Back to Journals » Journal of Hepatocellular Carcinoma » Volume 2
Type I insulin-like growth factor as a liver reserve assessment tool in hepatocellular carcinoma
Authors Abdel-Wahab R ![]() , Shehata S, Hassan M, Habra M, Eskandari G, Tinkey P, Mitchell J, Lee J
, Shehata S, Hassan M, Habra M, Eskandari G, Tinkey P, Mitchell J, Lee J ![]() , Amin H
, Amin H ![]() , Kaseb A
, Kaseb A
Received 21 January 2015
Accepted for publication 14 April 2015
Published 18 September 2015 Volume 2015:2 Pages 131—142
DOI https://doi.org/10.2147/JHC.S81309
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Junji Furuse
Reham Abdel-Wahab,1,2 Samir Shehata,2 Manal M Hassan,1 Mouhammed A Habra,3 Ghazaleh Eskandari,4 Peggy T Tinkey,5 Jennifer Mitchell,5 Ju-Seog Lee,6 Hesham M Amin,4,7 Ahmed O Kaseb1
1Department of Gastrointestinal Medical Oncology, The University of Texas MD Anderson Cancer Center, Houston, TX, USA; 2Department of Clinical Oncology, Assiut University Hospital, Assiut, Egypt; 3Department of Endocrinology, 4Department of Hematopathology, 5Department of Veterinary Medicine and Surgery, 6Department of Systems Biology, The University of Texas MD Anderson Cancer Center, 7Graduate School of Biomedical Sciences, Houston, TX, USA
Abstract: Chronic liver diseases (CLDs) encompass a wide range of illnesses, including nonalcoholic fatty liver disease, nonalcoholic steatohepatitis, and viral hepatitis. Deterioration of liver capacity, with subsequent progression into cirrhosis and hepatocellular carcinoma (HCC), ultimately leads to a further decrease in the hepatic reserve. The Child–Turcotte–Pugh scoring system is the standard tool for assessing underlying liver reserve capacity in routine practice and in clinical trials of CLD and HCC. In this review, we highlight the clinical significance of insulin-like growth factor-I (IGF-I) and the growth hormone (GH) signaling pathway in HCC. IGF-I could be a marker for liver reserve capacity in CLDs and HCC in clinical practice. This approach could improve the risk assessment and stratifications of patients on the basis of their underlying liver reserve, either before active treatment in routine practice or before they are enrolled in clinical trials.
Keywords: IGF-I, growth hormone, chronic liver disease
Insulin-like growth factors and binding proteins
Insulin-like growth factors (IGFs) were first described by Salmon and Daughaday in 1957.1 The IGF axis includes several molecules: two ligands (IGF-I [somatomedin C] and IGF-II [somatomedin A]), two transmembrane receptors (IGF-I receptor [IGF-IR] and IGF-II receptor [IGF-IIR]), and eight high-affinity IGF binding proteins (IGFBPs) (IGFBP-1 to -6, along with the lesser characterized IGFBP-7 and -8) (Figure 1).2–9 These factors stimulate musculoskeletal growth and differentiation, particularly during prenatal growth.1,10–12 Under normal physiological conditions, all IGF axis molecules work together in a harmonized manner to maintain cellular homeostasis.
 | Figure 1 Insulin-like growth factor-I (IGF-I) axis and variable sources of IGF-I production. |
IGF-I is a hormone with a small molecular weight; it contains 70 amino acids, and unlike other peptides, 99% of it is protein bound. As the biochemical structure of IGF-IR is similar to that of the insulin receptor, the free IGF-I possesses a high affinity to bind with IGF-IR compared to that of insulin receptor, inducing cell proliferation and inhibiting apoptosis. It also binds with a high affinity to hybrid receptors, which contain an alpha–beta IGF half-receptor paired with an alpha–beta insulin half-receptor. The physiologic significance of hybrid receptors is not well defined, but they may mediate the insulin-like actions of IGF-I (Figure 1). These effects can be primarily inhibited by IGFBP-3, which binds to and prevents IGF-I from binding to IGF-IR.4,7,13
Mechanism and sources of IGF-I synthesis
Growth hormone (GH) is secreted, with diurnal variation, from the anterior pituitary gland in a pulsatile manner. This occurs under hypothalamic control through the influence of hypothalamic neuropeptides, GH-releasing hormone, and GH-inhibitory hormone (somatostatin); in addition to the influence of both IGF-I and ghrelin (a gastric hormone).14,15
Released GH produced from the pituitary gland is transported by GH binding protein to bind to its receptors in different tissues, including the liver, which is considered the main target of GH. This binding upregulates IGF-I synthesis through stimulation of IGF-I gene transcription.8,16 Approximately 75% of circulating IGF synthesized by the liver is believed to perform an “endocrine” function as it is typically used remotely.17–19 In contrast, approximately 25% of IGF-I that is synthesized in the bones, cartilage, central nervous system, kidneys, ovaries, and erythroid cell precursors executes autocrine and paracrine functions (Figures 1 and 2).20–25
Factors affecting plasma levels of GH/IGF-I
GH/IGF secretion can be stimulated directly by the “push effect” or indirectly by the “pull effect” by reducing the negative feedback inhibitory effect.26 Normally, the circulating IGF level changes with age. During childhood, increase in the production of sex steroid hormones results in increased production of GH27–29 and subsequently IGF-I in both sexes.27,30–34 GH/IGF-I levels rapidly decline during the second decade of life, followed by a slow decline until the age of 60 years.35 However, the relationship between steroid hormones and GH/IGF is affected by sex. Several studies have shown that in men, regardless of age, testosterone centrally increases the GH level, followed by IGF-I production through the push effect.36–40 Studies were performed to determine whether testosterone enhances IGF-I directly or as a costimulatory factor to GH; testosterone alone had a very limited or no effect on circulating IGF-I levels except in the presence of GH.41–43 On the contrary, there is a debate concerning the relationship between estrogen and GH/IGF-I. Researchers have found that during menstruation, GH levels increase in response to an estrogen peak, with higher GH levels in premenopausal women than in postmenopausal women.44,45 However, most studies did not evaluate IGF-I levels and thus could not determine whether GH levels increased as the result of a pushing or pulling effect.39,46,47 Notably, recent studies showed that estrogen indirectly stimulates GH production by inhibiting the IGF-I “pulling effect”.48–51
Notably, elevated glucose, insulin, cortisol, and non-stratified free fatty acid could also inhibit GH production. Amino acids, sleep, and exercise increase GH secretion levels. In all of these conditions, IGF-I is influenced by changes in the GH level. The presence of all these factors complicates the GH/IGF-I secretion control process.52–56 Furthermore, IGF-I that is synthesized in peripheral tissues is influenced by several factors on the basis of the site of production:57,58 1) bone and cartilage (parathyroid hormone [PTH] regulates IGF-I gene transcription in the bone, while GH increases IGF-I synthesis from osteoblasts and chondrocytes); 2) erythroid cell precursors (which synthesize IGF-I under the influence of erythropoietin); 3) skeletal muscles (both muscle injury and hypertrophy stimulate IGF-I synthesis); and 4) kidneys (which are an important local source of IGF-I). Notably, unilateral nephrectomy induces compensatory growth of the contralateral kidney, with a subsequent increase in IGF-I expression.
