Back to Journals » Therapeutics and Clinical Risk Management » Volume 17
Unresponsive Thrombotic Thrombocytopenic Purpura (TTP): Challenges and Solutions
Authors Lemiale V, Valade S, Mariotte E
Received 23 March 2021
Accepted for publication 14 May 2021
Published 3 June 2021 Volume 2021:17 Pages 577—587
DOI https://doi.org/10.2147/TCRM.S205632
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Virginie Lemiale, Sandrine Valade, Eric Mariotte
Medical Intensive Care Unit, Saint Louis University Hospital, Assistance Publique des Hôpitaux de Paris, Paris, France
Correspondence: Eric Mariotte
Medical Intensive Care Unit, Saint Louis University Hospital, Assistance Publique des Hôpitaux de Paris, 1 Avenue Claude Vellefaux, Paris, 75010, France
Email [email protected]
Abstract: Thrombotic thrombocytopenic purpura (TTP) is a thrombotic microangiopathy secondary to a severely decreased A Disintegrin And Metalloprotease with ThromboSpondin type 1 repeats 13 (ADAMTS13) activity, resulting in the formation of widespread von Willebrand factor - and platelet-rich microthrombi. ADAMTS13 deficiency is mainly acquired through anti-ADAMTS13 autoantibodies in adults. With modern standards of care, unresponsive TTP has become rarer with a frequency of refractory/relapsing forms dropping from > 40% to < 10%. As patients with unresponsive TTP are at increased risk of mortality, prompt recognition and early therapeutic intensification are mandatory. Therapeutic options at the disposal of clinicians caring for patients with refractory TTP consist of increased ADAMTS13 supplementation, increased immunosuppression, and inhibition of von Willebrand factor adhesion to platelets. In this work, we focus on possible therapies for the management of patients with unresponsive TTP, and propose an algorithm for the management of these difficult cases.
Keywords: thrombotic thrombocytopenic purpura, refractory, relapsing, rituximab, caplacizumab
Introduction
Thrombotic thrombocytopenic purpura (TTP) is a thrombotic microangiopathy (TMA), characterized by the spontaneous formation of thrombi in the microcirculation.1 The diagnosis of TMA relies on the association of hemolytic mechanical anemia, peripheral thrombocytopenia and various signs of visceral ischemia ascribable to microvessel thrombosis. In TTP, a severe ADAMTS13 (A Disintegrin And Metalloprotease with ThromboSpondin type 1 repeats 13) activity deficiency is responsible for an excess of unusually large von Willebrand factor (vWF) multimers. Under high shear stress conditions associated with the microcirculation, these vWF multimers spontaneously aggregate with platelets, leading to the different signs of the disease.2,3 Of note vWF and ADAMTS13 play a key role in various other thrombotic conditions and in cardiovascular diseases.4–7
ADAMTS13 deficiency can be secondary to mutations or polymorphisms of the ADAMTS13 gene (9q34) in hereditary TTP (Upshaw Schulman syndrome, Online Mendelian Inheritance in Man number, 274150)8 or can be acquired through anti-ADAMTS13 antibodies in the majority of adult forms (immune TTP).9 TTP is a rare disease with an incidence rate estimated as 1.5–6/million/year, affecting mostly young women.10–12 It can be a devastating condition, leading to death in more than 80% of cases if not treated promptly with plasmatherapy.13
Modern standards of care for suspected cases of TTP include therapeutic plasma exchanges (TPE), systemic corticosteroids, anti-CD20 monoclonal antibody and anti-vWF domain A1 nanobody.14 With this therapeutic schedule, a vast majority of patients will reach TTP remission and mortality rate is less than 5%.15 However, in some patients, first line therapy fails and additional treatments are required in emergency.16 The aim of this review is to explore current options at the disposal of clinicians faced with cases of unresponsive TTP in adults.
Challenge 1: Recognize Unresponsive TTP
Refractory TTP was first defined in the 2003 British Committee for Standards in Haematology guidelines as cases who fail to reach a platelet rate above 150 G/L after 7 TPE sessions.17 In the 2017 consensus on standardization of terminology on TTP,18 response to therapy was defined as a platelet rate above 150 G/L and Lactate De Hydrogenase (LDH) level <1.5 the upper norm of the laboratory after 5 TPE sessions. Remission consists of a durable response 30 days after the end of therapy (at that time, last TPE session). The 2021 consensus introduced several new notions.19 First, clinical response now also requires the absence of signs of new or progressive signs of ischemic organ injury, in addition to platelet and LDH rates improvement. Second, it introduces the notion of biological recovery, based on ADAMTS13 activity normalization, underpinned by the fact that TTP patients failing to reach ADAMTS13 activity recovery at the end of treatment are at very high risk of clinical relapse.
Unresponsive TTP in this work will, though, include cases presenting with:
- Failure to reach clinical response after 5 TPE sessions (non-response or insufficient clinical response).
- Occurrence of any new clinical or biological sign of TMA while in therapy, or within 30 days following the end of treatment (TPE or caplacizumab) (exacerbation).
- Re-occurrence of TTP after a period of remission (relapse).
Figure 1 illustrates these definitions, according to the timeline of the disease.
 |
Figure 1 Timeline of acute TTP management with favorable (green) and unfavorable (red) outcomes. Non/insufficient response is defined as failure to reach platelet rate >150G/L and/or LDH rate <1.5N and/or occurrence of new/progressive signs of ischemic organ involvement after 5 TPE sessions; Exacerbation is defined as recurrence of any sign of TTP after a phase of clinical response, up to 30 days after the end of therapy; Remission is defined as clinical response maintained over 30 days after cessation of therapy; Relapse is defined as the recurrence of any sign of TTP after remission (ADAMTS13 activity decrease or clinical/biological signs of thrombotic microangiopathy). |
Challenge 2: Confirm TTP Refractoriness
Ascertain the Diagnosis of TTP
TTP is sometimes a difficult clinical diagnosis to make.20
First, TTP may be mistaken with other diseases that do not belong to the TMA syndrome. Examples of such diseases that may present as TMA and usually do not respond to standard TTP therapy are provided in Table 1.
