Back to Journals » Cancer Management and Research » Volume 12
Ultrasound-Targeted Microbubble Destruction Mediated si-CyclinD1 Inhibits the Development of Hepatocellular Carcinoma via Suppression of PI3K/AKT Signaling Pathway
Authors Yan W ![]() , Cheng L
, Cheng L ![]() , Zhang D
, Zhang D ![]()
Received 28 May 2020
Accepted for publication 1 September 2020
Published 29 October 2020 Volume 2020:12 Pages 10829—10839
DOI https://doi.org/10.2147/CMAR.S263590
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Bilikere Dwarakanath
Wei Yan,* Li Cheng,* Dongmei Zhang
Department of Electrical Diagnosis, Changchun University of Traditional Chinese Medicine Affiliated Hospital, Changchun 130021, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Wei Yan
Department of Electrical Diagnosis, Changchun University of Traditional Chinese Medicine Affiliated Hospital, Changchun 130021, People’s Republic of China
Tel/Fax +86-0431-85380511
Email [email protected]
Background and Aim: In our study, we aimed to investigate the effect of ultrasound-targeted microbubble destruction (UTMD)mediated si-CyclinD1 (CCND1) on the growth of hepatocellular carcinoma (HCC) cells.
Patients and Methods: Bioinformatics analysis was performed to detect the difference of CCND1 expression of HCC and normal liver tissues. After treatment with UTMDmediated si-CCND1, the growth and apoptosis of HepG2 cells were detected by flow cytometry, MTT, EdU staining, colony formation assay, Hoechst 33,258 staining and Western blot analysis. The growth of HepG2 cells in vivo was also studied via xenograft tumor in nude mice.
Results: CCND1 was highly expressed in HCC tissues and HCC cell lines. UTMDmediated si-CCND1 could inhibit the growth of HepG2 cells and promote apoptosis via suppression of the PI3K/AKT signaling pathway. UTMDmediated si-CCND1 could also suppress the growth of HepG2 cells in vivo.
Conclusion: Our study provided evidence that UTMDmediated si-CCND1 could inhibit the growth of HepG2 cells and promote apoptosis via suppression of the PI3K/AKT signaling pathway.
Keywords: hepatocellular carcinoma, ultrasound-targeted microbubble destruction, CCND1, PI3K/AKT signaling pathway
Introduction
Hepatocellular carcinoma (HCC) is the most common type of primary liver cancer in adults and is the leading cause of death.1,2 HCC ranks as the major histological subtype among primary liver cancers and accounts for 70 to 85% of the total burden of liver cancers all over the world.3 The incidence of HCC varies greatly among geographical regions with the highest incidence in Eastern Asia and sub-Saharan Africa.4 The difference in HCC incidence between different geographical regions and countries is mainly due to different potential risk factors including drinking, occupational exposure, viral hepatitis as well as non-alcoholic fatty liver.5 Treatment and management of HCC varies according to the stage of the disease. It is reported that surgery, tyrosine kinase inhibitor and image-guided tumor ablation are the optional approaches for patients with HCC.6,7 The prognosis of HCC remains poor, with only onethird of patients eligible to curative treatments.8 Thus, it is very urgent for us to find and develop new therapeutic targets for more effective treatments for HCC patients.
CyclinD1 (CCND1) is a member of the G1 cyclins and is a main positive mediator of the G1 restriction point, and the abnormal expression of CCND1 may result in the proliferation and invasion of tumor cells.9 CCND1 is often highly expressed in cancers and its overexpression can be attributed to multiple factors such as increased transcription, translation, and protein stability.10 A previous study has demonstrated that CCND1 plays an important role in regenerating HCC and many types of cancers.11 Gene therapy is a promising method to destroy tumor cells in various cancers, but delivery technology needs to be improved to achieve clinical application.12 Ultrasound, known as a diagnostic tool, disturbs cell membranes and enhances genes to enter into cells, while microbubbles (MBs) play important roles in enhancing gene delivery efficiency, without causing cell damage.13 Ultrasound-targeted microbubble destruction (UTMD) has been used to deliver genes to cells in vitro and in vivo to treat diabetes and cardiovascular disease in experimental animal models.14 Because this noninvasive and site-specific approach to gene delivery could have significant effect on treating tumors, we tested the general hypothesis that UTMD-mediated gene delivery could be used to treat solid tumors. Specifically, we investigated whether UTMDmediated-si-CCND1 retarded tumor growth in HCC.
Materials and Methods
Bioinformatics Analysis
The data of CCND1 expression from 367 HCC tissues were downloaded from the Cancer Genome Atlas (TCGA) database. The data of CCND1 expression from 110 normal liver tissues were downloaded from GTEX database. Data analysis was performed using online analysis website http://xena.ucsc.edu/.
Synthesis of CCND1 Overexpression Vector and Interfering Plasmid
CCND1 small interfering RNA plasmid (pGPU6) (si-CCND1-1 and si-CCND1-2) and the corresponding negative control plasmid (scramble siRNA) were synthesized by Shanghai Genepharma Company (Shanghai, China). By comparing the silencing efficiency of the two siRNAs, one with better efficiency was selected for subsequent experiment.
Preparation of MBs
Cationic lipid MBs were synthesized by ultrasonic dispersion of polyethylene glycol-40 stearate (1 mg/mL), 1-bisstearoyl phosphatidylcholine (2 mg/mL), 1.2-bisstearoyl-3-trifluoromethylpropane (0.4 mg/mL) and decafluorobutane (all from Avanti Polar Lipids Inc., Alabaster, AL, USA) in a water tank. Afterwards, the synthesized MBs were observed and detected using an inverted microscopy (Eclipse Ti microscope, Nikon, Japan) and Nanosight (Malvern, UK). The si-CCND1 plasmid was then mixed with the synthesized MBs at a ratio of 1:50 (w/w) and incubated at 37°C for 30 min. Then 10 times the volume of Dulbecco’s modified Eagle’s medium (DMEM) was added to dilute the MB-plasmid mixture.
