")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 15
Unusual Presentation Of Kawasaki Disease With Gastrointestinal And Renal Manifestations
Authors Lazea C , Man O , Sur LM, Serban R , Lazar C
Received 12 August 2019
Accepted for publication 17 October 2019
Published 5 December 2019 Volume 2019:15 Pages 1411—1416
DOI https://doi.org/10.2147/TCRM.S226624
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Cecilia Lazea,1,* Oana Man,2,* Lucia Maria Sur,1 Radu Serban,1 Calin Lazar1
1University of Medicine and Pharmacy, Emergency Clinic Hospital for Children, Department Pediatrics I, Cluj-Napoca, Romania; 2Emergency Clinic Hospital for Children, Department Pediatrics I, Cluj-Napoca, Romania
*These authors contributed equally to this work
Correspondence: Cecilia Lazea
University of Medicine and Pharmacy, Iuliu Hatieganu Cluj-Napoca, Department Pediatrics I, 68, Motilor Street, Cluj-Napoca 400370, Romania
Tel +40744353764
Fax +40264402539
Email [email protected]
Abstract: Diagnosis of Kawasaki disease (KD) is based on well-established clinical criteria. In incomplete or atypical KD, the diagnosis is challenging, because of the paucity of clinical signs or because of the presence of clinical manifestations that generally are not seen in KD. We describe the case of a 3-year-old female patient with persistent high fever, vomiting, watery diarrhea, metabolic acidosis and severe hypopotassemia. On the fourth day of fever, bilateral conjunctivitis, mucous and extremity changes were registered. Urine changes as glycosuria and proteinuria were also noticed. Echocardiography revealed ectasia of the left anterior descending coronary artery, and diagnosis of KD was established. The treatment consisted of intravenous immunoglobulin (IVIG) and oral aspirin. Recurrence of disease was recorded on the 23rd day of the disease, with favorable evolution after the second dose of IVIG was infused.
Keywords: atypical, incomplete, Kawasaki disease, gastrointestinal, renal
Introduction
Kawasaki disease (KD) is an acute, self-limited febrile illness of childhood. It is a pediatric vasculitis syndrome and is characterized by inflammation of predominantly medium-sized blood vessels, with a predilection for the coronary arteries. It mainly, but not exclusively, affects young children, with a higher susceptibility in boys.1–3 KD is considered the leading cause of acquired heart disease in children in most developed countries.4,5 Various pathogenic hypotheses have been postulated, but the current consensus agrees that an (infectious) trigger initiates an abnormal immune response involving innate and adaptive pathways in genetically predisposed children.3,6,7 Diagnosis of KD is based on a constellation of clinical findings that appear in a typical temporal sequence. “Classic KD” is defined by the patient experiencing ≥5 days of fever and showing signs of ≥4 of the five principal clinical features: a. erythema and cracking of lips, strawberry tongue, and/or erythema of oral and pharyngeal mucosa; b. bilateral bulbar conjunctival injection without exudate; c. rash: maculopapular, diffuse erythroderma, or erythema multiforme-like lesions; d. erythema and edema of the hands and feet in the acute phase and/or periungual desquamation in the subacute phase; e. cervical lymphadenopathy (≥1.5 cm diameter), usually unilateral. The term “atypical Kawasaki disease” describes patients who have clinical manifestations that generally are not seen, but have compatible laboratory findings and no other explanation for their illness. Incomplete KD should be considered in children with unexplained fever for ≥5 days associated with fewer than three of the clinical features, based on an algorithm that includes coronary artery abnormalities (CAA) on echocardiography and/or compatible laboratory findings.1–3,8 Unfortunately, these criteria are neither sufficiently sensitive nor sufficiently specific, and there are no pathognomonic tests that can help the clinician in confirming a diagnosis, so it may be delayed or overlooked. Laboratory findings are usually characteristic: leukocytosis with neutrophilia and immature forms, elevated acute-phase reactants, normocytic normochromic anemia, abnormal plasma lipids, hypoalbuminemia, hyponatremia, thrombocytosis after the first week, sterile pyuria, elevated serum transaminases, pleocytosis of the cerebrospinal fluid, leukocytosis in synovial fluid, etc. Two-dimensional echocardiography is a non-invasive imaging method and is considered the most useful test to evaluate coronary arteries, in the acute phase and during follow-up, as well as myocardial function.1–3,9–14 Treatment of acute KD consists of high-dose intravenous immunoglobulin (IVIG) associated with administration of acetylsalicylic acid.
