HHV-8-associated MCD have been undertaken. One study by Uldrick et al70 utilized high-dose zidovudine and valganciclovir in 14 patients, with 86% achieving major clinical response (defined as 50% or greater reduction in signs and symptoms) and 50% achieving major biochemical response (50% or greater improvement in disease-associated laboratory abnormalities). The median progression-free survival was 6 months, and the major toxicities were hematological, with five patients developing Grade 4 anemia and four patients developing Grade 4 neutropenia.
Ganciclovir has also been shown to induce clinical and virologic improvement in three patients, though one patient died from overwhelming fungal infection shortly after commencement and another patient had only short-term benefit.71 Cidofovir, another antiviral agent active against HHV-8, did not have meaningful effect in MCD.72
Based on these studies, it remains unclear how best to incorporate antiviral agents into MCD therapy. While hematological toxicity may prohibit the combination of high-dose zidovudine and valganciclovir, valganciclovir or ganciclovir could be combined with rituximab and/or chemotherapy, or used as maintenance therapy after more effective upfront regimens.
Other
Corticosteroids may offer effective symptom relief but, as the duration of response is typically limited, their main role is in combination with chemotherapy (such as CHOP) or other MCD treatments. Case reports and small series of interferon α,73–75 thalidomide,76–79 lenalidomide,80 bortezomib,81–85 and autologous stem cell transplantation86–88 have also been published (Table 4), providing grounds for larger trials of these therapies as well as supporting their potential use as salvage options in relapsed, refractory disease.
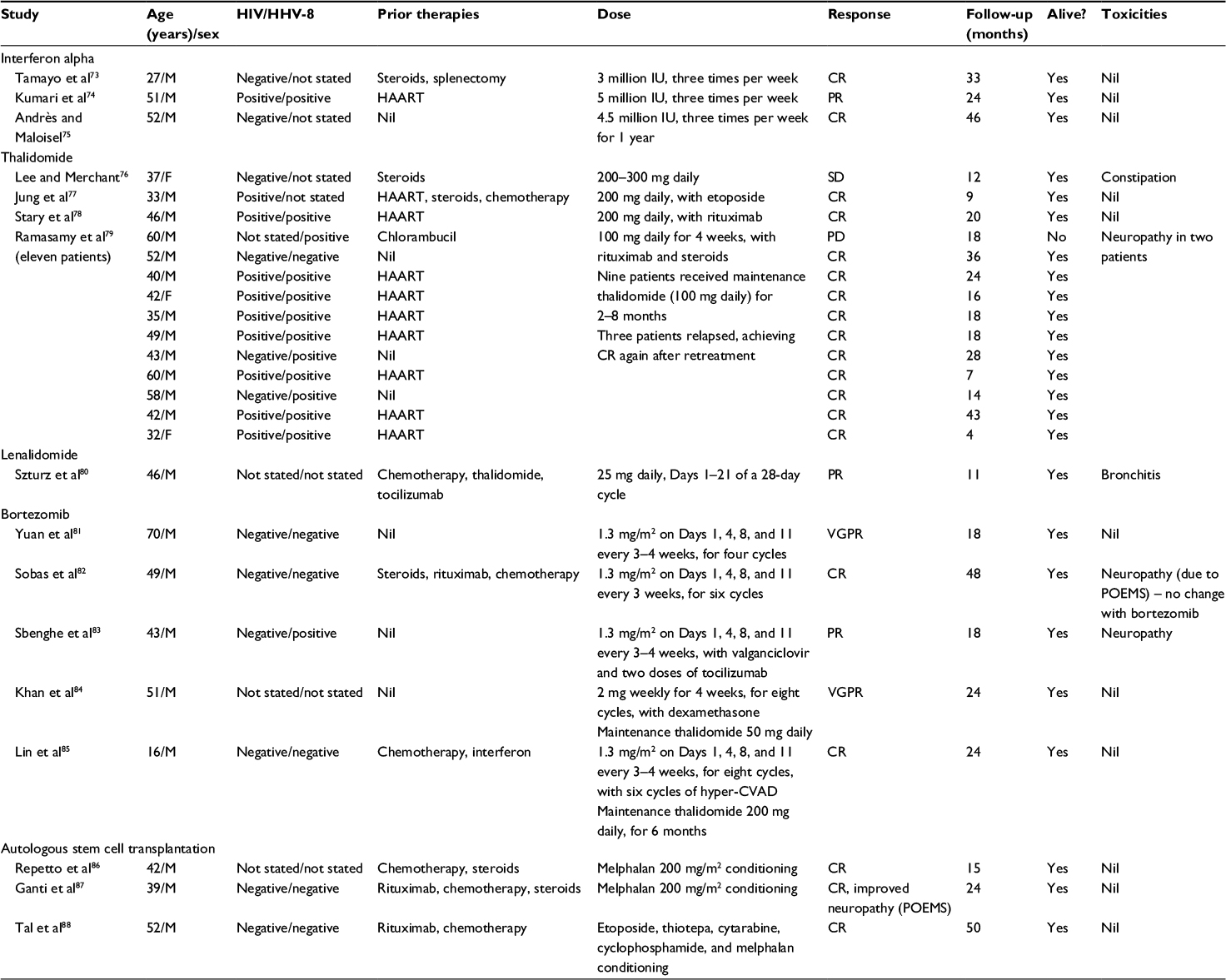 | Table 4 Summary of immunomodulatory drugs and autologous stem cell transplantation in MCD Abbreviations: MCD, multicentric Castleman disease; HIV, human immunodeficiency virus; HHV-8, human herpesvirus-8; M, male; F, female; IU, international units; HAART, highly active antiretroviral therapy; SD, stable disease; PR, partial response; VGPR, very good partial response; CR, complete response; hyper-CVAD, hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone; POEMS, polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes. |