Mechanism of action of IGF-I
Synthesized IGF-I is cleaved by protease enzymes before being released into the circulation. IGFBPs, which are present in all extracellular fluids, transport IGF-I by binding to approximately 99% of it with a higher affinity than IGF-IR.14,59 The bound form of IGF-I is mainly synthesized in the liver, while the free form, which is produced by other tissues, has a low affinity to IGFBPs and is responsible for its autocrine and paracrine effects.16,17
Notably, elevated serum levels of IGF-I induce a negative feedback effect on GH secretion, either directly through a local inhibitory effect on the pituitary gland or indirectly by stimulating somatostatin release. Thus, IGF-I and GH work cooperatively as IGF-I regulates GH effects, which in turn control the release of IGF-I.60–67
The role of GH receptor (GHR) in harmonizing the association between GH elevation68 and IGF-I suppression69 has been reported in previous studies. Chang et al70 studied the correlation between these changes and GHRs, which are present on hepatocyte cell membranes. They determined the presence of GHR in human HCC, cirrhosis, and normal tissue samples using radio-receptor assays and discovered that GHR was absent in both cirrhotic and HCC samples, which explains the persistent decrease in the serum level of IGF-I with elevated levels of GH.
Normally, both GH and IGF-I have an anabolic effect, promoting lipolysis and protein synthesis by stimulating amino acid uptake, stimulating cell growth and differentiation, increasing muscle mass through sarcomere hyperplasia, and stimulating the immune system by restoring a normal nitrogen balance and causing a 25% increase in GFR. IGF-I also decreases blood glucose levels, improves insulin resistance, decreases reactive oxygen species, and has an antifibrotic effect.71–73
Molecular role of IGF-I in cancer development
In 1990, the role of IGF-I in the process of tumorigenesis was revealed. Since then, a major research focus has been to better understand the nature and role of the IGF axis in the pathogenesis of various neoplasms.11
Recently, there has been renewed interest in the roles of GH/IGF-I in cancer development because of an increase in cancer incidence, including breast, thyroid, colon, and prostate cancers, in acromegalic cancer patients with elevated serum IGF-I secondary to GH-producing pituitary tumors.74 Elevated serum IGF-I and GH levels were reported in non-acromegalic cancer patients. GH enhances cancer development through several pathways:75–81 1) it binds to GHR and activates several intracellular signal pathways; 2) it stimulates IGF-I production from the liver; and 3) it induces peripheral tissue insulin resistance, with subsequent elevation of serum insulin levels.
Binding of IGF-I to the alpha subunit of IGF-IR leads to auto-activation of tyrosine kinase and the auto-phosphorylation of tyrosines, with subsequent phosphorylation of insulin receptor substrate-1 (IRS-I) and insulin receptor substrate 2 (IRS-II).82,83 IRS-I stimulates several kinase pathways, such as phosphatidylinositol 3 kinase (PI3K), SHC, and Src. Through the SHC pathway, Grb-2 forms a complex that activates son of sevenless (SOS) protein. This complex activates p21 Ras, which is a mitogen-activated protein kinase pathway. Activation of this pathway is important for stimulating cell growth.83 IRS-I also activates the PI3K/mitogen-activated protein kinase (mTOR) pathway, which is important for stimulating protein synthesis, glucose transportation, cell motility, and apoptosis inhibition.84
IGF-I plays an important role in cancer development by regulating angiogenesis, lymphangiogenesis, degradation of the extracellular matrix (ECM), tumor invasion into both the ECM and blood vessels, and maintenance of tumor cell survival and proliferation.7,85
Several basic science studies showed that IGF-I regulates angiogenesis and lymphangiogenesis by activating vascular endothelial growth factor and stimulating the expression of hypoxia-inducible factor 1 via the PI3K/Akt and Ras/mTOR pathways.86–89 IGF-I is transported across the vascular endothelial cell lining through a paracellular route where it binds to the subendothelial ECM to stimulate the migration and morphological differentiation of endothelial cells.90–92 Subsequently, IGF-I activates matrix metalloproteinase-9, which is a type IV collagenase.93 IGF-I also increases the binding of single-chain urokinase-type plasminogen activator (uPA) to the cell-surface uPA receptor (uPAR). This combination converts serum plasminogen to plasmin, which is a broad-spectrum serum protease enzyme. Both metalloproteinase-9 and uPAR/uPA are major molecular mediators that play a significant role in ECM proteolysis and degradation, followed by tumor invasion (Figure 3).94
 | Figure 2 Roles and regulations of IGF-I in relation to GH in both normal and reduced hepatic reserve. |
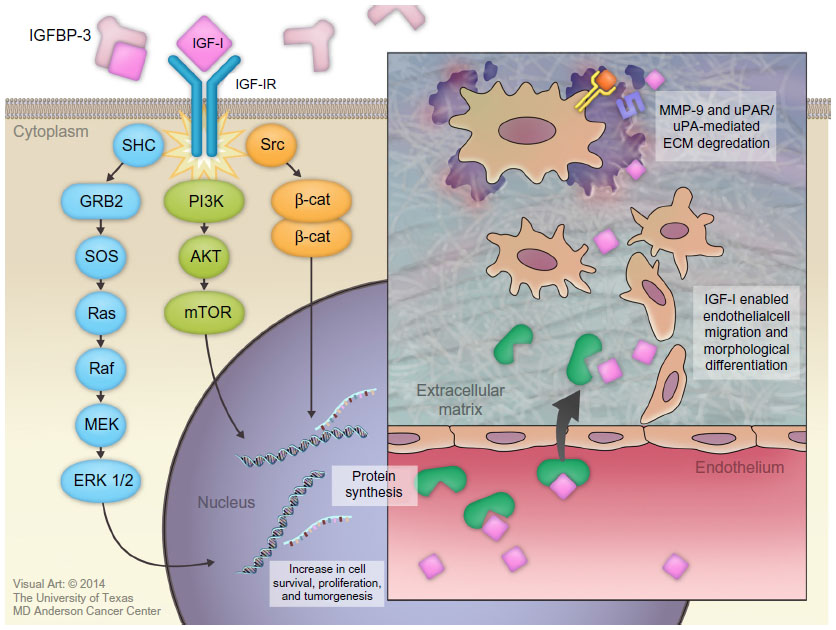 | Figure 3 Roles of insulin-like growth factor-I (IGF-I) in cancer development. |
GH/IGF-I as an indicator of hepatic reserve
A previous article reported an IGF-I deficiency in CLDs such as nonalcoholic fatty liver disease, nonalcoholic steatohepatitis, viral hepatitis, cirrhosis, and HCC; it occurs through several mechanisms, including insulin resistance, oxidative stress, mitochondrial dysfunction, and the inflammatory cascade.95
The correlation between GH and IGF-I levels in liver cirrhosis has been previously evaluated; elevated plasma levels of GH were found in cirrhosis patients, with a pulse frequency and plasma half-life that were more than twice those in the control group; this was partially explained by the associated hyperglycemia.96
In 1993, Buzzelli et al63 studied changes in the GH/IGF-I circadian rhythm in cirrhosis patients, regardless of the presence or absence of associated HCC. They concluded that, compared to the control group, patients with cirrhosis had lower serum IGF-I levels and higher GH levels. These changes remained stable for 24 hours, resulting in loss of GH/IGF-I circadian rhythm.63 This phenomenon was explained by the decrease in GHRs and their binding capacity in damaged liver tissue compared with in normal liver tissue.