Kawasaki disease is a vasculitis which can involve multiple organs. The renal involvement in KD is rarely described. Herein, we describe the case of a girl aged 3 years and 4 months who was diagnosed with incomplete KD and unusual presentation.
Written informed consent was obtained from the patient’s parents for this presentation. Institutional approval was not required to publish the case details.
Case Report
The patient reported here is a girl aged 3 years and 4 months who was admitted to our department for fever (39.2°C) associated with loss of appetite, vomiting, severe and diffuse abdominal pain and watery diarrhea during the last 24 hrs. Her past medical history was not significant. At the admission, the patient presented altered general condition, fever, clinical dehydration signs, watery rhinorrhea, and erythematous pharynx. A diagnosis of acute gastroenteritis and dehydration was established. Laboratory tests showed: leukocytosis (29,300/µL), neutrophilia (27,200/µL), elevated erythrocyte sedimentation rate (ESR=76 mm/h) and elevated C-reactive protein (CRP=13.3 mg/dL), normochromic normocytic anemia (Hemoglobin = 10.7 g/dL), metabolic acidosis (pH = 7.33; HCO3 = 14.3 mmol/L; BE = −13.3 mmol/L), hyponatremia (133 mmol/L), hypopotassemia (3.2 mmol/L) and hyperchloremia (119 mmol/L). Stool sample analysis did not identify any pathological aspects. Surgical examination and abdominal ultrasound excluded acute appendicitis. The patient was treated with intravenous rehydration therapy, antipyretics and empirical antibiotics (Ceftriaxone iv). On the fourth day of the fever, the patient had an altered general state, persistent high fever (39.4°C), tachycardia (150–160 bpm), normal blood pressure (102/60 mmHg), normal capillary refill time, skin pallor, fissured, dry and red lips, congested pharynx, strawberry tongue, bilateral conjunctivitis, erythema of the palms and soles, as well as diffuse and intense abdominal pain, vomiting and diarrhea (watery stools more than 10 times a day). The patient presented persistent decompensated metabolic acidosis and hypopotassemia (2.2 mmol/L) in spite of intravenous supplementation (total daily dose of potassium was 5–6.7 mmol/kg), hyponatremia (131 mmol/L) and hyperchloremia (118 mmol/L). In this situation, digestive sepsis was ruled out based on negative procalcitonin, and incomplete Kawasaki disease was suspected based on persistent fever associated with the following three principal clinical features: nonsuppurative bilateral conjunctivitis; erythema and cracking of lips, strawberry tongue and erythema of pharyngeal mucosa; erythema of the palms and soles. Confirmation of clinical features was completed with a laboratory test (elevated CRP and ESR, anemia and leukocytosis, hypoalbuminemia) and echocardiography, which revealed ectasia of the left anterior descending coronary artery (Z score +4.3), perivascular brightness of the left coronary artery, mild mitral regurgitation and small pericardial effusion (Table 1). A urine sample revealed reduced specific urine gravity, hematuria, glycosuria and mild proteinuria. Urine culture was negative. Urea and creatinine levels were normal (Table 1). Incomplete Kawasaki disease diagnosis was formulated according to the algorithm described by McCrindle et al.2 Blood serology tests of cytomegalovirus, Epstein–Barr virus, hepatitis B and C, enterovirus, adenovirus and coxsackie B virus reported a negative titer level. IgM antibodies for herpes simplex 1 virus (HSV-1) were detected. Electrocardiography showed tachycardia and non-specific ST and T-wave changes. The therapeutic approach consisted of administration of IVIG 2 g/kg, infusion over 12 hrs and oral aspirin of 90 mg/kg/day; intravenous rehydration therapy was continued over 24 hrs. Acyclovir orally therapy for 5 days was also associated. The patient had a favorable outcome: fever disappeared 36 hrs after IVIG therapy, oral mucous and extremity changes and conjunctivitis also disappeared after 3 days; echocardiography showed a slight decrease of the coronary dilatation and disappearance of pericardial effusion; progressive decrease of inflammation markers, normalization of the serum electrolyte levels 36 hrs after starting IVIG and also of the glycosuria and proteinuria were registered. Desquamation of palms appeared in the second week of the disease, and of soles in the third week of disease. Diuresis was normal at presentation, but polyuria was present starting the 5th day of illness (2.25 L/m2/24 hrs) and persisted for 2 weeks, associated with reduced urine gravity. Thrombocytosis (821,000/µL) appeared on the 14th day of the disease. Urine potassium determination was possible on the 7th day of the disease and was at the upper limit (Table 1).