Emerging use of biologics
Anti-IL-6 therapy
The advent of monoclonal antibody therapy targeting the IL-6 pathway has significantly impacted the treatment of patients with idiopathic MCD. Agents that have been studied include siltuximab, a chimeric monoclonal antibody to IL-6, and tocilizumab, a recombinant humanized monoclonal antibody that blocks the IL-6 receptor (Table 5). Siltuximab has regulatory approval in the USA and Europe for the treatment of HIV-negative and HHV-8-negative MCD, while tocilizumab has approval for use in Japan. Access to these therapies is currently obtained through clinical use agreements in their respective countries.
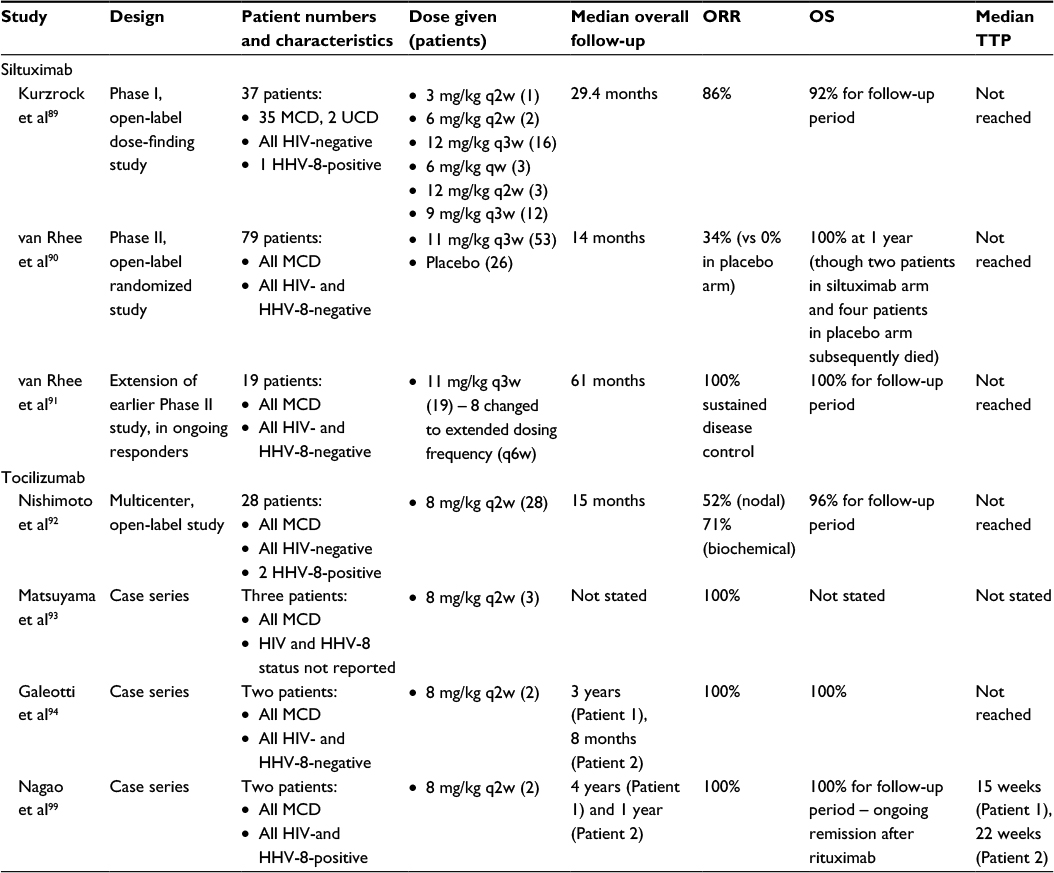 | Table 5 Summary of clinical trials and case series for siltuximab and tocilizumab in MCD Abbreviations: MCD, multicentric Castleman disease; ORR, overall response rate; OS, overall survival; TTP, time to progression; HIV, human immunodeficiency virus; HHV-8, human herpesvirus-8; UCD, unicentric Castleman disease; qw, weekly; q2w, every 2 weeks; q3w, every 3 weeks; q6w, every 6 weeks. |
Siltuximab
Siltuximab is a chimeric (human/murine) IgG1 monoclonal antibody that complexes with IL-6 and prevents it from binding to both soluble and membrane-bound IL-6 receptors. In the initial Phase I study by Kurzrock et al,89 siltuximab was trialed in patients with non-Hodgkin lymphoma, multiple myeloma, and idiopathic MCD, with escalating dose cohorts ranging from 3 to 12 mg/kg every 2 weeks. A total of 37 MCD patients were enrolled, with 32 patients (86%) deriving clinical benefit response, defined as improvement (and no worsening of the remaining categories) in anemia, fatigue, fever or night sweats, increase in weight, and reduction of lymphadenopathy. Overall survival was 92% during a median follow-up period of 2.4 years.