Cell Culture and Treatment
Human HCC cell lines HepG2, HHCC, PLC/PRF/5 and HCCLM3 and human normal hepatocyte (LO2) cell purchased from the Cell Resource Center of Shanghai Institute of Biology, Chinese Academy of Sciences (Shanghai, China) were seeded in cell culture dishes at 1 × 105 cells/cm2 in RPMI-1640 medium containing 10% fetal bovine serum (FBS), and DMEM culture medium (all from Gibco, Grand Island, NY, USA) in a 5% CO2 incubator at 37°C for 48 h. When the cell confluence reached about 80%–90%, the cells were detached using 0.25% trypsin (Gibco) and subcultured.
The constructed CCND1 small interfering RNA plasmid (pGPU6) (si-CCND1) and the corresponding negative control plasmid (scramble siRNA) were transfected into HepG2 cells using Lipofectamine2000 kit. At the same time, HepG2 cells with good growth condition were added with si-CCND1/MB mixture and pretreated for 30 min. The cells were then treated with ultrasonic therapy instrument (Fysiomed, Doornstraat, Belgium) with the frequency of 1 MHz and the sound intensity of 0.75w/cm2 for 45 s.
Reverse Transcription-Quantitative Polymerase Chain Reaction (RT-qPCR)
TRIzol (ThermoFisher, Waltham, USA) method was used to extract total RNA from cells with the ratio of optical density (OD)260/280 between 1.8 and 2.2. Afterwards, ABI High Capacity cDNA Reverse Transcription Kit (ThermoFisher) was used to synthetize cDNA. The preparation steps for quantitative PCR were carried out on ice. Primers (Table 1) and templates were added to the PCR tube. The reaction system was: Premix Taq (25 μL), cDNA template (2 μL), forward primer (1 μL; 20 μM), reverse primer (1 μL; 20 μM), and ultrapure water (21 μL). The PCR (CFX96 TouchTM, Bio-Rad, CA) reaction condition was: incubation at 94°C for 5 min, reaction at 94°C for 30 s, reaction at 56°C for 30 s, extending at 72°C for 3 min with a total of 40 cycles. Data analysis was performed using ABI SDS version 2.3 software. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as internal reference.
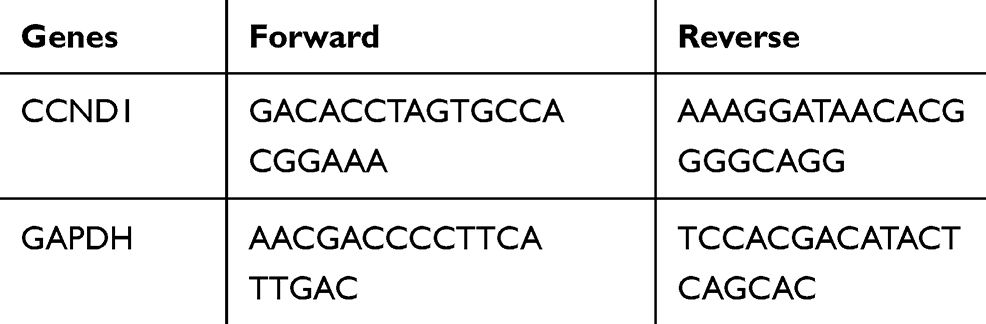 |
Table 1 Primer Sequences |
Western Blot Analysis
The total protein was extracted by a RIPA lysis buffer containing phenylmethylsulfonyl fluoride (Beytime, Bejing, China). Standard bicinchoninic acid method was applied to determine the protein level. The protein (50 mg) was loaded onto 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then transferred to the polyvinylidene fluoride membranes (Amersham Pharmacia) after electrophoresis. After that, PVDF membranes were incubated with sealing solution (Boster, Wuhan, China) at room temperature to block nonspecific binding. Subsequently, the membranes were incubated with primary antibodies (Table 2) at 4°C overnight, and then incubated with secondary antibody horseradish peroxidase-labeled goat-anti rabbit immunoglobulin G (IgG) (1:2000; ab205718; Abcam Inc., Cambridge, MA, USA) or goat-anti mouse IgG (1:2000; ab205719; Abcam) at room temperature for 1 h. The sample was developed by electrochemiluminescence, imaged using BioSpectrum gel imaging system (Bio-Rad, Hercules, CA, USA), and analyzed by ImageJ v1.48u software (National Institutes of Health, Bethesda, Maryland, USA).
 |
Table 2 Primary Antibodies Used in Western Blot Analysis |
Flow Cytometry
After the cells in good growth condition were made into cell suspension, the cells were cultured with DMEM containing 2 mmoL carboxyfluorescein diacetate succinimidyl ester (CFSE) (Invitrogen Inc., Carlsbad, CA, USA) for 15–30 min, and then the proliferation ability of cells in each group was detected; or the cells were added with 5 μL Annexin V-fluorescein isothiocyanate or 5 μL propidium iodide (PI) for incubation in the dark for 10 min after staining, cell apoptosis was detected.
3-(4, 5-Dimethylthiazol-2-Yl)-2, 5-Diphenyltetrazolium Bromide (MTT) Assay
The cell activity was tested using the MTT kit (Sigma-Aldrich, Shanghai, China) and all the operations were strictly performed in accordance with the instructions. Then microplate reader (Multiscan, Thermo, USA) was used to detect the OD value at 490 nm.
Colony Formation Assay
HepG2 cells with good growth in each group were detached with 0.25% trypsin and seeded in the six-well plates at the density of 1000 cells/well. After cells were incubated in a 5% CO2 incubator at 37°C for 14 days, the cells were treated with 75% methyl alcohol for 30 min, and stained with 0.2% crystal violet. The number of clones was calculated. The experiment was repeated for three times.
5-Ethynyl-2ʹ-Deoxyuridine (EdU) Assay
The DNA replication ability of HepG2 cells in good growth condition was detected by Cell-light EdU luminous detection kit (RiboBio Company, Guangzhou, China). The cells were treated according to the instructions of EdU kit.