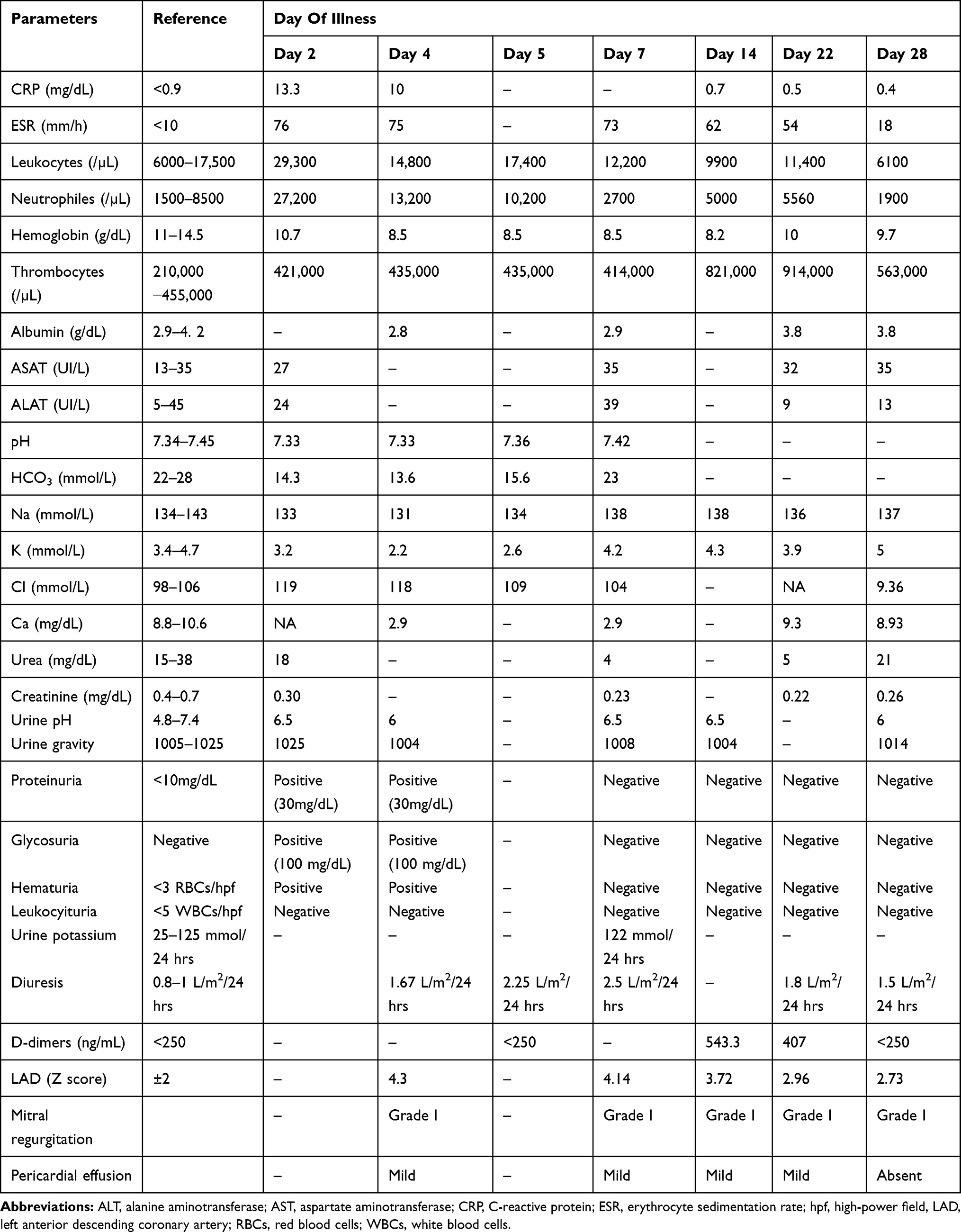 |
Table 1 Laboratory Tests And Echocardiographic Changes |
On the 23rd day, clinical recurrence appeared (fever 38.5°C, vomiting and watery diarrhea) and the second dose of IVIG was administrated, with favorable evolution. The patient was discharged from the hospital on the 29th day of disease with oral aspirin of 5 mg/kg/day. Repeated echocardiography after 2 months was normal. No relapses were recorded in the next 10 months.
