Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Ultrasound-targeted stromal cell-derived factor-1-loaded microbubble destruction promotes mesenchymal stem cell homing to kidneys in diabetic nephropathy rats
Authors Wu S, Li L, Wang G, Shen W, Xu Y, Liu Z, Zhuo Z, Xia H, Gao Y, Tan K
Received 8 September 2014
Accepted for publication 25 October 2014
Published 3 December 2014 Volume 2014:9(1) Pages 5639—5651
DOI https://doi.org/10.2147/IJN.S73950
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Thomas Webster
Shengzheng Wu,1 Lu Li,1 Gong Wang,1 Weiwei Shen,2 Yali Xu,1 Zheng Liu,1 Zhongxiong Zhuo,1 Hongmei Xia,1 Yunhua Gao,1 Kaibin Tan1
1Department of Ultrasound, 2Department of Orthopedics, Xinqiao Hospital, Third Military Medical University, Chongqing, People’s Republic of China
Abstract: Mesenchymal stem cell (MSC) therapy has been considered a promising strategy to cure diabetic nephropathy (DN). However, insufficient MSCs can settle in injured kidneys, which constitute one of the major barriers to the effective implementation of MSC therapy. Stromal cell-derived factor-1 (SDF-1) plays a vital role in MSC migration and involves activation, mobilization, homing, and retention, which are presumably related to the poor homing in DN therapy. Ultrasound-targeted microbubble destruction has become one of the most promising strategies for the targeted delivery of drugs and genes. To improve MSC homing to DN kidneys, we present a strategy to increase SDF-1 via ultrasound-targeted microbubble destruction. In this study, we developed SDF-1-loaded microbubbles (MBSDF-1) via covalent conjugation. The characterization and bioactivity of MBSDF-1 were assessed in vitro. Target release in the targeted kidneys was triggered with diagnostic ultrasound in combination with MBSDF-1. The related bioeffects were also elucidated. Early DN was induced in rats with streptozotocin. Green fluorescent protein-labeled MSCs were transplanted intravenously following the target release of SDF-1 in the kidneys of normal and DN rats. The homing efficacy was assessed by detecting the implanted exogenous MSCs at 24 hours. The in vitro results showed an impressive SDF-1 loading efficacy of 79% and a loading content of 15.8 µg/mL. MBSDF-1 remained bioactive as a chemoattractant. In the in vivo study, SDF-1 was successfully released in the targeted kidneys. The homing efficacy of MSCs to DN kidneys after the target release of SDF-1 was remarkably ameliorated at 24 hours compared with control treatments in normal rats and DN rats. In conclusion, ultrasound-targeted MBSDF-1 destruction could promote the homing of MSCs to early DN kidneys and provide a novel potential therapeutic approach for DN kidney repair.
Keywords: mesenchymal stem cell, ultrasound, microbubbles, homing, stromal cell-derived factor-1, diabetic nephropathy
Introduction
Diabetes mellitus is a common disease worldwide. The global number of adult patients is expected to increase from 285 million in 2010 to 439 million in 2030.1 Twenty-five to forty percent of patients will develop diabetic nephropathy (DN). DN is the most detrimental complication and greatly increases the occurrence of social and economic burdens because it progresses to end-stage chronic kidney disease, which results in the requirement of hemodialysis or even kidney transplantation.2 Mesenchymal stem cell (MSC) transplantation has been considered a promising strategy to manage patients with DN due to the renoprotective potential of MSCs and the safety of this procedure.3 Currently, MSC delivery strategies in preclinical models include intravenous injection, renal artery injection, and direct parenchymal injection. MSC therapy reportedly ameliorates kidney injury, accelerates tubule proliferation, and restores renal function.4,5 However, many difficulties must be overcome to successfully implement MSC therapy, one of which is the scarcity of MSCs in injured tissues after systematic infusion.6–8 The insufficient homing of MSCs and the inability to drive them to the damaged kidney tissues have become a major barrier to the effective implementation of MSC therapy.9 Effective MSC therapy requires that MSCs reach the injured site.10 Hence, strengthening the homing of MSCs to the kidney may facilitate the regeneration of the DN kidney. Our previous investigation revealed that ultrasound-targeted microbubble destruction (UTMD) increases the homing of transplanted MSCs to kidneys and promotes kidney repair in DN rats.11 However, the homing efficacy remains unsatisfactory and must urgently be improved.
Stromal cell-derived factor-1 (SDF-1), also named CXCL12, reportedly binds to two unique receptors, CXCR4 and CXCR7. This chemokine is well known for stem cell activation, mobilization, homing, and retention.12 Generally, the SDF-1/CXCR4 axis plays a vital role in MSC migration.13–16 This function significantly differs from the role of CXCR7, which is involved in angiogenesis in tumor development.17 When tissues are injured, the localized SDF-1 level increases due to the cellular expression of SDF-1,12,18,19 which aids the mobilization and homing of MSCs.
Microbubbles (MBs), which are known as diagnostic ultrasound contrast agents, have been developed to enhance the echogenicity of blood and to delineate the vasculature of tissues.20 They were also developed as a carrier of drugs and genes that can be released in a targeted manner for therapy using ultrasound irradiation.21–23 UTMD also reportedly increases renal interstitial permeability,24,25 which increases the intercellular gap and changes the microenvironment. In addition, the beneficial involvement of endocrine cytokines after UTMD improves the implantation of MSCs.11,26
Because the SDF-1/CXCR4 axis regulates the migration of MSCs, we sought to upregulate the SDF-1 level to improve the MSC homing efficacy in order to develop an innovative intervention that improves the limited MSC tropism for DN kidneys. In this study, we developed a lipid MB loaded with SDF-1 via covalent conjugation and established a rat model of early DN. Exogenous SDF-1 was released in the kidney in a targeted manner via diagnostic ultrasound-targeted SDF-1-loaded MB (MBSDF-1) destruction. Subsequently, green fluorescent protein (GFP)-labeled MSCs were intravenously administered, and confocal laser scanning microscopy was employed to determine the homing of exogenous MSCs to the DN kidney. The study aimed to potentiate the MSC homing to the DN kidney in order to provide an approach for effective MSC-based therapy for DN repair.
Materials and methods
Materials and animals
Dipalmitoyl phosphatidylglycerol and distearoyl phosphatidylcholine were purchased from Genzyme Pharmaceuticals (Liestal, Switzerland). Carboxyl-polyethylene glycol-4000-carboxyl (COOH-PEG4000-COOH) was purchased from YareBio Technology (Shanghai, People’s Republic of China). Recombinant rat SDF-1α with a purity exceeding 98% was purchased from PeproTech Technology (Rocky Hill, NJ, USA). 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), N-hydroxysulfosuccinimide sodium salt (sulfo-NHS), polybrene, AMD3100, and streptozotocin (STZ) were purchased from Sigma-Aldrich (St Louis, MO, USA). Sprague Dawley (SD) rat MSC growth medium (consists of Dulbecco’s Modified Eagle’s Medium/F12 as basal medium and 10% fetal bovine serum) and the GFP lentiviral control were purchased from Cyagen Biosciences (Guangzhou, People’s Republic of China). Rabbit polyclonal SDF-1 antibody and horseradish peroxidase-labeled goat anti-rabbit IgG were purchased from Boster (Wuhan, People’s Republic of China). Four-week-old male SD rats (weighed 60–80 g) and adult male SD rats (weighed 200–220 g) were supplied by the experimental animal center of Xinqiao Hospital, which is affiliated with the Third Military Medical University. All animal procedures were performed according to the protocol approved by the Animal Ethics Committee of Xinqiao Hospital.
