Back to Journals » Journal of Pain Research » Volume 13
Unusual Pain Disorders – What Can Be Learned from Them?
Authors Sachau J, Kersebaum D ![]() , Baron R, Dickenson AH
, Baron R, Dickenson AH
Received 30 October 2020
Accepted for publication 18 February 2021
Published 15 March 2021 Volume 2020:13 Pages 3539—3554
DOI https://doi.org/10.2147/JPR.S287603
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Krishnan Chakravarthy
Juliane Sachau,1 Dilara Kersebaum,1 Ralf Baron,1 Anthony H Dickenson2
1Division of Neurological Pain Research and Therapy, Department of Neurology, University Hospital Schleswig-Holstein, Campus Kiel, Kiel, 24105, Germany; 2Department of Neuroscience, Physiology and Pharmacology, University College London, London, WC1E 6BT, UK
Correspondence: Juliane Sachau
Division of Neurological Pain Research and Therapy, Department of Neurology, Universitätsklinikum Schleswig-Holstein, Campus Kiel, Arnold-Heller-Strasse 3, Haus D, Kiel, 24105, Germany
Tel +49 431 500 23911
Fax +49 431 500 23914
Email [email protected]
Abstract: Pain is common in many different disorders and leads to a significant reduction in quality of life in the affected patients. Current treatment options are limited and often result in insufficient pain relief, partly due to the incomplete understanding of the underlying pathophysiological mechanisms. The identification of these pathomechanisms is therefore a central object of current research. There are also a number of rare pain diseases, that are generally little known and often undiagnosed, but whose correct diagnosis and examination can help to improve the management of pain disorders in general. In some of these unusual pain disorders like sodium-channelopathies or sensory modulation disorder the underlying pathophysiological mechanisms have only recently been unravelled. These mechanisms might serve as pharmacological targets that may also play a role in subgroups of other, more common pain diseases. In other unusual pain disorders, the identification of pathomechanisms has already led to the development of new drugs. A completely new therapeutic approach, the gene silencing, can even stop progression in hereditary transthyretin amyloidosis and porphyria, ie in pain diseases that would otherwise be rapidly fatal if left untreated. Thus, pain therapists and researchers should be aware of these rare and unusual pain disorders as they offer the unique opportunity to study mechanisms, identify new druggable targets and finally because early diagnosis might save many patient lives.
Keywords: pain mechanisms, sodium channelopathies, hereditary pain diseases, sensory modulation disorder, central sensitization, gene silencing
Introduction
Pain arises from a complex interplay between peripheral processes and central circuits with interactions between sensory and affective CNS pathways. A broad classification of different pain states exists where nociceptive pain arises from chemical activation of peripheral nociceptors, neuropathic pain from a lesion or disease to somatosensory nervous system leading to abnormal electrical events in neurons and nociplastic pain occurring where there is no obvious peripheral drive.1 Complications to this schema are mixed nociceptive and neuropathic pains such as seen in some low back pain and some cancer pain patients and neuropathic components to the pain in some patients with osteoarthritis, an archetypical nociceptive pain.2
Quantitative sensory testing (QST), both as a research tool and for bedside characterisation of patients as well as a number of questionnaires have produced detailed descriptions of the sensory phenotypes of patients with neuropathic pain. Much less has been reported for other pain states. Importantly, where sensory phenotypes have been studied, it is clear that in neuropathic pain, fibromyalgia (FM) and persistent post-surgical pain, not all patients are the same, and several subgroups have been described.3,4 This leads to the idea that the subgroups are likely to have different underlying pain mechanisms and so may not respond to the same therapies. There is clinical and preclinical evidence to support this premise.5
There are a number of much less common pain conditions and although rare, may shed light on pain mechanisms that could be prevalent in other patients. In this account, we shine a light on the characteristics of a number of patients with pain from uncommon conditions where the pain may result from peripheral, spinal or central alterations.
We have compiled several examples of uncommon pain disorders based on two selection criteria. First, a deeper understanding of pathophysiological mechanisms was recently unravelled which led to the development of more refined diagnostic procedures and hypotheses for therapeutical targets. Second, and even more importantly, novel causative treatment options have been discovered in recent years which have the potential to extend the life expectancy of patients significantly.
Pain Disorders with Recently Unravelled Mechanisms
Sodium Channelopathies
Understanding pain mechanisms is key to the development of novel analgesics and to better use of existing agents. Translational research within this domain is vital and could lead to mechanism-based treatments, a laudable goal. Sodium channels are key to excitability. Importantly, studying rare inherited disorders can also shed light on underlying mechanisms if the aberrant protein has been identified. In pain, the inherited sodium channelopathies relating to Nav1.7 are a good example of this. Depending on the mutation, the clinical phenotype will either result in increased or absent pain sensation. Loss of function of Nav1.7 results in analgesia and anosmia (congenital insensitivity to pain, CIP).6 Other mutations – causing reduced thresholds or loss of inactivation – lead to pain syndromes in the absence of tissue or nerve damage: inherited erythromelalgia (IEM) (or primary EM) and paroxysmal extreme pain disorder (PEPD).6 Some of these mutations were also recently described in patients with painful diabetic neuropathy.7 IEM becomes clinically apparent through reddened, painful hands and feet accompanied by an increased temperature.8 While these symptoms initially appear intermittently and are associated with triggers like exercising and warmth, they might become persistent in the course of the disease. Changes in the nose tip, chin and earlobes as well as upper parts of the lower extremities are possible in an advanced state. Mind that the experiences of these triggers might lead to typical behaviours of patients, eg not wearing socks even in winter or sleeping with uncovered feet.9 Patients with PEPD suffer from episodes of ocular, mandibular and/or rectal pain, also accompanied by flushing of the skin. Again, there are triggers that might provoke the pain such as spicy/hot food.
This translates the preclinical identification of multiple sodium channels to humans and offers proof of concept of Nav1.7 as a key target for analgesia while we await the development of antagonists.6,10
The general efficacy of drugs for neuropathic pain is not very high on a population basis.11 One intriguing study has directly tested this premise.12 Using the sodium channel blocker, oxcarbazepine, patients with peripheral neuropathic pain were tested with the drug but there was no separation from placebo. However, there is a group of neuropathic pain patients who are characterised by hypersensitivity to evoked stimuli – the so-called irritable nociceptor group. Remarkably, these patients responded to the therapy. This study has huge implications for assessment of pain drugs since it suggests that a trial based simply on aetiology and the presumption that all patients are the same may fail if sensory phenotypes of subgroups are not considered.4 Many drugs may have been abandoned on this basis. And recently, a trial of a novel selective 1.7 blocker failed.13 There are a number of possible reasons for this failure to find an effect of a drug acting on a validated target. One is that the compound was peripherally restricted that would not allow actions on the central terminals of the nociceptive fibers where the channel plays an important role. Secondly, patients were not selected other than having ongoing pain so that the irritable nociceptor subgroup was not studied.
Small fiber neuropathy (SFN) is morphologically characterized by injury to the intraepidermal nerve fibers, specifically the unmyelinated C- and thinly myelinated Aδ-fibers, and in some patients, a gain of function of Nav1.7 can be found.14 The mutations in the channel vary depending on the particular amino-acid sequence. Using the sodium channel blocker, lacosamide, sensitivity to the drug was only seen predominantly in patients with a particular mutation of the channel.15
Others with alterations in other parts of the channel were weakly or not responsive. Genetic testing of the site of the mutation will not be generally possible so the idea of patient sensory phenotypes as a basis for drug actions will be more widely useful. Here, neuronal recordings in pain pathways in animals using various pain models can gauge drug effects on different modalities and so guide clinical assessment of pain drugs that target particular aspects of pain.16
Pain mechanisms are ancient and many receptors and channels predate mammals. It is perhaps not surprising but still fascinating to consider the impact of altered Nav1.7 channels over thousands of years. A recently published paper shows that some present-day humans carry a Neanderthal variant of the sodium channel Nav1.7 which is three amino acids different to the common variant.17 This change makes the channel more likely to open and so the Neanderthal variant leads to carriers likely to experience more pain. In total, 0.4% of present day Britons carry this mutated channel.
The very clear associations between gains and loss of function of sodium channels and pain states in humans is compelling evidence for the importance of the excitability of afferent fibers and pain.18 However, it is not always so simple as a change in function of a single gene product. In a family with a gain of function of Nav1.7, pain ratings by the mother are obviously lower than her son despite the same altered channel. This led to a search within the dorsal root ganglion for other changes molecular entities and it turned out that the mother had an upregulated potassium channel, KCNQ. This is the substrate for the m current, an important regulatory influence on neuronal excitability, including pain messages from animal studies.19 So the mother had less pain than her son as a consequence of her increased ability to hyperpolarise overactive nerves.
Very recently, another potassium channel has been identified, Kv6.4 that influences human labour pain by modulating the excitability of uterine nociceptors.20 There are no clinically useful drugs that are able to alter potassium channels in the clinic but clearly, they could be useful in the correct patients. Retigabine has been developed and perhaps needs re-assessment of its potential clinical analgesic actions.21
So even when a pain disorder can be attributed to a single sodium channel, the exact nature of the mutation and changes in other channels such as potassium channels can alter responses to treatment and the pain phenotype.
Hereditary Hyperekplexia
The bulk of the peripheral mechanisms of pain are excitatory events whereby impulses are produced in sensory nerves as a consequence of nociceptor activation or nerve dysfunction. At central sites, notably the spinal cord, inhibitory systems exist that are able to modulate incoming excitatory messages. The interplay and gating of low and high threshold sensory inputs into the spinal cord was a key premise of the “Gate Theory of Pain”, first published in 1965.22
Preclinical studies of models of neuropathy have provided many examples of spinal excitability such as neuronal wind-up and central sensitisation but there is also a loss of inhibitory function that contributes to the pain state. Many years ago, Tony Yaksh showed that blocking inhibitory function in a normal animal by spinal administration of strychnine, the glycine receptor blocker, or GABA antagonism caused rats to become agitated in response to light tactile stimulation.23 These altered sensory changes resemble sensory changes in patients such as sensory dysaesthesia following neuropathy. There is an inherited human condition, namely hereditary hyperekplexia, that relates to this. The syndrome is a consequence of a loss of glycine receptor function and its associated chloride channel, causing a reduction in the normal inhibitory control by this transmitter likely to be key at spinal cord levels.