In the follow-up Phase II randomized multicenter study by van Rhee et al,90 79 idiopathic MCD patients were randomized in a 2:1 fashion to siltuximab (11 mg/kg every 3 weeks) or placebo. The primary endpoint was durable tumor and symptomatic response, defined as a complete or partial nodal response, as well as stabilization of disease-related symptoms for at least 18 weeks. A total of 18 patients (34% – 1 CR, 17 PR) in the siltuximab arm achieved the primary endpoint, compared to 0% receiving placebo. Furthermore, 30 patients (57%) had a durable symptomatic response, compared to five patients (19%) in the placebo arm. The 1-year survival rate was 100% in the siltuximab group and 92% in the placebo group. In the extension study,91 all 19 patients continued to receive siltuximab for up to 7 years, without evidence of cumulative toxicity or treatment discontinuation.
Siltuximab appears to be well-tolerated, with no dose-limiting toxicities observed in its Phase I study. In the Phase II study, pruritus was the most common adverse event (42%), followed by upper respiratory tract infection (36%), fatigue (34%), maculopapular rash (34%), and peripheral edema (32%). Infusion reactions occurred in 8% of patients, with one being Grade 3 in severity (anaphylactic). Only one episode of sepsis was reported, which is notable given that patients on anti-IL-6 therapy may have delayed presentation with infection due to suppression of typical clinical features mediated by IL-6, such as fever or elevated inflammatory markers like CRP.32 No treatment-related deaths occurred.
Tocilizumab
Unlike siltuximab, tocilizumab is a recombinant humanized IgG1 monoclonal antibody that attaches to soluble and membrane-bound IL-6 receptors, blocking IL-6 binding and downstream intracellular signaling. The major multicenter, open-label study of tocilizumab enrolled 28 patients, of whom 26 had idiopathic MCD and two were HIV-negative but positive for HHV-8.92 All patients received 8 mg/kg of tocilizumab for 16 weeks, after which doses varied according to investigator discretion as part of a study extension. Tocilizumab therapy resulted in significant reduction in lymphadenopathy (30% reduction in the mean short axis of involved lymph nodes) for 52% of patients after 1 year of treatment. Marked improvement was also seen in biochemical measures such as CRP and fibrinogen in 64% and 71% of patients, respectively. Most patients (27, 96%) received tocilizumab for over 3 years, without evidence of disease progression.
Major reported adverse effects of tocilizumab from the previous study included upper respiratory tract infections (57%), pruritus (21%), malaise (21%), pharyngitis (18%), and diarrhea (18%). Fourteen patients (50%) experienced infusion reactions, though all were transient and mild. Two patients required admission for cellulitis, but no other serious infections were noted.
Two small case series of tocilizumab in MCD have also been published, with a total of five patients achieving 100% response rates in follow-up ranging up to 3 years.93,94 Single case reports have also suggested efficacy of tocilizumab on potential associated manifestations of MCD, including cardiomyopathy,95 autoimmune hemolytic anemia,96 uveitis,97 and cardiac amyloidosis,98 though these findings require confirmation. However, when used in two patients with HIV- and HHV-8-associated MCD, the response to tocilizumab was very short-lived, implying that IL-6 receptor blockade alone may be inadequate in this subpopulation, and that further studies in this subpopulation are clearly required.99
Anakinra
Anakinra, a recombinant interleukin-1 (IL-1) receptor antagonist, has theoretical benefit in MCD based on the ability of IL-1 to stimulate IL-6 production and the use of both IL-1 and IL-6 antagonists in the treatment of inflammatory disorders such as rheumatoid arthritis. Use of anakinra in MCD remains anecdotal and confined to two case reports, though both patients did achieve dramatic clinical and biochemical responses.100,101
Long-term outcome
The prognosis of CD is variable and depends predominantly on disease subtype. As previously mentioned, patients with UCD enjoy excellent long-term outcomes if complete resection can be achieved, with 10-year overall survival rates greater than 95%.33 Even in unresectable UCD cases, radiotherapy may offer good long-term response rates (82% overall survival at 20 months), though supporting data is limited. The HIV- and HHV-8-associated subtype of MCD has the worst prognosis, with the majority of patients in early studies dying within 2 years of diagnosis.7,44,102 Introduction of rituximab appears to have ameliorated these outcomes somewhat, with 2-year overall survival in rituximab-treated cases improving to 94% in a larger case series.6 Finally, although there is a paucity of survival data for the idiopathic MCD cohort prior to anti-IL-6 therapy, a systematic literature review identified a 3-year disease-free survival rate of 45.7% in HIV-negative patients (though HHV-8 status is unknown).4 The high rates of durable response seen in the early siltuximab and tocilizumab studies provide considerable promise for idiopathic MCD patients, though larger studies are needed to confirm these findings.