Preparation of MBSDF-1
The unloaded MBs (with no SDF-1) were prepared according to the protocols of Liu et al27 with slight modification. Briefly, dipalmitoyl phosphatidylglycerol, distearoyl phosphatidylcholine, and COOH-PEG4000-COOH (mass ratio of 30:30:3,000) were mixed with ultrapure water. The mixture was dispersed into 3 mL vials (1 mL/vial) and then lyophilized. The lyophilized powder was dissolved in 1 mL of a mixed solution consisting of 50% glucose, propylene glycol, and glycerin (volume ratio of 8:1:1) to obtain the unloaded MB solution. To produce the final unloaded MBs, the vial was mechanically vibrated for 45 seconds at a frequency of 4,300 min−1 and amplitude of 15 mm after substituting the air with perfluoropropane.
To obtain MBSDF-1, SDF-1 was firmly cross-linked with COOH-PEG4000-COOH via covalent conjugation using EDC/sulfo-NHS as coupling agents. The COOH groups of COOH-PEG4000-COOH were activated with EDC/sulfo-NHS. Briefly, EDC (5 μL, 50 mg/mL) and sulfo-NHS (5 μL, 100 mg/mL) were added to the unloaded MB solution mentioned earlier for 15 minutes at room temperature. Subsequently, 20 μL of SDF-1 solution (20 μg SDF-1 dissolved in 20 μL of water) was added and allowed to react with the activated COOH groups for 2 hours at room temperature. The resultant solution was treated following the same protocol as that used for unloaded MBs to form the primary MBSDF-1 (Figure 1). After washing with phosphate-buffered saline (PBS) and centrifugation at 1,000 rpm for 5 minutes, they were separated into two layers. The clear underlayer solution was discarded, by which the free SDF-1 (not loaded to the MBs) as well as the coupling agents were separated and removed. The milky suspension was acquired and the MBSDF-1 was ultimately obtained. The SDF-1 lipid solution was obtained after the sonication of MBSDF-1 to further characterize the MBs and conduct a cell migration assay.
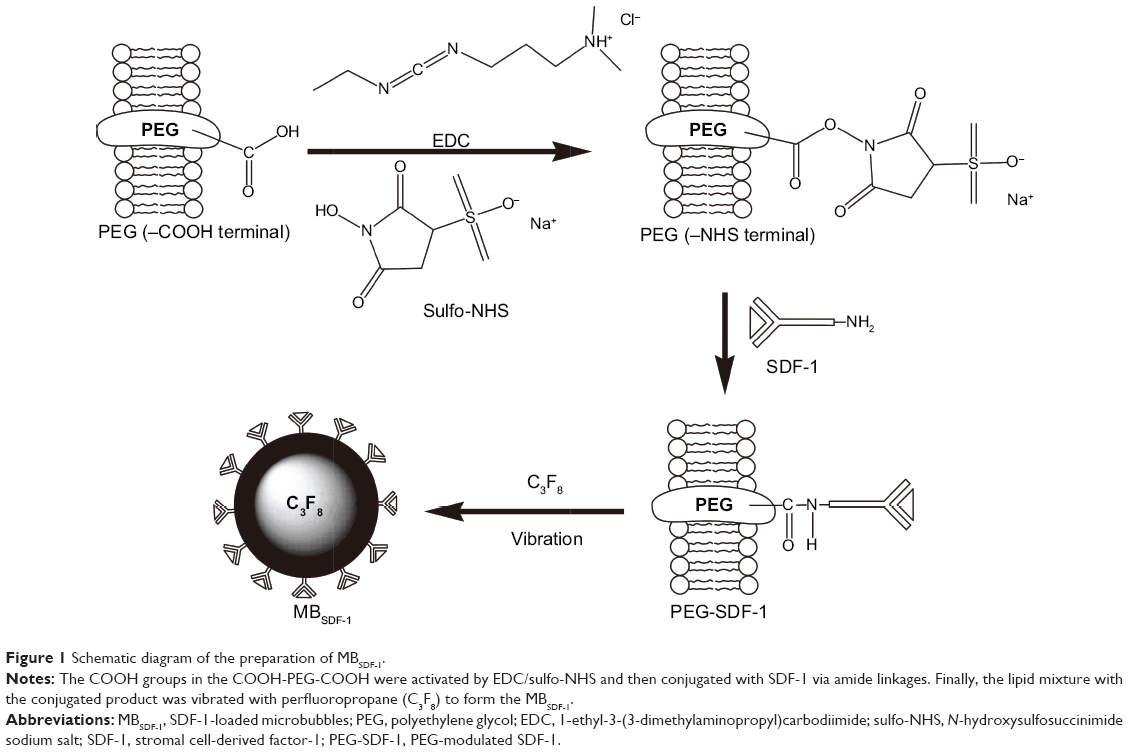 | Figure 1 Schematic diagram of the preparation of MBSDF-1. |
To determine the conjunction rate of SDF-1 and track the biodistribution after SDF-1 release by UTMD in vivo, fluorescent MBs were prepared following the same procedure as that used to prepare MBSDF-1, except that SDF-1 was replaced with fluorescein isothiocyanate (FITC)-labeled SDF-1.
For all in vivo studies, the animals received 0.2 mL of MBs, which was diluted to a final volume of 1.0 mL with PBS.
MB characterization
The MB concentration and size were determined with a Coulter particle counting instrument (Omec Technology Co, Ltd, Zhuhai, People’s Republic of China). Both the unloaded MBs and FITC-labeled MBSDF-1 (FITC-MBSDF-1) were observed and photographed with a confocal laser scanning microscope (CLSM) (Leica Microsystems, Wetzlar, Germany). Flow cytometry was used to detect the conjunction rate. The loaded SDF-1 encapsulated in MBSDF-1 was quantitatively determined with an enzyme-linked immunosorbent assay (ELISA) (R&D Systems, Inc, Minneapolis, MN, USA) according to the manufacturer’s instructions. The SDF-1 encapsulation efficiency and loading content were calculated according to Equations 1 and 2, respectively.
| (1) |

| (2) |

Mesenchymal stem cells
MSCs were isolated from the femurs and tibias of 4-week-old SD rats. Then, they were cultured and expanded in vitro. The phenotypic properties of MSCs were identified with flow cytometry, resulting in the positive surface markers for CD44, CD29, and CD90, as well as the negative surface markers for CD34, CD45, and CD11b. The MSCs could differentiate into osteogenic or adipogenic cells following a standard protocol from Cyagen Biosciences. The third or fourth passage of MSCs was used for the in vitro and in vivo study. All cells were cultured in SD rat MSC growth medium and incubated at 37°C in 5% CO2.
For in vivo tracking, we labeled MSCs with GFP via GFP lentiviral transduction according to the manufacturer’s protocol. Briefly, MSCs were seeded at 1×105 cells in six-well plates. After 24 hours of incubation, 1 mL MSC growth medium per well containing 5 μg/mL polybrene and GFP lentiviral were added to the cells at ten multiplicities of infection. The cells were incubated for 6 hours after lentiviral addition. Then, they were washed with PBS and the medium was replaced by fresh growth medium. Seventy-two hours after transfection, the transfected MSCs were observed by fluorescent microscope. To determine the GFP labeling efficiency, the transfected MSCs were collected, resuspended in PBS, and then analyzed by flow cytometry. MSCs without transduction served as negative controls. The results were reported as percentages of positive cells among the gated cells.