Hereditary hyperekplexia is characterised by exaggerated startle responses to a variety of stimuli and neonatal hypertonia that declines with age whilst the enhanced startle responses remain.
In this rare disorder, Vuilleumier et al recruited and tested seven patients who had been clinically and genetically verified as having hyperekplexia.24 A meticulous sensory testing approach was taken using mechanical, thermal and other modalities. The patients had lower thresholds for all test stimuli, including pressure, cold pain, and electrical pain to a single shock and to repeated shocks, the latter being a test for central spinal hypersensitivity, evoking wind-up. The withdrawal reflex threshold to applied stimuli was also reduced. The data showing that the patients had a wide range of similarly augmented responses to many modalities of painful stimuli indicates that glycine acts broadly to control many modalities and is suggestive of a lack of control of incoming spinal information. However, and surprisingly given the enhanced pain responses, conditioned pain modulation (CPM), a measure of activity in descending pain modulatory systems from the brain to the spinal cord, where one pain inhibits another was reduced.
It could be concluded that glycine plays a generalised role in modulating sensory inputs and loss of this inhibitory system in otherwise normal patients is sufficient to enhance pain.
Sensory Modulation Disorder (SMD)
Patients with this syndrome often report non-painful stimuli as unpleasant or bothersome. In a recent comprehensive review the authors suggested allodynia to mirror sensory over-responsivity (SOR), a subtype of SMD, so that non-painful sensations are perceived as irritating, unpleasant or painful.25,26 Many patients with neuropathic pains have tactile or cold allodynia that suggest some overlap of mechanisms. Investigation of patients with SOR with regard to thresholds measured using QST revealed no difference between children and adults compared to controls using light touch, vibration, warm and cool stimuli. However, when stimuli above threshold were studied, there were enhanced responses in those with SOR. Furthermore, there were after sensations that lingered. The patients had normal CPM that fails in many other pain conditions such as peripheral neuropathy, fibromyalgia, musculoskeletal pain, etc.27 Thus, it would appear that the sufferers are in a state of pro-nociception as a consequence of excess excitation rather than loss of inhibition. It has been suggested that SMD is due to atypical neural processing of both non-painful sensory stimulus of different modalities such as somatosensory or auditory and abnormal integration of simultaneous multi-sensory stimulation. Studies using electrophysiology have revealed greater and prolonged early event-related potentials (ERPs; a brain response to a specific external event) in response to tactile and auditory stimuli. It is interesting to note how migraine patients have triggers such as light, odours, hunger, etc., so this multi-sensory hypersensitivity may point to an abnormal central multisensory integration in migraine as well. SOR appears to result from a cortical hypersensitivity coupled with a loss of brain modulation of sensory signals.
But that’s not all. FM is characterized by a global disturbance in sensory responsiveness where patients have a greater sensitivity to various non-painful multimodal sensory stimuli (tactile, thermal, electrical, auditory)28–33 It has been suggested that this is a result of a generalized hypervigilance and is considered as one of the pathophysiological mechanisms of FM alongside the diffuse hyperalgesia, particularly to deep tissue stimuli.34,35 The widely reported pro-nociceptive profile of these patients differs from SMD since the sensory over-responsiveness in FM appears to be due to both a decrease in inhibitory and an increase in facilitatory activity in the CNS as evidenced by human fMRI imaging of activity in the descending pathways from the brain to spinal cord in FM patients.36–39 These pathways are controlled by regions of the brain involved in the affective and aversive responses to pain so dysfunction in these zones may explain the common comorbidities of mood as well as altered pain sensitivity seen in these patients since pain is a multidimensional system.
Ehlers–Danlos Syndrome
Hypermobile Ehlers–Danlos syndrome (hEDS) is an inherited disorder of connective tissue producing generalized joint hypermobility, various connective tissue disorders and consequent musculoskeletal problems. At first sight, it could be thought to involve altered peripheral function consequent to tissue damage in sensory pathways and a morphological study even suggested a pure SFN.40 However, hEDS-related pain has a diffuse nature and patients can suffer from sleep problems, fatigue and mood disorders, suggestive of central changes resembling FM. In this recent study, 22 patients were recruited with pain due to Ehlers–Danlos syndrome and compared to the same number of matched healthy participants using a range of sensory tests using QST to measure thermal and mechanical pain thresholds and the wind-up ratio, a measure of spinal hyperexcitability – central sensitisation.41 Finally, descending controls from the brain were gauged using CPM. All of the patients had widespread pain, no small fiber deficit and so unlikely neuropathy but in the area of greatest pain, wind-up was increased.
In the healthy controls, descending inhibition triggered by a second stimulus was present but in the hEDS group, the painful test stimulus rating increased during conditioning, revealing not only a lack of inhibition but a gain of excitation. Thus, the painful syndrome likely involves abnormal peripheral inputs generating enhanced spinal activity, loss of descending inhibitions and gain of excitations.40
For reference, a study of 400 pain patients looked at two of these measures, wind-up (expressed as temporal summation (TSP)) and CPM to examine if subgroups with different clinical and experimental pain sensitivity existed in patients with persistent pain of non-malignant origins.42 Four groups of patients could be distinguished. Group 1 had impaired CPM and facilitated TSP and made up 21% of the total and formed the worst pain condition. This group had the most widespread pain, more pain areas, higher pain ratings and hyperalgesia than group 4 who had normal CPM and normal TSP and made up 30%. Group 2 (37%) had a loss of normal descending inhibitions but normal TSP and again, more hyperalgesia than 4. The smallest group (11%), Group 3, had normal CPM and facilitated TSP. This survey suggests that spinal and descending modulation changes variously can be gauged in these patients and that the worst outcome is loss of brainstem inhibition and gain of spinal facilitation which appears to be the case in these patients with hEDS.
New Therapeutic Options for Unusual Pain Disorders
Systemic Amyloidosis
In the past, systemic transthyretin (ATTR) amyloidosis was classified as a poorly or completely untreatable, rapidly progressive disease. Painful polyneuropathy is an early symptom that significantly reduces the quality of life in the affected patients. Extensive research within the last years has led to the identification of new – also non-invasive – diagnostic tools and promising therapeutic options to improve the overall treatment of patients, making it critical and relevant to raise awareness of this disease.
Systemic ATTR amyloidosis results from 1) a mutation causing a change in the TTR protein structure, ie hereditary ATTR (ATTRv) amyloidosis or 2) a process during ageing, ie wild-type ATTR (ATTwt) amyloidosis. Physiologically, transthyretin (TTR) acts as a carrier protein to transport thyroxin and retinol (vitamin A) via associating with its binding protein and is mostly synthesized by the liver. In ATTR amyloidosis, the TTR protein loses its stable tetrameric conformation, dissociates into monomers that are misfolded, aggregate and finally deposit as amyloid into tissues and organs (Figure 1).43 Since amyloid deposition can theoretically affect every organ, the clinical manifestation is very heterogenous. This clinical chameleon complicates the correct diagnosis resulting in delayed start of therapy and hence rapid disease progression.
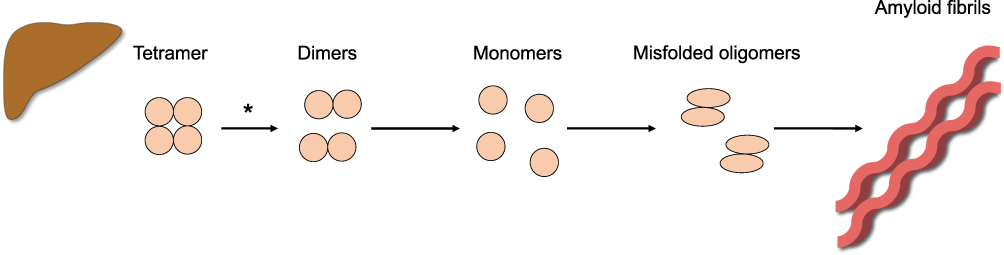 |
Figure 1 Model of amyloid fibril formation in hereditary transthyretin (ATTRv) amyloidosis. Mutations in the TTR protein lead to a loss of its stable conformation. TTR dissociates into monomers that are misfolded, aggregate and deposit as amyloid in the extracellular spaces of various organs. *The rate-limiting step is the dissociation of the tetrameric TTR protein to dimers. |
Pain is a common clinical symptom in ATTRv amyloidosis and led to the first description of the disease by Corino Andrade in 1952 who observed an increased incidence of a rapid progressive painful foot disease in families from north of Portugal, that was associated with symptoms of a peripheral neuropathy and a high mortality rate.44 In a study by Lozeron et al, pain was present in more than half of the patients with demyelinating ATTRv amyloid polyneuropathy.45 Different pathophysiological mechanisms have been proposed that lead to nerve fiber damage in ATTRv amyloidosis.46 For example, examination of sural nerve biopsies showed a disruption of blood-nerve barrier, ie loss of tight junctions and endothelial fenestration, regardless of the presence of amyloid position that may lead to nerve fiber damage via entry of TTR into the endoneurial space.47
Neuropathy symptoms occur early in the disease course and present with length-dependent sensory abnormalities starting distally in the feet. In addition, the hands can also be affected leading to an impaired cold perception and mechanical hyperalgesia, which can be helpful in discriminating ATTRv amyloidosis from other polyneuropathies.48 A positive history of a bilateral carpal tunnel syndrome (CTS) is also common in ATTRv amyloidosis and has therefore been declared as one red flag symptom (see below).49 The damage of small nerve fibers leads to an impaired temperature sensation, eg a reduced warm and heat sensitivity or paradoxical heat sensations, and neuropathic pain symptoms like burning pain, painful dysesthesia or shooting pain attacks. This can be accompanied by an autonomic dysfunction that includes orthostatic hypotension leading to dizziness, dysuria, gastrointestinal symptoms like constipation/diarrhea, early satiety and nausea. Large fiber involvement also occurs in a length-dependent manner starting in the feet and rapidly extending to the thigh and upper limbs. Patients report progressive difficulties in walking, muscle weakness, step gait and impairment of fine motor skills.