Conclusion and future directions
In the 60 years since Castleman’s initial report, substantial advances have been made in terms of characterizing the pathophysiology of CD, defining distinct clinicopathological subtypes and developing more effective, targeted treatment strategies. However, in some respects, these revelations raise more questions than answers – particularly in MCD, where much is known about the pathophysiology of HIV- and HHV-8-associated cases, but treatment options remain limited. Conversely, the emerging use of biologic therapies (including monoclonal antibodies targeting the IL-6 pathway) has great potential in treating HIV- and HHV-8-negative cases, but the underlying disease mechanisms in this subgroup are still poorly understood.
Given the rarity and heterogeneity of CD, a high index of clinical suspicion, consistent case definition, and international collaboration – through organizations like the Castleman Disease Collaborative Network103 – will be integral factors in facilitating ongoing discoveries for this fascinating disease, hopefully translating into continued improvements in patient care and more favorable survival outcomes.
Disclosure
SJH has received funding from Janssen-Cilag for the conduct of clinical trials and participation in advisory boards in relation to Castleman disease. The authors report no other conflicts of interest in this work.
References
Case records of the Massachusetts general hospital weekly clinicopathological exercises: case 40011. N Engl J Med. 1954;250(1): | ||
Munshi N, Mehra M, van de Velde H, Desai A, Potluri R, Vermeulen J. Use of a claims database to characterize and estimate the incidence rate for Castleman disease. Leuk Lymphoma. 2015;56(5): | ||
Powles T, Stebbing J, Bazeos A, et al. The role of immune suppression and HHV-8 in the increasing incidence of HIV-associated multicentric Castleman’s disease. Ann Oncol. 2009;20(4):775–779. | ||
Talat N, Schulte KM. Castleman’s disease: systematic analysis of 416 patients from the literature. Oncologist. 2011;16(9):1316–1324. | ||
Dispenzieri A, Armitage JO, Loe MJ, et al. The clinical spectrum of Castleman’s disease. Am J Hematol. 2012;87(11):997–1002. | ||
Bower M, Newsom-Davis T, Naresh K, et al. Clinical features and outcome in HIV-associated multicentric Castleman’s disease. J Clin Oncol. 2011;29(18):2481–2486. | ||
Oksenhendler E, Duarte M, Soulier J, et al. Multicentric Castleman’s disease in HIV infection: a clinical and pathological study of 20 patients. | ||
Robinson D, Reynolds M, Casper C, et al. Clinical epidemiology and treatment patterns of patients with multicentric Castleman disease: results from two US treatment centres. Br J Haematol. 2014;165(1):39–48. | ||
Dong Y, Wang M, Nong L, et al. Clinical and laboratory characterization of 114 cases of Castleman disease patients from a single centre: paraneoplastic pemphigus is an unfavourable prognostic factor. Br J Haematol. | ||
Dossier A, Meignin V, Fieschi C, Boutboul D, Oksenhendler E, Galicier L. Human herpesvirus 8-related Castleman disease in the absence of HIV infection. Clin Infect Dis. 2013;56(6):833–842. | ||
Soulier J, Grollet L, Oksenhendler E, et al. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman’s disease. Blood. 1995;86(4):1276–1280. | ||
Zhai S, Simpson D. Polynesian variant of idiopathic multicentric Castleman disease. Blood. 2013;122(21):5127. | ||
Oksenhendler E, Carcelain G, Aoki Y, et al. High levels of human herpesvirus 8 viral load, human interleukin-6, interleukin-10, and C reactive protein correlate with exacerbation of multicentric Castleman disease in HIV-infected patients. Blood. 2000;96(6):2069–2073. | ||
Fajgenbaum DC, van Rhee F, Nabel CS. HHV-8-negative, idiopathic multicentric Castleman disease: novel insights into biology, pathogenesis, and therapy. Blood. 2014;123(19):2924–2933. | ||
Chang Y, Cesarman E, Pessin MS, et al. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science. 1994;266(5192):1865–1869. | ||
Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Med. 1995;332(18):1186–1191. | ||