In vitro bioactivity assay
A cell migration assay was utilized to detect the bioactivity of PEG-modulated SDF-1 encapsulated in MBs. The migration assay was designed using transwell plates (Corning Costar, Cambridge, MA, USA) suitable for six-well plates with 8 μm pore filters. The concentration of MSCs was adjusted to 2×105 mL−1 in basal medium. For the chemotaxis assay, three upper chambers were seeded with 500 μL of MSCs. One lower chamber was filled with 1.5 mL of medium containing 10% fetal bovine serum, which served as a control. In addition, the other two lower chambers were filled with SDF-1 to a final concentration of 100 ng/mL and MBSDF-1 solution at the same concentration of SDF-1, which served as the SDF-1 group and MBSDF-1 group, respectively. For the chemotaxis inhibition assay, the MSCs were preincubated with AMD3100 (5 μg/mL) for 30 minutes to block CXCR4 and then adjusted to a concentration of 2×105 mL−1 in basal medium. The following procedure was consistent with the chemotaxis assay, and the three groups considered were the control + AMD3100 group, SDF-1 + AMD3100 group, and MBSDF-1 + AMD3100 group. After incubation for 6 hours, the cells in the upper chamber were gently removed, and the membranes were fixed in 4% paraformaldehyde for 30 minutes. The cells that migrated to the lower side of the filter were stained with 0.1% crystal violet for 20 minutes and then washed with water. They were observed under a light microscope, and the number of migrated cells was counted in ten randomly selected fields from each group.
Animal preparation
To establish the early DN rat model, 16 adult male rats were fasted overnight without water deprivation and received a single intraperitoneal injection of STZ (60 mg/kg). After 3 days of feeding, the rats were considered to have developed diabetes when the blood glucose increased to a stable level >16.7 mmol/L for 3 consecutive days, at which point they were used for the subsequent study. Four weeks after the confirmation of diabetes, the early DN rat model was established upon the emergence of mild microalbuminuria, which is an early sign of DN.
For in vivo handling, all rats were anesthetized using an intraperitoneal injection of pentobarbital (35 mg/kg), placed in left lateral position with the left abdominal region shaved, and the left kidneys were treated.
Immunohistochemistry
Immunohistochemistry was performed to compare the SDF-1 expression between normal and DN rats. Five normal rats and five DN rats were sacrificed with pentobarbital (120 mg/kg), and the kidneys were harvested, fixed in 10% formaldehyde, embedded in paraffin, and then sectioned into 4 μm slices. The slices were dewaxed and hydrated in 70% ethanol, incubated in 3% hydrogen peroxide in methanol for 15 minutes, and washed in PBS. For antigen retrieval, they were microwaved in 0.1 M Tris–HCl for 10 minutes and washed in PBS. Then they were incubated with 1:100 rabbit polyclonal antibody against SDF-1 or a negative control reagent for 1 hour. After that, they were washed in PBS, and incubated with horseradish peroxidase-labeled goat anti-rabbit IgG for 30 minutes. The immunostaining was visualized with diaminobenzidine, which resulted in a brown precipitate at the antigen site. After counterstaining with hematoxylin, the slices were observed under a light microscope. Ten random fields of each section were photographed and a semiquantitative evaluation of SDF-1 expression was performed. Integrated optical density (IOD) of SDF-1 expression was analyzed using Image-Pro plus 6.0 software.
In vivo imaging and UTMD
Three normal rats were used to assess the contrast enhancement of MBSDF-1 in the kidney. Contrast-enhanced ultrasound was performed using the 9L4 linear probe of an Acuson S2000 diagnostic ultrasound device (Siemens Medical Solutions, Mountain View, CA, USA). The contrast pulse sequence imaging mode was applied with identical settings: a transmission frequency of 4 MHz, a mechanical index (MI) of 0.05, and a single ultrasound focus at 1.5 cm. MBs were infused into the tail vein of the rats to monitor the improvement in the kidneys in real time.
For UTMD, MBSDF-1 were continuously infused during 10 minutes of ultrasound exposure. MB destruction (MBD) was activated with 4 seconds of destruction and 4 seconds of replenishment of the MBs. The MI was set to 1.5 during MBD and 0.05 during MB replenishment. The destruction–replenishment circle was repeated. The probe was positioned at the left kidney, moving right and left along the longitudinal axis view of the kidney to assure that the entire kidney was sonicated. Clips were captured for later analysis using a contrast dynamic software installed in the device.
Ultrastructural examination
The UTMD-treated kidneys and untreated kidneys were harvested from the three rats mentioned earlier. The cortices of the kidneys, which were approximately 1 mm3 in size, were fixed in 2.5% glutaraldehyde solution at 4°C overnight, postfixed in 1% osmium tetroxide, dehydrated with a serial alcohol gradient, and embedded in resin. They were subsequently cut into ultrathin sections of 60–80 nm and then stained with uranyl acetate and lead citrate. Ultrastructural changes in the blood vessels were studied with a transmission electron microscope (Philips TECNAI 10, USA).
In vivo target release
Three normal rats were used to visualize the target release of SDF-1 in the left kidneys. FITC-MBSDF-1 was infused instead when UTMD was applied. The untreated right kidneys, hearts, lungs, livers, and spleens served as a control. Immediately after the treatment, the rats were sacrificed, the tested organs were harvested to be frozen, and then sectioned into 8 μm tissue slices. After nuclear counterstaining with 4′,6-diamidino-2-phenylindole (DAPI), the biodistribution of FITC-labeled SDF-1 in the tested organs was detected using a CLSM. The quantitative fluorescence of FITC, represented by a gray-scale value (ranging from 0 to 255), was assessed using the LAS AF Lite software from Leica Microsystems. Ten random fields within each slice were photographed, while avoiding possible perturbation from the fluorescence of major blood vessels, and the mean gray-scale values of the entire image were measured.
Group assignment and MSC transplantation
Fifteen normal rats and 15 DN rats were divided into three groups: the control group (n=5), the UTMD group (n=5), and the UTMD + SDF-1 group (n=5). The UTMD + SDF-1 group received UTMD using MBSDF-1, while the UTMD group received UTMD using unloaded MBs. The control group only received the same dose of PBS. After group treatment, all rats received an intravenous injection of GFP-labeled MSCs into the tail vein. The concentration of MSCs was adjusted to 1×106 mL−1 in a volume of 1.0 mL.
Detection of implanted MSCs
All rats were sacrificed 24 hours after the treatment, and their left kidneys were frozen and sectioned into 8 μm slices to detect the implanted MSCs. After nuclei counterstaining with DAPI, CLSM was employed to observe the GFP-labeled MSCs that had homed to the kidney. Ten randomly selected fields were photographed, and the exogenous MSCs were calculated.
Statistical analysis
All continuous data are expressed as the mean ± standard deviation. Statistical analysis was carried out using SPSS 13.0 (Chicago, IL, USA). One-way analysis of variance and post hoc comparisons were used for the statistical evaluation. A P-value <0.05 was considered statistically significant.