In general, the symptom pattern of ATTRv amyloidosis depends on the mutation. So far, about 140 mutations have been identified, whereby the Val30Met mutation is the most frequent one (http://amyloidosismutations.com/mut-attr.php). Some mutations are associated with a sensorimotor polyneuropathic course (Val30Met) whereas others cause a mainly cardiac phenotype (Val122Ile). However, other tissues may be affected as well, resulting in an overall diverse clinical picture, that often leads to misdiagnosis. A retrospective analysis has shown that about 32% of ATTRv amyloidosis patients were initially misdiagnosed, most commonly as CIDP.50
In order to facilitate early diagnosis of the disease, red flags were defined by Conceicao et al (Figure 2)49 and more recently Gertz et al recommended an algorithm for general practitioners.51
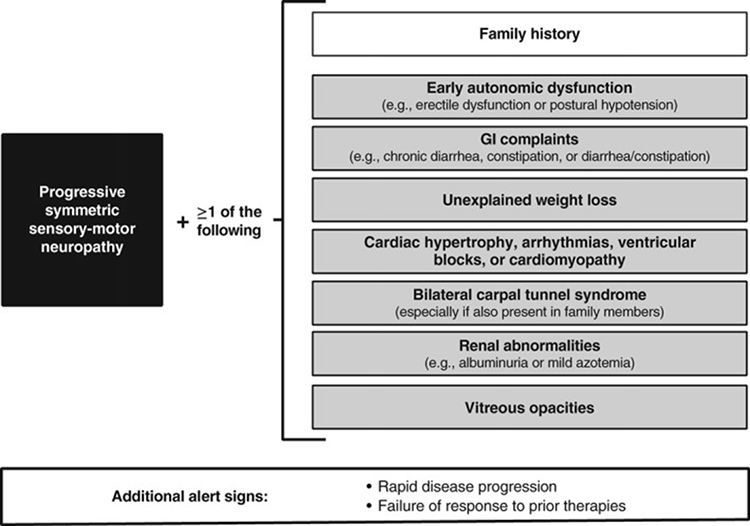 |
Figure 2 Potential red‐flag symptom clusters that may warn of a diagnosis of hereditary transthyretin (ATTRv) amyloidosis with polyneuropathy (Reused from Conceição et al49 with permission of John Wiley and Sons, Copyright © 2016 Conceição I, González-Duarte A, Obici L, et al. “Red-flag” symptom clusters in transthyretin familial amyloid polyneuropathy. Journal of the Peripheral Nervous System published by Wiley Periodicals, Inc. on behalf of Peripheral Nerve Society). |
In contrast to the hereditary form, the wild type ATTR amyloidosis presents predominantly with cardiomyopathy, although data from the THAOS (Transthyretin-Associated Amyloidosis Outcome Survey) registry show that about 20% of the patients complain about accompanying or solely polyneuropathic symptoms.52 Affected patients are most frequently males of older age. A bilateral CTS is a common comorbidity in patients with ATTRwt amyloidosis, eg retrospective analyses report a prevalence of 25% to 60%.53,54 CTS occurs early in the disease course, preceding the diagnosis of a cardiac amyloidosis by 5–9 years, and also seems to be a prognostic marker in ATTR amyloidosis.53 Overall, a cardiac involvement, ie cardiac mutations or ATTRwt amyloidosis, is associated with a lower survival rate.52
Since recently, several drugs have become available that can stop disease progression and improve the quality of life of ATTR amyloidosis affected patients. Thus, an early diagnosis is essential.
The rate-limiting step in the formation of amyloid fibrils is the dissociation of the TTR tetramer into dimers (Figure 1). Thus, stabilization of the tetrameric TTR conformation is one possibility to prevent degradation and hence disease progression.55 Tafamidis, a small molecule TTR stabilizer, is approved for stage 1 ATTRv amyloidosis with polyneuropathy (according to Couthino stages with 0: asymptomatic, 1: symptomatic, walking without assistance, 2: need for walking assistance, 3: wheelchair or bedridden).56
Approval was based on results of a randomized, double-blind, placebo-controlled trial, where tafamidis lead to an improvement in the neuropathy impairment score of the lower limb (NIS-LL) and an improvement of quality of life in the efficacy-evaluable population.57 Long-term efficacy and safety was shown in an up to 6 years follow-up study depending on the early start of treatment and TTR mutation, ie progression of the disease was observed in some patients with a delayed start of treatment and non-Val30Met mutations.58 More recently, tafamidis was also approved for treatment of ATTR-related cardiomyopathy.59
Furthermore, a completely new treatment approach, namely gene-silencing, has been successfully applied in treatment of ATTRv amyloidosis. Since 2018, patisiran and inotersen are approved for the treatment of ATTRv amyloidosis with polyneuropathy stages 1 and 2. Both drugs degrade TTR messenger RNA (mRNA) via two slightly different mechanisms of action, leading to a substantial reduction of both mutant and variant TTR protein levels. Patisiran is the first-ever RNA interference-based drug. RNA interference (RNAi) is a physiological cellular process that enables targeted gene-silencing. After endocytosis, doubled-strained small interfering RNA (siRNA) is incorporated into the RNA-induced silencing complex (RISC) where it dissociates into single strands.60 The complex of RISC and single-strained siRNA binds to the complementary mRNA, leading to a degradation of the mRNA and thus reduction in protein translation. In order to be delivered to the hepatocytes, patisiran is formulated as a lipid nanoparticle. In the APPOLO Phase 3 study in 225 ATTRv amyloidosis patients with polyneuropathy, patisiran reduced TTR protein levels by 81% and led to a significant improvement in modified neuropathy +7 (mNIS+7), quality of life, walking, nutritional status and autonomic symptoms at 18 months after start of therapy. The disease-stabilizing effect of patisiran was shown to be maintained in an open-label extension phase over 24 months with overall acceptable safety profile.61
Inotersen is a single-strained antisense oligonucleotide, which is complementary to the TTR mRNA. Inotersen binds to the mRNA, this mRNA/DNA heteroduplex is then cleaved by the nuclear ribonuclease H1 (RNase H1) leading to reduced TTR protein levels. Efficacy of subcutaneous inotersen has been shown in the NEURO-TTR trial, a phase 3, randomized, double-blind placebo-controlled trial investigating 173 patients with ATTRv amyloidosis with polyneuropathy.62 Compared to placebo, inotersen led to a significant improvement of mNIS+7 and quality of life. TTR protein levels were reduced by about 79%. Effects were maintained in an open-label extension study after two years of follow-up.63
Although ATTR amyloidosis is declared as a “rare” disease, estimations about the overall prevalence, especially for the wild-type form, have increased in the last few years. This acceleration could be mainly referred to an improvement or broader availability of diagnostic approaches and a higher disease awareness. Recently, recommendations have been proposed by expert groups in order to improve diagnosis and monitor disease progression and treatment effects in patients with ATTRv amyloidosis.64,65
Fabry Disease
Fabry disease is an X-linked inherited lysosomal storage disorder caused by mutations in the GLA gene encoding the lysosomal α-galactosidase A. The mutations lead to a diminished or completely absent enzyme function which results in a cellular accumulation of glycosphingolipids, in particular Gb3 (globotriaosylceramide), within lysosomes of various cell types and hence progressive organ dysfunction. So far, more than 900 mutations have been described, most of them with a sporadic occurrence in single families (http://fabry-database.org).66
As accumulation of Gb3 can occur in many different organs affected patients can present with a wide range of symptoms. One common and quality of life-impacting symptom, that already occurs during childhood, is pain, which is reported by about 60–70% of male patients and somewhat less frequently also by females (40–60%).67 Pain in Fabry disease is assumed to be mainly neuropathic, since Gb3 deposits have been detected in dorsal root ganglia and reduced intraepidermal nerve fiber density as well as alteration of thermal perception threshold indicate small fiber pathology.68,69 In addition, a recently published study showed a decreased endothelial NO release in Fabry patients compared to healthy controls and polyneuropathy patients, which may also affect perfusion of vasa nervorum and could therefore contribute to pain.70 Nevertheless, there are also hints for a nociceptive component as some patients report a reduction of their pain due to anti-inflammatory drug usage.71 Neuropathic pain is reported to be the most frequent symptom during childhood (41% of females and 59% of males) and is mainly localized in hands and feet, in particular in the fingertips, palms and soles, although other body areas (abdomen, joints, back, etc.) can be affected as well.71,72 Patients report about four different pain phenotypes, that can occur alone or in combination: evoked pain (hyperalgesia, allodynia), permanent pain, pain attacks and pain crisis.71 Typically, patients complain about unbearable, extreme severe acute pain crisis, so-called ‘Fabry crises’ that start acral locally and can spread over the entire body including colic-like abdominal pain. These crises are triggered by an increased body temperature due to fever/infections or physical activity, but also by a high ambient temperature, can last for several days and are resistant to analgesic therapy. In addition, pain can also present as burning pain attacks, that are provoked by heat, fever or physical activity, but in contrast to the ‘Fabry crisis’ that ceases after trigger elimination, this can manifest as permanent burning, stabbing, tingling or shooting pain that is independent of provoking factors. It has been reported that pain decreases with age in some patients.71,73 Instead, damage to large nerve fibers can occur in the course of the disease leading to hypoesthesia and paresthesia.
Other symptoms that contribute to the clinical picture of Fabry disease at the beginning or during the course of the disease are angiokeratomas, gastrointestinal symptoms (nausea/vomiting, diarrhea, abdominal pain), tinnitus and loss of hearing as well as an impaired sudomotor function.72,74,75 Progressive nephropathy, cardiomyopathy and cerebrovascular complications, like stroke, result in a reduction of life expectancy. Based on data of a prospective study it has been estimated that about 1.2% of stroke patients aged between 18 and 55 years suffer from an underlying Fabry disease.76
Since the GLA gene is located on the X-chromosome, hemizygous males mainly develop a classic severe phenotype with earlier occurrence of symptoms and more rapid disease progression compared to heterogenous female carriers, who demonstrate a phenotype ranging from completely asymptomatic to substantially affected.77
Due to the fact that the Fabry disease is a rare disease with an estimated prevalence of 1:40.000 for the classic form presenting with a variety of symptoms, diagnosis is often delayed by up to 20 years.78,79 Common misdiagnoses are rheumatoid arthritis, rheumatic fever or growing pains.