A low serum IGF-I level leads to several metabolic changes induced by reduced peripheral glucose and lipid uptake, increased liver glucose production, increased stored triglyceride hydrolysis, and subsequently elevated circulating glucose and free fatty acid levels (Figure 2).62 Furthermore, a few studies showed that, under normal conditions, IGF-I stimulates hepatocyte growth factor (HGF) production from hepatic stellate cells, and that administration of human recombinant HGF suppressed the onset of liver fibrosis/cirrhosis in animal models.97–99 Thus, low levels of free IGF-I lead to a loss of antifibrotic effects. Reactive oxygen species, different cytokines, and inflammatory mediators can easily activate hepatic stellate cells and induce fibrosis.100
Further studies showed a lower rate of IGF-I expression in patients diagnosed with either nonalcoholic fatty liver disease or nonalcoholic steatohepatitis (Table 1).101,102
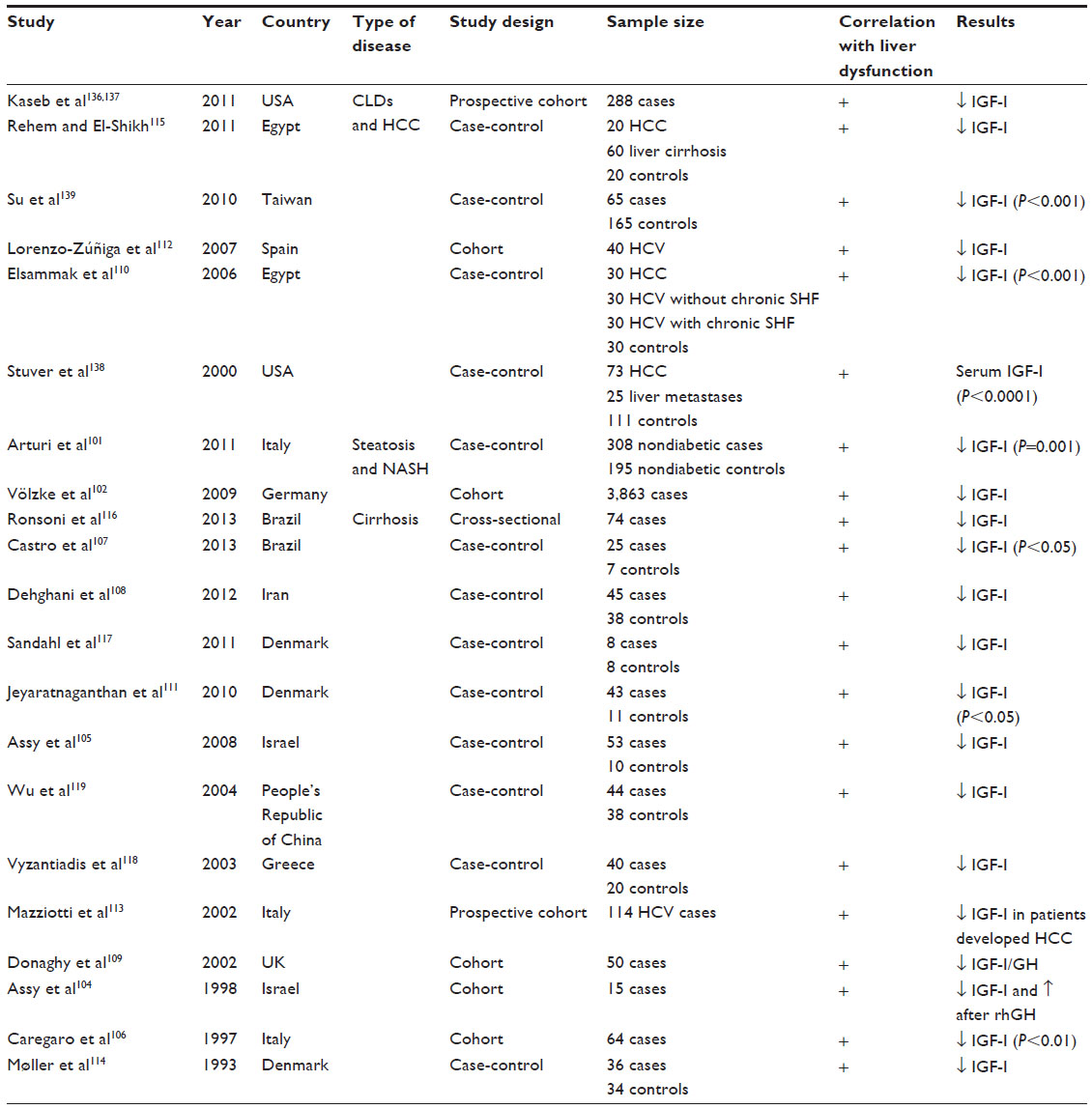 | Table 1 Clinical studies of circulating IGF-I in CLDs |
Notably, liver cirrhosis, which is a chronic disease in which the liver tissue is irreversibly replaced by fibrous tissue, necrosis, and regenerating nodules, leads to the deterioration of normal hepatic function.103 Systematically, cirrhosis patients experience several clinical manifestations of their decreased metabolic liver capacity and subsequent IGF-I deficiency and GH elevation.
Several studies reported decreased serum IGF-I levels in patients with diseased liver compared to normal population. This suggests that circulating levels of IGF-I are a surrogated marker for assessment of liver dysfunction (Table 1).101,102,104–119
Collectively, these findings support the hypothesis that plasma IGF-I levels reflect hepatic synthetic function and hence should be considered a surrogate marker for determining the hepatic reserve.
IGF-I as an assessment tool for liver reserve capacity in HCC
Currently, surgical resection and liver transplantation are the only curative treatments for HCC.120,121 Unfortunately, most patients are not surgical candidates because of an advanced tumor stage at presentation or advanced underlying CLDs.122,123 These factors have a significant effect on treatment decisions and outcomes (including overall survival [OS]) and prognostic stratification for clinical trial enrollment.
Several HCC prognostic systems are used to assess underlying CLD status, predict treatment outcome and OS, and stratify patients in clinical trials. However, the standard system for assessing hepatic reserve in HCC staging systems is the Child-Turcotte-Pugh (CTP) score, which depends on two subjective parameters (encephalopathy and ascites) and three objective parameters (serum albumin, serum bilirubin, and prothrombin time or the international normalized ratio).107,111,113,116,117,119 Despite its limitations, the CTP score has remained the standard tool for predicting the degree of underlying CLD in HCC patients before active therapy or trial entry, using CTP class A (CTP-A) as the standard treatable patient population.124–131 Several studies have concluded that patients classified as CTP-B or CTP-C have significantly shorter survival duration than do those classified as CTP-A because of deterioration in their hepatic function. Therefore, CTP-A patients are the only patients who are eligible for active treatment and clinical trials122,123,132 and are the only population approved by the US Food and Drug Administration (FDA) for sorafenib therapy on the basis of the results of the first international, randomized, double-blind, placebo-controlled, multicenter Phase III study.121
The CTP-A group is heterogeneous (especially nonsurgical patients, who constitute the main pool in clinical practice and clinical trials, as described at the most recent international expert consensus conference),123 and post-therapeutic decline in liver functions is still a major challenge to outcome prediction. Furthermore, the survival benefit of sorafenib was associated with only a 2.3% objective response rate, as defined by Response Evaluation Criteria in Solid Tumors. Sorafenib is an expensive treatment; it is not affordable to many patients and may increase health care costs, especially in low- and middle-income countries. National and international guidelines recommend sorafenib only in patients with CTP-A to avoid potential severe adverse effects and death due to hepatic failure.133–135 However, hepatic failure and sorafenib intolerance still occur in patients with CTP-A. Thus, there is a critical and immediate need for more sensitive tools than the CTP score to predict survival duration in HCC patients undergoing treatment and in selected patients who are not eligible for enrollment in clinical trials.