Results
Characterization of MBs
The MBSDF-1 were stable at room temperature. They were milky in appearance, round, homogeneously distributed, and well dispersed. The concentration and diameter were 2–9×109 mL−1 and 1–5 μm, respectively. The SDF-1 encapsulation efficiency and loading content were 79% and 15.8 μg/mL, respectively. According to the CLSM images, green fluorescence was not found in unloaded MBs (Figure 2A), whereas FITC-labeled SDF-1 could bind to the MBs (Figure 2B) at a binding rate of 84.8%.
 | Figure 2 FITC-labeled SDF-1-loaded MBs by confocal laser scanning microscopy. |
In vitro bioactivity assay
A remarkable number of MSCs migrated in the SDF-1 group and the MBSDF-1 group compared with the control group. The number of migrated MSCs did not significantly differ between the MBSDF-1 group and the SDF-1 group (Figure 3A). After blocking CXCR4 with AMD3100, the migrated cells in the SDF-1 + AMD3100 group and the MBSDF-1 + AMD3100 group decreased compared with those in the SDF-1 group and the MBSDF-1 group, respectively (Figure 3A and B).
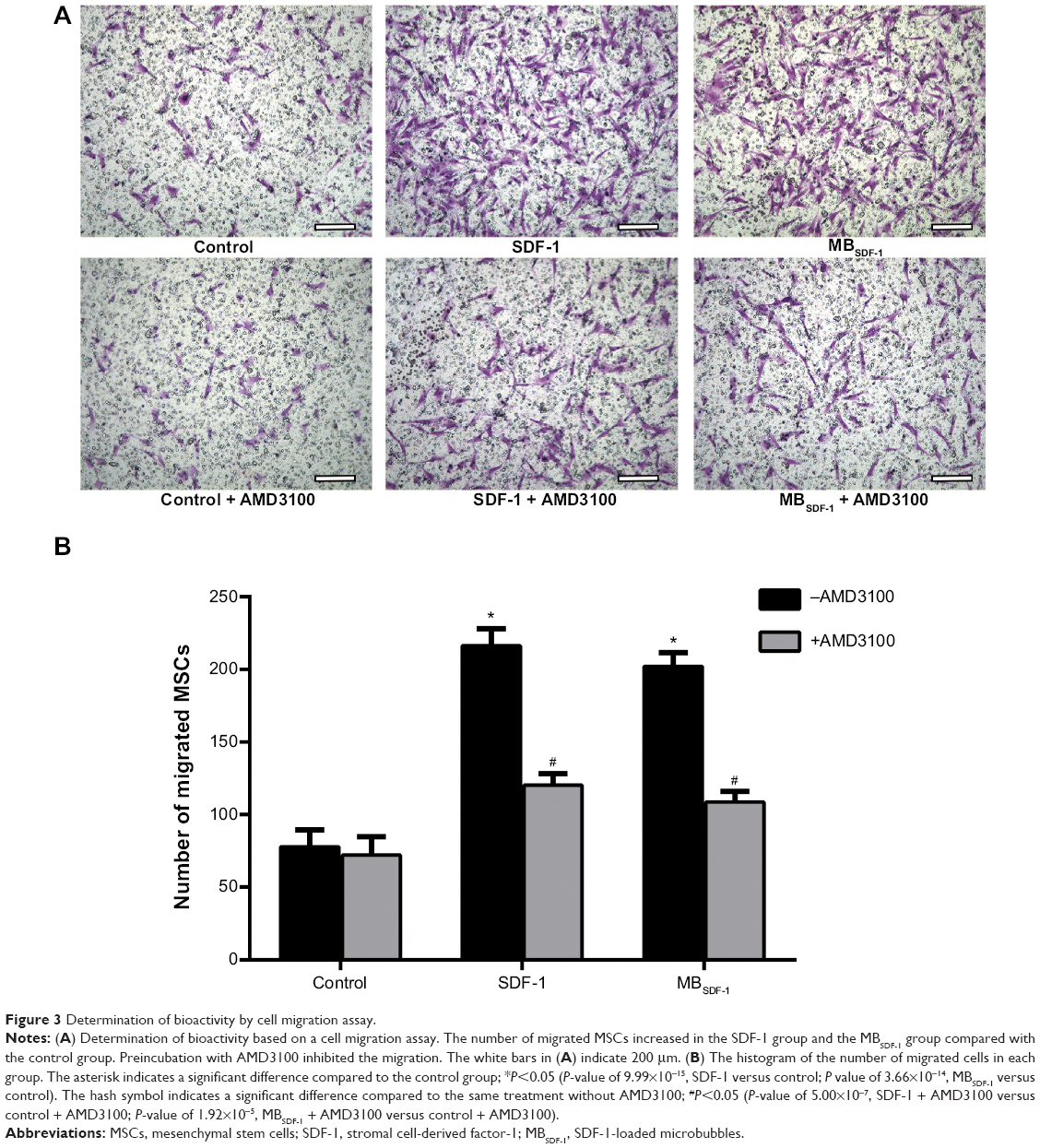 | Figure 3 Determination of bioactivity by cell migration assay. |
In vivo imaging and UTMD
After MBSDF-1 infusion, the echogenicity of renal parenchyma was markedly enhanced, with an explicit border at a low MI of 0.05 (Figure 4A). During 4 seconds of MBD at a high MI of 1.5, the MBs were destroyed from the near acoustic field to the far acoustic field (Figure 4B). Immediately after MBD, the MBs in the parenchyma almost vanished, leaving a hypo-echoic and blurred renal outline (Figure 4C). Within 4 seconds, the circulating MBs gradually and smoothly replenished until the perfusion of renal MBs completely recovered (Figure 4C–F). The time–intensity curve obtained from contrast dynamic analysis indicated a relative hyper-echo before MBD, a sudden decrease during MBD, and a gradually increasing recovery process (Figure 4G and H).
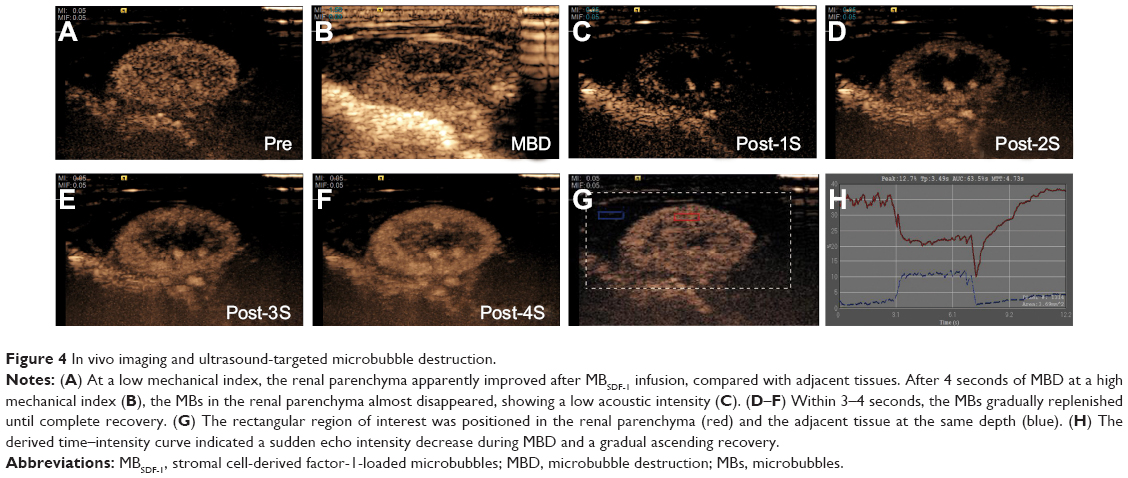 | Figure 4 In vivo imaging and ultrasound-targeted microbubble destruction. |
Ultrastructural alterations
The ultrastructural alterations mainly occurred to the micro-blood vessels after UTMD. Partial vessel walls suffered severe injury, while the contralateral vessels suffered mild injury. Peg-shaped tubers originated from the body of endothelial cells, some of which detached and fell into the vessel lumen. The vessel walls roughened and became thinner than usual (Figure 5A). Part of the vessel walls dissolved to generate several pores, allowing direct communication between the inside vascular lumen and extravascular interstitium through the discontinuous walls (Figure 5B). Large vessels survived with an intact wall structure. In comparison, the vessel wall remained continuous in normal renal tissues (Figure 5C).