Hoechst 33,258 Staining
Chromatin dye Hoechst 33,258 was used to observe chromosome condensation and morphological changes in nucleus by fluorescence microscope (BX50-FLA; Olympus, Tokyo, Japan). The cells were fixed in 4% paraformaldehyde and stained with Hoechst 33,258 at the final concentration of 10 μg/mL for 10 min. Living cells showed normal nuclear size and uniform fluorescence. Apoptotic cells showed condensed nucleus or nuclear concentration.
Xenograft Tumor in Nude Mice
Twenty BALB/c mice (4–6 weeks of age, weight 18–25 g) purchased from the Medical Laboratory Animal Center (Guangdong Province, China) were stored without specific pathogens. Then the HepG2 cells with stable silenced CCND1, scramble siRNA and UTMD-mediated CCND1 were re-suspended in 50 μL phosphate buffer saline, and then 50 μL matrix glue with the concentration of 5 × 106 cells/mL was added and subcutaneously injected into each mouse. The growth of human liver cancer xenografts in mice was monitored every 5 days, and the tumor growth was monitored every 3 days after 20 days. The mice were euthanized by carbon dioxide asphyxiation 35 days after injection. Animals were operated according to the guidelines for the care and use of experimental animals and the experiment was approved by the institutional ethics guidelines for Changchun University of Traditional Chinese Medicine Affiliated Hospital.
Immunohistochemistry Staining
The tumor sections of liver cancer xenografts from nude mice (5 μm thick) were stained with anti-PI3K antibody (1:1000; ab182651; Abcam), anti-p-AKT (1:500; ab8933; Abcam), Cleaved Cas-3 (1:20; ab2302, Abcam) and anti-Ki67 antibody (1:500; ab15580; Abcam) at 4°C overnight, and then reacted with anti-IgG secondary antibody (1:1000; ab6721; Abcam) for 30 min. 3,3-diaminodiphenylamine (DAB, DA1010, Solarbio, Beijing, China) was used for visualization. Five fields of 200× magnification were randomly captured at each repeat using an inverted microscope (Nikon, Tokyo, Japan).
Statistical Analysis
SPSS 21.0 (IBM Corp., Armonk, NY, USA) was applied for data analysis. Kolmogorov–Smirnov test showed whether the data were in normal distribution. The results were expressed as mean ± standard deviation. Comparison between the two groups was analyzed by unpaired t test, and comparison among multiple groups was analyzed by one-way or two-way analysis of variance (ANOVA), and pairwise comparison after ANOVA was conducted by Tukey’s multiple comparisons test. p was obtained by two-tailed test and p < 0.05 indicated significant difference.
Results
CCND1 Was Highly Expressed in HCC Tissues and Cells
Firstly, the data of 367 HCC tissues from TCGA and 110 normal liver tissues from GTEX database were analyzed by online tumor data analysis website (http://xena.ucsc.edu/),15 we found that CCND1 was highly expressed in HCC tissues (p < 0.01) (Figure 1A). Then, the expression of CCND1 in HepG2, HHCC, PLC/PRF/5 and HCCLM3 cell lines and human normal hepatocytes LO2 was detected by RT-qPCR and Western blot analysis. The results showed that the expression of CCND1 in HCC cells was significantly higher than that in LO2 cells (Figure 1B and C). Because the relative expression of CCND1 in HepG2 cells was the highest, HepG2 was selected for the following experiments.
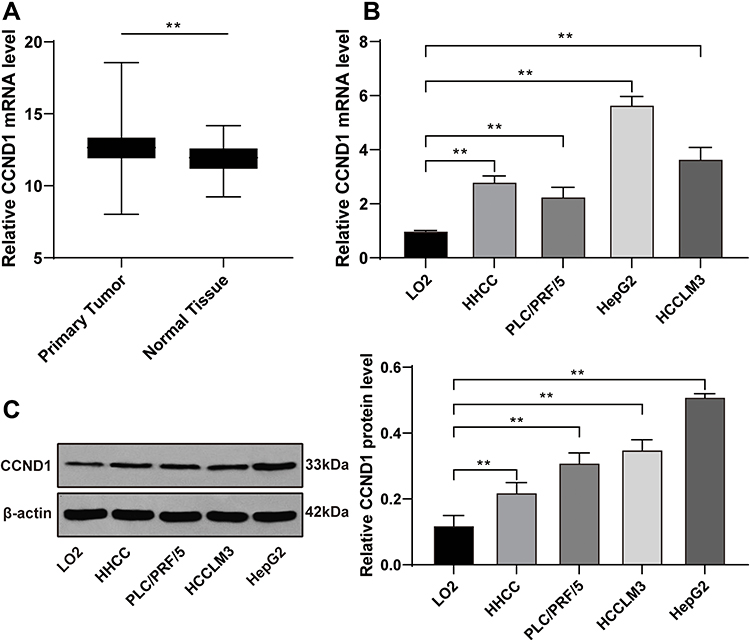 |
Figure 1 CCND1 is abnormally upregulated in HCC tissues and cell lines. (A) The CCND1 expression level from 367 HCC patients and 110 healthy normal hepatic tissues were analyzed by online database. The datasets were obtained from TCGA and GTEX database respectively. (B) The CCND1 mRNA level from HCC cell lines and normal hepatocytes LO2 was detected by RT-qPCR, normalized to GAPDH expression. (C) The CCND1 protein level from HCC cell lines and normal hepatocytes LO2 was detected by Western blot analysis, normalized to GAPDH expression. Data are expressed as mean ± standard deviation, representative of three independent experiments. One-way ANOVA was used to determine statistical significance. **p < 0.01. |
UTMD-Mediated si-CCND1 Was Successfully Transfected
The particle size of the MBs (1.35 ~ 4.62 microns) was observed by transmission electron microscope and detected by Nasosight (Figure 2A and B). Firstly, two synthetic CCND1-siRNAs were transfected into HepG2 cells by Lipofectamine2000, and then the mRNA level of CCND1 was detected by RT-qPCR. The results showed that si-CCND1-1 had higher silencing efficiency (Figure 2C), so si-CCND1-1 was selected for subsequent experiments. CCND1-siRNA, Scramble siRNA, and MBs containing si-CCND1 were then added to the well-grown HepG2 cells at a dose of 10 μg/mL, followed by an ultrasonic treatment of 10 MHz frequency for 45 s. After 48 h, the cells were observed with green fluorescence under a fluorescence microscope (Figure 2D). The expression of CCND1 in the cells detected by RT-qPCR and Western blot analysis was significantly lower than that in the si-NC group, indicating that the transfection was successful (Figure 2E and F).