Integrating IGF-I levels into HCC management
Since the liver produces >75% of circulating IGF-I, and IGF-I’s role is documented in other CLDs, several studies have investigated whether IGF-I levels can be used to assess hepatic capacity in HCC patients and detect its correlation with HCC prognosis and survival outcome (Table 1).136–139 The decline in serum levels of IGF-I in HCC is likely mediated through the decreased synthetic capacity of normal liver cells, which have been replaced by tumor cells.109,140–142
We recently reported the utility of plasma IGF-I as a molecular biomarker for assessing liver reserve in HCC patients.136,137,143 In addition, two recent studies reported that low pretreatment IGF-I levels independently correlated with poor outcome in the form of a shorter TTP and OS in patients with HCC who underwent TACE.144,145
On the basis of the widely adopted American Association for the Study of Liver Diseases guidelines, HCC can be diagnosed using a noninvasive imaging approach.123,146 There is a critical need to develop a blood-based biomarker strategy to assess hepatic reserve and predict patients’ survival and treatment outcomes. This approach will improve the personalization of HCC treatment by allowing us to select the best candidates for specific therapeutic modalities and avoid unnecessary harm and health care expenses.
Developing the IGF-I score
The CTP score is the standard tool currently used for assessing hepatic reserve in HCC staging systems. Recently, our research group studied the value of incorporating IGF-I into the CTP system to replace the two subjective parameters, ascites and encephalopathy (Table 2).143 Our results indicated that the IGF-CTP score significantly improved OS prediction and patient risk stratification compared to the CTP score in both the training and validation cohorts (P=0.003 and P=0.005, respectively, when measured by the C-index) (Table 3). Differences between the C-indices were not large but were statistically significant as the C-index computes the ability to predict OS for all patients in the cohort, including those whose CTP and IGF-CTP scores are different and those whose scores are the same. Interestingly, patients with CTP-A that was reclassified as IGF-CTP-B had significantly shorter OS than did patients whose IGF-CTP-A classification remained unchanged in both the training and validation cohorts (P=0.03 and P<0.001, respectively) (Figure 4 and Table 4).
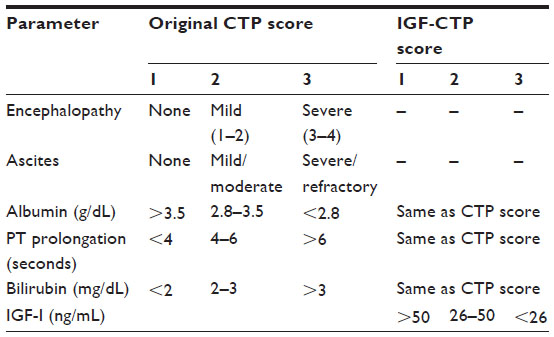 | Table 2 Original CTP scoring system replaced by the new IGF-I CTP scoring system |
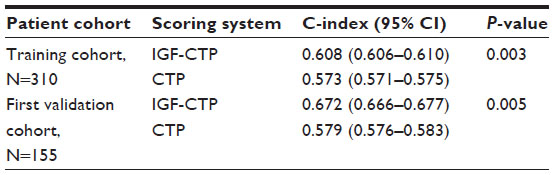 | Table 3 Ranking of scoring systems by C-index |
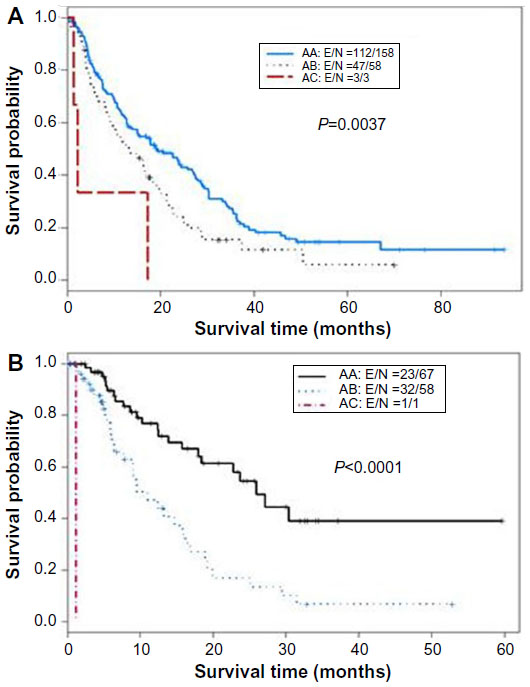 | Figure 4 Kaplan–Meier curves for IGF classification of Child–Turcotte–Pugh (CTP) class A hepatocellular carcinoma patients. |
 | Table 4 Rearrangement of originally CTP class A according to the IGF-CTP score |
Conclusion
Classification of the degree of liver reserve is critical to HCC management and for selecting patients for clinical trials. CTP is the most commonly used clinical tool to assess hepatic reserve, but it has multiple limitations, including the use of two subjective variables (ascites and encephalopathy) that are difficult to assess and may change daily under the influence of medications, nutritional status, and comorbidities. In addition, these subjective variables and their arbitrary cutoff points have been randomly selected. Emerging data about the GH/IGF-I axis in HCC by our research group and others indicate that plasma IGF-I should be incorporated in assessment of the liver reserve capacity. In our recent studies, we incorporated plasma IGF-I into the objective parameters of CTP to create an exclusively objective blood-based score and reported the results from two independent cohorts at our institution. The score is currently undergoing independent multicenter and international validation. We anticipate that IGF-I use will enhance the accuracy of selecting appropriate patients for active therapy in routine practice and for enrollment in clinical trials. Importantly, a rigorous analysis of the interactions and correlations between the GH/IGF-I axis in HCC will help advance our current understanding of the complex pathogenesis of HCC development and progression. The emerging data on upregulating GH in patients with cirrhosis and HCC is intriguing, given the potential to target this pathway for HCC prevention and treatment. Future validation studies of this approach are warranted.
Acknowledgments
The current study is supported, in part, by National Institutes of Health through grants CA170035-01 (to AOK), CA106458-01 (to MMH), and CA151533 (to HMA). The contents of this paper are solely the responsibility of the authors and do not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health. RA is supported by a scholarship from the Egyptian Ministry of Higher Education.
Disclosure
The authors report no conflicts of interest in this work.