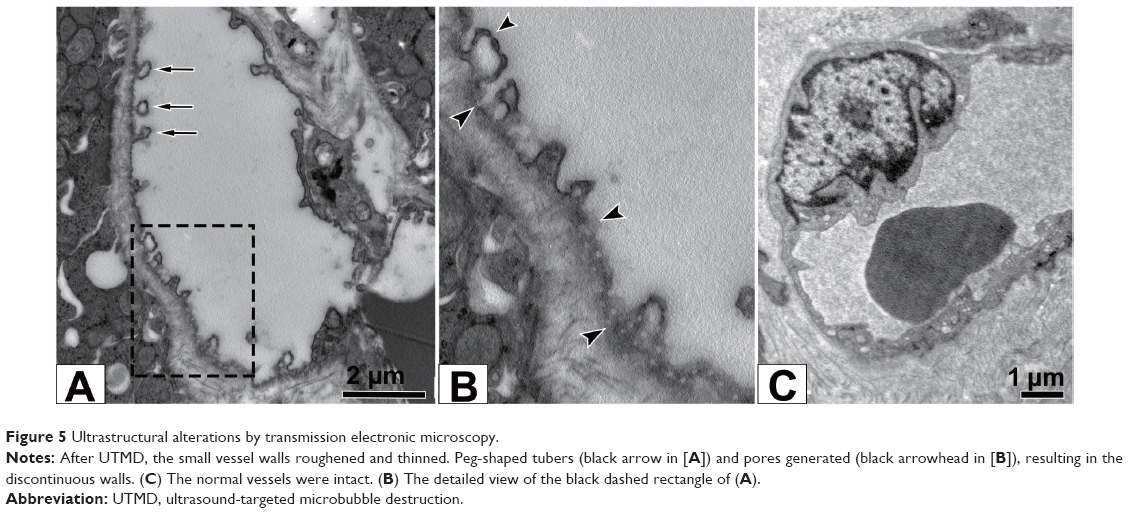 | Figure 5 Ultrastructural alterations by transmission electronic microscopy. |
In vivo target release
Immediately after UTMD using FITC-MBSDF-1, frozen sections of the kidneys exhibited sporadic but bright green fluorescence (Figure 6A). The fluorescence mainly distributed around the small vessels and into the adjacent renal interstitium. In comparison, fluorescence was not visualized in the kidneys without UTMD treatment as well as in the spleens, and fluorescence was rare in the hearts, lungs, and livers. The mean gray-scale value of the targeted kidney was significantly higher than those of any other control organs (Figure 6B), indicating that the loaded SDF-1 was successfully released in the targeted kidney tissues.
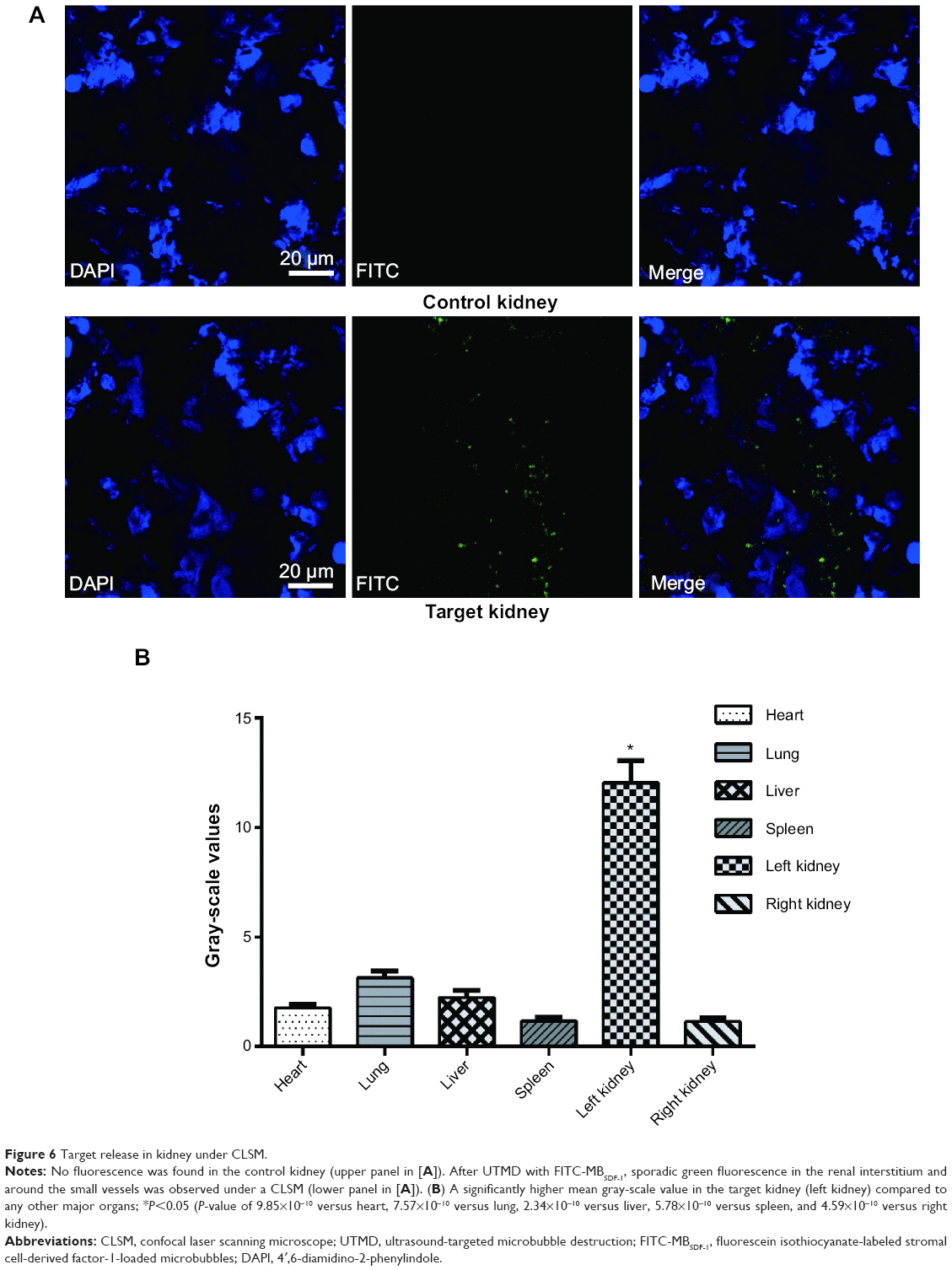 | Figure 6 Target release in kidney under CLSM. |
SDF-1 expression
The brown area was the positive expression of SDF-1. The expression localized to the cytoplasm. SDF-1 was slightly expressed in tubules and rarely expressed in the glomeruli of DN kidneys. In comparison, SDF-1 expression was occasionally visualized in normal kidneys (Figure 7). The IOD of SDF-1expression was obviously higher in DN kidneys (IOD value of 35,762±4,541) than that of normal kidneys (IOD value of 2,994±854).