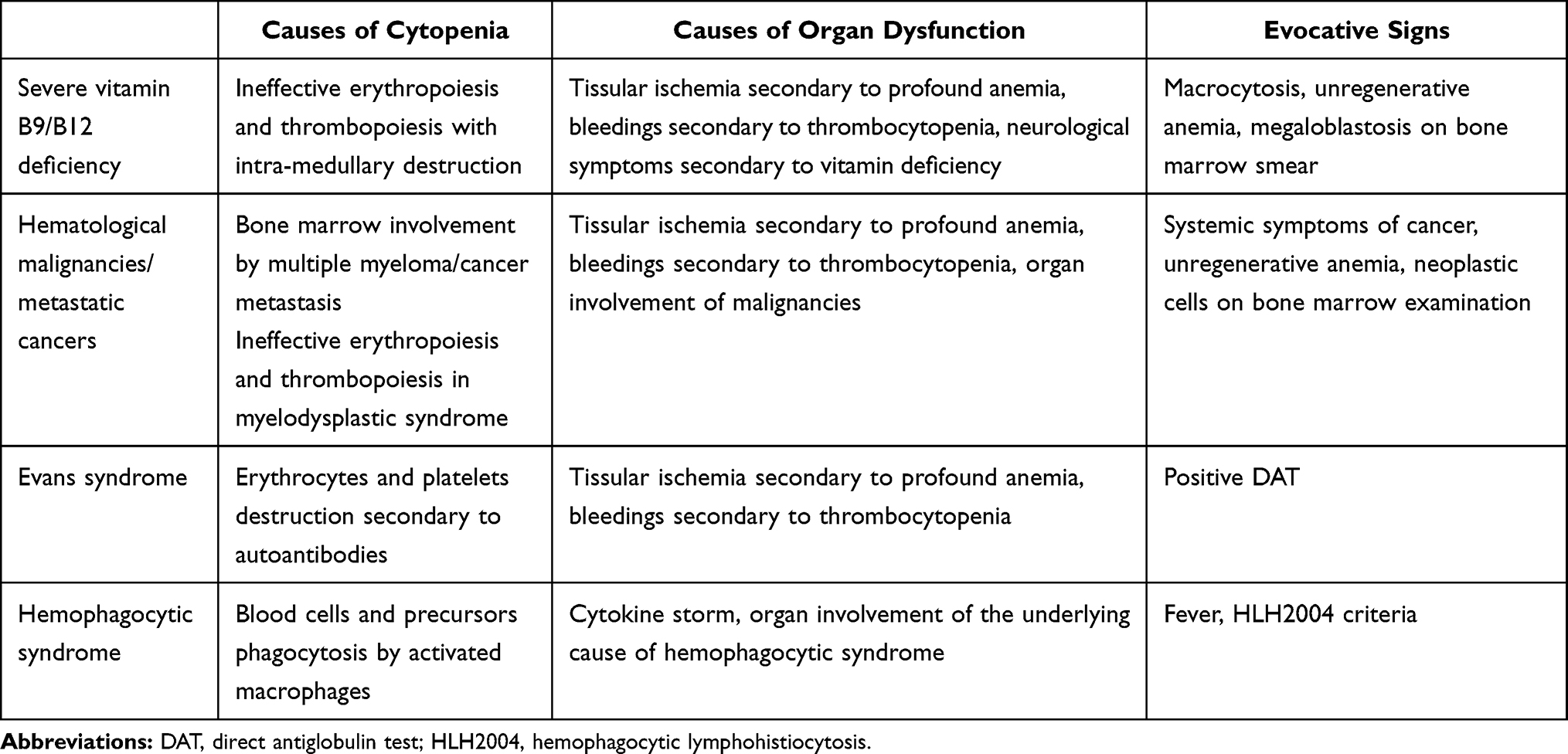 |
Table 1 TMA Differential Diagnosis |
TMA syndrome consists of a number of different diseases and TTP is far from being the most frequent (estimated frequency being 3% of all TMA syndromes).21 ADAMTS13 activity study is of critical importance to differentiate TTP from other TMA syndromes with potentially similar clinical presentations (secondary TMA, complement-mediated hemolytic uremic syndrome (HUS), shigatoxin associated HUS, etc). Laboratory tests should be performed on blood samples retrieved before the onset of plasmatherapy. This way, a diagnosis of refractory TTP can readily be excluded if patients displayed a detectable activity at onset of therapy and eventually fail first line therapy.22,23
Search for Confounders
Dissociated clinical and biological evolution is unusual in TTP patients. In the case of persistent thrombocytopenia and/or anemia in patients with satisfying clinical evolution on first-line therapy, clinicians should look for alternate diagnoses others than refractory TTP. Alternative causes of persistent or new organ dysfunctions should also systematically be sought, especially if hematological involvement is improving. Examples of confounders are provided in Table 2.
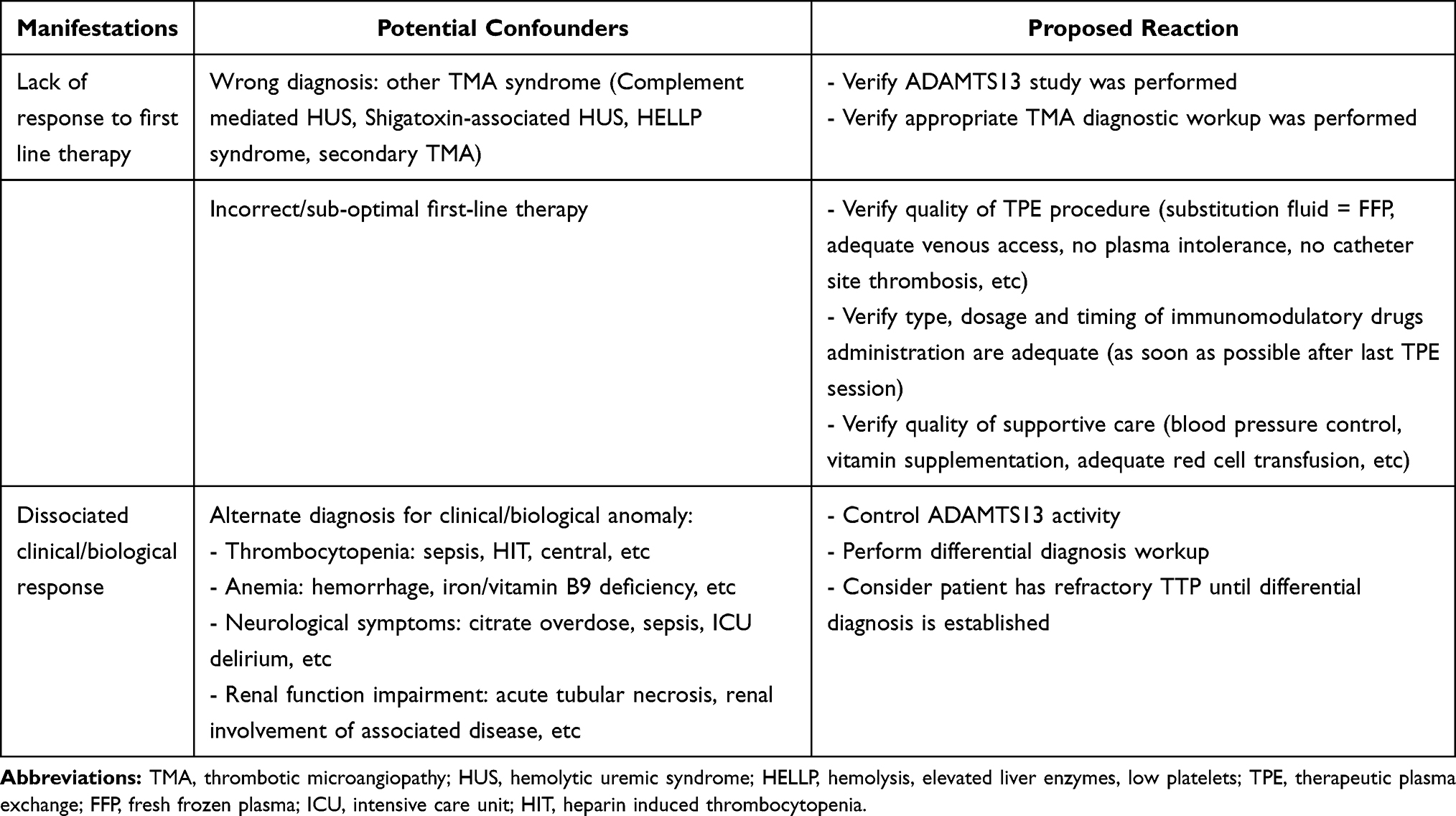 |
Table 2 Confounders for TTP Refractoriness |
Nevertheless, clinicians should always keep a high index of suspicion and if the cause of clinical or biological degradation is uncertain, patients must be considered as having refractory TTP and prompt therapeutic intensification must be performed.23
ADAMTS13 activity control in patients on therapy may also prove useful, as organ dysfunctions appearing/persisting in patients with normalized ADAMTS13 activity are very unlikely to be related to refractory TTP.
Challenge 3: Choose Therapeutic Targets for Unresponsive TTP
First-Line Treatment
Figure 2 displays the different therapeutic targets available for the management of TTP.
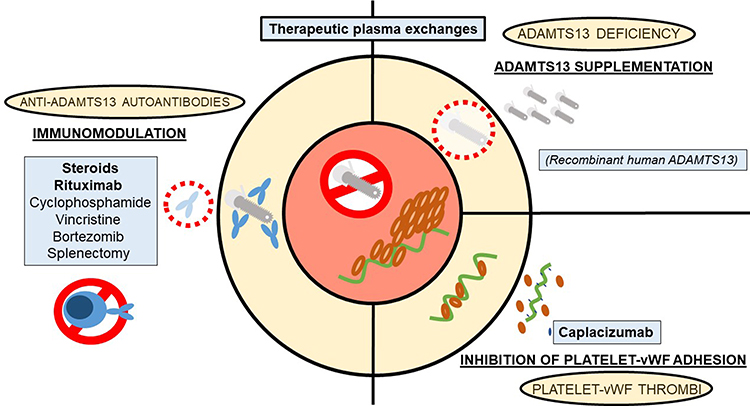 |
Figure 2 Pathophysiology, potential therapeutic targets and available therapy for the management of TTP. |
ADAMTS13 Supplementation: Therapeutic Plasma Exchanges
TPE still are the cornerstone of therapy for patients with acquired TTP. As part of first line therapy they are performed at a volume of 1.5 estimated plasma volume (EPV), daily, with healthy donor plasma or derived products (ie cryosupernatant) until patients reach clinical remission.17,23,24 The main mechanisms of action of TPE in TTP are thought to be both the administration of massive amounts of functional ADAMTS13 and the removal of anti-ADAMTS13 autoantibodies and unusually large vWF multimers.25 TPE should be preferred to plasma infusions in patients suspected of having TTP, as they are associated with a better outcome.26 EPV may be calculated with the following formula:
EPV = Total Blood Volume (TBV) x (1-Hematocrit)
TBV is estimated using either Nadler’s formula (based on gender, height and weight) or Gilcher’s rule of 5 (based on gender and morphotype).27 With simplifications, EPV is often considered 40 mL/kg in critical care patients.
Immunomodulation
Steroids
Corticosteroids were the first immunomodulating drugs used in patients with TTP and they are part of the first line therapeutic schedule of patients with a suspicion of acquired TTP since 2003.17,28 In one study, the use of corticosteroids (prednisone 200 mg/d) as standalone therapy in mild forms of TTP with no sign of neurological involvement (hematological signs only) induced durable remission in 52% of cases (28/54).29
Anti-CD20 Monoclonal Antibodies