References
Salmon WD Jr, Daughaday WH. A hormonally controlled serum factor which stimulates sulfate incorporation by cartilage in vitro. J Lab Clin Med. 1957;49(6):825–836. | |
Baxter RC. Insulin-like growth factor (IGF)-binding proteins: interactions with IGFs and intrinsic bioactivities. Am J Physiol Endocrinol Metab. 2000;278(6):E967–E976. | |
Clemmons DR. Use of mutagenesis to probe IGF-binding protein structure/function relationships. Endocr Rev. 2001;22(6):800–817. | |
Kostecka Y, Blahovec J. Insulin-like growth factor binding proteins and their functions (minireview). Endocr Regul. 1999;33(2):90–94. | |
LeRoith D, Werner H, Beitner-Johnson D, Roberts CT Jr. Molecular and cellular aspects of the insulin-like growth factor I receptor. Endocr Rev. 1995;16(2):143–163. | |
Murphy LJ. Insulin-like growth factor-binding proteins: functional diversity or redundancy? J Mol Endocrinol. 1998;21(2):97–107. | |
Samani AA, Yakar S, LeRoith D, Brodt P. The role of the IGF system in cancer growth and metastasis: overview and recent insights. Endocr Rev. 2007;28(1):20–47. | |
Sridhar SS, Goodwin PJ. Insulin-insulin-like growth factor axis and colon cancer. J Clin Oncol. 2009;27(2):165–167. | |
Zarrilli R, Bruni CB, Riccio A. Multiple levels of control of insulin-like growth factor gene expression. Mol Cell Endocrinol. 1994;101(1–2):R1–R14. | |
Daughaday WH. The possible autocrine/paracrine and endocrine roles of insulin-like growth factors of human tumors. Endocrinology. 1990;127(1):1–4. | |
Maki RG. Small is beautiful: insulin-like growth factors and their role in growth, development, and cancer. J Clin Oncol. 2010;28(33):4985–4995. | |
Masoodi M, Aghazadeh R, Somi MH, Shavakhi H, Shabestari AA, Zali MR. Serum insulin-like growth factor-I and tumor size in patients with metastatic liver cancer. Hepat Mon. 2008;8(3):179–183. | |
LeRoith D, Roberts CT. The insulin-like growth factor system and cancer. Cancer Lett. 2003;195(2):127–137. | |
Rosenbloom AL, Guevara-Aguirre J, Rosenfeld RG, Pollock BH. Growth in growth hormone insensitivity. Trends Endocrinol Metab. 1994;5(7):296–303. | |
Smith RG, Jiang H, Sun Y. Developments in ghrelin biology and potential clinical relevance. Trends Endocrinol Metab. 2005;16(9):436–442. | |
Vajdos FF, Ultsch M, Schaffer ML, et al. Crystal structure of human insulin-like growth factor-1: detergent binding inhibits binding protein interactions. Biochemistry. 2001;40(37):11022–11029. | |
Ohlsson C, Mohan S, Sjögren K, et al. The role of liver-derived insulin-like growth factor-I. Endocr Rev. 2009;30(5):494–535. | |
Froesch ER, Schmid C, Schwander J, Zapf J. Actions of insulin-like growth factors. Annu Rev Phsiol. 1985;47:443–467. | |
Sjögren K, Liu JL, Blad K, et al. Liver-derived insulin-like growth factor I (IGF-I) is the principal source of IGF-I in blood but is not required for postnatal body growth in mice. Proc Natl Acad Sci U S A. 1999;96(12):7088–7092. | |
Baserga R, Peruzzi F, Reiss K. The IGF-1 receptor in cancer biology. Int J Cancer. 2003;107(6):873–877. | |
Baserga R, Porcu P, Rubini M, Sell C. Cell cycle control by the IGF-1 receptor and its ligands. Adv Exp Med Biol. 1993;343:105–112. | |
Cosman D, Lyman SD, Idzerda RL, et al. A new cytokine receptor superfamily. Trends Biochem Sci. 1990;15(7):265–270. | |
Cruickshank J, Grossman DI, Peng RK, Famula TR, Oberbauer AM. Spatial distribution of growth hormone receptor, insulin-like growth factor-I receptor and apoptotic chondrocytes during growth plate development. J Endocrinol. 2005;184(3):543–553. | |
Schwander JC, Hauri C, Zapf J, Froesch ER. Synthesis and secretion of insulin-like growth factor and its binding protein by the perfused rat liver: dependence on growth hormone status. Endocrinology. 1983;113(1):297–305. | |
Werner H, LeRoith D. The role of the insulin-like growth factor system in human cancer. Adv Cancer Res. 1996;68:183–223. | |
Ho KY, Veldhuis JD, Johnson ML, et al. Fasting enhances growth hormone secretion and amplifies the complex rhythms of growth hormone secretion in man. J Clin Invest. 1988;81(4):968–975. | |
Giustina A, Scalvini T, Tassi C, et al. Maturation of the regulation of growth hormone secretion in young males with hypogonadotropic hypogonadism pharmacologically exposed to progressive increments in serum testosterone. J Clin Endocrinol Metab. 1997;82(4):1210–1219. | |
Keenan BS, Richards GE, Ponder SW, Dallas JS, Nagamani M, Smith ER. Androgen-stimulated pubertal growth: the effects of testosterone and dihydrotestosterone on growth hormone and insulin-like growth factor-I in the treatment of short stature and delayed puberty. J Clin Endocrinol Metab. 1993;76(4):996–1001. | |
Loche S, Colao A, Cappa M, et al. The growth hormone response to hexarelin in children: reproducibility and effect of sex steroids. J Clin Endocrinol Metab. 1997;82(3):861–864. | |
Kerrigan JR, Rogol AD. The impact of gonadal steroid hormone action on growth hormone secretion during childhood and adolescence. Endocr Rev. 1992;13(2):281–298. | |
Jospe N, Orlowski CC, Furlanetto RW. Comparison of transdermal and oral estrogen therapy in girls with Turner’s syndrome. J Pediatr Endocrinol Metab. 1995;8(2):111–116. | |
Harris DA, Van Vliet G, Egli CA, et al. Somatomedin-C in normal puberty and in true precocious puberty before and after treatment with a potent luteinizing hormone-releasing hormone agonist. J Clin Endocrinol Metab. 1985;61(1):152–159. | |
Mansfield MJ, Rudlin CR, Crigler JF Jr, et al. Changes in growth and serum growth hormone and plasma somatomedin-C levels during suppression of gonadal sex steroid secretion in girls with central precocious puberty. J Clin Endocrinol Metab. 1988;66(1):3–9. | |
Blum WF, Albertsson-Wikland K, Rosberg S, Ranke MB. Serum levels of insulin-like growth factor I (IGF-I) and IGF binding protein 3 reflect spontaneous growth hormone secretion. J Clin Endocrinol Metab. 1993;76(6):1610–1616. | |
Brabant G, von zur Mühlen A, Wüster C, et al. Serum insulin-like growth factor I reference values for an automated chemiluminescence immunoassay system: results from a multicenter study. Horm Res. 2003;60(2):53–60. | |
Giusti M, Torre R, Cavagnaro P, Attanasio R, Traverso L, Giordano G. The effect of long-term pulsatile GnRH administration on the 24-hour integrated concentration of GH in hypogonadotropic hypogonadic patients. Acta Endocrinol (Copenh). 1989;120(6):724–728. | |
Weissberger AJ, Ho KK. Activation of the somatotropic axis by testosterone in adult males: evidence for the role of aromatization. J Clin Endocrinol Metab. 1993;76(6):1407–1412. | |
Bondanelli M, Ambrosio MR, Margutti A, Franceschetti P, Zatelli MC, degli Uberti EC. Activation of the somatotropic axis by testosterone in adult men: evidence for a role of hypothalamic growth hormone-releasing hormone. Neuroendocrinology. 2003;77(6):380–387. | |
Liu L, Merriam GR, Sherins RJ. Chronic sex steroid exposure increases mean plasma growth hormone concentration and pulse amplitude in men with isolated hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 1987;64(4):651–656. | |