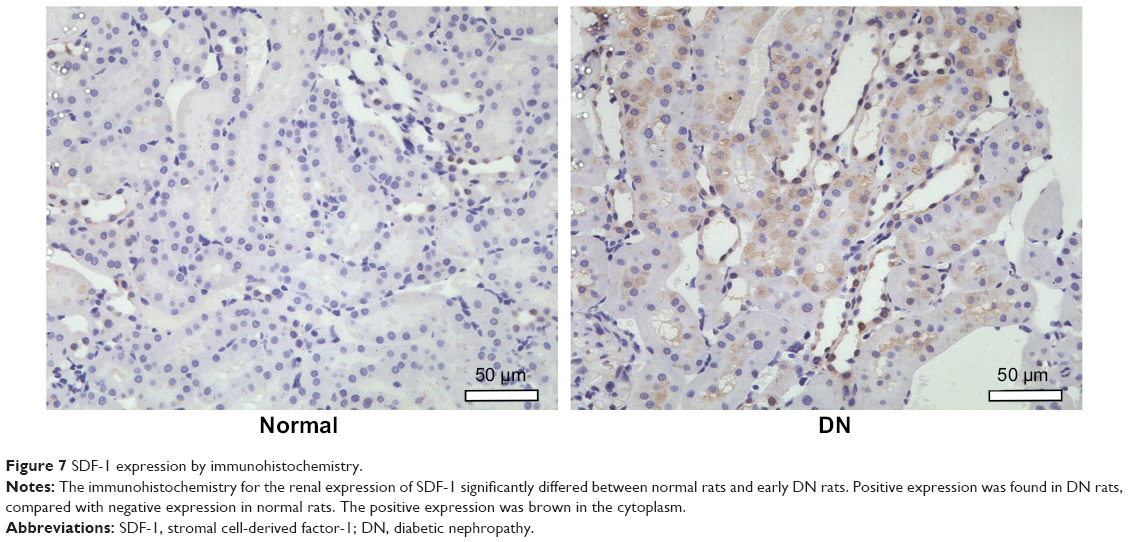 | Figure 7 SDF-1 expression by immunohistochemistry. |
Detection of exogenous MSCs
The cytoplasm and nuclei of GFP-labeled MSCs cultured in vitro exhibited bright green fluorescence, and the labeling rate was 72.2% according to flow cytometry. Twenty-four hours after group treatments and the transplantation of GFP-labeled MSCs, implanted exogenous MSCs were scarce in the control and UTMD groups in normal rats, while implanted exogenous MSCs were occasionally visualized in the UTMD + SDF-1 group (Figure 8A). In DN rats, implanted MSCs (number of 3.6±2.1) were rare in the control group. The MSCs significantly increased after UTMD (number of 13.4±3.1), and a much higher amount of implanted MSCs was found in the UTMD + SDF-1 group (number of 23.8±3.6) (Figure 8B). The implanted MSCs were located mainly around the small blood vessels and in the peritubular interstitium. Few MSCs were detected in the glomeruli.
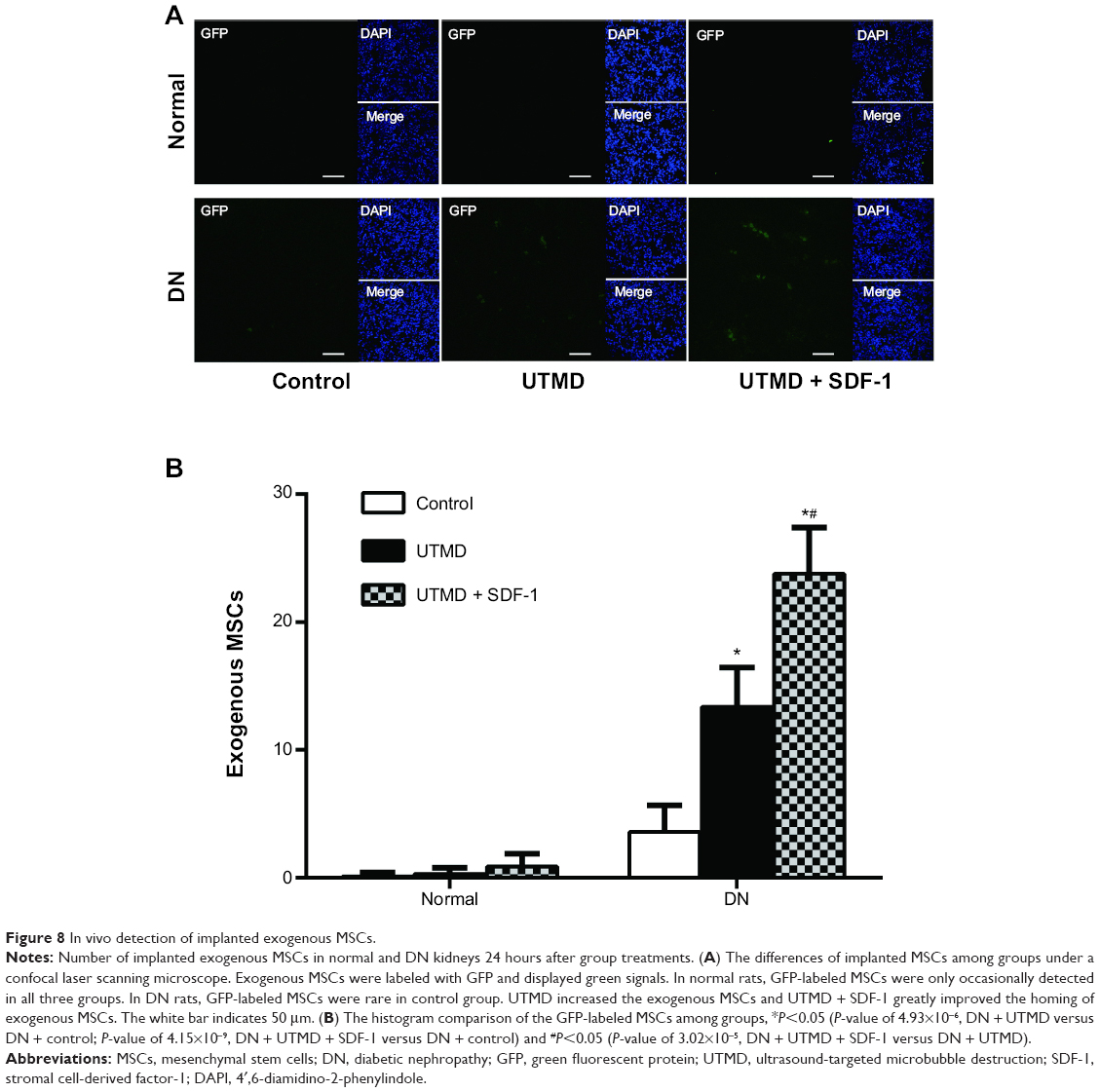 | Figure 8 In vivo detection of implanted exogenous MSCs. |
Discussion
Although the exact mechanism of MSC therapy has been controversial, its curative effects in kidney injury have been recognized.28 Current MSC delivery strategies include local injection and systemic injection. Direct parenchymal injection allows efficient local delivery of MSCs and promotes the recovery of renal fuction.29 However, the locally delivered MSCs tend to be confined to the injection site without diffusion to the whole kidney. Furthermore, this invasive method may cause bleeding and tissue destruction leading to secondary kidney damage and adverse effect on renal function recovery. Systemic administration is by far the most popular route for MSC delivery. Studies indicate that systemically administered MSCs can indeed home to the injured kidney.30 Nevertheless, low uptake of MSCs in the injured site hinders the therapeutic progress. Effective therapy requires sufficient MSCs participating in repair, and most importantly, the arrival of MSCs at the injured site across the endothelium. To overcome the difficulty of the poor homing efficacy of MSCs to the DN kidney, we developed a novel therapeutic strategy based on two points, ie, the regulatory role of the SDF-1/CXCR4 axis in MSC migration and the noninvasive delivery system of UTMD.