 |
Figure 2 CCND1 transfection mediated by UTMD was feasible. (A) Size of MBs was measured by Z2 Coultercounter (Beckman Coulter, Sharon Hill, PA). (B) The morphology of MBs was observed using transmission electron microscopy. (C) The transfection efficiency of siRNA was detected by RT-qPCR. (D) Microscopy of MBs was collected by fluorescence microscope (Eclipse Ti microscope, Nikon, Japan). HepG2 cells in the si-NC and Lipo-si group were transfected with scramble siRNA and CCND1 siRNA by Lipofectamine 2000, while HepG2 cells in the UTMD-si group were treated with 10 MHz ultrasound for 45 s after co-culture with synthetic MBs containing CCND1-siRNA. HepG2 cells in the blank group were treated with equal saline as control; representative fluorescent micrograph (magnification, x100) represented the transfection by UTMD successfully. (E) RT-qPCR and (F) Western blot analysis was utilized for measurement of CCND1 mRNA and protein level, mRNA level and protein level normalized to GAPDH and β-actin expression, respectively. Data are expressed as mean ± standard deviation, representative of three independent experiments. One-way ANOVA was used to determine statistical significance. *p < 0.05, **p < 0.01. |
UTMD-Mediated si-CCND1 Inhibited Proliferation of HepG2 Cells
Previous study demonstrated that abnormal expression of CCND1 could lead to abnormal cell proliferation.16 Therefore, the activity of HepG2 cells was detected by MTT kit. The results showed that UTMD-mediated si-CCND1 inhibited proliferation of HepG2 cells (Figure 3A and B). EdU staining can be used for the specific detection of proliferative cells. The cells in the proliferation stage are stained in red (EdU), and the other cells are stained in blue (DAPI). The results revealed that EdU positive cells decreased significantly after si-CCND1 transfection (Figure 3C). Colony formation assay results presented that the number of cell clones decreased significantly after treatment of si-CCND1 (Figure 3D).
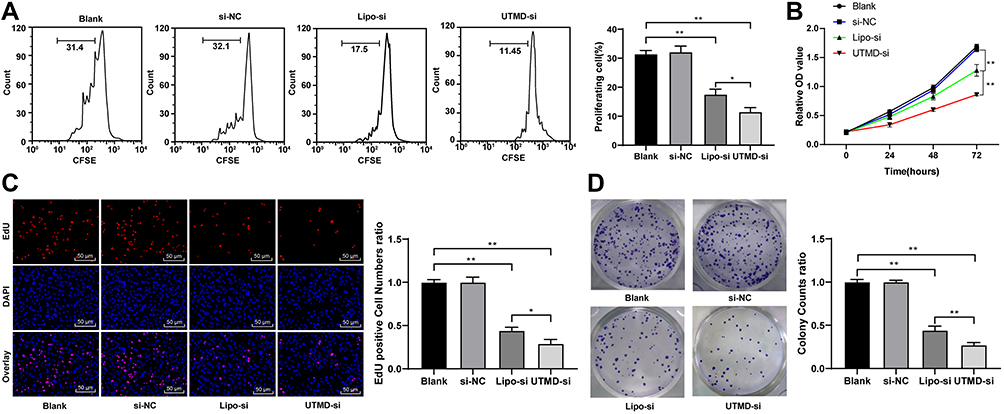 |
Figure 3 UTMD-mediated si-CCND1 inhibited HepG2 cell proliferation. HepG2 cells in the si-NC and Lipo-si group were transfected with scramble siRNA and CCND1 siRNA by Lipofectamine 2000, while HepG2 cells in the UTMD-si group were treated with 10 MHz ultrasound for 45 s after co-culture with synthetic MBs containing CCND1-siRNA. HepG2 cells in the blank group were treated with equal saline as control. (A) HepG2 cell was labeled with CFSE for 15 min, and then determined by flow cytometry. (B) HepG2 cell viability was measured by MTT analysis. (C) EdU staining and (D) colony formation assays were performed to determine HepG2 cell proliferation. For EdU staining, positive cell was stained with red. Data are expressed as mean ± standard deviation, representative of three independent experiments. In panels A, C and D, one-way ANOVA was used to determine statistical significance. In panel B, two-way ANOVA was used to determine statistical significance. *p < 0.05, **p < 0.01. |
UTMD-Mediated si-CCND1 Promoted Apoptosis of HepG2 Cells
In order to further detect the effect of UTMD-mediated si-CCND1 on the growth of HepG2 cells, we detected the apoptosis of HepG2 cells using flow cytometry and Hoechst 33,258 staining. The results showed that UTMD-mediated si-CCND1 enhanced the apoptosis of HepG2 cells (Figure 4A and B). Besides, apoptosis-related proteins were detected using Western blot analysis. The results showed that the expression of apoptosis-related proteins PUMA, Cleaved Caspase-3 and Cleaved PARP was significantly increased after the treatment of UTMD-mediated si-CCND1 (Figure 4C).