Anti-CD20 monoclonal antibodies target B-lymphocytes implicated in the production of anti-ADAMTS13 autoantibodies in a vast majority of adult patients with acquired TTP.30,31 They were developed for the treatment of B-cell lymphomas and secondarily used in many autoimmune diseases. Most of the literature has described the use of rituximab, a chimeric mouse-human monoclonal anti-CD20 antibody, in these indications.32,33 Severe adverse side-effects include infusional reactions, serum sickness, hypogammaglobulinemia and infections including Hepatitis virus B reactivations and progressive multifocal leukoencephalopathy (due to JC-BK virus central nervous system infection). These manifestations were mostly described in patients with lymphomas receiving cytotoxic polychemotherapy along with rituximab.34 In patients with TTP, rituximab seems to present a very favorable security profile.30
Anti-CD20 monoclonal antibodies are recommended in case of refractory or relapsing TTP.35 They are also increasingly used as part of first-line therapy, during the first 3 days after diagnosis.15,28,35 The dose varies from 375 mg/m2 to 100–1000 mg fixed doses, administered weekly36 or on days 1-4-8-15,37 considering TPE will remove a significant part of the monoclonal antibodies.38
This recommendation is not based on randomized controlled studies but on case-control studies and case series.30,39 In TTP, authors have shown rituximab may improve time to clinical remission,37 improve time to intensive care discharge and decrease the risk of TTP relapse during the year following administration.40 There also seems to be a benefit with early use of rituximab (≤3 days after diagnosis) in comparison to late administration in terms of time to clinical remission.41
As the efficacy of rituximab is expected around 2 weeks after the drug’s administration, early rituximab infusions are now recommended as a first line therapy in TTP, even before the results of ADAMTS13 studies are known.28 If rituximab is used as second-line therapy, it is mandatory to continue first-line therapy (at least daily TPE) and adjunct fast-acting therapies may also be required. The number of rituximab injections may be adapted according to B-cell depletion monitored in blood.42
Rituximab can also be used in patients that present with an ADAMTS13 activity decrease while in clinical remission of TTP. In this context, rituximab, as a single agent therapy in patients with no clinical or biological signs of TMA, may induce ADAMTS13 activity normalization and prevent progression to TTP clinical relapse (preemptive treatment).43–46 In this setting, sub-cutaneous administration is possible.47
Clinicians should be cautious about the apparition of anti-rituximab antibodies in patients treated with multiple courses of rituximab. These autoantibodies have been associated with infusional reactions, serum sickness and loss of rituximab efficiency.48 Patients who fail or become intolerant to rituximab may benefit from treatment with other anti-CD20 antibodies (eg obinutuzumab49 or ofatumumab).50
VWF Targeting Agents: Anti-vWF Domain A1 Nanobodies
Caplacizumab is an anti-vWF domain A1 nanobody, impairing the adhesion of vWF to platelet receptor gp Ib-IX-V. The therapeutic schedule includes a 10 mg intravenous loading dose administered before a TPE session followed by subcutaneous 10 mg injections after each TPE and up to 30 days after the end of plasmatherapy. In two randomized controlled studies, the addition of caplacizumab to standard of care shortened time to clinical remission in TTP patients.51,52 Real world data are also in favor of caplacizumab's early administration in patients suspected of having TTP.15,53 Caplacizumab is approved for the treatment of acquired TTP28 but the cost-effectiveness of frontline caplacizumab administration in all TTP patients remains uncertain.54,55 Adverse side effects consist of hemorrhage, usually non severe.