Nador RG, Cesarman E, Chadburn A, et al. Primary effusion lymphoma: a distinct clinicopathologic entity associated with the Kaposi’s sarcoma-associated herpes virus. Blood. 1996;88(2):645–656. | ||
Bhutani M, Polizzotto MN, Uldrick TS, Yarchoan R. Kaposi sarcoma-associated herpesvirus-associated malignancies: epidemiology, pathogenesis, and advances in treatment. Semin Oncol. 2015;42(2):223–246. | ||
Flore O, Rafii S, Ely S, O’Leary JJ, Hyjek EM, Cesarman E. Transformation of primary human endothelial cells by Kaposi’s sarcoma-associated herpesvirus. Nature. 1998;394(6693):588–592. | ||
Hassman LM, Ellison TJ, Kedes DH. KSHV infects a subset of human tonsillar B cells, driving proliferation and plasmablast differentiation. J Clin Invest. 2011;121(2):752–768. | ||
Ganem D. KSHV infection and the pathogenesis of Kaposi’s sarcoma. Annu Rev Pathol. 2006;1:273–296. | ||
Yoshizaki K, Matsuda T, Nishimoto N, et al. Pathogenic significance of interleukin-6 (IL-6/BSF-2) in Castleman’s disease. Blood. 1989; | ||
Brandt SJ, Bodine DM, Dunbar CE, Nienhuis AW. Dysregulated interleukin 6 expression produces a syndrome resembling Castleman’s disease in mice. J Clin Invest. 1990;86(2):592–599. | ||
Suthaus J, Stuhlmann-Laeisz C, Tompkins VS, et al. HHV-8-encoded viral IL-6 collaborates with mouse IL-6 in the development of multicentric Castleman disease in mice. Blood. 2012;119(22):5173–5181. | ||
Burger R, Neipel F, Fleckenstein B, et al. Human herpesvirus type 8 interleukin-6 homologue is functionally active on human myeloma cells. | ||
Schwarz M, Murphy PM. Kaposi’s sarcoma-associated herpesvirus G protein-coupled receptor constitutively activates NF-kappa B and induces proinflammatory cytokine and chemokine production via a C-terminal signaling determinant. J Immunol. 2001;167(1): | ||
An J, Lichtenstein AK, Brent G, Rettig MB. The Kaposi sarcoma-associated herpesvirus (KSHV) induces cellular interleukin 6 expression: role of the KSHV latency-associated nuclear antigen and the AP1 response element. Blood. 2002;99(2):649–654. | ||
Mori Y, Nishimoto N, Ohno M, et al. Human herpesvirus 8-encoded interleukin-6 homologue (viral IL-6) induces endogenous human IL-6 secretion. J Med Virol. 2000;61(3):332–335. | ||
Aoki Y, Jaffe ES, Chang Y, et al. Angiogenesis and hematopoiesis induced by Kaposi’s sarcoma-associated herpesvirus-encoded interleukin-6. Blood. 1999;93(12):4034–4043. | ||
Polizzotto MN, Uldrick TS, Wang V, et al. Human and viral interleukin-6 and other cytokines in Kaposi sarcoma herpesvirus-associated multicentric Castleman disease. Blood. 2013;122(26):4189–4198. | ||
Kawabata H, Tomosugi N, Kanda J, Tanaka Y, Yoshizaki K, Uchiyama T. Anti-interleukin 6 receptor antibody tocilizumab reduces the level of serum hepcidin in patients with multicentric Castleman’s disease. Haematologica. 2007;92(6):857–858. | ||
Rossi JF, Lu ZY, Jourdan M, Klein B. Interleukin-6 as a therapeutic target. Clin Cancer Res. 2015;21(6):1248–1257. | ||
Talat N, Belgaumkar AP, Schulte KM. Surgery in Castleman’s disease: a systematic review of 404 published cases. Ann Surg. 2012;255(4):677–684. | ||
Marsh JH, Colbourn DS, Donovan V, Staszewski H. Systemic Castleman’s disease in association with Evan’s syndrome and vitiligo. Med Pediatr Oncol. 1990;18(2):169–172. | ||
Marietta M, Pozzi S, Luppi M, et al. Acquired haemophilia in HIV negative, HHV-8 positive multicentric Castleman’s disease: a case report. Eur J Haematol. 2003;70(3):181–182. | ||
Dispenzieri A, Kyle RA, Lacy MQ, et al. POEMS syndrome: definitions and long-term outcome. Blood. 2003;101(7):2496–2506. | ||
Kawabata H, Takai K, Kojima M, et al. Castleman–Kojima disease (TAFRO syndrome): a novel systemic inflammatory disease characterized by a constellation of symptoms, namely, thrombocytopenia, ascites (anasarca), microcytic anemia, myelofibrosis, renal dysfunction, and organomegaly: a status report and summary of Fukushima (6 June, 2012) and Nagoya meetings (22 September, 2012). J Clin Exp Hematop. 2013; | ||
D’Souza A, Hayman SR, Buadi F, et al. The utility of plasma vascular endothelial growth factor levels in the diagnosis and follow-up of patients with POEMS syndrome. Blood. 2011;118(17): | ||