Since 2001 intravenous enzyme replacement therapy with recombinant agalsidase α and β is available, that can help limiting the irreversible organ damage and control disease progression.80,81 Another treatment approach is based on pharmacological chaperon proteins, ie small molecules, that stabilize mutant forms of the α-galactosidase A and hence restore its correct functionality. In 2016, migalastat, the first oral chaperone protein, has been approved for the treatment of Fabry’s disease patients, with a migalastat-amenable GLA mutation.82 Overall, the multi-systemic character of the disease requires an individualized organ-specific treatment approach.83
However, these therapy approaches have only a limited effect in late stages with severe organ damage. In addition, Fabry disease has a clear influence on the quality of life and well-being of affected children and adolescents with a negative impact especially on social development.84 Thus, early diagnosis is essential in order to start treatment as soon as possible and improve quality of life.
Certain symptoms from the patient’s medical history can give an indication of the presence of the disease. In addition, disease-specific tools can be used to confirm the suspicion of Fabry disease and, if necessary, to initiate further diagnostic steps.67 The FabryScan is a screening tool consisting of 10 questionnaires related to clinical characteristics of the disease and three simple bedside-tests, that can be used to identify affected patients.85,86
Porphyrias
A group of rare, underdiagnosed conditions whose leading symptom is an acute, severe pain are the acute porphyrias.87 The porphyrias are a group of metabolic disorders. Several enzymatic steps are involved in the heme biosynthesis pathway (Figure 3). A malfunctioning of any of them leads to an accumulation of heme precursors like 5-aminolaevulinic acid (5-ALA) or porphobilinogen (PBG), predominantly in either the liver or bone marrow. Depending on the specific enzyme that is altered in its activity, eight different hereditary types of porphyrias are known. They can be classified as “hepatic” or “erythropoietic” (describing the location primarily affected from the precursor-accumulation) or as “acute” or “cutaneous” (based on clinical manifestations). Two types of porphyrias, the hereditary coproporphyria (HCP) and the variegate porphyria (VP), are classified as both acute and cutaneous.
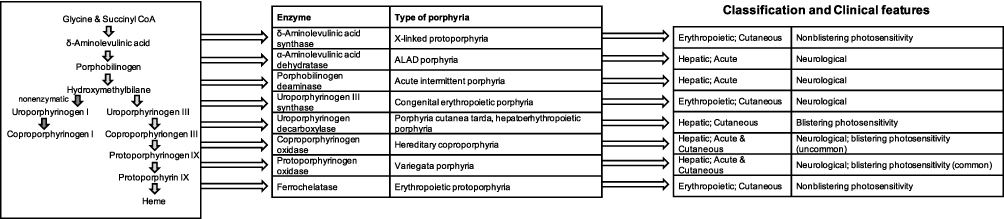 |
Figure 3 The heme biosynthetic pathway showing intermediates, enzymes and types of porphyria associated with each enzyme (Adapted from Ramanujam and Anderson, Porphyria diagnostics-part 1: a brief overview of the porphyrias. Current Protocols in Human Genetics89 with permission of John Wiley and Sons, Copyright © 2015 John Wiley & Sons, Inc.). |
The most common acute porphyria, the acute intermittent porphyria (AIP), is reported to affect 1:75,000 people in European countries. Sweden, where 1:1000 people are affected, is an exception due to a founder effect.88 The most common porphyria, porphyria cutanea tarda (PCT), has a prevalence of 1:10,000 according to European studies.51
It is of utmost importance to consider porphyrias when related symptoms and findings appear—not least due to the fact that the wrong medication can lead to a dramatic exacerbation of the disease. Clinical features are dependent on the type of porphyria. The striking, principle symptom of patients with acute porphyrias is severe abdominal pain that might present itself as an “acute abdomen” but remains without peritonism. Other manifestations are predominantly neurological, psychological and autonomic. Cutaneous porphyrias on the other hand lead to either blistering or nonblistering photosensitivity (overlap of clinical symptoms in HCP and VP).
The following section will focus on pain in acute porphyrias.
Acute porphyrias – ie AIP, HCP, VP and delta-aminolevulinic acid dehydratase deficiency porphyria (ADP) – can be life-threatening and occur episodically in the form of attacks. Among abdominal pain, patients show nausea, vomiting and constipation.89 Motor neuropathies as well as neurovisceral and neuropathic pain are among the neurological manifestations.87,90,91 The affection of proximal muscles (mostly of the upper extremities) is common but tetraplegia, lethal respiratory and bulbar paralysis do also occur.88 Neuropathic symptoms like tickling or numbness were found to be characteristic for the chronic state of the condition in a study with 19 patients.91 In a review on AIP with focus on pain, it is reported that pain in the chest, head, neck and limbs were among the most common clinical manifestations affecting 50–52% of the patients.92 The involvement of cranial nerves was reported in 35–55% of the patients and peripheral neuropathy in 10–40%. Sensory loss was present in 9–38%. The authors discussed several mechanisms that, secondary to heme deficiency or to the accumulation of neurotoxic substances such as ALA, might contribute to the development of central and peripheral sensitisation and neuropathic and neurovisceral pain. For instance, a demyelination due to the release of free radicals is discussed. An affection of central and peripheral nociceptive transmission by increased levels of brain tryptophan and increased turnover of the neurotransmitter 5-hydroxytryptamine (resulting from a decreased activity of the heme-dependent enzyme liver tryptophan pyrrolase) was another hypothesis posed by the authors.
Lin et al conducted excitability measurements of peripheral motor axons in 20 patients with AIP.93 These were then combined with the findings of genetic screening and biochemical and conventional nerve conduction studies. They proposed that porphyrin neurotoxicity may be the cause of a subclinical reduction in the hyperpolarization-activated, cyclic nucleotide-dependent current in the axons of patients with acute porphyric episodes without clinical neuropathy. On the other side, it was suggested that the development of porphyric neuropathy might occur when a reduced activity of the Na(+)/K(+) pump results in membrane depolarization.
Treating the acute attack appropriately is key to achieving a satisfactory analgesia and warding off life-threatening neurological symptoms (Table 1). For patients with known porphyria, though, the priority should always be the prevention and avoidance of possible, attack-precipitating factors. Those can be starvation and infections, hormonal fluctuations (eg during menstrual cycle), alcohol, smoking and the usage of certain drugs (including hormonal contraceptives).94–96 P450-inducing drugs, for instance, increase the hepatic heme turnover and thus act as porphyrinogenic.
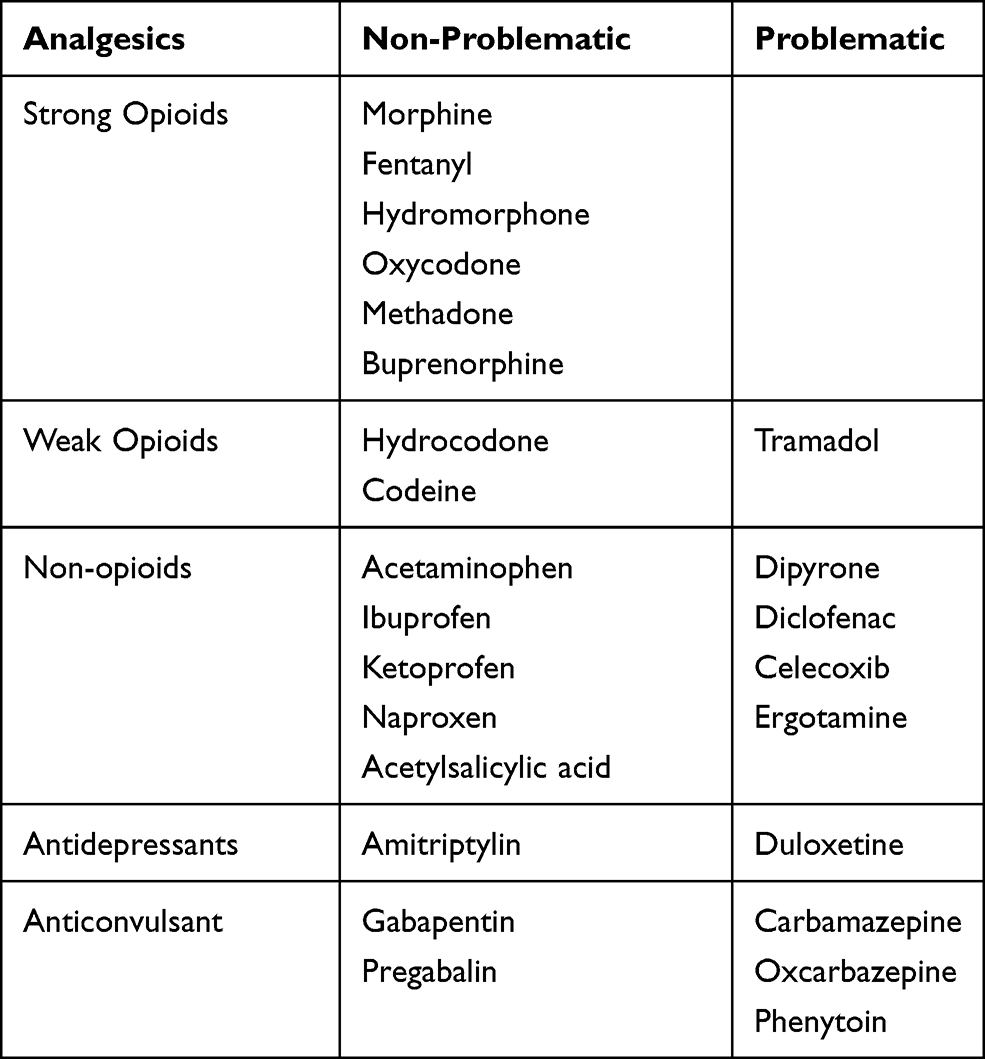 |
Table 1 Problematic and Non-Problematic Analgesics for the Treatment of Patients with Acute Intermittent Porphyria |
The characteristic symptoms are accompanied by an increased urinary excretion of PBG and 5-ALA (https://porphyria.eu/pt/content/laboratory-diagnosis).97 It should be noted that patients with the ALA dehydrase deficiency porphyria (ADP) have a normal PBG excretion. If the acute attack has been diagnosed and other potentially life-threatening and intervention-requiring diagnoses are excluded, treatment must begin in a timely manner. If present, precipitating factors should be eliminated (eg treatment of possible infections, correction of hypocaloric diets).