Discussion
The diagnosis of incomplete or atypical KD is challenging especially in children who do not fulfill the clinical criteria. Despite growing knowledge of its etiology, many aspects remain unknown or unclear, and diagnosis still depends on clinical features of persistent fever in combination with other characteristic signs and symptoms, thus remaining an enormous challenge for the clinician.1–3
First, particular feature of this case is the presentation with digestive clinical manifestations that had been gradually associated with high fever. In evolution, the clinical presentation containing some specific mucocutaneous signs associated with left anterior descending coronary artery dilatation (Z score 4.3), and laboratory findings (elevated CRP and ESR, anemia, leukocytosis and hypoalbuminemia) supported the diagnosis of incomplete KD.
According to AHA recommendations, incomplete or atypical KD should be considered in patients with fever ≥5 days, and 2 or 3 compatible clinical criteria associated with cardiac findings on echocardiography and supplemental laboratory criteria (anemia, thrombocytosis >450,000 after the 7th day of fever, decreased albumin level < 3 g/dL, elevated alanine aminotransferase, leukocytosis ≥ 15,000/mm3 and pyuria ≥ 10 WBC/hpf).2 In our case, coronary artery abnormality was present and these findings represented the diagnosis clue. However, CAA are not generally detected by echocardiography in the first week of illness, but a normal echocardiogram does not rule out the diagnosis and repeated echocardiograms are mandatory. The delayed recognition of incomplete KD acts as an important risk factor for CAA.14–16 The presence of CAA is considered a specific criterion for the diagnosis of KD, particularly in patients who do not meet the full clinical criteria.2,10–13 The following echocardiographic findings are considered criteria supportive of the diagnosis of incomplete KD: score of left anterior descending coronary artery or right coronary artery ≥2.5 or coronary artery aneurysm; or ≥3 other suggestive features including decreased left ventricular function, mitral regurgitation, pericardial effusion, or Z scores in left anterior descending coronary artery or right coronary artery of 2 to 2.5.2
Role of several markers was evaluated for the diagnosis of incomplete KD. NT-proBNP and interleukin 17 (IL-17) have been found elevated in the acute phase of KD, but none of them has been validated for clinical practice.8,17
There are various initial gastrointestinal symptoms which may confuse the clinical pictures of KD and delay diagnosis especially in patients with incomplete form of disease. Ohnishi et al reported a 4-year-old boy with KD presenting as sigmoid colitis and Rosencrantz et al reported a 2.5-year-old boy with KD presenting as sclerosing cholangitis.18,19 Chen et al have demonstrated that sonographic gallbladder abnormalities are associated with IVIG resistance in KD.20 Bagrul et al presented a 3-year-old boy who was diagnosed with atypical KD, with coronary arteries dilatation, sterile pyuria and progressive bowel oedema, severe abdominal pain, hepatosplenomegaly and hydrops of the gallbladder.16 A computerized search conducted by Colomba et al described 48 cases of KD with intestinal involvement and recommended to consider Kawasaki disease in the differential diagnosis when a child has high fever and abdominal pain.21 Atypical forms of KD have a higher frequency of coronary ectasia, vomiting, anemia, thrombocytosis and a higher serum alanine aminotransferase level.22,23
Secondly, another aspect of our case is represented by renal damage as a result of vasculitis at this level, represented by severe dyselectrolytemia (K = 2.2 mmol/L), proteinuria (30mg/dL) and glycosuria, persisted reduced urine gravity (1004–1008) and polyuria (2.25–2.5L/m2/24 hrs) interpreted as part of renal tubular abnormalities. Severe hypopotassemia could also attribute to gastrointestinal manifestation, but persisted dyselectrolytemia despite large quantities of potassium solutions infused, associated with other manifestations of renal tubular disorders (polyuria, low urine specific gravity, hyperchloremic metabolic acidosis, mild proteinuria and glycosuria) and rapid clinical and laboratory findings improvement after immunoglobulin administration suggests