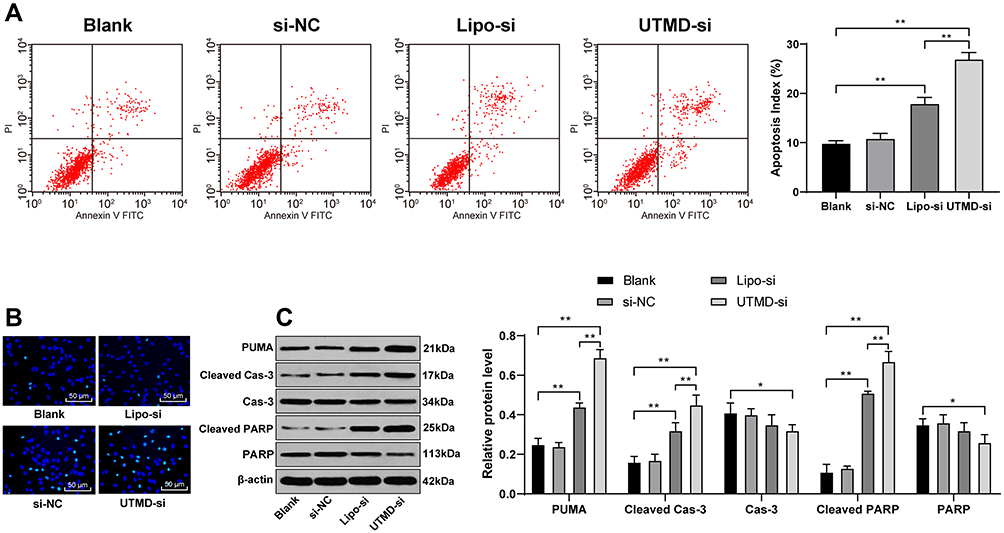 |
Figure 4 UTMD-mediated si-CCND1 enhanced HepG2 cell apoptosis. HepG2 cells in the si-NC and Lipo-si group were transfected with scramble siRNA and CCND1 siRNA by Lipofectamine 2000, while HepG2 cells in the UTMD-si group were treated with 10 MHz ultrasound for 45 s after co-culture with synthetic MBs containing CCND1-siRNA. HepG2 cells in the blank group were treated with equal saline as control. (A) HepG2 cells were labeled with PI and Annexin-V, and then determined by flow cytometry for apoptosis index. (B) Representative views of apoptotic cells. (C) Apoptosis-related factors measured by Western blot analysis and protein level normalized to β-actin expression. Data are expressed as mean ± standard deviation, representative of three independent experiments. In panel A, one-way ANOVA was used to determine statistical significance. In panel C, two-way ANOVA was used was used to determine statistical significance. *p < 0.05, **p < 0.01. |
UTMD-Mediated si-CCND1 Inhibited Proliferation of HepG2 Cells by Suppression of the PI3K/AKT Signaling Pathway
According to the literature, CCND1 upregulated the PI3K/AKT signaling pathway in non-small cell lung cancer to promote the malignant degree of the tumor.17 The PI3K/AKT signaling pathway has been involved, and is related to the proliferation and apoptosis of HCC cells.18–20 Therefore, we speculated that there may have the same results in HCC cell HepG2. The content and phosphorylation level of AKT/PI3K were detected by Western blot analysis. The results showed that silencing CCND1 inhibited the activation of the AKT/PI3K signaling pathway (Figure 5).
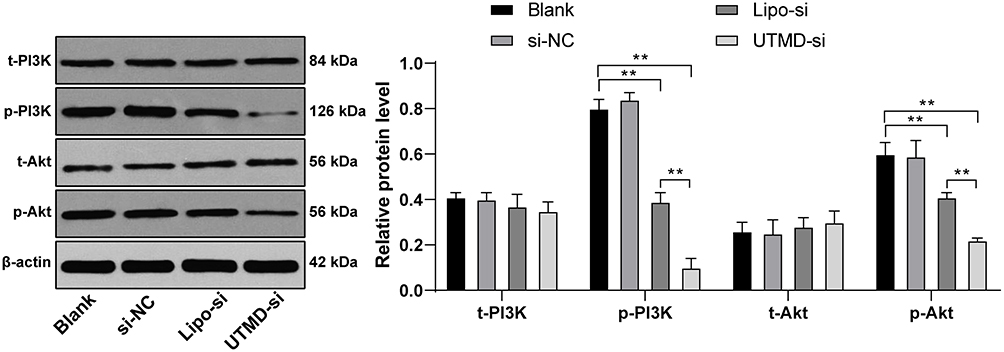 |
Figure 5 UTMD-mediated si-CCND1 inhibited HepG2 cell growth by suppression of PI3K/AKT signaling pathway. HepG2 cells in the si-NC and Lipo-si group were transfected with scramble siRNA and CCND1 siRNA by Lipofectamine 2000, while HepG2 cells in the UTMD-si group were treated with 10 MHz ultrasound for 45 s after co-culture with synthetic MBs containing CCND1-siRNA. HepG2 cells in the blank group were treated with equal saline as control. Western blot analysis was utilizing for determining PI3K/AKT signaling pathway-related content. For AKT activation, the phosphorylation site was T308, whereas PI3K phosphorylation site was Y607. Data are expressed as mean ± standard deviation, representative of three independent experiments. Two-way ANOVA was used to determine statistical significance. **p < 0.01. |
UTMD-Mediated si-CCND1 Inhibited Proliferation in vivo
In order to further evaluate the effect of UTMD-mediated si-CCND1 on the growth of HepG2 cells in vivo, we constructed xenograft tumor in nude mice. The growth trend and weight of HepG2 cells were evaluated by detecting and recording tumor growth trend. The results showed that UTMD-mediated si-CCND1 could inhibit the growth of HepG2 cells in vivo (Figure 6A and B). The expression of Ki67, p-PI3K and p-AKT was reduced, while cleaved Cas-3 expression was elevated after treatment of UTMD-mediated si-CCND1 (Figure 6C).