The prime principle must be the avoidance of porphyrinogenic drugs, whenever possible. The Norwegian Porphyria Centre (NAPOS) drug database for acute porphyria can be consulted to learn about the potential of drugs to provoke attacks of acute porphyria (see http://www.drugs-porphyria.org). As a specific therapy, intravenous hematin has shown to suppress hepatic ALA synthase by negative feedback, thus reducing the excretion of aminolaevulinic acid and PBG.98,99 In a review article published in 2017, a daily dose of 3 to 4 mg Panhematin per kg of body weight infused over a period of 30 to 40 min is recommended.100
Recently new gene therapy strategies, such as the use of a viral vector to deliver the normal hydroxymethylbilane synthase gene to hepatocytes and the use of a siRNA directed against ALA synthase 1 have been discussed the latter being very promising:
Balwani et al successfully conducted a phase 3 trial of givosiran (iRNA therapy) for AIP and have thus put forth a therapy for the treatment of acute hepatic porphyria in adults approved by the Food and Drug Administration in November 2019 and by the European Medicines Agency (EMA) in March 2020.101 Patients who received givosiran were shown to have a significantly lower rate of porphyria attacks than those who received placebo. The subcutaneously administered agent prevents the accumulation of ALA and PBG through the targeting of hepatic ALAS1 messenger RNA.102 The targeted delivery to hepatocytes is ensured by a conjugation of givosiran to a trivalent N-acetyl-galactosamine ligand specifically binding to the asialoglycoprotein receptor.103–105 Compared to the placebo group, patients who received givosiran showed less use of analgesics and better daily pain sores. A clinical relevance of the latter was implied through the improvements of in physical health status and in the bodily pain domain of the SF-12. Concerning safety profile, a higher frequency of hepatic and renal adverse events was observed in patients who received givosiran.
Liver transplantations are reported as a therapeutic option for AIP patients for severely affected patients who suffer from recurrent attacks.106,107
For further management of the acute attack, fluid and calorie intake must be ensured, though it is important to consider that hypotonic dextrose fluids might aggravate hyponatraemia. Patients should be provided with at least 300 mg carbohydrates/day.97
Schwannomatosis
Schwannomatosis, a form of neurofibromatosis, is one of the many conditions primarily manifesting themselves clinically through chronic pain.108
Patients suffering from this disorder have the predisposition to develop multiple nonvestibular, noninterdermal schwannomas (nerve sheath tumours) and, rarely, meningiomas. While peripheral nerves (89%, mainly arms and leg) and the spine (74%, mainly lumbar) are most commonly affected, cranial nerves might also be involved (8%, mainly the trigeminal nerve).109,110 Approximately a third of the patients display a so-called segmental schwannomatosis that results from genetic mosaicism.111,112 Germline mutations of the SMARCB1 or the LZTR1 tumour suppressor genes have been detected in 86% of familial and 40% of sporadic schwannomatosis cases. The univestibular affection in schwannomatosis has been reported and linked to LZTR1.113 It is reported that this tumour predisposition syndrome is associated with a concomitant mutational inactivation of two or more tumour suppressor genes.114 While the tumours are mostly benign, malignant transformation remains as a risk.115–117 In a retrospective analysis with 87 patients, additional tumours to schwannomas (lipomas11% and angiolipomas 3%) have been found.110
Pain in schwannomatosis patients has been categorised as neuropathic or inflammatory and is the most prominent symptom of patients with schwannomatosis and its expression can vary (acute or chronic and localized or widespread).110,118 There are contradictory reports on whether the severity of the pain correlates with the tumour burden. Apart from a diffuse or localized pain, this disorder might also become apparent through an asymptomatic mass, more rarely through numbness or weakness.110 It has repeatedly been reported that patients experience a serious impairment of quality of life and psychological interventions should be included in the therapeutic concept.119,120
The mechanism of pain in schwannomatosis is not entirely clear. In a study with 20 patients where QST, laser evoked potentials (LEPs), whole-body magnetic resonance imaging (wbMRI), magnetic resonance neurography (MRN) and skin biopsies were conducted, the authors discussed SFN, more specifically, C-fiber loss appeared to be causal to the pain in most schwannomatosis patients.120 Schwann cells are described to interact with their associated axons, possibly promoting the generation of neuropathic pain through cytokines like tumor necrosis factor alpha (TNF-α).121–123 It has also been hypothesised that dysfunctional sprouting might occur in an environment with neoplastic (thus faulty) Schwann cells, given that they physiologically play a key role during neuronal regeneration for the sprouting of axons.121 Accordingly, Ostrow et al conducted examinations on the secretomes of painful vs nonpainful schwannomatosis tumour cells and posed the hypothesis that painful tumours secrete proteins that sensitise neurons in vitro.118 A genetic involvement has been discussed by Jordan et al who suggested that schwannomatosis-associated pain might be related to the germline mutation LZTR1.124
In terms of pain management, the primary treatment option is the surgical resection of the tumour.112 Though potentially effective, it harbours the risks of any other surgical intervention, might itself cause pain or nerve damage and is not always an option due to possibly inaccessible tumour locations. Unfortunately, the removal will neither guarantee the pain to disappear, nor prevent a possible return of the tumour.125
Conventional pain therapy does not achieve satisfactory analgesic effect on patients although co-analgesics (eg gabapentin, pregabalin) have been shown to be effective in the treatment of the neuropathic pain.108
There have been therapeutic advances. Blakeley and Plotkin et al published a case report where the usage of Bevacizumab, the monoclonal antibody to vascular endothelial growth factor (VEGF-A), has resulted in a clinical improvement of a 23-year old, male patient whose schwannoma had previously been confirmed to express tumor VEGF.111 The rationale was the earlier implication of VEGF signalling in preclinical injury models of neuropathic pain and the fact that both VEGF and its primary receptors were known to be expressed in schwannomas.126,127 Ahmed et al have published the results of a gene-therapeutic approach by adeno-associated virus delivery of the pore-forming protein Gasdermin-D into intra-sciatic schwannomas.128 They were able to achieve a reduction of tumour growth and tumour-associated pain, thus presenting promising data for future therapy options.
Conclusion
The correct diagnosis and precise examination of rare or unusual pain disorders has a potential to improve the management of pain disorders in general. First, pathophysiological mechanisms and new pharmacological targets identified in rare disorders may also operate in other pain condition at least in subgroups of these. These targets can then be validated in clinical trials. Second, the knowledge of pain mechanisms and targets derived from rare conditions can be back-translated into animal research in order to study their behavioural and electrophysiological consequences in more detail. Third, development of new therapeutic approaches, in particular new gene therapies, can help to stop disease progression and improve quality of life in affected patients. Thus, awareness of and screening for these rare pain disorders are of particular importance to establish early effective treatment. Furthermore, new therapeutic strategies may also have an impact on other fields in medicine.
In summary, pain doctors and researchers should also be aware of rare and unusual pain disorders because they offer the unique opportunity to study mechanisms, identify new druggable targets and because early diagnosis might save many patient lives.
Acknowledgments
AHD acknowledges research funding through the Wellcome Trust Pain Consortium.
Disclosure