the renal tubular damage. Urine potassium revealed an upper limit level. A preexistent tubular disease was also suspected, but repeated laboratory tests made few weeks after the febrile period did not identify any pathologic changes of renal function. The most frequent renal involvements described in KD are sterile pyuria (reported by Watanabe in 30–80% of cases, mainly in infants and associated with sub-clinical renal injuries) and trace proteinuria.24 In some rare instances, acute kidney injuries (AKI) and urinary tract involvement have been reported in patients with KD. Saviour et al reported a 2-year-old child presenting bloody diarrhea, mucocutaneous clinical findings and laboratory findings suggesting hemolytic uremic syndrome (15). Chuang et al reported an incidence of 28% of acute kidney injuries associated with hepatic dysfunction in KD children with age less than 2 years.25 Nephrotic syndrome in KD was also reported in three children by Krug et al.26 A computerized search conducted by Watanabe et al described 39 patients with KD who developed AKI and have been reported in 28 publications as case reports.23 Other kidney and urinary tract involvement reported in KD are: prerenal AKI, intrinsic AKI caused by tubulointerstitial nephritis, acute nephritic syndrome, immune complex mediated nephropathy and renal AKI associated with KD shock syndrome. The precise pathogenesis is not completely understood, but several possible mechanisms have been proposed: immune-complex mediated kidney injuries, T-cell immune-regulatory abnormalities, renal and glomerular endothelial injury resulting from vasculitis and capillary leak, and an increased release of cytokines.27,28
Kawasaki disease shock syndrome could be also suspected in this case, but the child had normal blood pressure values over the entire acute illness, and no signs of poor peripheral perfusion, so the requirements for concurrent KD shock syndrome proposed by Kanegaye et al in 2009 were not met.29
The main objectives of KD are to control the systemic inflammatory response, to reduce the prevalence of coronary aneurysms and to prevent coronary thrombosis. In the acute phase, IVIG (2 g/kg over 8–12 hrs) within 10 days, and ideally within 7 days, of illness, are recommended. Delayed IVIG treatment is considered to be an independent risk factor for the development of CALs, especially in patients with high levels of CRP and ESR.14,30–32 Nevertheless, about 10–20% of patients receiving IVIG are refractory to this therapy and they are also at higher risk for developing CALs. In this situation, adjunctive therapies include retreatment with IVIG, a tapering course of corticosteroids, infliximab, cyclosporine, cyclophosphamide and other immunomodulatory therapies. Risk factors associated with IVIG resistance include: male sex, young age, high CRP, high neutrophil count, and KD shock syndrome.14,30–34 In our case, a second febrile episode associated with diarrhea and vomiting appeared on the 23rd day of the disease, and a second dose of IVIG was administrated with rapid resolution of fever. A persistent elevated level of IL-6 on the 20th day of the disease (31.22 pg/mL; NV <7 pg/mL) was correlated with a recrudescent fever episode. Serum IL-6 increases in the acute phase of KD, whereas it declines to a normal level after IVIG therapy, being positively correlated with CRP and ESR. However, whether IL-6 can serve as a novel biomarker for predicting incomplete KD is still to be elucidated.35
Conclusions
The diagnosis of incomplete or atypical KD is challenging and very often a definitive diagnosis is not possible upon the initial presentation. Clinical presentation with gastrointestinal manifestation makes the diagnosis more complex for clinicians especially in children who do not fulfill the clinical criteria for KD. Echocardiography showing coronary artery abnormalities can support the diagnosis in a child who does not meet the case definition based on principal clinical findings, to prompt immunoglobulin intravenous administration. Although the renal involvement in KD is rare, urinary changes can be signs of atypical KD presentation.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Son MBF, Newburger JW. Kawasaki disease. In: Kliegman RM, Stanton BF, St Geme JW, Schor NF, editors. Nelson Textbook of Pediatrics.