Robinson H, Prince HM, Ramdave S, Seymour JF, Elliott P, Hicks R. Preliminary experience of 18F-fluorodeoxyglucose positron emission tomography in Castleman’s disease. Leuk Lymphoma. 2006;47(12): | ||
Barker R, Kazmi F, Stebbing J, et al. FDG-PET/CT imaging in the management of HIV-associated multicentric Castleman’s disease. Eur J Nucl Med Mol Imaging. 2009;36(4):648–652. | ||
Polizzotto MN, Millo C, Uldrick TS, et al. 18F-fluorodeoxyglucose positron emission tomography in kaposi sarcoma herpesvirus-associated multicentric Castleman disease: correlation with activity, severity, inflammatory and virologic parameters. J Infect Dis. 2015;212(8): | ||
Castleman B, Iverson L, Menendez VP. Localized mediastinal lymphnode hyperplasia resembling thymoma. Cancer. 1956;9(4): | ||
Keller AR, Hochholzer L, Castleman B. Hyaline-vascular and plasma-cell types of giant lymph node hyperplasia of the mediastinum and other locations. Cancer. 1972;29(3):670–683. | ||
Dupin N, Diss TL, Kellam P, et al. HHV-8 is associated with a plasmablastic variant of Castleman disease that is linked to HHV-8-positive plasmablastic lymphoma. Blood. 2000;95(4):1406–1412. | ||
Cronin DM, Warnke RA. Castleman disease: an update on classification and the spectrum of associated lesions. Adv Anat Pathol. 2009;16(4): | ||
Bandera B, Ainsworth C, Shikle J, Rupard E, Roach M. Treatment of unicentric Castleman disease with neoadjuvant rituximab. Chest. 2010;138(5):1239–1241. | ||
Fitzpatrick PJ, Brown TC. Angiofollicular lymph node hyperplasia. Can Med Assoc J. 1968;99(25):1259–1262. | ||
Nordstrom DG, Tewfik HH, Latourette HB. Plasma cell giant lymph node hyperplasia responding to radiation therapy. AJR Am J Roentgenol. | ||
Stokes SH, Griffith RC, Thomas PR. Angiofollicular lymph node hyperplasia (Castleman’s disease) associated with vertebral destruction. Cancer. 1985;56(4):876–879. | ||
Weisenburger DD, DeGowin RL, Gibson P, Armitage JO. Remission of giant lymph node hyperplasia with anemia after radiotherapy. Cancer. 1979;44(2):457–462. | ||
Massey GV, Kornstein MJ, Wahl D, Huang XL, McCrady CW, Carchman RA. Angiofollicular lymph node hyperplasia (Castleman’s disease) in an adolescent female. Clinical and immunologic findings. Cancer. 1991;68(6):1365–1372. | ||
Veldhuis GJ, van der Leest AH, de Wolf JT, de Vries EG, Vellenga E. A case of localized Castleman’s disease with systemic involvement: treatment and pathogenetic aspects. Ann Hematol. 1996;73(1):47–50. | ||
Bowne WB, Lewis JJ, Filippa DA, et al. The management of unicentric and multicentric Castleman’s disease: a report of 16 cases and a review of the literature. Cancer. 1999;85(3):706–717. | ||
Chronowski GM, Ha CS, Wilder RB, Cabanillas F, Manning J, Cox JD. Treatment of unicentric and multicentric Castleman disease and the role of radiotherapy. Cancer. 2001;92(3):670–676. | ||
Neuhof D, Debus J. Outcome and late complications of radiotherapy in patients with unicentric Castleman disease. Acta Oncol. 2006;45(8):1126–1131. | ||
Li YM, Liu PH, Zhang YH, et al. Radiotherapy of unicentric mediastinal Castleman’s disease. Chin J Cancer. 2011;30(5):351–356. | ||
Matthiesen C, Ramgopol R, Seavey J, Ahmad S, Herman T. Intensity modulated radiation therapy (IMRT) for the treatment of unicentric Castlemans disease: a case report and review of the use of radiotherapy in the literature. Radiol Oncol. 2012;46(3):265–270. | ||
Miranda FA, Faria VH, Arruda GV, Silva LG. Radiation therapy in the treatment of unicentric Castleman’s disease. J Bras Pneumol. 2013;39(1): | ||
Scott D, Cabral L, Harrington WJ. Treatment of HIV-associated multicentric Castleman’s disease with oral etoposide. Am J Hematol. 2001;66(2): | ||
Bordeleau L, Bredeson C, Markman S. 2-Chloro-deoxyadenosine therapy for giant lymph node hyperplasia. Br J Haematol. 1995;91(3):668–670. | ||
Colleoni GW, Duarte LC, Kerbauy FR, et al. 2-Chloro-deoxyadenosine induces durable complete remission in Castleman’s disease but may accelerate its transformation to non-Hodgkin’s lymphoma. Acta Oncol. 2003;42(7):784–787. | ||