JS reports consultant fees from Pfizer Pharma GmbH, speaker fees from Grünenthal GmbH and travel support from Alnylam Pharmaceuticals and Pfizer, outside the submitted work. DK reports no conflicts of interest. RB reports grants and research support from EU Projects: “Europain” (115007). DOLORisk (633491). IMI Paincare (777500). German Federal Ministry of Education and Research (BMBF): Verbundprojekt: Frühdetektion von Schmerzchronifizierung (NoChro) (13GW0338C). German Research Network on Neuropathic Pain (01EM0903). Pfizer Pharma GmbH, Genzyme GmbH, Grünenthal GmbH, Mundipharma Research GmbH und Co. KG., Novartis Pharma GmbH, Alnylam Pharmaceuticals Inc., Zambon GmbH, Sanofi-Aventis Deutschland GmbH, speaker fees from Pfizer Pharma GmbH, Genzyme GmbH, Grünenthal GmbH, Mundipharma, Sanofi Pasteur, Medtronic Inc. Neuromodulation, Eisai Co. Ltd., Lilly GmbH, Boehringer Ingelheim Pharma GmbH & Co. KG, Astellas Pharma GmbH, Desitin Arzneimittel GmbH, Teva GmbH, Bayer-Schering, MSD GmbH, Seqirus Australia Pty. Ltd, Novartis Pharma GmbH, TAD Pharma GmbH, Grünenthal SA Portugal, Sanofi-Aventis Deutschland GmbH, Agentur Brigitte Süss, Grünenthal Pharma AG Schweiz, Grünenthal B.V. Niederlande, Evapharma, Takeda Pharmaceuticals International AG Schweiz, Ology Medical Education Netherlands, consultant fees from Pfizer Pharma GmbH, Genzyme GmbH, Grünenthal GmbH, Mundipharma Research GmbH und Co. KG, Allergan, Sanofi Pasteur, Medtronic, Eisai, Lilly GmbH, Boehringer Ingelheim Pharma GmbH&Co.KG, Astellas Pharma GmbH, Novartis Pharma GmbH, Bristol-Myers Squibb, Biogenidec, AstraZeneca GmbH, Merck, Abbvie, Daiichi Sankyo, Glenmark Pharmaceuticals S.A., Seqirus Australia Pty. Ltd, Teva Pharmaceuticals Europe Niederlande, Teva GmbH, Genentech, Mundipharma International Ltd. UK, Astellas Pharma Ltd. UK, Galapagos NV, Kyowa Kirin GmbH, Vertex Pharmaceuticals Inc., Biotest AG, Celgene GmbH, Desitin Arzneimittel GmbH, Regeneron Pharmaceuticals Inc. USA, Theranexus DSV CEA Frankreich, Abbott Products Operations AG Schweiz, Bayer AG, Grünenthal Pharma AG Schweiz, Mundipharma Research Ltd. UK, Akcea Therapeutics Germany GmbH, Asahi Kasei Pharma Corporation, AbbVie Deutschland GmbH & Co. KG, Air Liquide Sante International Frankreich, Alnylam Germany GmbH, Lateral Pharma Pty Ltd, Hexal AG, Angelini, Janssen, SIMR Biotech Pty Ltd Australien, Confo Therapeutics N. V. Belgium outside the submitted work. AHD reports personal fees from Grunenthal, personal fees from Teva, personal fees from Allergan, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Kosek E, Cohen M, Baron R, et al. Do we need a third mechanistic descriptor for chronic pain states? Pain. 2016;157(7):1382–1386. doi:10.1097/j.pain.0000000000000507
2. Thakur M, Dickenson AH, Baron R. Osteoarthritis pain: nociceptive or neuropathic? Nat Rev Rheumatol. 2014;10(6):374–380. doi:10.1038/nrrheum.2014.47
3. Hurtig IM, Raak RI, Kendall SA, Gerdle B, Wahren LK. Quantitative sensory testing in fibromyalgia patients and in healthy subjects: identification of subgroups. Clin J Pain. 2001;17(4):316–322. doi:10.1097/00002508-200112000-00005
4. Baron R, Maier C, Attal N, et al. Peripheral neuropathic pain: a mechanism-related organizing principle based on sensory profiles. Pain. 2017;158(2):261–272. doi:10.1097/j.pain.0000000000000753
5. Bannister K, Sachau J, Baron R, Dickenson AH. Neuropathic pain: mechanism-based therapeutics. Annu Rev Pharmacol Toxicol. 2020;60:257–274. doi:10.1146/annurev-pharmtox-010818-021524
6. Dib-Hajj SD, Geha P, Waxman SG. Sodium channels in pain disorders: pathophysiology and prospects for treatment. Pain. 2017;158(Suppl 1):S97–S107. doi:10.1097/j.pain.0000000000000854
7. Blesneac I, Themistocleous AC, Fratter C, et al. Rare NaV1.7 variants associated with painful diabetic peripheral neuropathy. Pain. 2018;159(3):469–480. doi:10.1097/j.pain.0000000000001116
8. McDonnell A, Schulman B, Ali Z, et al. Inherited erythromelalgia due to mutations in SCN9A: natural history, clinical phenotype and somatosensory profile. Brain. 2016;139(4):1052–1065. doi:10.1093/brain/aww007
9. Drenth JPH, Waxman SG. Mutations in sodium-channel gene SCN9A cause a spectrum of human genetic pain disorders. J Clin Invest. 2007;117(12):3603–3609. doi:10.1172/JCI33297
10. Bennett DL, Clark AJ, Huang J, Waxman SG, Dib-Hajj SD. The role of voltage-gated sodium channels in pain signaling. Physiol Rev. 2019;99(2):1079–1151. doi:10.1152/physrev.00052.2017
11. Finnerup NB, Attal N, Haroutounian S, et al. Pharmacotherapy for neuropathic pain in adults: a systematic review and meta-analysis. Lancet Neurol. 2015;14(2):162–173. doi:10.1016/S1474-4422(14)70251-0
12. Demant DT, Lund K, Vollert J, et al. The effect of oxcarbazepine in peripheral neuropathic pain depends on pain phenotype: a randomised, double-blind, placebo-controlled phenotype-stratified study. Pain. 2014;155(11):2263–2273. doi:10.1016/j.pain.2014.08.014
13. McDonnell A, Collins S, Ali Z, et al. Efficacy of the Nav1.7 blocker PF-05089771 in a randomised, placebo-controlled, double-blind clinical study in subjects with painful diabetic peripheral neuropathy. Pain. 2018;159(8):1465–1476. doi:10.1097/j.pain.0000000000001227
14. Labau JIR, Estacion M, Tanaka BS, et al. Differential effect of lacosamide on Nav1.7 variants from responsive and non-responsive patients with small fibre neuropathy. Brain. 2020;143(3):771–782. doi:10.1093/brain/awaa016
15. de Greef BTA, Hoeijmakers JGJ, Geerts M, et al. Lacosamide in patients with Nav1.7 mutations-related small fibre neuropathy: a randomized controlled trial. Brain. 2019;142(2):263–275. doi:10.1093/brain/awy329
16. Dickenson AH, Patel R. Sense and sensibility-logical approaches to profiling in animal models. Pain. 2018;159(7):1426–1428. doi:10.1097/j.pain.0000000000001245
17. Zeberg H, Dannemann M, Sahlholm K, et al. A neanderthal sodium channel increases pain sensitivity in present-day humans. Curr Biol. 2020;30(17):3465–3469.e4. doi:10.1016/j.cub.2020.06.045
18. Mis MA, Yang Y, Tanaka BS, et al. Resilience to pain: a peripheral component identified using induced pluripotent stem cells and dynamic clamp. J Neurosci. 2019;39(3):382–392. doi:10.1523/JNEUROSCI.2433-18.2018
19. Passmore GM, Selyanko AA, Mistry M, et al. KCNQ/M currents in sensory neurons: significance for pain therapy. J Neurosci. 2003;23(18):7227–7236. doi:10.1523/JNEUROSCI.23-18-07227.2003
20. Lee MC, Nahorski MS, Hockley JRF, et al. Human labor pain is influenced by the voltage-gated potassium channel KV6.4 subunit. Cell Rep. 2020;32(3):107941. doi:10.1016/j.celrep.2020.107941
21. Blackburn-Munro G, Jensen BS. The anticonvulsant retigabine attenuates nociceptive behaviours in rat models of persistent and neuropathic pain. Eur J Pharmacol. 2003;460(2–3):109–116. doi:10.1016/s0014-2999(02)02924-2
22. Wall PD. The gate control theory of pain mechanisms. A re-examination and re-statement. Brain. 1978;101(1):1–18. doi:10.1093/brain/101.1.1
23. Yaksh TL. Behavioral and autonomic correlates of the tactile evoked allodynia produced by spinal glycine inhibition: effects of modulatory receptor systems and excitatory amino acid antagonists. Pain. 1989;37(1):111–123. doi:10.1016/0304-3959(89)90160-7
24. Vuilleumier PH, Fritsche R, Schliessbach J, et al. Mutations affecting glycinergic neurotransmission in hyperekplexia increase pain sensitivity. Brain. 2018;141(1):63–71. doi:10.1093/brain/awx289
25. Bar-Shalita T, Granovsky Y, Parush S, Weissman-Fogel I. Sensory modulation disorder (SMD) and pain: a new perspective. Front Integr Neurosci. 2019;13:27. doi:10.3389/fnint.2019.00027
26. Miller LJ, Anzalone ME, Lane SJ, Cermak SA, Osten ET. Concept evolution in sensory integration: a proposed nosology for diagnosis. Am J Occup Ther. 2007;61(2):135–140. doi:10.5014/ajot.61.2.135
27. Yarnitsky D. Conditioned pain modulation (the diffuse noxious inhibitory control-like effect): its relevance for acute and chronic pain states. Curr Opin Anaesthesiol. 2010;23(5):611–615. doi:10.1097/ACO.0b013e32833c348b
28. Lautenbacher S, Rollman GB, McCain GA. Multi-method assessment of experimental and clinical pain in patients with fibromyalgia. Pain. 1994;59(1):45–53. doi:10.1016/0304-3959(94)90046-9
29. Geisser ME, Glass JM, Rajcevska LD, et al. A psychophysical study of auditory and pressure sensitivity in patients with fibromyalgia and healthy controls. J Pain. 2008;9(5):417–422. doi:10.1016/j.jpain.2007.12.006
30. Montoya P, Sitges C, García-Herrera M, et al. Reduced brain habituation to somatosensory stimulation in patients with fibromyalgia. Arthritis Rheum. 2006;54(6):1995–2003. doi:10.1002/art.21910
31. Hollins M, Harper D, Gallagher S, et al. Perceived intensity and unpleasantness of cutaneous and auditory stimuli: an evaluation of the generalized hypervigilance hypothesis. Pain. 2009;141(3):215–221. doi:10.1016/j.pain.2008.10.003
32. Rehm SE, Koroschetz J, Gockel U, et al. A cross-sectional survey of 3035 patients with fibromyalgia: subgroups of patients with typical comorbidities and sensory symptom profiles. Rheumatology (Oxford). 2010;49(6):1146–1152. doi:10.1093/rheumatology/keq066
33. Koroschetz J, Rehm SE, Gockel U, et al. Fibromyalgia and neuropathic pain–differences and similarities. A comparison of 3057 patients with diabetic painful neuropathy and fibromyalgia. BMC Neurol. 2011;11:55. doi:10.1186/1471-2377-11-55
34. McDermid AJ, Rollman GB, McCain GA. Generalized hypervigilance in fibromyalgia: evidence of perceptual amplification. Pain. 1996;66(2):133–144. doi:10.1016/0304-3959(96)03059-x