Caplacizumab may be used in patients with refractory TTP who did not receive this drug frontline or who present with relapses after caplacizumab withdrawal.
Due to the mechanism of action of caplacizumab, that is the inhibition of the early stage of thrombi formation, this drug should be administered as soon as possible after the diagnosis of TTP.28 It is a “time buying” or suspensive therapy that has no effect on anti-ADAMTS13 autoantibodies production, explaining a relatively high rate of relapses in patients that keep a severely decreased ADAMTS13 activity at the end of treatment. In these patients, intensification of the immunosuppressive regimen and caplacizumab continuation are recommended until ADAMTS13 activity is restored.
Unresponsive TTP Treatment
Therapeutic options for patients with unresponsive TTP are summarized in Table 3.
 |
Table 3 Therapeutic Options Currently Available for Unresponsive TTP Management |
First-Line Treatment Adaptation
ADAMTS13 Supplementation
In patients that do not respond to daily TPE or clinically deteriorate while on first line therapy, twice-daily TPE have been proposed, with a volume of 1 to 1.5 EPV per 12 hours.56,57 This costly and time-consuming procedure has not been evaluated in controlled studies but may prove helpful in patients with very severe forms of TTP, high titles of anti-ADAMTS13 autoantibodies and in whom immunosuppressive interventions have not yet reached their efficacy (during the first days of treatment). Clinicians must anticipate significant removal of therapeutic drugs with high molecular weights (ie monoclonal antibodies) and/or important albumin binding in patients receiving this therapeutic schedule.25
Immunomodulation
As high dose pulses of steroids may be associated with improved outcomes in comparison with lower doses,58 their use as second-line therapy in unresponsive cases of TTP may be proposed in patients not already undergoing treatment with corticosteroids or who received lower doses of steroids.
Second Line Therapy
Vincristine
Vincristine is a vinca-alcaloid used as anti-cancer chemotherapy or immunomodulating agents in various indications. In TTP, vincristine may also play an inhibitory role on platelets glycoprotein receptors to vWF.59
Patients with refractory TTP may respond to second-line therapy with 1.4 mg/m2 (maximum dose 2 mg) vincristine intravenously. A single injection is usually administered, with an efficacy expected in less than a week and toxicities including cytopenias due to bone marrow depletion and peripheral neuropathy.22,59,60
Cyclophosphamide
Cyclophosphamide is an alkylating agent used as anti-cancer chemotherapy and as an immunosuppressive agent. It can be used in patients with refractory TTP with a single parenteral injection of 500 mg/m2 or 750 mg/m2, with an efficacy expected from one to two weeks after treatment and toxicities including bone marrow depletion.61,62 Cyclophosphamide may be particularly indicated in TTP cases associated with other autoimmune diseases, like systemic lupus erythematosus and Sicca syndrome.
Bortezomib
Bortezomib is a proteasome inhibitor approved for the treatment of multiple myeloma and other plasma cells disorders.63 It may be a part of refractory TTP therapy at a dose of 1–1.3 mg/m2, administered intravenously or subcutaneously, from 1 to 4 times (days 1-4-8-11) depending on response which can be expected in days following administration.64,65 Side-effects described in patients with refractory TTP are peripheral neuropathy, pulmonary and cardiac toxicities.
Other Immunomodulating Agents
Many other immunomodulating drugs have been tried in patients with refractory/relapsing TTP, including cyclosporine A,66 azathioprine,67 intravenous polyvalent human immunoglobulin68 and eculizumab.69,70 Data concerning these therapies is scant, sometimes contradictory and limited to case reports or small case series, often pre-dating the advent of rituximab use.
N-Acetyl Cysteine
In clinical practice, N-acetyl cysteine is principally used as an antidote for acetaminophen poisoning.71 It has the capacity to disrupt the di-sulfite bonds linking vWF monomers, to disrupt vWF domain A1 and display an inhibitory effect on ADP- and collagen-induced platelet aggregation in vitro and in animal models.72–74 By these mechanisms, N-acetylcysteine could reduce the amount of unusually large vWF multimers found in TTP patients and reduce vWF-platelets adhesion.75 It can be administered using the same protocol as in acetaminophen poisoning (continuous infusion of 300 mg/day), for an indeterminate duration (usually less than 1 week). Efficacy is expected in the days following N-acetylcysteine infusion. There is no significant side-effect of N-acetylcysteine described in TTP patients.