2. McCrindle BW, Rowley AH, Newburger JW, et al. Diagnosis, treatment and long-term management of Kawasaki disease: a scientific statement for health professionals from the American Heart Association. Circulation. 2017;135:e1–e73. doi:10.1161/CIR.0000000000000484
3. Dietz SM, van Stijn D, Burgner D, et al. Dissecting Kawasaki disease: a state-of-the-art review. Eur J Pediatr. 2017;176:995–1009. doi:10.1007/s00431-017-2937-5
4. Kawasaki T, Singh S. Kawasaki disease – the journey over 50 years: 1967–2017. Int J Rheum Dis. 2018;21:7–9. doi:10.1111/apl.2018.21.issue-1
5. Nakamura Y. Kawasaki disease: epidemiology and the lessons from it. Int J Rheum Dis. 2018;21:16–19. doi:10.1111/apl.2018.21.issue-1
6. Rowley AH, Shulman ST. The epidemiology and pathogenesis of Kawasaki disease. Front Pediatr. 2018;6:1–4. doi:10.3389/fped.2018.00374
7. Marani E, Burns JC, Cimaz R. How should we classify Kawasaki disease? Front Immunol. 2018;9:1–7. doi:10.3389/fimmu.2018.00001
8. Singh S, Jindal AK, Pilania RK. Diagnosis of Kawasaki disease. Int J Rheum Dis. 2018;21:36–44. doi:10.1111/apl.2018.21.issue-1
9. Ronai C, Hamaoka-Okamoto A, Baker AL, et al. Coronary artery aneurysm measurement and Z score variability in Kawasaki disease. J Am Soc Echocardiogr. 2016;29:150–157. doi:10.1016/j.echo.2015.08.013
10. Satoh K, Wakejima J, Gau M, Kiguchi T, Matsuda T, Takasawa R. Risk of coronary artery lesions in young infants with Kawasaki disease: need for a new diagnostic method. Int J Rheum Dis. 2017;21:746–754. doi:10.1111/1756-185X.13223
11. Na J-H, Kim S, Eun LY. Utilization of coronary artery to aorta for early detection of Kawasaki disease. Pediatr Cardiol. 2019;40:461–467. doi:10.1007/s00246-018-1985-6
12. McCrindle BW, Cifra B. The role of echocardiography in Kawasaki disease. Int J Rheum Dis. 2018;21:50–55. doi:10.1111/apl.2018.21.issue-1
13. Dionne A, Dahdah N. Myocarditis and Kawasaki disease. Int J Rheum Dis. 2018;21:45–49. doi:10.1111/apl.2018.21.issue-1
14. Toole KP, Frank C. Atypical or incomplete kawasaki disease in a young child: a case report. J Pediatr Health Care. 2018;33:485–488. doi:10.1016/j.pedhc.2018.10.004
15. Saviour MJ, Hassan S. Kawasaki disease presenting with bloody diarrhea and acute renal failure: first case. Pediatr Rep. 2017;9:7163. doi:10.4081/pr.2017.7163
16. Bagrul D, Karadeniz EG, Koca S. Gastrointestinal involvement in Kawasaki disease: a case report. Cardiol Young. 2018;28:1070–1073. doi:10.1017/S1047951118000847
17. Wu L, Chen Y, Zhong S, Li Y, Dai X, Di Y. Blood N-terminal pro-brain natriuretic peptide and interleukin-17 for distinguishing incomplete Kawasaki disease from infectious diseases. Indian Pediatr. 2015;52:477–480. doi:10.1007/s13312-015-0659-1
18. Ohnishi Y, Mori K, Inoue M, Satake N, Yano M. A case of Kawasaki disease presenting as sigmoid colitis. J Med Ultrasonics. 2018;45:381–384. doi:10.1007/s10396-017-0808-3