Veldhuis JD, Keenan DM, Mielke K, Miles JM, Bowers CY. Testosterone supplementation in healthy older men drives GH and IGF-I secretion without potentiating peptidyl secretagogue efficacy. Eur J Endocrinol. 2005;153(4):577–586. | |
Mauras N, Rini A, Welch S, Sager B, Murphy SP. Synergistic effects of testosterone and growth hormone on protein metabolism and body composition in prepubertal boys. Metabolism. 2003;52(8):964–969. | |
Saggese G, Cesaretti G, Franchi G, Startari L. Testosterone-induced increase of insulin-like growth factor I levels depends upon normal levels of growth hormone. Eur J Endocrinol. 1996;135(2):211–215. | |
Gibney J, Wolthers T, Johannsson G, Umpleby AM, Ho KK. Growth hormone and testosterone interact positively to enhance protein and energy metabolism in hypopituitary men. Am J Physiol Endocrinol Metab. 2005;289(2):E266–E271. | |
Ovesen P, Vahl N, Fisker S, Veldhuis JD, Christiansen JS, Jørgensen JO. Increased pulsatile, but not basal, growth hormone secretion rates and plasma insulin-like growth factor I levels during the periovulatory interval in normal women. J Clin Endocrinol Metab. 1998;83(5):1662–1667. | |
Weissberger AJ, Ho KK, Lazarus L. Contrasting effects of oral and transdermal routes of estrogen replacement therapy on 24-hour growth hormone (GH) secretion, insulin-like growth factor I, and GH-binding protein in postmenopausal women. J Clin Endocrinol Metab. 1991;72(2):374–381. | |
Moll GW Jr, Rosenfield RL, Fang VS. Administration of low-dose estrogen rapidly and directly stimulates growth hormone production. Am J Dis Child. 1986;140(2):124–127. | |
Schober E, Frisch H, Waldhauser F, Bieglmayr C. Influence of estrogen administration on growth hormone response to GHRH and L-Dopa in patients with Turner’s syndrome. Acta Endocrinol (Copenh). 1989;120(4):442–446. | |
Leung KC, Johannsson G, Leong GM, Ho KK. Estrogen regulation of growth hormone action. Endocr Rev. 2004;25(5):693–721. | |
Friend KE, Hartman ML, Pezzoli SS, Clasey JL, Thorner MO. Both oral and transdermal estrogen increase growth hormone release in postmenopausal women – a clinical research center study. J Clin Endocrinol Metab. 1996;81(6):2250–2256. | |
Gibney J, Johannsson G, Leung KC, Ho KK. Comparison of the metabolic effects of raloxifene and oral estrogen in postmenopausal and growth hormone-deficient women. J Clin Endocrinol Metab. 2005;90(7):3897–3903. | |
Liu PY, Hoey KA, Mielke KL, Veldhuis JD, Khosla S. A randomized placebo-controlled trial of short-term graded transdermal estradiol in healthy gonadotropin-releasing hormone agonist-suppressed pre- and postmenopausal women: effects on serum markers of bone turnover, insulin-like growth factor-I, and osteoclastogenic mediators. J Clin Endocrinol Metab. 2005;90(4):1953–1960. | |
Roth J, Glick SM, Yalow RS, Berson SA. Secretion of human growth hormone: physiologic and experimental modification. Metabolism. 1963;12:577–579. | |
Imaki T, Shibasaki T, Shizume K, et al. The effect of free fatty acids on growth hormone (GH)-releasing hormone-mediated GH secretion in man. J Clin Endocrinol Metab. 1985;60(2):290–293. | |
Merimee TJ, Rabinowtitz D, Fineberg SE. Arginine-initiated release of human growth hormone. Factors modifying the response in normal man. N Engl J Med. 1969;280(26):1434–1438. | |
Melmed S. Insulin suppresses growth hormone secretion by rat pituitary cells. J Clin Invest. 1984;73(5):1425–1433. | |
Vahl N, Jørgensen JO, Skjaerbaek C, Veldhuis JD, Orskov H, Christiansen JS. Abdominal adiposity rather than age and sex predicts mass and regularity of GH secretion in healthy adults. Am J Physiol. 1997;272(6 Pt 1):E1108–E1116. | |
Butler AA, Le Roith D. Control of growth by the somatropic axis: growth hormone and the insulin-like growth factors have related and independent roles. Annu Rev Physiol. 2001;63:141–164. | |
Philippou A, Halapas A, Maridaki M, Koutsilieris M. Type I insulin-like growth factor receptor signaling in skeletal muscle regeneration and hypertrophy. J Musculoskelet Neuronal Interact. 2007;7(3):208–218. | |
Nyomba BL, Berard L, Murphy LJ. Free insulin-like growth factor I (IGF-I) in healthy subjects: relationship with IGF-binding proteins and insulin sensitivity. J Clin Endocrinol Metab. 1997;82(7):2177–2181. | |
Berelowitz M, Szabo M, Frohman LA, Firestone S, Chu L, Hintz RL. Somatomedin-C mediates growth hormone negative feedback by effects on both the hypothalamus and the pituitary. Science. 1981;212(4500):1279–1281. | |
Bertherat J, Bluet-Pajot MT, Epelbaum J. Neuroendocrine regulation of growth hormone. Eur J Endocrinol. 1995;132(1):12–24. | |
Bonefeld K, Møller S. Insulin-like growth factor-I and the liver. Liver Int. 2011;31(7):911–919. | |
Buzzelli G, Dattolo P, Pinzani M, Brocchi A, Romano S, Gentilini P. Circulating growth hormone and insulin-like growth factor-I in nonalcoholic liver cirrhosis with or without superimposed hepatocarcinoma: evidence of an altered circadian rhythm. Am J Gastroenterol. 1993;88(10):1744–1748. | |
Daughaday WH, Rotwein P. Insulin-like growth factors I and II. Peptide, messenger ribonucleic acid and gene structures, serum, and tissue concentrations. Endocr Rev. 1989;10(1):68–91. | |
Holly JM, Wass JA. Insulin-like growth factors; autocrine, paracrine or endocrine? New perspectives of the somatomedin hypothesis in the light of recent developments. J Endocrinol. 1989;122(3):611–618. | |
Tannenbaum GS, Guyda HJ, Posner BI. Insulin-like growth factors: a role in growth hormone negative feedback and body weight regulation via brain. Science. 1983;220(4592):77–79. | |
Underwood LE, D’Ercole AJ, Clemmons DR, Van Wyk JJ. Paracrine functions of somatomedins. Clin Endocrinol Metab. 1986;15(1):59–77. | |
Muggeo M, Tiengo A, Fedele D, Crepaldi G. Altered control of growth hormone secretion in patients with cirrhosis of the liver. Arch Intern Med. 1979;139(10):1157–1160. | |
Schimpff RM, Lebrec D, Donnadieu M. Somatomedin production in normal adults and cirrhotic patients. Acta Endocrinol (Copenh). 1977;86(2):355–362. | |
Chang TC, Lin JJ, Yu SC, Chang TJ. Absence of growth-hormone receptor in hepatocellular carcinoma and cirrhotic liver. Hepatology. 1990;11(1):123–126. | |
Stratikopoulos E, Szabolcs M, Dragatsis I, Klinakis A, Efstratiadis A. The hormonal action of IGF1 in postnatal mouse growth. Proc Natl Acad Sci U S A. 2008;105(49):19378–19383. | |
Kupfer SR, Underwood LE, Baxter RC, Clemmons DR. Enhancement of the anabolic effects of growth hormone and insulin-like growth factor I by use of both agents simultaneously. J Clin Invest. 1993;91(2):391–396. | |
Clemmons DR. Metabolic actions of insulin-like growth factor-I in normal physiology and diabetes. Endocrinol Metab Clin North Am. 2012;41(2):425–443, vii–viii. | |
Renehan AG, Brennan BM. Acromegaly, growth hormone and cancer risk. Best Pract Res Clin Endocrinol Metab. 2008;22(4):639–657. | |
Bougen NM, Steiner M, Pertziger M, et al. Autocrine human GH promotes radioresistance in mammary and endometrial carcinoma cells. Endocr Relat Cancer. 2012;19(5):625–644. | |
Bougen NM, Yang T, Chen H, Lobie PE, Perry JK. Autocrine human growth hormone reduces mammary and endometrial carcinoma cell sensitivity to mitomycin C. Oncol Rep. 2011;26(2):487–493. | |