Herrada J, Cabanillas F, Rice L, Manning J, Pugh W. The clinical behavior of localized and multicentric Castleman disease. Ann Intern Med. 1998;128(8):657–662. | ||
Zhu SH, Yu YH, Zhang Y, Sun JJ, Han DL, Li J. Clinical features and outcome of patients with HIV-negative multicentric Castleman’s disease treated with combination chemotherapy: a report on 10 patients. Med Oncol. 2013;30(1):492. | ||
Gérard L, Bérezné A, Galicier L, et al. Prospective study of rituximab in chemotherapy-dependent human immunodeficiency virus associated multicentric Castleman’s disease: ANRS 117 CastlemaB Trial. J Clin Oncol. 2007;25(22):3350–3356. | ||
Bower M, Powles T, Williams S, et al. Brief communication: rituximab in HIV-associated multicentric Castleman disease. Ann Intern Med. 2007;147(12):836–839. | ||
Ide M, Kawachi Y, Izumi Y, Kasagi K, Ogino T. Long-term remission in HIV-negative patients with multicentric Castleman’s disease using rituximab. Eur J Haematol. 2006;76(2):119–123. | ||
Uldrick TS, Polizzotto MN, Aleman K, et al. Rituximab plus liposomal doxorubicin in HIV-infected patients with KSHV-associated multicentric Castleman disease. Blood. 2014;124(24):3544–3552. | ||
Hoffmann C, Schmid H, Müller M, et al. Improved outcome with rituximab in patients with HIV-associated multicentric Castleman disease. | ||
Gérard L, Michot JM, Burcheri S, et al. Rituximab decreases the risk of lymphoma in patients with HIV-associated multicentric Castleman disease. Blood. 2012;119(10):2228–2233. | ||
Uldrick TS, Polizzotto MN, Aleman K, et al. High-dose zidovudine plus valganciclovir for Kaposi sarcoma herpesvirus-associated multicentric Castleman disease: a pilot study of virus-activated cytotoxic therapy. Blood. 2011;117(26):6977–6986. | ||
Casper C, Nichols WG, Huang ML, Corey L, Wald A. Remission of HHV-8 and HIV-associated multicentric Castleman disease with ganciclovir treatment. Blood. 2004;103(5):1632–1634. | ||
Berezne A, Agbalika F, Oksenhendler E. Failure of cidofovir in HIV-associated multicentric Castleman disease. Blood. 2004;103(11):4368–4369; author reply 4369. | ||
Tamayo M, Gonzalez C, Majado MJ, Candel R, Ramos J. Long-term complete remission after interferon treatment in a case of multicentric Castelman’s disease. Am J Hematol. 1995;49(4):359–360. | ||
Kumari P, Schechter GP, Saini N, Benator DA. Successful treatment of human immunodeficiency virus-related Castleman’s disease with interferon-alpha. Clin Infect Dis. 2000;31(2):602–604. | ||
Andrès E, Maloisel F. Interferon-alpha as first-line therapy for treatment of multicentric Castleman’s disease. Ann Oncol. 2000;11(12):1613–1614. | ||
Lee FC, Merchant SH. Alleviation of systemic manifestations of multicentric Castleman’s disease by thalidomide. Am J Hematol. 2003;73(1):48–53. | ||
Jung CP, Emmerich B, Goebel FD, Bogner JR. Successful treatment of a patient with HIV-associated multicentric Castleman disease (MCD) with thalidomide. Am J Hematol. 2004;75(3):176–177. | ||
Stary G, Kohrgruber N, Herneth AM, Gaiger A, Stingl G, Rieger A. Complete regression of HIV-associated multicentric Castleman disease treated with rituximab and thalidomide. AIDS. 2008;22(10):1232–1234. | ||
Ramasamy K, Gandhi S, Tenant-Flowers M, et al. Rituximab and thalidomide combination therapy for Castleman disease. Br J Haematol. 2012;158(3):421–423. | ||
Szturz P, Adam Z, Chovancová J, et al. Lenalidomide: a new treatment option for Castleman disease. Leuk Lymphoma. 2012;53(10): | ||
Yuan ZG, Dun XY, Li YH, Hou J. Treatment of multicentric Castleman’s disease accompanying multiple myeloma with bortezomib: a case report. J Hematol Oncol. 2009;2:19. | ||
Sobas MA, Alonso Vence N, Diaz Arias J, Bendaña Lopez A, Fraga Rodriguez M, Bello Lopez JL. Efficacy of bortezomib in refractory form of multicentric Castleman disease associated to poems syndrome (MCD-POEMS variant). Ann Hematol. 2010;89(2):217–219. | ||
Sbenghe MM, Besa E, Mahipal A, Dulau Florea A, Bray P, Caro J. HHV-8-associated multicentric Castleman’s disease in HIV-negative patient: a novel therapy for an orphan disease. Oncologist. 2012;17(1):145–146; author reply 147–148. | ||