Splenectomy
Splenectomy has been proposed to patients with refractory or relapsing TTP, mainly before the rituximab era.61,76,77 The presumed mechanisms of action of splenectomy in TTP are unclear, probably related to the removal of a major site of anti-ADAMTS13 autoantibodies production as well as a major site of platelet consumption. This procedure usually leads to prompt remission of the disease but can be difficult to perform in severely thrombocytopenic patients. Adverse side effects include surgical complications and thromboembolic disease.76 Due to the risk of hemorrhagic complications, laparoscopic procedures should probably be preferred. Prophylactic platelet transfusion should be avoided whenever possible in the context of unresponsive TTP.77
Unresponsive TTP: Management Algorithm Proposal
In our institution, first line therapy for suspected TTP includes daily TPE, corticosteroids, caplacizumab (despite doubts about its cost-effectiveness) and early rituximab administration. With this schedule, we observe a response rate over 90% and a mortality rate less than 5%.15 Considering the highly variable clinical presentations and first-line therapeutic management protocols of acquired TTP patients, it is difficult to recommend a universal management algorithm for refractory forms of the disease. In most of the literature on refractory TTP, patients receive multiple second-line therapies, sequentially at best, making it difficult to draw any conclusions about which drug the patients ultimately responded to.
Patients with a suspicion of TTP undergo a diagnostic workup, including ADAMTS13 study, before the onset of first-line emergency treatment. Frontline therapy includes TPE (60 mL/kg/day with fresh frozen plasma), corticosteroids (1 mg/kg/day prednisone equivalent, administered after TPE sessions), early rituximab (375 mg/m2 administered after TPE sessions at day 1 and 4) and caplacizumab (10 mg/kg IV before first TPE then 10 mg/day SC after TPE sessions). In cases where patients respond to first-line treatment, TPE are stopped when platelet rate is measured above 150 G/L for 2 consecutive days. Corticosteroids are maintained at the same dose for 3 weeks and then tapered and stopped on 1 to 2 weeks. If remission of TTP occurs before day 8, B-cells depletion is monitored in the blood and patients may skip scheduled rituximab infusions on days 8 and 15 if CD-20 cells are not detectable anymore. Caplacizumab is administered for 4 weeks after the last TPE. Shorter or longer durations of caplacizumab are discussed on a case by case basis, depending on the results of ADAMTS13 activity dosages performed weekly during that period.
In case of a refractory course of TTP, patients are thoroughly evaluated for confounding factors. If refractory TTP cannot be ruled out, second-line therapy is promptly started. According to our local protocols of care, the choice of therapeutic intensification depends on the kind of symptoms revealing refractory TTP.
In patients with persisting biological anomalies only (thrombocytopenia, hemolysis markers, etc), our local practice is to add vincristine infusions (1.4 mg/m2, one single dose) and/or high dose steroid pulses (methylprednisolone 1000 mg/day for 3 days, administered after TPE sessions) to first-line therapy. In our experience, this schedule is sufficient to lead most patients to remission.
Patients with minor clinical events ascribable to TTP (headaches, brisk reflexes, angina, etc) while on first-line therapy receive vincristine and/or high dose steroid pulses and twice-daily TPE for 1 to 3 days depending on symptoms resolution.
Patients with major clinical events (seizures, coma, unstable angina, etc) receive vincristine, high dose steroid pulses, twice-daily TPE and additional immunosuppressive drugs (cyclophosphamide, bortezomib, etc).
In patients not responding to all previous therapies, splenectomy is used as salvage therapy.
Figure 3 illustrates the management of refractory TTP at our institution.
 |
Figure 3 Unresponsive TTP management algorithm. |
As TTP is a rare disease and a refractory course even rarer, management of these cases still relies mostly on expert clinicians' experience. Patients with refractory TTP should at least be evaluated by a physician used to TTP management, and in some cases be transferred to an expert team.
Future Perspective: Human Recombinant ADAMTS13
Human recombinant ADAMTS13 (rhADAMTS13) is not yet available in routine practice.
A Phase 1 study on rhADAMTS13 (BAX930) in patients with congenital TTP demonstrated safety and a pharmacokinetic profile comparable to fresh frozen plasma.78 A Phase 3 study on rhADAMTS13 use as prophylaxis or on demand treatment in patients with congenital TTP is ongoing (NCT03393975).
There is limited data suggesting the efficacy of rhADAMTS13 in acquired TTP, the mechanism of action being an overriding of anti-ADAMTS13 autoantibodies (ex-vivo and animal model).79
A Phase 2 study aiming at determining the potential role of rhADAMTS13 (SHP655) in addition to the standard of care in patients with acquired TTP is also ongoing (NCT03922308).
Conclusion
With modern standards of care, including TPE, corticosteroids, rituximab and caplacizumab, the mortality of acquired TTP has dropped from 10–20% to less than 5% and refractory/relapsing course from over 40% to less than 5%. As TTP is a rare disease, there will probably never be high quality data concerning the optimal management of these difficult cases. The algorithm for therapeutic escalation in unresponsive forms will ultimately depend on expert opinions and each center experience/preference. New therapeutic options, such as recombinant human ADAMTS13 (NCT03922308) and anfibatide (NCT04021173), are under evaluation and may hopefully lead to the disappearance of unresponsive TTP.
Disclosure
Dr Virginie Lemiale reports they belong to a research group which received fees from gilead, MSD, Alexion, celgene, and baxter, and that biomerieux pay travel for congress, outside the submitted work. Dr Sandrine Valade reports personal fees from Sanofi and Gilead/Kite, and non-financial support from Pfizer, outside the submitted work. Dr Eric Mariotte reports personal fees from Sanofi, outside the submitted work. The authors reported no other potential conflicts of interest for this work.