Mazzoccoli G, Sothern RB, Pazienza V, et al. Circadian aspects of growth hormone-insulin-like growth factor axis function in patients with lung cancer. Clin Lung Cancer. 2012;13(1):68–74. | |
Minoia M, Gentilin E, Molè D, et al. Growth hormone receptor blockade inhibits growth hormone-induced chemoresistance by restoring cytotoxic-induced apoptosis in breast cancer cells independently of estrogen receptor expression. J Clin Endocrinol Metab. 2012;97(6):E907–E916. | |
Perry JK, Emerald BS, Mertani HC, Lobie PE. The oncogenic potential of growth hormone. Growth Horm IGF Res. 2006;16(5–6):277–289. | |
Wu ZS, Yang K, Wan Y, et al. Tumor expression of human growth hormone and human prolactin predict a worse survival outcome in patients with mammary or endometrial carcinoma. J Clin Endocrinol Metab. 2011;96(10):E1619–E1629. | |
Zatelli MC, Minoia M, Molè D, et al. Growth hormone excess promotes breast cancer chemoresistance. J Clin Endocrinol Metab. 2009;94(10):3931–3938. | |
Werner H, Weinstein D, Bentov I. Similarities and differences between insulin and IGF-I: structures, receptors, and signalling pathways. Arch Physiol Bochem. 2008;114(1):17–22. | |
Lee YH, White MF. Insulin receptor substrate proteins and diabetes. Arch Pharm Res. 2004;27(4):361–370. | |
Clemmons D, Maile L, Xi G, Shen X, Radhakrishnan Y. Igf-I signaling in response to hyperglycemia and the development of diabetic complications. Curr Diabetes Rev. 2011;7(4):235–245. | |
Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. 2002;2(8):563–572. | |
Feldser D, Agani F, Iyer NV, Pak B, Ferreira G, Semenza GL. Reciprocal positive regulation of hypoxia-inducible factor 1alpha and insulin-like growth factor 2. Cancer Res. 1999;59(16):3915–3918. | |
Zelzer E, Levy Y, Kahana C, Shilo BZ, Rubinstein M, Cohen B. Insulin induces transcription of target genes through the hypoxia-inducible factor HIF-1alpha/ARNT. EMBO J. 1998;17(17):5085–5094. | |
Fukuda R, Hirota K, Fan F, Jung YD, Ellis LM, Semenza GL. Insulin-like growth factor 1 induces hypoxia-inducible factor 1-mediated vascular endothelial growth factor expression, which is dependent on MAP kinase and phosphatidylinositol 3-kinase signaling in colon cancer cells. J Biol Chem. 2002;277(41):38205–382011. | |
Clarke K, Smith K, Gullick WJ, Harris AL. Mutant epidermal growth factor receptor enhances induction of vascular endothelial growth factor by hypoxia and insulin-like growth factor-1 via a PI3 kinase dependent pathway. Br J Cancer. 2001;84(10):1322–1329. | |
Shigematsu S, Yamauchi K, Nakajima K, Iijima S, Aizawa T, Hashizume K. IGF-1 regulates migration and angiogenesis of human endothelial cells. Endocr J. 1999;46 Suppl:S59–S62. | |
Lee OH, Bae SK, Bae MH, et al. Identification of angiogenic properties of insulin-like growth factor II in in vitro angiogenesis models. Br J Cancer. 2000;82(2):385–391. | |
Grulich-Henn J, Ritter J, Mesewinkel S, Heinrich U, Bettendorf M, Preissner KT. Transport of insulin-like growth factor-I across endothelial cell monolayers and its binding to the subendothelial matrix. Exp Clin Endocrinol Diabetes. 2002;110(2):67–73. | |
Mira E, Mañes S, Lacalle RA, Márquez G, Martínez-AC. Insulin-like growth factor I-triggered cell migration and invasion are mediated by matrix metalloproteinase-9. Endocrinology. 1999;140(4):1657–1664. | |
Dunn SE, Torres JV, Nihei N, Barrett JC. The insulin-like growth factor-1 elevates urokinase-type plasminogen activator-1 in human breast cancer cells: a new avenue for breast cancer therapy. Mol Carcinog. 2000;27(1):10–17. | |
Smith BW, Adams LA. Non-alcoholic fatty liver disease. Crit Rev Clin Lab Sci. 2011;48(3):97–113. | |
Cuneo RC, Hickman PE, Wallace JD, et al. Altered endogenous growth hormone secretory kinetics and diurnal GH-binding protein profiles in adults with chronic liver disease. Clin Endocrinol (Oxf). 1995;43(3):265–275. | |
Skrtic S, Wallenius K, Gressner AM, Jansson JO. Insulin-like growth factor signaling pathways in rat hepatic stellate cells: importance for deoxyribonucleic acid synthesis and hepatocyte growth factor production. Endocrinology. 1999;140(12):5729–5735. | |
Skrtic S, Wallenius V, Ekberg S, Brenzel A, Gressner AM, Jansson JO. Insulin-like growth factors stimulate expression of hepatocyte growth factor but not transforming growth factor beta1 in cultured hepatic stellate cells. Endocrinology. 1997;138(11):4683–4689. | |
Matsuda Y, Matsumoto K, Yamada A, et al. Preventive and therapeutic effects in rats of hepatocyte growth factor infusion on liver fibrosis/cirrhosis. Hepatology. 1997;26(1):81–89. | |
Puche JE, Castilla- Cortázar I. Human conditions of insulin-like growth factor-I (IGF-I) deficiency. J Transl Med. 2012;10:224. | |
Arturi F, Succurro E, Procopio C, et al. Nonalcoholic fatty liver disease is associated with low circulating levels of insulin-like growth factor-I. J Clin Endocrinol Metab. 2011;96(10):E1640–E1644. | |
Völzke H, Nauck M, Rettig R, et al. Association between hepatic steatosis and serum IGF1 and IGFBP-3 levels in a population-based sample. Eur J Endocrinol. 2009;161(5):705–713. | |
Anthony PP, Ishak KG, Nayak NC, Poulsen HE, Scheuer PJ, Sobin LH. The morphology of cirrhosis: definition, nomenclature, and classification. Bull World Health Organ. 1977;55(4):521–540. | |
Assy N, Hochberg Z, Enat R, Baruch Y. Prognostic value of generation of growth hormone-stimulated insulin-like growth factor-I (IGF-I) and its binding protein-3 in patients with compensated and decompensated liver cirrhosis. Dig Dis Sci. 1998;43(6):1317–1321. | |
Assy N, Pruzansky Y, Gaitini D, Shen Orr Z, Hochberg Z, Baruch Y. Growth hormone-stimulated IGF-1 generation in cirrhosis reflects hepatocellular dysfunction. J Hepatol. 2008;49(1):34–42. | |
Caregaro L, Alberino F, Amodio P, et al. Nutritional and prognostic significance of insulin-like growth factor 1 in patients with liver cirrhosis. Nutrition. 1997;13(3):185–190. | |
Castro GR, Coelho JC, Parolin MB, Matias JE, de Freitas AC. Insulin-like growth factor I correlates with MELD and returns to normal level after liver transplantation. Ann Transplant. 2013;18:57–62. | |
Dehghani SM, Karamifar H, Hamzavi SS, Haghighat M, Malek-Hosseini SA. Serum insulinlike growth factor-1 and its binding protein-3 levels in children with cirrhosis waiting for a liver transplant. Exp Clin Transplant. 2012;10(3):252–257. | |
Donaghy AJ, Delhanty PJ, Ho KK, Williams R, Baxter RC. Regulation of the growth hormone receptor/binding protein, insulin-like growth factor ternary complex system in human cirrhosis. J Hepatol. 2002; 36(6):751–758. | |
Elsammak MY, Amin GM, Khalil GM, Ragab WS, Abaza MM. Possible contribution of serum activin A and IGF-1 in the development of hepatocellular carcinoma in Egyptian patients suffering from combined hepatitis C virus infection and hepatic schistosomiasis. Clin Biochem. 2006;39(6):623–629. | |
Jeyaratnaganthan N, Grønbaek H, Holland-Fischer P, et al. Ascites from patients with alcoholic liver cirrhosis contains higher IGF-I bioactivity than serum. Clin Endocrinol (Oxf). 2010;72(5):625–632. | |