In this study, we successfully used covalent conjugation to develop a lipid MB that carried SDF-1. The resultant MBs were impressive, with an appreciable encapsulation efficiency and loading content of SDF-1. Up to 84.8% of the MBs exhibited green fluorescence due to FITC-SDF-1, which was firmly attached to the lipid coat. In the in vitro bioactivity assay, the MBSDF-1 solution promoted the migration of MSCs, playing the same chemoattractant role as SDF-1. The migration was restrained after the perturbation of the SDF-1/CXCR4 axis by AMD3100, a highly specific CXCR4 receptor antagonist. The results indicated that the PEG-modified SDF-1 retained its bioactivity as a chemoattractant in the SDF-1/CXCR4 axis. We used homo-bifunctional PEG derivatives that were linear with double terminal COOH groups in this study; thus, they were not likely to shelter the functional domain or binding sites when cross-linked with SDF-1. The recognition of the antigen in the ELISA assay proved that the antigen-binding sites were not sheltered or disrupted.
In contrast ultrasound imaging mode with a low MI, the acoustic output was relatively low within the kidney tissues. When introduced into circulation, the MBs remained mostly intact, and no obvious bioeffect was induced under this condition. In addition, they were limited to circulating only within blood vessels and reflected much more echo information. Thus, they could act as contrast agents to enhance ultrasonic imaging and as tracers for the real-time monitoring of blood perfusion. Given its rich blood supply, the kidney is the ideal target organ for UTMD, where sufficient MBSDF-1 may accumulate to provide a precondition for effective SDF-1 release. At the start of UTMD, the MI was set at a high level and the acoustic output was relatively high. A burst of MBs occurred under this condition resulting in acoustic cavitation, which produced several mechanical effects, such as high pressure, shock waves, and microjets.31 In vivo, UTMD was targeted to vascular areas containing MBs. In particular, microvessels are more vulnerable to attack by acoustic cavitation due to the relatively fragile wall structure, narrow space, and slow blood flow. Sonoporation may result from MBD under proper conditions, as revealed in many studies. Transient and non-lethal porosity may occur in biological membranes, which increases the capillary permeability.32 In our study, the porosity of the micro-blood vessels was confirmed by transmission electronic microscopy, and this porosity was presumably associated with the generation of shock waves and microjets.33 The acoustic radiation force may also be involved, which pushes the circulating MBs away from acoustic source along the ultrasound propagation route and tends to aggregate or repulse MBs. Thus, sonoporation was generated more unilaterally compared to the contralateral intact vessel walls (Figure 5A). The acoustic radiation force also reportedly enhances the target release of therapeutic agents.34 Ultrasound-targeted MBSDF-1 destruction allows the spatiotemporal target release of encapsulated SDF-1, and sonoporation as well as the acoustic radiation force noninvasively facilitates SDF-1 transport into interstitium across the endothelial barrier, which locally increases the SDF-1 level in the kidney. In addition, the intermittent working mode of MBD replenishment enables sufficient levels of MBSDF-1 to access renal perfusion following the complete destruction of MBs, which improves the effectiveness of SDF-1 release.
In early DN rats, SDF-1 expression in the kidneys is weak and presumably responsible for the poor homing efficacy of transplanted MSCs. The target release increased the levels of SDF-1 in the targeted kidney tissues. As previously revealed, UTMD noninvasively increases the homing of MSCs to kidneys and promotes renal repair in DN rats.11 Our study confirmed the role of UTMD in the homing of MSCs, and the increase in the levels of chemoattractants attracts more administered exogenous MSCs, as theoretically speculated. After 24 hours, the number of MSCs implanted in the DN rats was significantly higher in the kidneys of the UTMD + SDF-1 group than in the control group and the UTMD group. However, in normal rats, whose kidneys do not express SDF-1, notable implanted MSCs were not detected in any of the groups. Given the difference in renal SDF-1 expression between DN rats and normal rats, these results support the role of SDF-1 in MSC homing. The ameliorated homing efficacy of MSCs may be due to the increase in SDF-1 and the bioeffects caused by UTMD. Notably, the increased capillary permeability induced by sonoporation may benefit the migration of MSCs across the endothelial barrier.
The increase in SDF-1 reported herein ameliorated the problem of poor MSC homing to DN kidneys. Moreover, in addition to the exogenous MSCs (GFP-labeled MSCs) we observed, endogenous MSCs may act via the same SDF-1/CXCR4 axis. However, the investigation of endogenous MSC homing is beyond the scope of this study. The improved homing of implanted exogenous MSCs and the supposed homing of endogenous MSCs may facilitate the effective repair of chronic kidney injury in DN. Given the tropism of MSCs for the injured tissue, as well as their paracrine and endocrine action, including mitogenic, antiapoptotic, anti-inflammatory, and angiogenic influences,28,35 the implantation of sufficient MSCs may serve as an effective early intervention of DN to prevent or even reverse renal deterioration. The results of this approach encourage the potential treatment of DN or other pathologies to repair chronic kidney injury.
This preliminary study features several limitations. We only detected the homing efficacy at 24 hours posttreatment. Neither the long-term survival and differentiation of implanted MSCs nor the long-term therapeutic effects were investigated. The correlated mechanism also needs to be detailed in a further study.
Conclusion
We prepared a lipid MB carrying SDF-1, a chemoattractant for homing MSCs. The MBSDF-1 can be used to monitor the real-time renal perfusion and can act as a component of the delivery system for target release. UTMD, as well as the related bioeffects, such as increased capillary permeability induced by sonoporation, resulted in the successful target release of SDF-1 and increase in the SDF-1 level in the kidney. Implanted exogenous MSCs remarkably targeted the kidneys of early DN. The improved MSC homing efficacy could be translated into therapeutic benefits to repair DN kidneys. The ultrasound-targeted MBSDF-1 destruction in the present study may provide a novel therapeutic approach for the repair of DN kidneys or even other chronic kidney injuries.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant Numbers 81071160 and 81101062). The authors gratefully acknowledge Dr Ping Liu (Department of Ultrasound, 22nd Hospital of People’s Liberation Army), Dr Wugui Chen (Department of Orthopedics, Xinqiao Hospital), and Dr Baoshang Zhou (Department of Nephrology, Xinqiao Hospital) for their assistance and facilities in the experiment.
Disclosure
The authors have no conflicts of interest to disclose.