19. Rosencrantz R, Huang T, Sonke PY, Tewari D, Chander P. Autoimmune sclerosing cholangitis: an atypical association with Kawasaki disease. Hepatology. 2016;64:2256–2263. doi:10.1002/hep.28694
20. Chen CH, Hung FC, Tiao MM, et al. Sonographic gallbladder abnormality is associated with intravenous immunoglobulin resistance in Kawasaki disease. Sci World J. 2012;2012:485758. doi:10.1100/2012/485758
21. Colomba C, La Placa S, Saporito L, et al. Intestinal involvement in Kawasaki disease. J Pediatr. 2018;202:16–193. doi:10.1016/j.jpeds.2018.06.034
22. Behmadi M, Alizadeh B, Malek A. Comparison of clinical symptoms and cardiac lesions in children with typical and atypical Kawasaki disease. Med Sci (Basel). 2019;7:63. doi:10.3390/medsci7040063.
23. Watanabe T. Pyuria in patients with Kawasaki diseases. World J Clin Pediatr. 2015;4:25–29. doi:10.5409/wjcp.v4.i2.25
24. Chuang GT, Tsai IJ, Lin MT, Chang LY. Acute kidney injury in patients with Kawasaki disease. Pediatr Res. 2016;80:224–227. doi:10.1038/pr.2016.81
25. Krug P, Boyer O, Balzamo E, Sidi D, Lehnert A, Niaudet P. Nephrotic syndrome in Kawasaki disease: a report of three cases. Pediatr Nephrol. 2012;27:1547–1550. doi:10.1007/s00467-012-2172-2
26. De La Harpe M, Di Bernardo S, Hofel M, Sekarski N. Thirty years of Kawasaki disease: a single center study at the University Hospital of Lausanne. Front Pediatr. 2019;7:E63. doi:10.3389/fped.2019.00011
27. Watanabe T. Clinical features of acute kidney injury in patients with Kawasaki disease. World J Clin Pediatr. 2018;7:83–88. doi:10.5409/wjcp.v7.i3.83
28. Watanabe T. Kidney and urinary tract involvement in Kawasaki disease: review article. Int J Pediatr. 2013;2013:831834. doi:10.1155/2013/841360
29. Kanegaye J, Wilder M, Molkara D, et al. Recognition of Kawasaki diseases shock syndrome. Pediatrics. 2009;123:e783–e789. doi:10.1542/peds.2008-1871
30. Hang NS, Singh R. A case report of refractory Kawasaki disease. Med J Malaysia. 2018;73:410–412.
31. Newburger JW. Kawasaki disease: medical therapies. Congenit Heart Dis. 2017;12:641–643. doi:10.1111/chd.12502
32. Lo MS, Newburger JW. Role of intravenous immunoglobulin in the treatment of Kawasaki disease. Int J Rheum Dis. 2018;21:64–69. doi:10.1111/apl.2018.21.issue-1
33. Qiu H, He Y, Rong X, et al. Delayed intravenous immunoglobulin treatment increased the risk of coronary artery lesions in children with Kawasaki disease at different status. Postgrad Med. 2018;130:442–447. doi:10.1080/00325481.2018.1468712
34. Takeuchi M, Imuzuka R, Hayashi T, et al. Novel risk assessment tool for immunoglobulin resistance in Kawasaki disease: application using a random forest classifier. J Pediatr Infect Dis. 2017;36:821–826. doi:10.1097/INF.0000000000001621
35. Wu Y, Liu FL, Xu Y, et al. Interleukin-6 is prone to be a candidate biomarker for predicting incomplete and IVIG nonresponsive Kawasaki disease rather than coronary artery aneurysm. Clin Exp Med. 2019;19:173–181. doi:10.1007/s10238-018-00544-5
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.