Lorenzo-Zúñiga V, Bartolí R, Masnou H, Montoliu S, Morillas RM, Planas R. Serum concentrations of insulin-like growth factor-I (igf-I) as a marker of liver fibrosis in patients with chronic hepatitis C. Dig Dis Sci. 2007;52(11):3245–3250. | |
Mazziotti G, Sorvillo F, Morisco F, et al. Serum insulin-like growth factor I evaluation as a useful tool for predicting the risk of developing hepatocellular carcinoma in patients with hepatitis C virus-related cirrhosis: a prospective study. Cancer. 2002;95(12):2539–2545. | |
Møller S, Grønbaek M, Main K, Becker U, Skakkebaek NE. Urinary growth hormone (U-GH) excretion and serum insulin-like growth factor 1 (IGF-1) in patients with alcoholic cirrhosis. J Hepatol. 1993; 17(3):315–320. | |
Rehem RN, El-Shikh WM. Serum IGF-1, IGF-2 and IGFBP-3 as parameters in the assessment of liver dysfunction in patients with hepatic cirrhosis and in the diagnosis of hepatocellular carcinoma. Hepatogastroenterology. 2011;58(107–108):949–954. | |
Ronsoni MF, Lazzarotto C, Fayad L, et al. IGF-I and IGFBP-3 serum levels in patients hospitalized for complications of liver cirrhosis. Ann Hepatol. 2013;12(3):456–463. | |
Sandahl TD, Aagaard NK, Thomsen KL, et al. Effects of insulin-like growth factor-I administration on in vivo regulation of urea synthesis in normal subjects and patients with cirrhosis. Liver Int. 2011;31(1):132–137. | |
Vyzantiadis T, Theodoridou S, Giouleme O, Harsoulis P, Evgenidis N, Vyzantiadis A. Serum concentrations of insulin-like growth factor-I (IGF-I) in patients with liver cirrhosis. Hepatogastroenterology. 2003; 50(51):814–816. | |
Wu YL, Ye J, Zhang S, Zhong J, Xi RP. Clinical significance of serum IGF-I, IGF-II and IGFBP-3 in liver cirrhosis. World J Gastroenterol. 2004;10(18):2740–2743. | |
Cheng AL, Kang YK, Chen Z, et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009;10(1):25–34. | |
Llovet JM, Ricci S, Mazzaferro V, et al; SHARP Investigators Study Group. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378–390. | |
Llovet JM, Di Bisceglie AM, Bruix J, et al; Panel of Experts in HCC-Design Clinical Trials. Design and endpoints of clinical trials in hepatocellular carcinoma. J Natl Cancer Inst. 2008;100(10):698–711. | |
Wilson SR, Greig P, Kaseb AO. Pretreatment assessment of hepatocellular cancer: expert consensus conference. HPB (Oxford). 2010;12(5):300–301. | |
Chevret S, Trinchet JC, Mathieu D, Rached AA, Beaugrand M, Chastang C. A new prognostic classification for predicting survival in patients with hepatocellular carcinoma. Groupe d’Etude et de Traitement du Carcinome Hepatocellulaire. J Hepatol. 1999;31(1):133–141. | |
Kudo M, Chung H, Osaki Y. Prognostic staging system for hepatocellular carcinoma (CLIP score): its value and limitations, and a proposal for a new staging system, the Japan Integrated Staging Score (JIS score). J Gastroenterol. 2003;38(3):207–215. | |
Leung TW, Tang AM, Zee B, et al. Construction of the Chinese University Prognostic Index for hepatocellular carcinoma and comparison with the TNM staging system, the Okuda staging system, and the Cancer of the Liver Italian Program staging system: a study based on 926 patients. Cancer. 2002;94(6):1760–1769. | |
Llovet JM, Brú C, Bruix J. Prognosis of hepatocellular carcinoma: the BCLC staging classification. Semin Liver Dis. 1999;19(3):329–338. | |
Okuda K, Ohtsuki T, Obata H, et al. Natural history of hepatocellular carcinoma and prognosis in relation to treatment. Study of 850 patients. Cancer. 1985;56(4):918–928. | |
Pons F, Varela M, Llovet JM. Staging systems in hepatocellular carcinoma. HPB (Oxford). 2005;7(1):35–41. | |
Tateishi R, Yoshida H, Shiina S, et al. Proposal of a new prognostic model for hepatocellular carcinoma: an analysis of 403 patients. Gut. 2005;54(3):419–425. | |
[No authors listed]. A new prognostic system for hepatocellular carcinoma: a retrospective study of 435 patients: the Cancer of the Liver Italian Program (CLIP) investigators. Hepatology. 1998;28(3):751–755. | |
Jørgensen JO, Blum WF, Møller N, Ranke MB, Christiansen JS. Circadian patterns of serum insulin-like growth factor (IGF) II and IGF binding protein 3 in growth hormone-deficient patients and age- and sex-matched normal subjects. Acta Endocrinol (Copenh). 1990;123(3):257–262. | |
National Comprehensive Cancer Network clinical practice guidelines: Hepatobiliary Cancers. http://www.nccn.org/professionals/physician_gls/pdf/hepatobiliary.pdf. 2015 version 2. Accessed July 24, 2015. | |
Thomas MB, Jaffe D, Choti MM, et al. Hepatocellular carcinoma: consensus recommendations of the National Cancer Institute Clinical Trials Planning Meeting. J Clin Oncol. 2010;28(25):3994-4005. | |
Verslype C, Rosmorduc O, Rougier P; ESMO Guidelines Working Group. Hepatocellular carcinoma: ESMO-ESDO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2012;23(7):vii41–48. | |
Kaseb AO, Abbruzzese JL, Vauthey JN, et al. I-CLIP: improved stratification of advanced hepatocellular carcinoma patients by integrating plasma IGF-1 into CLIP score. Oncology. 2011;80(5–6):373–381. | |
Kaseb AO, Morris JS, Hassan MM, et al. Clinical and prognostic implications of plasma insulin-like growth factor-1 and vascular endothelial growth factor in patients with hepatocellular carcinoma. J Clin Oncol. 2011;29(29):3892–3899. | |
Stuver SO, Kuper H, Tzonou A, et al. Insulin-like growth factor 1 in hepatocellular carcinoma and metastatic liver cancer in men. Int J Cancer. 2000;87(1):118–121. | |
Su WW, Lee KT, Yeh YT, et al. Association of circulating insulin-like growth factor 1 with hepatocellular carcinoma: one cross-sectional correlation study. J Clin Lab Anal. 2010;24(3):195–200. | |
Assy N, Hochberg Z, Amit T, Shen-Orr Z, Enat R, Baruch Y. Growth hormone-stimulated insulin-like growth factor (IGF) I and IGF-binding protein-3 in liver cirrhosis. J Hepatol. 1997;27(5):796–802. | |
De Benedetti F, Alonzi T, Moretta A, et al. Interleukin 6 causes growth impairment in transgenic mice through a decrease in insulin-like growth factor-I. A model for stunted growth in children with chronic inflammation. J Clin Invest. 1997;99(4):643–650. | |
Jones JI, Clemmons DR. Insulin-like growth factors and their binding proteins: biological actions. Endocr Rev. 1995;16(1):3–34. | |
Kaseb AO, Xiao L, Hassan MM, et al. Development and validation of insulin-like growth factor-1 score to assess hepatic reserve in hepatocellular carcinoma. J Natl Cancer Inst. 2014;106(5). | |
Elmashad N, Ibrahim WS, Mayah WW, et al. Predictive value of serum insulin-like growth factor-1 in hepatocellular carcinoma. Asian Pac J Cancer Prev. 2015;16(2):613–619. | |
Cho E, Kim HC, Lee JH, et al. Serum insulin-like growth factor-1 predicts disease progression and survival in patients with hepatocellular carcinoma who undergo transarterial chemoembolization. PLoS One. 2014;9(3):e90862. | |
Bruix J, Sherman M; Practice Guidelines Committee, American Association for the Study of Liver Diseases. Management of hepatocellular carcinoma. Hepatology. 2005;42(5):1208–1236. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.