References
Shaw JE, Sicree RA, Zimmet PZ. Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res Clin Pract. 2010;87(1):4–14. | ||
Reutens AT, Atkins RC. Epidemiology of diabetic nephropathy. Contrib Nephrol. 2011;170:1–7. | ||
Ezquer ME, Ezquer FE, Arango-Rodriguez ML, Conget PA. MSC transplantation: a promising therapeutic strategy to manage the onset and progression of diabetic nephropathy. Biol Res. 2012;45(3):289–296. | ||
Baer PC, Geiger H. Mesenchymal stem cell interactions with growth factors on kidney repair. Curr Opin Nephrol Hypertens. 2010;19(1):1–6. | ||
Singaravelu K, Padanilam BJ. In vitro differentiation of MSC into cells with a renal tubular epithelial-like phenotype. Ren Fail. 2009;31(6):492–502. | ||
Lange C, Tögel F, Ittrich H, et al. Administered mesenchymal stem cells enhance recovery from ischemia/reperfusion-induced acute renal failure in rats. Kidney Int. 2005;68(4):1613–1617. | ||
Fang TC, Otto WR, Rao J, et al. Haematopoietic lineage-committed bone marrow cells, but not cloned cultured mesenchymal stem cells, contribute to regeneration of renal tubular epithelium after HgCl2-induced acute tubular injury. Cell Prolif. 2008;41(4):575–591. | ||
Schrepfer S, Deuse T, Reichenspurner H, Fischbein MP, Robbins RC, Pelletier MP. Stem cell transplantation: the lung barrier. Transplant Proc. 2007;39(2):573–576. | ||
Xinaris C, Morigi M, Benedetti V, et al. A novel strategy to enhance mesenchymal stem cell migration capacity and promote tissue repair in an injury specific fashion. Cell Transplant. 2013;22(3):423–436. | ||
Reinders ME, Fibbe WE, Rabelink TJ. Multipotent mesenchymal stromal cell therapy in renal disease and kidney transplantation. Nephrol Dial Transplant. 2010;25(1):17–24. | ||
Zhang Y, Ye C, Wang G, et al. Kidney-targeted transplantation of mesenchymal stem cells by ultrasound-targeted microbubble destruction promotes kidney repair in diabetic nephropathy rats. Biomed Res Int. 2013;2013:526367. | ||
Lau TT, Wang DA. Stromal cell-derived factor-1 (SDF-1): homing factor for engineered regenerative medicine. Expert Opin Biol Ther. 2011;11(2):189–197. | ||
Togel F, Isaac J, Hu Z, Weiss K, Westenfelder C. Renal SDF-1 signals mobilization and homing of CXCR4-positive cells to the kidney after ischemic injury. Kidney Int. 2005;67(5):1772–1784. | ||
Kucia M, Jankowski K, Reca R, et al. CXCR4-SDF-1 signalling, locomotion, chemotaxis and adhesion. J Mol Histol. 2004;35(3):233–245. | ||
Marquez-Curtis LA, Janowska-Wieczorek A. Enhancing the migration ability of mesenchymal stromal cells by targeting the SDF-1/CXCR4 axis. Biomed Res Int. 2013;2013:561098. | ||
Liu X, Duan B, Cheng Z, et al. SDF-1/CXCR4 axis modulates bone marrow mesenchymal stem cell apoptosis, migration and cytokine secretion. Protein Cell. 2011;2(10):845–854. | ||
Burns JM, Summers BC, Wang Y, et al. A novel chemokine receptor for SDF-1 and I-TAC involved in cell survival, cell adhesion, and tumor development. J Exp Med. 2006;203(9):2201–2213. | ||
Rosenkranz K, Kumbruch S, Lebermann K, et al. The chemokine SDF-1/CXCL12 contributes to the ‘homing’ of umbilical cord blood cells to a hypoxic-ischemic lesion in the rat brain. J Neurosci Res. 2010;88(6):1223–1233. | ||
Salcedo L, Sopko N, Jiang HH, Damaser M, Penn M, Zutshi M. Chemokine upregulation in response to anal sphincter and pudendal nerve injury: potential signals for stem cell homing. Int J Colorectal Dis. 2011;26(12):1577–1581. | ||
Miller DL, Averkiou MA, Brayman AA, et al. Bioeffects considerations for diagnostic ultrasound contrast agents. J Ultrasound Med. 2008;27(4):611–632. quiz 633–636. | ||
Tinkov S, Coester C, Serba S, et al. New doxorubicin-loaded phospholipid microbubbles for targeted tumor therapy: in vivo characterization. J Control Release. 2010;148(3):368–372. | ||
Li P, Zheng Y, Ran H, et al. Ultrasound triggered drug release from 10-hydroxycamptothecin-loaded phospholipid microbubbles for targeted tumor therapy in mice. J Control Release. 2012;162(2):349–354. | ||
Florinas S, Kim J, Nam K, Janat-Amsbury MM, Kim SW. Ultrasound-assisted siRNA delivery via arginine-grafted bioreducible polymer and microbubbles targeting VEGF for ovarian cancer treatment. J Control Release. 2014;183:1–8. | ||
Zhang Y, Ye C, Xu Y, et al. Ultrasound-mediated microbubble destruction increases renal interstitial capillary permeability in early diabetic nephropathy rats. Ultrasound Med Biol. 2014;40(6):1273–1281. | ||
Li P, Gao Y, Zhang J, et al. Renal interstitial permeability changes induced by microbubble-enhanced diagnostic ultrasound. J Drug Target. 2013;21(5):507–514. | ||
Xu YL, Gao YH, Liu Z, et al. Myocardium-targeted transplantation of mesenchymal stem cells by diagnostic ultrasound-mediated microbubble destruction improves cardiac function in myocardial infarction of New Zealand rabbits. Int J Cardiol. 2010;138(2):182–195. | ||
Liu P, Wang X, Zhou S, Hua X, Liu Z, Gao Y. Effects of a novel ultrasound contrast agent with long persistence on right ventricular pressure: comparison with SonoVue. Ultrasonics. 2011;51(2):210–214. | ||
Morigi M, Benigni A. Mesenchymal stem cells and kidney repair. Nephrol Dial Transplant. 2013;28(4):788–793. | ||
Alfarano C, Roubeix C, Chaaya R, et al. Intraparenchymal injection of bone marrow mesenchymal stem cells reduces kidney fibrosis after ischemia-reperfusion in cyclosporine-immunosuppressed rats. Cell Transplant. 2012;21(9):2009–2019. | ||
Ittrich H, Lange C, Tögel F, et al. In vivo magnetic resonance imaging of iron oxide-labeled, arterially-injected mesenchymal stem cells in kidneys of rats with acute ischemic kidney injury: detection and monitoring at 3T. J Magn Reson Imaging. 2007;25(6):1179–1191. | ||
Prentice P, Cuschieri A, Dholakia K, Prausnitz M, Campbell P. Membrane disruption by optically controlled microbubble cavitation. Nat Phys. 2005;1(2):107–110. | ||
Delalande A, Kotopoulis S, Postema M, Midoux P, Pichon C. Sonoporation: mechanistic insights and ongoing challenges for gene transfer. Gene. 2013;525(2):191–199. | ||
Ohl CD, Arora M, Ikink R, et al. Sonoporation from jetting cavitation bubbles. Biophys J. 2006;91(11):4285–4295. | ||
Lum AF, Borden MA, Dayton PA, Kruse DE, Simon SI, Ferrara KW. Ultrasound radiation force enables targeted deposition of model drug carriers loaded on microbubbles. J Control Release. 2006;111(1–2):128–134. | ||
Humphreys BD, Bonventre JV. Mesenchymal stem cells in acute kidney injury. Annu Rev Med. 2008;59:311–325. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.