 |
Figure 6 UTMD-mediated si-CCND1 inhibited HepG2 cell proliferation in vivo. HepG2 cells with stable CCND1-siRNA and scramble siRNA were inoculated subcutaneously into BALB/c nude mice at a dose of 5 × 106 per mouse (n = 3 in each group). Tumor growth was measured continuously every 5 days, and 20 days later, tumor growth was monitored every 3 days. At 35 days post-implantation, the mice were euthanized by carbon dioxide asphyxiation. (A), tumor size; (B), tumor weight. (C) Tumor sections were obtained and stained with anti-p-PI3K, anti-p-AKT, anti-cleaved Cas-3 and anti-Ki67 antibodies; representative views of p-PI3K, p-AKT, cleaved Cas-3 and Ki67-positive tumor cells and quantification of immunohistochemistry staining. N=3, Data are expressed as mean± standard deviation. In panel A, two-way ANOVA was used to determine statistical significance, whereas in panels B and C, one-way ANOVA was used to determine statistical significance. *p < 0.05, **p < 0.01. |
Discussion
As one of the most common and fatal cancers in human beings, the 5-year survival rate of patients with HCC is very low due to the lack of diagnostic indicators at an early stage.21 Importantly, it had been highlighted that CCND1 was involved in the progression of HCC, and its overexpression may be an early event in hepatocarcinogenesis.22 It is reported that UTMD-mediated Foxp3-miRNA/shRNA could suppress tumor growth of HCC.23 In the present study, we assumed that there might be roles of UTMD-mediated si-CCND1 in the growth of HepG2 cells in vitro and in vivo. Collectively, our findings demonstrated that UTMD-mediated si-CCND1 could inhibit the growth and promote apoptosis of HepG2 cells via suppression of the PI3K/AKT signaling pathway.
Deregulation of cell cycle progression is a well-known and common feature of cancer.24 CCND1 protein acts as a regulator of cell cycle progression through G1 and S phases.25 Previous evidences noted that overexpression or disordered regulation of CCND1 was a driving force in some human cancers, such as non-small-cell lung cancer and breast cancer.26,27 In our study, we pointed out that HCC tissues and cells showed high CCND1 expression. In line with our study, Deane et al also revealed that the increased levels of CCND1 have been associated with aggressive forms of human HCC.28
Besides, we also provided evidence that UTMD-mediated si-CCND1 inhibited proliferation and promoted apoptosis of HepG2 cells. CCND1 allows cells to accelerate through promoting the G1/S cell cycle transition and can thus promote cell proliferation.29 Additionally, CCND1 can also indirectly inhibit cell migration by suppression of epithelial-mesenchymal transition.30 Moreover, CCND1 overexpression is a common event in many cancers. Therefore, we could assume that CCND1 downregulation might be an effective way to slow down cancer progression. On previous report clarified that CCND1 downregulation could lead to the inhibited cell proliferation and enhanced cell apoptosis in glioblastoma cells.31 Further, Xia et al also noted that overexpression of CCND1 could promote proliferation and self-renewal of liver cancer stem cells through activation of the TGF-beta/Smad signaling pathway.32 Ultrasound MBs are well-known nanobubbles and have the advantages of high safety, stability and transfection efficiency.33 Accumulating evidence confirmed that UTMD can effectively promote gene delivery and has the practical value of transfection in vivo at the same time.34–37 At present, a large number of studies have confirmed that UTMD can effectively mediated gene transfer, which has the advantages of safety, practicability and targeting, and is one of the main trends of gene therapy.38–41 Consistent with our study, one report focused on UTMD-mediated suicide gene and hepatic cancer also demonstrated that the tumor inhibition rate in the treatment group was markedly higher compared with that in the control group.42 Furthermore, the present study also noted that the in vitro findings were further confirmed via in vivo study.
The above antitumor effects were achieved through suppression of the PI3K/AKT signaling pathway. In this study, Western blot analysis was conducted to detect the expression of the PI3K/AKT signaling pathway-related proteins. Interestingly, we found that si-CCND1 could inhibit the activation of the PI3K/AKT signaling pathway. Presently, many signaling pathways have been proven to be involved in the modulation of HCC cell invasion and metastasis, the PI3K/AKT signaling pathway is one of the classical pathways.43 PI3K, an intracellular phosphatidylinositol kinase, is involved in the development of many malignant tumors, including nasopharyngeal cancer, breast cancer, and HCC.44–46 Accumulating evidence showed that PI3K could decrease the expression of cell apoptosis-related proteins through the phosphorylation of AKT, which acts as an important regulator of cell proliferation.47 Therefore, the suppression of the PI3K/AKT signaling pathway can arrest more cells in the G2/M phase related to DNA damage, then result in cell apoptosis.48
Conclusion
All in all, our study supported that UTMD-mediated si-CCND1 could repressed HCC cell growth in vitro and in vivo through inhibition of the PI3K/AKT signaling pathway. These results indeed revealed a novel therapeutic target for tough treatment against HCC. Though our findings offer therapeutic implication in the treatment for HCC, the effective application into clinic practice still need future validation. Other signaling pathways, such as the MAPK pathway, will be further studied in our future study.
Data Sharing Statement
All the data generated or analyzed during this study are included in this published article.
Ethics Statement
This study was approved and supervised by the animal ethics committee of Changchun University. Significant efforts were made in order to minimize both the number of animals and their suffering. All procedures were strictly conducted in accordance with the code of ethics and with the recommendations of the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (NIH Pub. No. 85-23, revised 1996).
Disclosure
All authors declare that they have no conflicts of interest in this study.