35. Rollman GB. Perspectives on hypervigilance. Pain. 2009;141(3):183–184. doi:10.1016/j.pain.2008.12.030
36. Staud R. Brain imaging in fibromyalgia syndrome. Clin Exp Rheumatol. 2011;29(6 Suppl 69):S109–117.
37. Staud R, Spaeth M. Psychophysical and neurochemical abnormalities of pain processing in fibromyalgia. CNS Spectr. 2008;13(3 Suppl 5):12–17. doi:10.1017/s109285290002678x
38. Harper DE, Ichesco E, Schrepf A, et al. Resting functional connectivity of the periaqueductal gray is associated with normal inhibition and pathological facilitation in conditioned pain modulation. J Pain. 2018;19(6):
39. O’Brien AT, Deitos A, Triñanes Pego Y, Fregni F, Carrillo-de-la-peña MT. Defective endogenous pain modulation in fibromyalgia: a meta-analysis of temporal summation and conditioned pain modulation paradigms. J Pain. 2018;19(8):819–836. doi:10.1016/j.jpain.2018.01.010
40. Leone CM, Celletti C, Gaudiano G, et al. Pain due to Ehlers-Danlos syndrome is associated with deficit of the endogenous pain inhibitory control. Pain Med. 2020;21(9):1929–1935. doi:10.1093/pm/pnaa038
41. Baron R, Hans G, Dickenson AH. Peripheral input and its importance for central sensitization. Ann Neurol. 2013;74(5):630–636. doi:10.1002/ana.24017
42. Vaegter HB, Graven-Nielsen T. Pain modulatory phenotypes differentiate subgroups with different clinical and experimental pain sensitivity. Pain. 2016;157(7):1480–1488. doi:10.1097/j.pain.0000000000000543
43. Adams D, Koike H, Slama M, Coelho T. Hereditary transthyretin amyloidosis: a model of medical progress for a fatal disease. Nat Rev Neurol. 2019;15(7):387–404. doi:10.1038/s41582-019-0210-4
44. Andrade C. A peculiar form of peripheral neuropathy; familiar atypical generalized amyloidosis with special involvement of the peripheral nerves. Brain. 1952;75(3):408–427. doi:10.1093/brain/75.3.408
45. Lozeron P, Mariani -L-L, Dodet P, et al. Transthyretin amyloid polyneuropathies mimicking a demyelinating polyneuropathy. Neurology. 2018;91(2):e143–e152. doi:10.1212/WNL.0000000000005777
46. Koike H, Katsuno M. Ultrastructure in transthyretin amyloidosis: from pathophysiology to therapeutic insights. Biomedicines. 2019;7(1). doi:10.3390/biomedicines7010011
47. Koike H, Ikeda S, Takahashi M, et al. Schwann cell and endothelial cell damage in transthyretin familial amyloid polyneuropathy. Neurology. 2016;87(21):2220–2229. doi:10.1212/WNL.0000000000003362
48. Escolano-Lozano F, Barreiros AP, Birklein F, Geber C. Transthyretin familial amyloid polyneuropathy (TTR-FAP): parameters for early diagnosis. Brain Behav. 2018;8(1):e00889. doi:10.1002/brb3.889
49. Conceição I, González-Duarte A, Obici L, et al. “Red-flag” symptom clusters in transthyretin familial amyloid polyneuropathy. J Peripher Nerv Syst. 2016;21(1):5–9. doi:10.1111/jns.12153
50. Cortese A, Lozza A, Vegezzi E, et al. Misdiagnoses of transthyretin amyloidosis: a clinical and electrodiagnostic study. Orphanet J Rare Dis. 2015;10(S1):O13. doi:10.1186/1750-1172-10-S1-O13
51. Gertz M, Adams D, Ando Y, et al. Avoiding misdiagnosis: expert consensus recommendations for the suspicion and diagnosis of transthyretin amyloidosis for the general practitioner. BMC Fam Pract. 2020;21(1):198. doi:10.1186/s12875-020-01252-4
52. Damy T, Kristen AV, Suhr OB, et al. Transthyretin cardiac amyloidosis in continental Western Europe: an insight through the Transthyretin Amyloidosis Outcomes Survey (THAOS). Eur Heart J. 2019. doi:10.1093/eurheartj/ehz173
53. Milandri A, Farioli A, Gagliardi C, et al. Carpal tunnel syndrome in cardiac amyloidosis: implications for early diagnosis and prognostic role across the spectrum of aetiologies. Eur J Heart Fail. 2020;22(3):507–515. doi:10.1002/ejhf.1742
54. Aus Dem Siepen F, Hein S, Prestel S, et al. Carpal tunnel syndrome and spinal canal stenosis: harbingers of transthyretin amyloid cardiomyopathy? Clin Res Cardiol. 2019;108(12):1324–1330. doi:10.1007/s00392-019-01467-1
55. Foss TR, Wiseman RL, Kelly JW. The pathway by which the tetrameric protein transthyretin dissociates. Biochemistry. 2005;44(47):15525–15533. doi:10.1021/bi051608t
56. Coutinho P, Martins da Silva A, Lopes Lima J, Resende Barbosa A. Forty years of experience with type I amyloid neuropathy: review of 483 cases. Amyloid Amyloidosis. 1980.
57. Coelho T, Maia LF, Martins da Silva A, et al. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology. 2012;79(8):785–792. doi:10.1212/WNL.0b013e3182661eb1
58. Barroso FA, Judge DP, Ebede B, et al. Long-term safety and efficacy of tafamidis for the treatment of hereditary transthyretin amyloid polyneuropathy: results up to 6 years. Amyloid. 2017;24(3):194–204. doi:10.1080/13506129.2017.1357545
59. Maurer MS, Schwartz JH, Gundapaneni B, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med. 2018;379(11):1007–1016. doi:10.1056/NEJMoa1805689
60. Kristen AV, Ajroud-Driss S, Conceição I, Gorevic P, Kyriakides T, Obici L. Patisiran, an RNAi therapeutic for the treatment of hereditary transthyretin-mediated amyloidosis. Neurodegener Dis Manag. 2019;9(1):5–23. doi:10.2217/nmt-2018-0033
61. Coelho T, Adams D, Conceição I, et al. A Phase II, open-label, extension study of long-term patisiran treatment in patients with hereditary transthyretin-mediated (hATTR) amyloidosis. Orphanet J Rare Dis. 2020;15(1):179. doi:10.1186/s13023-020-01399-4
62. Benson MD, Waddington-Cruz M, Berk JL, et al. Inotersen treatment for patients with hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(1):22–31. doi:10.1056/NEJMoa1716793
63. Brannagan TH, Wang AK, Coelho T, et al. Early data on long-term efficacy and safety of inotersen in patients with hereditary transthyretin amyloidosis: a 2-year update from the open-label extension of the NEURO-TTR trial. Eur J Neurol. 2020;27(8):1374–1381. doi:10.1111/ene.14285
64. Adams D, Ando Y, Beirão JM, et al. Expert consensus recommendations to improve diagnosis of ATTR amyloidosis with polyneuropathy. J Neurol. 2020. doi:10.1007/s00415-019-09688-0
65. Dohrn MF, Auer-Grumbach M, Baron R, et al. Chance or challenge, spoilt for choice? New recommendations on diagnostic and therapeutic considerations in hereditary transthyretin amyloidosis with polyneuropathy: the German/Austrian position and review of the literature. J Neurol. doi:10.1007/s00415-020-09962-6
66. Saito S, Ohno K, Sakuraba H. Fabry-database.org: database of the clinical phenotypes, genotypes and mutant α-galactosidase A structures in Fabry disease. J Hum Genet. 2011;56(6):467–468. doi:10.1038/jhg.2011.31
67. Politei JM, Bouhassira D, Germain DP, et al. Pain in fabry disease: practical recommendations for diagnosis and treatment. CNS Neurosci Ther. 2016;22(7):568–576. doi:10.1111/cns.12542
68. Biegstraaten M, Hollak CEM, Bakkers M, Faber CG, Aerts JMFG, van Schaik IN. Small fiber neuropathy in Fabry disease. Mol Genet Metab. 2012;106(2):135–141. doi:10.1016/j.ymgme.2012.03.010
69. Maag R, Binder A, Maier C, et al. Detection of a characteristic painful neuropathy in Fabry disease: a pilot study. Pain Med. 2008;9(8):1217–1223. doi:10.1111/j.1526-4637.2008.00470.x
70. Forstenpointner J, Sendel M, Moeller P, et al. Bridging the gap between vessels and nerves in fabry disease. Front Neurosci. 2020;14:448. doi:10.3389/fnins.2020.00448
71. Üçeyler N, Ganendiran S, Kramer D, Sommer C. Characterization of pain in fabry disease. Clin J Pain. 2014;30(10):915–920. doi:10.1097/AJP.0000000000000041
72. Hopkin RJ, Bissler J, Banikazemi M, et al. Characterization of Fabry disease in 352 pediatric patients in the Fabry Registry. Pediatr Res. 2008;64(5):550–555. doi:10.1203/PDR.0b013e318183f132
73. MacDermot KD, Holmes A, Miners AH. Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 98 hemizygous males. J Med Genet. 2001;38(11):750–760. doi:10.1136/jmg.38.11.750
74. Keilmann A, Hajioff D, Ramaswami U, Investigators FOS. Ear symptoms in children with Fabry disease: data from the Fabry outcome survey. J Inherit Metab Dis. 2009;32(6):739. doi:10.1007/s10545-009-1290-x
75. Lidove O, Jaussaud R, Aractingi S. Dermatological and soft-tissue manifestations of Fabry disease: characteristics and response to enzyme replacement therapy. In: Mehta A, Beck M, Sunder-Plassmann G, editors. Fabry Disease: Perspectives from 5 Years of FOS. Oxford PharmaGenesis; 2006.
76. Rolfs A, Böttcher T, Zschiesche M, et al. Prevalence of Fabry disease in patients with cryptogenic stroke: a prospective study. Lancet. 2005;366(9499):1794–1796. doi:10.1016/S0140-6736(05)67635-0