References
1. Crawley JTB, Scully MA. Thrombotic thrombocytopenic purpura: basic pathophysiology and therapeutic strategies. Hematology Am Soc Hematol Educ Program. 2013;2013:292–299. doi:10.1182/asheducation-2013.1.292
2. Sadler JE. Pathophysiology of thrombotic thrombocytopenic purpura. Blood. 2017;130(10):1181–1188. doi:10.1182/blood-2017-04-636431
3. Furlan M, Robles R, Galbusera M, et al. von Willebrand factor-cleaving protease in thrombotic thrombocytopenic purpura and the hemolytic-uremic syndrome. N Engl J Med. 1998;339(22):1578–1584. doi:10.1056/NEJM199811263392202
4. Calabrò P, Gragnano F, Golia E, Grove EL. von Willebrand factor and venous thromboembolism: pathogenic link and therapeutic implications. Semin Thromb Hemost. 2018;44(3):249–260. doi:10.1055/s-0037-1605564
5. Gragnano F, Golia E, Natale F, et al. Von Willebrand factor and cardiovascular disease: from a biochemical marker to an attractive therapeutic target. Curr Vasc Pharmacol. 2017;15(5):404–415. doi:10.2174/1570161115666170201114835
6. Gragnano F, Sperlongano S, Golia E, et al. The role of von Willebrand factor in vascular inflammation: from pathogenesis to targeted therapy. Mediators Inflamm. 2017;2017:5620314. doi:10.1155/2017/5620314
7. Levi M, Scully M, Singer M. The role of ADAMTS-13 in the coagulopathy of sepsis. J Thromb Haemost. 2018;16(4):646–651. doi:10.1111/jth.13953
8. Kremer Hovinga JA, George JN, Longo DL. Hereditary thrombotic thrombocytopenic purpura. N Engl J Med. 2019;381(17):1653–1662. doi:10.1056/NEJMra1813013
9. Tsai HM, Lian EC. Antibodies to von Willebrand factor-cleaving protease in acute thrombotic thrombocytopenic purpura. N Engl J Med. 1998;339(22):1585–1594. doi:10.1056/NEJM199811263392203
10. Mariotte E, Azoulay E, Galicier L, et al. Epidemiology and pathophysiology of adulthood-onset thrombotic microangiopathy with severe ADAMTS13 deficiency (thrombotic thrombocytopenic purpura): a cross-sectional analysis of the French national registry for thrombotic microangiopathy. Lancet Haematol. 2016;3(5):e237–245. doi:10.1016/S2352-3026(16)30018-7
11. Scully M, Yarranton H, Liesner R, et al. Regional UK TTP registry: correlation with laboratory ADAMTS 13 analysis and clinical features. Br J Haematol. 2008;142(5):819–826. doi:10.1111/j.1365-2141.2008.07276.x
12. Miesbach W, Menne J, Bommer M, et al. Incidence of acquired thrombotic thrombocytopenic purpura in Germany: a hospital level study. Orphanet J Rare Dis. 2019;14(1):260. doi:10.1186/s13023-019-1240-0
13. Dane K, Chaturvedi S. Beyond plasma exchange: novel therapies for thrombotic thrombocytopenic purpura. Hematology Am Soc Hematol Educ Program. 2018;2018(1):539–547. doi:10.1182/asheducation-2018.1.539
14. Zheng XL, Vesely SK, Cataland SR, et al. ISTH guidelines for the diagnosis of thrombotic thrombocytopenic purpura. J Thromb Haemost. 2020;18(10):2486–2495. doi:10.1111/jth.15006
15. Coppo P, Bubenheim M, Azoulay E, et al. A regimen with caplacizumab, immunosuppression, and plasma exchange prevents unfavorable outcomes in immune-mediated TTP. Blood. 2021;137(6):733–742. doi:10.1182/blood.2020008021
16. Mariotte E, Blet A, Galicier L, et al. Unresponsive thrombotic thrombocytopenic purpura in critically ill adults. Intensive Care Med. 2013;39(7):1272–1281. doi:10.1007/s00134-013-2873-4
17. Allford SL, Hunt BJ, Rose P, Machin SJ; Haemostasis and Thrombosis Task Force, British Committee for Standards in Haematology. Guidelines on the diagnosis and management of the thrombotic microangiopathic haemolytic anaemias. Br J Haematol. 2003;120(4):556–573. doi:10.1046/j.1365-2141.2003.04049.x
18. Scully M, Cataland S, Coppo P, et al. Consensus on the standardization of terminology in thrombotic thrombocytopenic purpura and related thrombotic microangiopathies. J Thromb Haemost. 2017;15(2):312–322. doi:10.1111/jth.13571
19. Cuker A, Cataland SR, Coppo P, et al. Redefining outcomes in immune TTP: an International Working Group Consensus Report. Blood. 2021;137:1855–1861. doi:10.1182/blood.2020009150
20. George JN. The remarkable diversity of thrombotic thrombocytopenic purpura: a perspective. Blood Adv. 2018;2(12):1510–1516. doi:10.1182/bloodadvances.2018018432
21. Bayer G, von Tokarski F, Thoreau B, et al. Etiology and outcomes of thrombotic microangiopathies. Clin J Am Soc Nephrol. 2019;14(4):557–566. doi:10.2215/CJN.11470918
22. Zheng XL, Vesely SK, Cataland SR, et al. Good practice statements (GPS) for the clinical care of patients with thrombotic thrombocytopenic purpura. J Thromb Haemost. 2020;18(10):2503–2512. doi:10.1111/jth.15009
23. Azoulay E, Bauer PR, Mariotte E, et al. Expert statement on the ICU management of patients with thrombotic thrombocytopenic purpura. Intensive Care Med. 2019;45(11):1518–1539. doi:10.1007/s00134-019-05736-5
24. Padmanabhan A, Connelly-Smith L, Aqui N, et al. Guidelines on the use of therapeutic apheresis in clinical practice - evidence-based approach from the writing committee of the American Society for Apheresis: the eighth special issue. J Clin Apher. 2019;34(3):171–354. doi:10.1002/jca.21705
25. Piedrafita A, Ribes D, Cointault O, Chauveau D, Faguer S, Huart A. Plasma exchange and thrombotic microangiopathies: from pathophysiology to clinical practice. Transfus Apher Sci. 2020;59(6):102990. doi:10.1016/j.transci.2020.102990
26. Rock GA, Shumak KH, Buskard NA, et al. Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. Canadian Apheresis Study Group. N Engl J Med. 1991;325(6):393–397. doi:10.1056/NEJM199108083250604
27. Neyrinck MM, Vrielink H; Joint Task Force for Education and Certification. Calculations in apheresis. J Clin Apher. 2015;30(1):38–42. doi:10.1002/jca.21347
28. Zheng XL, Vesely SK, Cataland SR, et al. ISTH guidelines for treatment of thrombotic thrombocytopenic purpura. J Thromb Haemost. 2020;18(10):2496–2502. doi:10.1111/jth.15010