References
1. Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet. 2012;379(9822):1245–1255. doi:10.1016/S0140-6736(11)61347-0
2. Khemlina G, Ikeda S, Kurzrock R. The biology of Hepatocellular carcinoma: implications for genomic and immune therapies. Mol Cancer. 2017;16(1):149. doi:10.1186/s12943-017-0712-x
3. Jiang C, Wu Y, Zhou J, Zhao J. Novel targets and small molecular interventions for liver cancer. Biomed Res Int. 2014;2014:148783. doi:10.1155/2014/148783
4. Tang A, Hallouch O, Chernyak V, Kamaya A, Sirlin CB. Epidemiology of hepatocellular carcinoma: target population for surveillance and diagnosis. Abdom Radiol (NY). 2018;43(1):13–25. doi:10.1007/s00261-017-1209-1
5. Aljumah AA, Kuriry H, AlZunaitan M, et al. Clinical presentation, risk factors, and treatment modalities of hepatocellular carcinoma: a single tertiary care center experience. Gastroenterol Res Pract. 2016;2016:1989045. doi:10.1155/2016/1989045
6. Lencioni R, Crocetti L. Local-regional treatment of hepatocellular carcinoma. Radiology. 2012;262(1):43–58. doi:10.1148/radiol.11110144
7. Villanueva A, Llovet JM. Targeted therapies for hepatocellular carcinoma. Gastroenterology. 2011;140(5):1410–1426. doi:10.1053/j.gastro.2011.03.006
8. Calderaro J, Rousseau B, Amaddeo G, et al. Programmed death ligand 1 expression in hepatocellular carcinoma: relationship with clinical and pathological features. Hepatology. 2016;64(6):2038–2046. doi:10.1002/hep.28710
9. Ao R, Zhang DR, Du YQ, Wang Y. Expression and significance of Pin1, beta-catenin and cyclin D1 in hepatocellular carcinoma. Mol Med Rep. 2014;10(4):1893–1898. doi:10.3892/mmr.2014.2456
10. Kim JK, Diehl JA. Nuclear cyclin D1: an oncogenic driver in human cancer. J Cell Physiol. 2009;220(2):292–296. doi:10.1002/jcp.21791
11. Chen J, Li X, Cheng Q, et al. Effects of cyclin D1 gene silencing on cell proliferation, cell cycle, and apoptosis of hepatocellular carcinoma cells. J Cell Biochem. 2018;119(2):2368–2380. doi:10.1002/jcb.26400
12. Thomas SM, Grandis JR. The current state of head and neck cancer gene therapy. Hum Gene Ther. 2009;20(12):1565–1575. doi:10.1089/hum.2009.163
13. Suzuki R, Oda Y, Utoguchi N, Maruyama K. Progress in the development of ultrasound-mediated gene delivery systems utilizing nano- and microbubbles. J Control Release. 2011;149(1):36–41. doi:10.1016/j.jconrel.2010.05.009
14. Carson AR, McTiernan CF, Lavery L, et al. Gene therapy of carcinoma using ultrasound-targeted microbubble destruction. Ultrasound Med Biol. 2011;37(3):393–402. doi:10.1016/j.ultrasmedbio.2010.11.011
15. Barajas JM, Reyes R, Guerrero MJ, Jacob ST, Motiwala T, Ghoshal K. The role of miR-122 in the dysregulation of glucose-6-phosphate dehydrogenase (G6PD) expression in hepatocellular cancer. Sci Rep. 2018;8(1):9105. doi:10.1038/s41598-018-27358-5
16. Le Marchand L, Seifried A, Lum-Jones A, Donlon T, Wilkens LR. Association of the cyclin D1 A870G polymorphism with advanced colorectal cancer. JAMA. 2003;290(21):2843–2848. doi:10.1001/jama.290.21.2843
17. Zhao M, Xu P, Liu Z, et al. Dual roles of miR-374a by modulated c-Jun respectively targets CCND1-inducing PI3K/AKT signal and PTEN-suppressing Wnt/beta-catenin signaling in non-small-cell lung cancer. Cell Death Dis. 2018;9(2):78. doi:10.1038/s41419-017-0103-7
18. Zhu P, Liu Z, Zhou J, et al. Tanshinol inhibits the growth, migration and invasion of hepatocellular carcinoma cells via regulating the PI3K-AKT signaling pathway. Onco Targets Ther. 2019;12:87–99. doi:10.2147/OTT.S185997
19. Wu T, Dong X, Yu D, et al. Natural product pectolinarigenin inhibits proliferation, induces apoptosis, and causes G2/M phase arrest of HCC via PI3K/AKT/mTOR/ERK signaling pathway. Onco Targets Ther. 2018;11:8633–8642. doi:10.2147/OTT.S186186
20. Lin Z, Li S, Guo P, et al. Columbamine suppresses hepatocellular carcinoma cells through down-regulation of PI3K/AKT, p38 and ERK1/2 MAPK signaling pathways. Life Sci. 2019;218:197–204. doi:10.1016/j.lfs.2018.12.038
21. Torre LA, Siegel RL, Jemal A. Lung cancer statistics. Adv Exp Med Biol. 2016;893:1–19. doi:10.1007/978-3-319-24223-1_1
22. Joo M, Kang YK, Kim MR, Lee HK, Jang JJ. Cyclin D1 overexpression in hepatocellular carcinoma. Liver. 2001;21(2):89–95.
23. Shi C, Zhang Y, Yang H, et al. Ultrasound-targeted microbubble destruction-mediated Foxp3 knockdown may suppress the tumor growth of HCC mice by relieving immunosuppressive Tregs function. Exp Ther Med. 2018;15(1):31–38. doi:10.3892/etm.2017.5421
24. Lu C, He Y, Duan J, et al. Expression of Wnt3a in hepatocellular carcinoma and its effects on cell cycle and metastasis. Int J Oncol. 2017;51(4):1135–1145. doi:10.3892/ijo.2017.4112
25. Saikawa Y, Kubota T, Otani Y, Kitajima M, Modlin IM. Cyclin D1 antisense oligonucleotide inhibits cell growth stimulated by epidermal growth factor and induces apoptosis of gastric cancer cells. Jpn J Cancer Res. 2001;92(10):1102–1109. doi:10.1111/j.1349-7006.2001.tb01065.x
26. Myong NH. Cyclin D1 overexpression, p16 loss, and pRb inactivation play a key role in pulmonary carcinogenesis and have a prognostic implication for the long-term survival in non-small cell lung carcinoma patients. Cancer Res Treat. 2008;40(2):45–52. doi:10.4143/crt.2008.40.2.45
27. Villegas SL, Darb-Esfahani S, von Minckwitz G, et al. Expression of Cyclin D1 protein in residual tumor after neoadjuvant chemotherapy for breast cancer. Breast Cancer Res Treat. 2018;168(1):179–187. doi:10.1007/s10549-017-4581-1
28. Deane NG, Parker MA, Aramandla R, et al. Hepatocellular carcinoma results from chronic cyclin D1 overexpression in transgenic mice. Cancer Res. 2001;61(14):5389–5395.