Khan AA, Siraj F, Bhargava M, Aggarwal S. Successful treatment of multicentric Castleman’s disease accompanying myeloma with bortezomib. BMJ Case Rep. 2012;2012:pii:bcr2012007646. | ||
Lin Q, Fang B, Huang H, et al. Efficacy of bortezomib and thalidomide in the recrudescent form of multicentric mixed-type Castleman’s disease. Blood Cancer J. 2015;5:e298. | ||
Repetto L, Jaiprakash MP, Selby PJ, Gusterson BA, Williams HJ, McElwain TJ. Aggressive angiofollicular lymph node hyperplasia (Castleman’s disease) treated with high dose melphalan and autologous bone marrow transplantation. Hematol Oncol. 1986;4(3):213–217. | ||
Ganti AK, Pipinos I, Culcea E, Armitage JO, Tarantolo S. Successful hematopoietic stem-cell transplantation in multicentric Castleman disease complicated by POEMS syndrome. Am J Hematol. 2005;79(3):206–210. | ||
Tal Y, Haber G, Cohen MJ, et al. Autologous stem cell transplantation in a rare multicentric Castleman disease of the plasma cell variant. Int J Hematol. 2011;93(5):677–680. | ||
Kurzrock R, Voorhees PM, Casper C, et al. A phase I, open-label study of siltuximab, an anti-IL-6 monoclonal antibody, in patients with B-cell non-Hodgkin lymphoma, multiple myeloma, or Castleman disease. Clin Cancer Res. 2013;19(13):3659–3670. | ||
van Rhee F, Wong RS, Munshi N, et al. Siltuximab for multicentric Castleman’s disease: a randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2014;15(9):966–974. | ||
van Rhee F, Casper C, Voorhees PM, et al. A Phase 2, open-label, multicenter study of the long-term safety of siltuximab (an anti-interleukin-6 monoclonal antibody) in patients with multicentric Castleman disease. Oncotarget. 2015;6(30):30408–30419. | ||
Nishimoto N, Kanakura Y, Aozasa K, et al. Humanized anti-interleukin-6 receptor antibody treatment of multicentric Castleman disease. | ||
Matsuyama M, Suzuki T, Tsuboi H, et al. Anti-interleukin-6 receptor antibody (tocilizumab) treatment of multicentric Castleman’s disease. Intern Med. 2007;46(11):771–774. | ||
Galeotti C, Boucheron A, Guillaume S, Koné-Paut I. Sustained remission of multicentric Castleman disease in children treated with tocilizumab, an anti-interleukin-6 receptor antibody. Mol Cancer Ther. 2012;11(8): | ||
Kanda J, Kawabata H, Yamaji Y, et al. Reversible cardiomyopathy associated with Multicentric Castleman disease: successful treatment with tocilizumab, an anti-interleukin 6 receptor antibody. Int J Hematol. | ||
Yuzuriha A, Saitoh T, Koiso H, et al. Successful treatment of autoimmune hemolytic anemia associated with multicentric Castleman disease by anti-interleukin-6 receptor antibody (tocilizumab) therapy. Acta Haematol. 2011;126(3):147–150. | ||
Oshitari T, Kajita F, Tobe A, et al. Refractory uveitis in patient with castleman disease successfully treated with tocilizumab. Case Rep Ophthalmol Med. 2012;2012:968180. | ||
Shirakawa K, Egashira T, Ieda M, et al. Multidisciplinary approach to the treatment of cardiac AA amyloidosis and aortic stenosis due to Castleman’s disease: a hybrid therapy with tocilizumab and aortic valve replacement. Int J Cardiol. 2014;173(2):e9–e11. | ||
Nagao A, Nakazawa S, Hanabusa H. Short-term efficacy of the IL6 receptor antibody tocilizumab in patients with HIV-associated multicentric Castleman disease: report of two cases. J Hematol Oncol. 2014; | ||
Galeotti C, Tran TA, Franchi-Abella S, Fabre M, Pariente D, Koné-Paut I. IL-1RA agonist (anakinra) in the treatment of multifocal castleman disease: case report. J Pediatr Hematol Oncol. 2008;30(12): | ||
El-Osta H, Janku F, Kurzrock R. Successful treatment of Castleman’s disease with interleukin-1 receptor antagonist (Anakinra). Mol Cancer Ther. 2010;9(6):1485–1488. | ||
Zietz C, Bogner JR, Goebel FD, Löhrs U. An unusual cluster of cases of Castleman’s disease during highly active antiretroviral therapy for AIDS. N Engl J Med. 1999;340(24):1923–1924. | ||
Newman SK, Jayanthan RK, Mitchell GW, et al. Taking control of Castleman disease: leveraging precision medicine technologies to accelerate rare disease research. Yale J Biol Med. 2015;88(4):383–388. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.