77. Germain DP. Fabry disease. Orphanet J Rare Dis. 2010;5:30. doi:10.1186/1750-1172-5-30
78. Scriver CR, Beaud AL, Sly WS, et al. The Metabolic and Molecular Bases of Inherited Disease.
79. Mehta A, Beck M, Eyskens F, et al. Fabry disease: a review of current management strategies. QJM. 2010;103(9):641–659. doi:10.1093/qjmed/hcq117
80. Mehta A, Beck M, Elliott P, et al. Enzyme replacement therapy with agalsidase alfa in patients with Fabry’s disease: an analysis of registry data. Lancet. 2009;374(9706):1986–1996. doi:10.1016/S0140-6736(09)61493-8
81. Schiffmann R, Kopp JB, Austin HA, et al. Enzyme replacement therapy in Fabry disease: a randomized controlled trial. JAMA. 2001;285(21):2743–2749. doi:10.1001/jama.285.21.2743
82. Germain DP, Hughes DA, Nicholls K, et al. Treatment of Fabry’s disease with the pharmacologic chaperone migalastat. N Engl J Med. 2016;375(6):545–555. doi:10.1056/NEJMoa1510198
83. Wanner C, Arad M, Baron R, et al. European expert consensus statement on therapeutic goals in Fabry disease. Mol Genet Metab. 2018;124(3):189–203. doi:10.1016/j.ymgme.2018.06.004
84. Bouwman MG, Maurice-Stam H, Linthorst GE, Hollak CEM, Wijburg FA, Grootenhuis MA. Impact of growing up with Fabry disease on achievement of psychosocial milestones and quality of life. Mol Genet Metab. 2011;104(3):308–313. doi:10.1016/j.ymgme.2011.07.006
85. Arning K, Naleschinski D, Maag R, et al. FabryScan: a screening tool for early detection of Fabry disease. J Neurol. 2012;259(11):2393–2400. doi:10.1007/s00415-012-6619-y
86. Forstenpointner J, Moeller P, Sendel M, Reimer M, Hüllemann P, Baron R. Stratification of patients with unclassified pain in the FabryScan database. J Pain Res. 2019;12:2223–2230. doi:10.2147/JPR.S206223
87. Wang B, Rudnick S, Cengia B, Bonkovsky HL. Acute hepatic porphyrias: review and recent progress. Hepatol Commun. 2019;3(2):193–206. doi:10.1002/hep4.1297
88. Puy H, Gouya L, Deybach J-C. Porphyrias. Lancet. 2010;375(9718):924–937. doi:10.1016/S0140-6736(09)61925-5
89. Ramanujam V-MS, Anderson KE. Porphyria diagnostics-part 1: a brief overview of the porphyrias. Curr Protoc Hum Genet. 2015;86:
90. Wu C-L, Ro L-S, Jung S-M, et al. Clinical presentation and electrophysiological findings of porphyric neuropathies: a follow-up study. Muscle Nerve. 2015;51(3):363–369. doi:10.1002/mus.24327
91. Simon A, Pompilus F, Querbes W, et al. Patient perspective on acute intermittent porphyria with frequent attacks: a disease with intermittent and chronic manifestations. Patient. 2018;11(5):527–537. doi:10.1007/s40271-018-0319-3
92. Cardenas JL, Guerrero C. Acute intermittent porphyria: general aspects with focus on pain. Curr Med Res Opin. 2018;34(7):1309–1315. doi:10.1080/03007995.2018.1435521
93. Lin CS-Y, Krishnan AV, Lee M-J, et al. Nerve function and dysfunction in acute intermittent porphyria. Brain. 2008;131(9):2510–2519. doi:10.1093/brain/awn152
94. Delaby C, To-Figueras J, Deybach JC, Casamitjana R, Puy H, Herrero C. Role of two nutritional hepatic markers (insulin-like growth factor 1 and transthyretin) in the clinical assessment and follow-up of acute intermittent porphyria patients. J Intern Med. 2009;266(3):277–285. doi:10.1111/j.1365-2796.2009.02118.x
95. Lip GY, McColl KE, Goldberg A, Moore MR. Smoking and recurrent attacks of acute intermittent porphyria. BMJ. 1991;302(6775):507. doi:10.1136/bmj.302.6775.507
96. Andersson C, Innala E, Bäckström T. Acute intermittent porphyria in women: clinical expression, use and experience of exogenous sex hormones. A population-based study in northern Sweden. J Intern Med. 2003;254(2):176–183. doi:10.1046/j.1365-2796.2003.01172.x
97. Pischik E, Kauppinen R. An update of clinical management of acute intermittent porphyria. Appl Clin Genet. 2015;8:201–214. doi:10.2147/TACG.S48605
98. Bonkovsky HL, Maddukuri VC, Yazici C, et al. Acute porphyrias in the USA: features of 108 subjects from porphyrias consortium. Am J Med. 2014;127(12):1233–1241. doi:10.1016/j.amjmed.2014.06.036
99. Elder GH, Hift RJ, Meissner PN. The acute porphyrias. Lancet. 1997;349(9065):1613–1617. doi:10.1016/S0140-6736(96)09070-8
100. Bissell DM, Anderson KE, Bonkovsky HL, Longo DL. Porphyria. N Engl J Med. 2017;377(9):862–872. doi:10.1056/NEJMra1608634
101. Balwani M, Sardh E, Ventura P, et al. Phase 3 trial of RNAi therapeutic givosiran for acute intermittent porphyria. N Engl J Med. 2020;382(24):2289–2301. doi:10.1056/NEJMoa1913147
102. Chan A, Liebow A, Yasuda M, et al. Preclinical development of a subcutaneous ALAS1 RNAi therapeutic for treatment of hepatic porphyrias using circulating RNA quantification. Mol Ther Nucleic Acids. 2015;4:e263. doi:10.1038/mtna.2015.36
103. Nair JK, Willoughby JLS, Chan A, et al. Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J Am Chem Soc. 2014;136(49):16958–16961. doi:10.1021/ja505986a
104. Setten RL, Rossi JJ, Han S-P. The current state and future directions of RNAi-based therapeutics. Nat Rev Drug Discov. 2019;18(6):421–446. doi:10.1038/s41573-019-0017-4
105. Sardh E, Harper P, Balwani M, et al. Phase 1 trial of an RNA interference therapy for acute intermittent porphyria. N Engl J Med. 2019;380(6):549–558. doi:10.1056/NEJMoa1807838
106. Singal AK, Parker C, Bowden C, Thapar M, Liu L, McGuire BM. Liver transplantation in the management of porphyria. Hepatology. 2014;60(3):1082–1089. doi:10.1002/hep.27086
107. Soonawalla ZF, Orug T, Badminton MN, et al. Liver transplantation as a cure for acute intermittent porphyria. Lancet. 2004;363(9410):705–706. doi:10.1016/S0140-6736(04)15646-8
108. Farschtschi S, Mautner V-F, McLean ACL, Schulz A, Friedrich RE, Rosahl SK. The neurofibromatoses. Dtsch Arztebl Int. 2020;117(20):354–360. doi:10.3238/arztebl.2020.0354
109. Li P, Zhao F, Zhang J, et al. Clinical features of spinal schwannomas in 65 patients with schwannomatosis compared with 831 with solitary schwannomas and 102 with neurofibromatosis type 2: a retrospective study at a single institution. J Neurosurg Spine. 2016;24(1):145–154. doi:10.3171/2015.3.SPINE141145
110. Merker VL, Esparza S, Smith MJ, Stemmer-Rachamimov A, Plotkin SR. Clinical features of schwannomatosis: a retrospective analysis of 87 patients. Oncologist. 2012;17(10):1317–1322. doi:10.1634/theoncologist.2012-0162
111. Blakeley JO, Plotkin SR. Therapeutic advances for the tumors associated with neurofibromatosis type 1, type 2, and schwannomatosis. Neuro-Oncology. 2016;18(5):624–638. doi:10.1093/neuonc/nov200
112. Alaidarous A, Parfait B, Ferkal S, Cohen J, Wolkenstein P, Mazereeuw-Hautier J. Segmental schwannomatosis: characteristics in 12 patients. Orphanet J Rare Dis. 2019;14(1):207. doi:10.1186/s13023-019-1176-4
113. Smith MJ, Bowers NL, Bulman M, et al. Revisiting neurofibromatosis type 2 diagnostic criteria to exclude LZTR1-related schwannomatosis. Neurology. 2017;88(1):87–92. doi:10.1212/WNL.0000000000003418
114. Kehrer-Sawatzki H, Farschtschi S, Mautner V-F, Cooper DN. The molecular pathogenesis of schwannomatosis, a paradigm for the co-involvement of multiple tumour suppressor genes in tumorigenesis. Hum Genet. 2017;136(2):129–148. doi:10.1007/s00439-016-1753-8
115. Evans DGR, Huson SM, Birch JM. Malignant peripheral nerve sheath tumours in inherited disease. Clin Sarcoma Res. 2012;2(1):17. doi:10.1186/2045-3329-2-17
116. Gonzalvo A, Fowler A, Cook RJ, et al. Schwannomatosis, sporadic schwannomatosis, and familial schwannomatosis: a surgical series with long-term follow-up. Clinical article. J Neurosurg. 2011;114(3):756–762. doi:10.3171/2010.8.JNS091900
117. Deiller C, Van-gils J, Zordan C, et al. Coexistence of schwannomatosis and glioblastoma in two families. Eur J Med Genet. 2019;62(8):103680. doi:10.1016/j.ejmg.2019.103680
118. Ostrow KL, Donaldson KJ, Caterina MJ, Belzberg A, Hoke A. The secretomes of painful versus nonpainful human schwannomatosis tumor cells differentially influence sensory neuron gene expression and sensitivity. Sci Rep. 2019;9(1):13098. doi:10.1038/s41598-019-49705-w
119. Merker VL, Bredella MA, Cai W, et al. Relationship between whole-body tumor burden, clinical phenotype, and quality of life in patients with neurofibromatosis. Am J Med Genet A. 2014;164(6):1431–1437. doi:10.1002/ajmg.a.36466
120. Farschtschi SC, Mainka T, Glatzel M, et al. C-fiber loss as a possible cause of neuropathic pain in schwannomatosis. Int J Mol Sci. 2020;21(10):3569. doi:10.3390/ijms21103569
121. Sughrue ME, Levine J, Barbaro NM. Pain as a symptom of peripheral nerve sheath tumors: clinical significance and future therapeutic directions. J Brachial Plex Peripher Nerve Inj. 2008;3:6. doi:10.1186/1749-7221-3-6
122. Koltzenburg M, Scadding J. Neuropathic pain. Curr Opin Neurol. 2001;14(5):641–647. doi:10.1097/00019052-200110000-00014
123. Wagner R, Myers RR. Endoneurial injection of TNF-alpha produces neuropathic pain behaviors. Neuroreport. 1996;7(18):2897–2901. doi:10.1097/00001756-199611250-00018
124. Jordan JT, Smith MJ, Walker JA, et al. Pain correlates with germline mutation in schwannomatosis. Medicine (Baltimore). 2018;97(5):e9717. doi:10.1097/MD.0000000000009717
125. Halvorsen CM, Rønning P, Hald J, et al. The long-term outcome after resection of intraspinal nerve sheath tumors: report of 131 consecutive cases. Neurosurgery. 2015;77(4):585–593. doi:10.1227/NEU.0000000000000890
126. Plotkin SR, Stemmer-Rachamimov AO, Barker FG, et al. Hearing improvement after bevacizumab in patients with neurofibromatosis type 2. N Engl J Med. 2009;361(4):358–367. doi:10.1056/NEJMoa0902579
127. Kiguchi N, Kobayashi Y, Kadowaki Y, Fukazawa Y, Saika F, Kishioka S. Vascular endothelial growth factor signaling in injured nerves underlies peripheral sensitization in neuropathic pain. J Neurochem. 2014;129(1):169–178. doi:10.1111/jnc.12614
128. Ahmed SG, Abdelanabi A, Doha M, Brenner GJ. Correction to: schwannoma gene therapy by adeno-associated virus delivery of the pore-forming protein Gasdermin-D. Cancer Gene Ther. 2020;27(3–4):265. doi:10.1038/s41417-019-0096-8
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.