29. Zhang Y, Wang S, Li L. EF hand protein IBA2 promotes cell proliferation in breast cancers via transcriptional control of cyclin D1. Cancer Res. 2016;76(15):4535–4545. doi:10.1158/0008-5472.CAN-15-2927
30. Tobin NP, Sims AH, Lundgren KL, Lehn S, Landberg G. Cyclin D1, Id1 and EMT in breast cancer. BMC Cancer. 2011;11:417. doi:10.1186/1471-2407-11-417
31. Wang J, Wang Q, Cui Y, et al. Knockdown of cyclin D1 inhibits proliferation, induces apoptosis, and attenuates the invasive capacity of human glioblastoma cells. J Neurooncol. 2012;106(3):473–484. doi:10.1007/s11060-011-0692-4
32. Xia W, Lo CM, Poon RYC, et al. Smad inhibitor induces CSC differentiation for effective chemosensitization in cyclin D1- and TGF-beta/Smad-regulated liver cancer stem cell-like cells. Oncotarget. 2017;8(24):38811–38824. doi:10.18632/oncotarget.16402
33. Guo X, Guo S, Pan L, Ruan L, Gu Y, Lai J. Anti-microRNA-21/221 and microRNA-199a transfected by ultrasound microbubbles induces the apoptosis of human hepatoma HepG2 cells. Oncol Lett. 2017;13(5):3669–3675. doi:10.3892/ol.2017.5910
34. Chen S, Grayburn PA. Ultrasound-targeted microbubble destruction for cardiac gene delivery. Methods Mol Biol. 2017;1521:205–218. doi:10.1007/978-1-4939-6588-5_14
35. Escoffre JM, Zeghimi A, Novell A, Bouakaz A. In-vivo gene delivery by sonoporation: recent progress and prospects. Curr Gene Ther. 2013;13(1):2–14. doi:10.2174/156652313804806606
36. Shapiro G, Wong AW, Bez M, et al. Multiparameter evaluation of in vivo gene delivery using ultrasound-guided, microbubble-enhanced sonoporation. J Control Release. 2016;223:157–164. doi:10.1016/j.jconrel.2015.12.001
37. Sirsi SR, Borden MA. Advances in ultrasound mediated gene therapy using microbubble contrast agents. Theranostics. 2012;2(12):1208–1222. doi:10.7150/thno.4306
38. Chen ZY, Liang K, Sheng XJ, et al. Optimization and apoptosis induction by RNAi with UTMD technology in vitro. Oncol Lett. 2012;3(5):1030–1036. doi:10.3892/ol.2012.610
39. Chen ZY, Yang F, Lin Y, et al. New development and application of ultrasound targeted microbubble destruction in gene therapy and drug delivery. Curr Gene Ther. 2013;13(4):250–274. doi:10.2174/15665232113139990003
40. Yoon CS, Park JH. Ultrasound-mediated gene delivery. Expert Opin Drug Deliv. 2010;7(3):321–330. doi:10.1517/17425241003596329
41. Yu BF, Wu J, Zhang Y, Sung HW, Xie J, Li RK. Ultrasound-targeted HSVtk and Timp3 gene delivery for synergistically enhanced antitumor effects in hepatoma. Cancer Gene Ther. 2013;20(5):290–297. doi:10.1038/cgt.2013.19
42. Tang Q, He X, Liao H, et al. Ultrasound microbubble contrast agent-mediated suicide gene transfection in the treatment of hepatic cancer. Oncol Lett. 2012;4(5):970–972. doi:10.3892/ol.2012.845
43. Li H, Shen S, Chen X, Ren Z, Li Z, Yu Z. miR-450b-5p loss mediated KIF26B activation promoted hepatocellular carcinoma progression by activating PI3K/AKT pathway. Cancer Cell Int. 2019;19:205. doi:10.1186/s12935-019-0923-x
44. Delaloge S, DeForceville L. Targeting PI3K/AKT pathway in triple-negative breast cancer. Lancet Oncol. 2017;18(10):1293–1294. doi:10.1016/S1470-2045(17)30514-4
45. Pellegrino R, Calvisi DF, Neumann O, et al. EEF1A2 inactivates p53 by way of PI3K/AKT/mTOR-dependent stabilization of MDM4 in hepatocellular carcinoma. Hepatology. 2014;59(5):1886–1899. doi:10.1002/hep.26954
46. Zhao M, Luo R, Liu Y, et al. miR-3188 regulates nasopharyngeal carcinoma proliferation and chemosensitivity through a FOXO1-modulated positive feedback loop with mTOR-p-PI3K/AKT-c-JUN. Nat Commun. 2016;7:11309. doi:10.1038/ncomms11309
47. Ho L, Tan SY, Wee S, et al. ELABELA is an endogenous growth factor that sustains hESC self-renewal via the PI3K/AKT pathway. Cell Stem Cell. 2015;17(4):435–447. doi:10.1016/j.stem.2015.08.010
48. Chen J, Chen Z, Huang Z, Yu H, Li Y, Huang W. Formiminotransferase cyclodeaminase suppresses hepatocellular carcinoma by modulating cell apoptosis, DNA damage, and phosphatidylinositol 3-kinases (PI3K)/Akt signaling pathway. Med Sci Monit. 2019;25:4474–4484. doi:10.12659/MSM.916202
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.