29. Bell WR, Braine HG, Ness PM, Kickler TS. Improved survival in thrombotic thrombocytopenic purpura-hemolytic uremic syndrome. Clinical experience in 108 patients. N Engl J Med. 1991;325(6):398–403. doi:10.1056/NEJM199108083250605
30. Froissart A, Veyradier A, Hié M, Benhamou Y, Coppo P; French Reference Center for Thrombotic Microangiopathies. Rituximab in autoimmune thrombotic thrombocytopenic purpura: a success story. Eur J Intern Med. 2015;26(9):659–665. doi:10.1016/j.ejim.2015.07.021
31. Scully M. Rituximab in the treatment of TTP. Hematology. 2012;17(Suppl 1):S22–24. doi:10.1179/102453312X13336169155178
32. Salles G, Barrett M, Foà R, et al. Rituximab in B-cell hematologic malignancies: a review of 20 years of clinical experience. Adv Ther. 2017;34(10):2232–2273. doi:10.1007/s12325-017-0612-x
33. Taylor RP, Lindorfer MA. Drug insight: the mechanism of action of rituximab in autoimmune disease–the immune complex decoy hypothesis. Nat Clin Pract Rheumatol. 2007;3(2):86–95. doi:10.1038/ncprheum0424
34. Hua Q, Zhu Y, Liu H. Severe and fatal adverse events risk associated with rituximab addition to B-cell non-Hodgkin’s lymphoma (B-NHL) chemotherapy: a meta-analysis. J Chemother. 2015;27(6):365–370. doi:10.1179/1973947815Y.0000000025
35. Lim W, Vesely SK, George JN. The role of rituximab in the management of patients with acquired thrombotic thrombocytopenic purpura. Blood. 2015;125(10):1526–1531. doi:10.1182/blood-2014-10-559211
36. George JN. Corticosteroids and rituximab as adjunctive treatments for thrombotic thrombocytopenic purpura. Am J Hematol. 2012;87(Suppl 1):S88–91. doi:10.1002/ajh.23126
37. Froissart A, Buffet M, Veyradier A, et al. Efficacy and safety of first-line rituximab in severe, acquired thrombotic thrombocytopenic purpura with a suboptimal response to plasma exchange. Experience of the French Thrombotic Microangiopathies Reference Center. Crit Care Med. 2012;40(1):104–111. doi:10.1097/CCM.0b013e31822e9d66
38. Puisset F, White-Koning M, Kamar N, et al. Population pharmacokinetics of rituximab with or without plasmapheresis in kidney patients with antibody-mediated disease. Br J Clin Pharmacol. 2013;76(5):734–740. doi:10.1111/bcp.12098
39. Tun NM, Villani GM. Efficacy of rituximab in acute refractory or chronic relapsing non-familial idiopathic thrombotic thrombocytopenic purpura: a systematic review with pooled data analysis. J Thromb Thrombolysis. 2012;34(3):347–359. doi:10.1007/s11239-012-0723-9
40. Scully M, McDonald V, Cavenagh J, et al. A phase 2 study of the safety and efficacy of rituximab with plasma exchange in acute acquired thrombotic thrombocytopenic purpura. Blood. 2011;118(7):1746–1753. doi:10.1182/blood-2011-03-341131
41. Westwood J-P, Webster H, McGuckin S, McDonald V, Machin SJ, Scully M. Rituximab for thrombotic thrombocytopenic purpura: benefit of early administration during acute episodes and use of prophylaxis to prevent relapse. J Thromb Haemost. 2013;11(3):481–490. doi:10.1111/jth.12114
42. Benhamou Y, Paintaud G, Azoulay E, et al. Efficacy of a rituximab regimen based on B cell depletion in thrombotic thrombocytopenic purpura with suboptimal response to standard treatment: results of a Phase II, multicenter noncomparative study. Am J Hematol. 2016;91(12):1246–1251. doi:10.1002/ajh.24559
43. Reddy MS, Hofmann S, Shen YM, et al. Comparison of low fixed dose versus standard-dose rituximab to treat thrombotic thrombocytopenic purpura in the acute phase and preemptively during remission. Transfus Apher Sci. 2020;59(6):102885. doi:10.1016/j.transci.2020.102885
44. Joly BS, Coppo P, Veyradier A. An update on pathogenesis and diagnosis of thrombotic thrombocytopenic purpura. Expert Rev Hematol. 2019;12(6):383–395. doi:10.1080/17474086.2019.1611423
45. Bresin E, Gastoldi S, Daina E, et al. Rituximab as pre-emptive treatment in patients with thrombotic thrombocytopenic purpura and evidence of anti-ADAMTS13 autoantibodies. Thromb Haemost. 2009;101(2):233–238. doi:10.1160/TH07-12-0753
46. Hie M, Gay J, Galicier L, et al. Preemptive rituximab infusions after remission efficiently prevent relapses in acquired thrombotic thrombocytopenic purpura. Blood. 2014;124(2):204–210. doi:10.1182/blood-2014-01-550244
47. Delrue M, Baylatry MT, Joly A-C, et al. Efficacy of subcutaneous preemptive rituximab in immune-mediated thrombotic thrombocytopenic purpura: experience from the first 12 cases. Am J Hematol. 2021;96(1):E26–E29. doi:10.1002/ajh.26022
48. Vendramin C, Thomas M, Westwood J-P, McGuckin S, Scully M. Rituximab-induced acute and delayed serum sickness in thrombotic thrombocytopenic purpura: the role of anti-rituximab antibodies. Br J Haematol. 2019;184(5):858–861. doi:10.1111/bjh.15177
49. Robertz J, Andres M, Mansouri Taleghani B, Koneth I, Binet I, Kremer Hovinga JA. Obinutuzumab in two patients suffering from immune-mediated thrombotic thrombocytopenic purpura intolerant to rituximab. Am J Hematol. 2019;94(10):E259–E261. doi:10.1002/ajh.25583
50. Al-Samkari H, Grace RF, Connors JM. Ofatumumab for acute treatment and prophylaxis of a patient with multiple relapses of acquired thrombotic thrombocytopenic purpura. J Thromb Thrombolysis. 2018;46(1):81–83. doi:10.1007/s11239-018-1647-9
51. Scully M, Cataland SR, Peyvandi F, et al. Caplacizumab treatment for acquired thrombotic thrombocytopenic purpura. N Engl J Med. 2019;380(4):335–346. doi:10.1056/NEJMoa1806311
52. Peyvandi F, Scully M, Kremer Hovinga JA, et al. Caplacizumab for acquired thrombotic thrombocytopenic purpura. N Engl J Med. 2016;374(6):511–522. doi:10.1056/NEJMoa1505533
53. Völker LA, Kaufeld J, Miesbach W, et al. Real-world data confirm the effectiveness of caplacizumab in acquired thrombotic thrombocytopenic purpura. Blood Adv. 2020;4(13):3085–3092. doi:10.1182/bloodadvances.2020001973
54. Picod A, Veyradier A, Coppo P. Should all patients with immune-mediated thrombotic thrombocytopenic purpura receive caplacizumab? J Thromb Haemost. 2021;19(1):58–67. doi:10.1111/jth.15194
55. Goshua G, Sinha P, Hendrickson JE, Tormey C, Bendapudi PK, Lee AI. Cost effectiveness of caplacizumab in acquired thrombotic thrombocytopenic purpura. Blood. 2021;137(7):969–976. doi:10.1182/blood.2020006052
56. Nguyen L, Li X, Duvall D, Terrell DR, Vesely SK, George JN. Twice-daily plasma exchange for patients with refractory thrombotic thrombocytopenic purpura: the experience of the Oklahoma Registry, 1989 through 2006. Transfusion. 2008;48(2):349–357. doi:10.1111/j.1537-2995.2007.01530.x
57. Soucemarianadin M, Benhamou Y, Delmas Y, et al. Twice-daily therapeutical plasma exchange-based salvage therapy in severe autoimmune thrombotic thrombocytopenic purpura: the French TMA Reference Center experience. Eur J Haematol. 2016;97(2):183–191. doi:10.1111/ejh.12706
58. Balduini CL, Gugliotta L, Luppi M, et al. High versus standard dose methylprednisolone in the acute phase of idiopathic thrombotic thrombocytopenic purpura: a randomized study. Ann Hematol. 2010;89(6):591–596. doi:10.1007/s00277-009-0877-5
59. Bobbio-Pallavicini E, Porta C, Centurioni R, et al. Vincristine sulfate for the treatment of thrombotic thrombocytopenic purpura refractory to plasma-exchange. The Italian Cooperative Group for TTP. Eur J Haematol. 1994;52(4):222–226. doi:10.1111/j.1600-0609.1994.tb00649.x
60. O’Connor NT, O’Shea MJ, Hill LF. Vincristine for thrombotic thrombocytopenic purpura. Lancet. 1992;340(8817):490. doi:10.1016/0140-6736(92)91815-p
61. Beloncle F, Buffet M, Coindre J-P, et al. Splenectomy and/or cyclophosphamide as salvage therapies in thrombotic thrombocytopenic purpura: the French TMA Reference Center experience. Transfusion. 2012;52(11):2436–2444. doi:10.1111/j.1537-2995.2012.03578.x
62. Zheng X, Pallera AM, Goodnough LT, Sadler JE, Blinder MA. Remission of chronic thrombotic thrombocytopenic purpura after treatment with cyclophosphamide and rituximab. Ann Intern Med. 2003;138(2):105–108. doi:10.7326/0003-4819-138-2-200301210-00011
63. Robak P, Robak T. Bortezomib for the treatment of hematologic malignancies: 15 years later. Drugs R D. 2019;19(2):73–92. doi:10.1007/s40268-019-0269-9
64. Eskazan AE. Bortezomib therapy in patients with relapsed/refractory acquired thrombotic thrombocytopenic purpura. Ann Hematol. 2016;95(11):1751–1756. doi:10.1007/s00277-016-2804-x
65. Patriquin CJ, Thomas MR, Dutt T, et al. Bortezomib in the treatment of refractory thrombotic thrombocytopenic purpura. Br J Haematol. 2016;173(5):779–785. doi:10.1111/bjh.13993
66. Cataland SR, Kourlas PJ, Yang S, et al. Cyclosporine or steroids as an adjunct to plasma exchange in the treatment of immune-mediated thrombotic thrombocytopenic purpura. Blood Adv. 2017;1(23):2075–2082. doi:10.1182/bloodadvances.2017009308
67. Moake JL, Rudy CK, Troll JH, et al. Therapy of chronic relapsing thrombotic thrombocytopenic purpura with prednisone and azathioprine. Am J Hematol. 1985;20(1):73–79. doi:10.1002/ajh.2830200110
68. Ding J, Baer MR, Hess JR, Zimrin AB. Intravenous immunoglobulin as salvage therapy for refractory thrombotic thrombocytopenic purpura. Am J Hematol. 2018;93(3):E77–E79. doi:10.1002/ajh.25007
69. Chapin J, Weksler B, Magro C, Laurence J. Eculizumab in the treatment of refractory idiopathic thrombotic thrombocytopenic purpura. Br J Haematol. 2012;157(6):772–774. doi:10.1111/j.1365-2141.2012.09084.x
70. Vigna E, Petrungaro A, Perri A, et al. Efficacy of eculizumab in severe ADAMTS13-deficient thrombotic thrombocytopenic purpura (TTP) refractory to standard therapies. Transfus Apher Sci. 2018;57(2):247–249. doi:10.1016/j.transci.2018.03.005
71. Samuni Y, Goldstein S, Dean OM, Berk M. The chemistry and biological activities of N-acetylcysteine. Biochim Biophys Acta. 2013;1830(8):4117–4129. doi:10.1016/j.bbagen.2013.04.016
72. Turner N, Nolasco L, Moake J. Generation and breakdown of soluble ultralarge von Willebrand factor multimers. Semin Thromb Hemost. 2012;38(1):38–46. doi:10.1055/s-0031-1300950
73. Chen J, Reheman A, Gushiken FC, et al. N-acetylcysteine reduces the size and activity of von Willebrand factor in human plasma and mice. J Clin Invest. 2011;121(2):593–603. doi:10.1172/JCI41062
74. Anfossi G, Russo I, Massucco P, Mattiello L, Cavalot F, Trovati M. N-acetyl-L-cysteine exerts direct anti-aggregating effect on human platelets. Eur J Clin Invest. 2001;31(5):452–461. doi:10.1046/j.1365-2362.2001.00815.x
75. Rottenstreich A, Hochberg-Klein S, Rund D, Kalish Y. The role of N-acetylcysteine in the treatment of thrombotic thrombocytopenic purpura. J Thromb Thrombolysis. 2016;41(4):678–683. doi:10.1007/s11239-015-1259-6
76. Kappers-Klunne MC, Wijermans P, Fijnheer R, et al. Splenectomy for the treatment of thrombotic thrombocytopenic purpura. Br J Haematol. 2005;130(5):768–776. doi:10.1111/j.1365-2141.2005.05681.x
77. Dubois L, Gray DK. Case series: splenectomy: does it still play a role in the management of thrombotic thrombocytopenic purpura? Can J Surg. 2010;53(5):349–355.
78. Scully M, Knöbl P, Kentouche K, et al. Recombinant ADAMTS-13: first-in-human pharmacokinetics and safety in congenital thrombotic thrombocytopenic purpura. Blood. 2017;130(19):2055–2063. doi:10.1182/blood-2017-06-788026
79. Plaimauer B, Kremer Hovinga JA, Juno C, et al. Recombinant ADAMTS13 normalizes von Willebrand factor-cleaving activity in plasma of acquired TTP patients by overriding inhibitory antibodies. J Thromb Haemost. 2011;9(5):936–944. doi:10.1111/j.1538-7836.2011.04224.x
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.